Design and Synthesis of Novel 1,3-Thiazole and 2-Hydrazinyl-1,3-Thiazole Derivatives as Anti-Candida Agents: In Vitro Antifungal Screening, Molecular Docking Study, and Spectroscopic Investigation of their Binding Interaction with Bovine Serum Albumin


 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
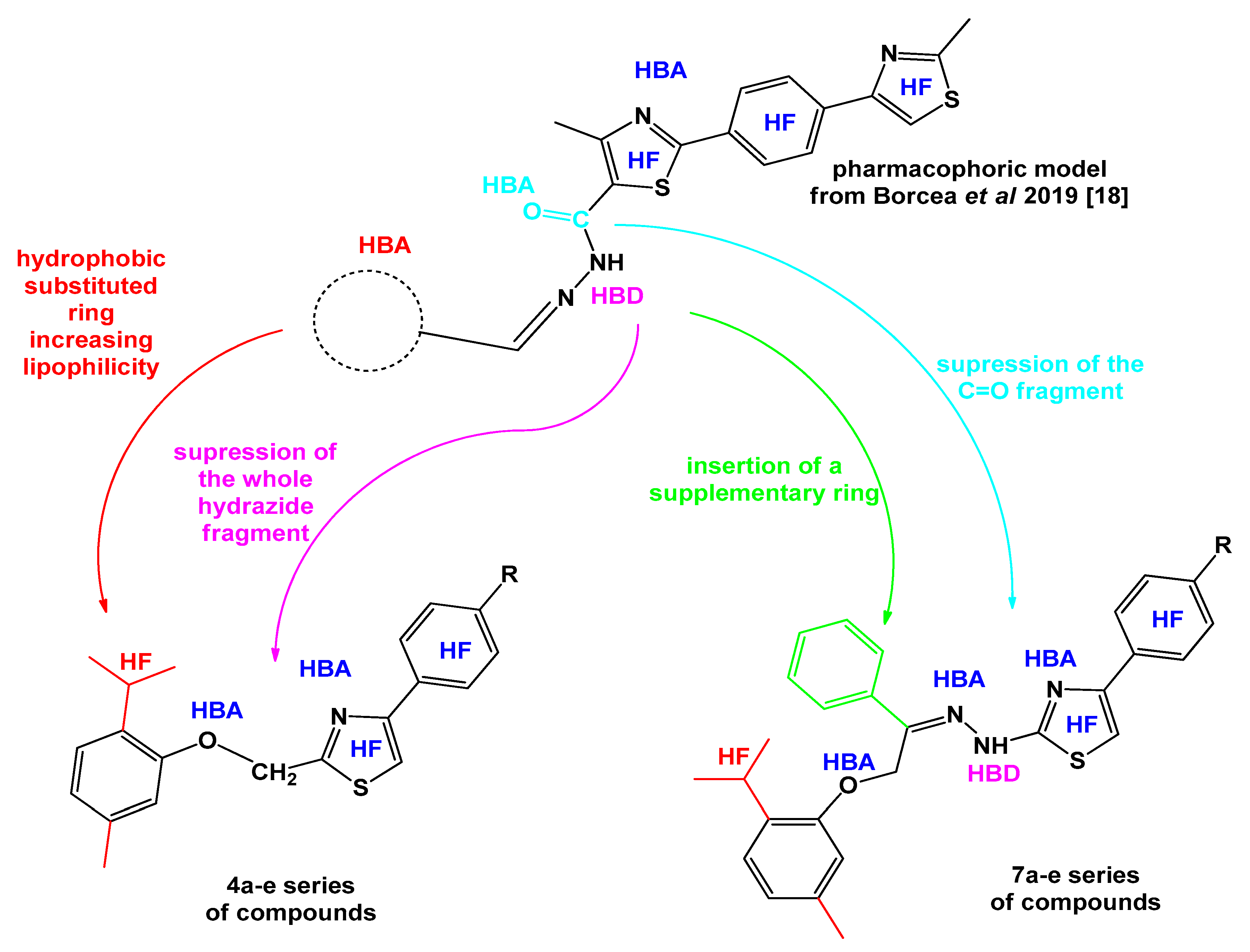
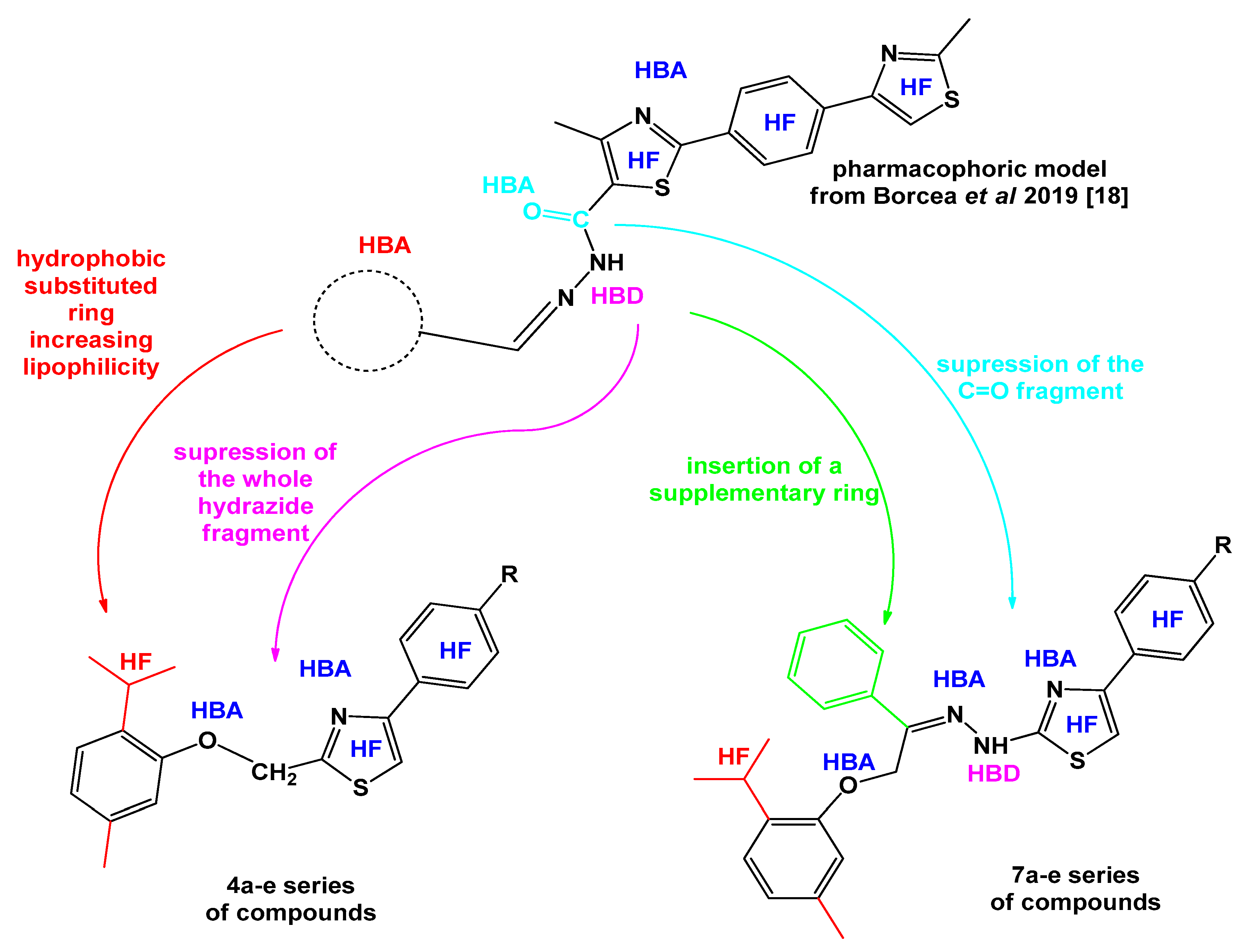
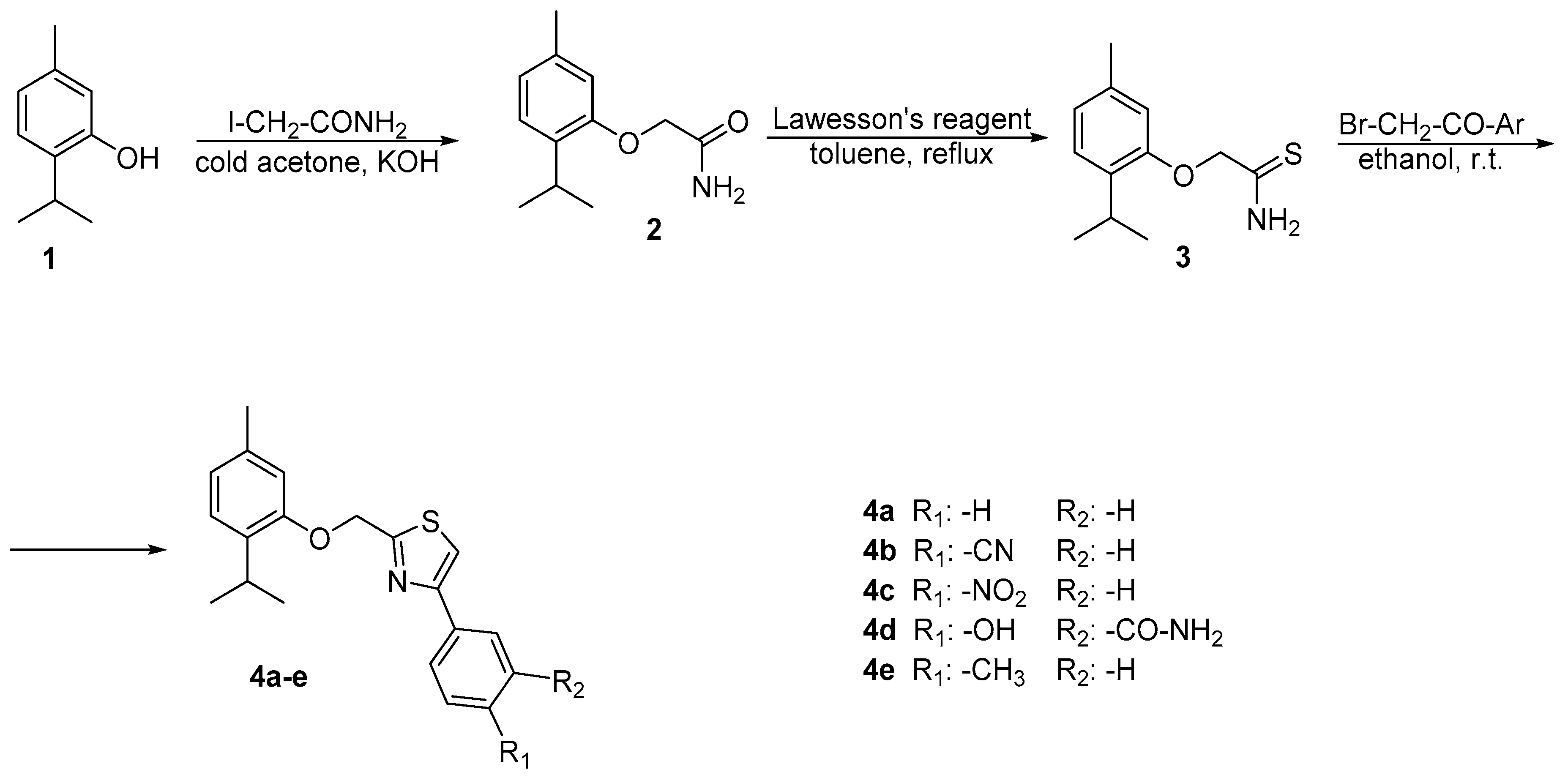
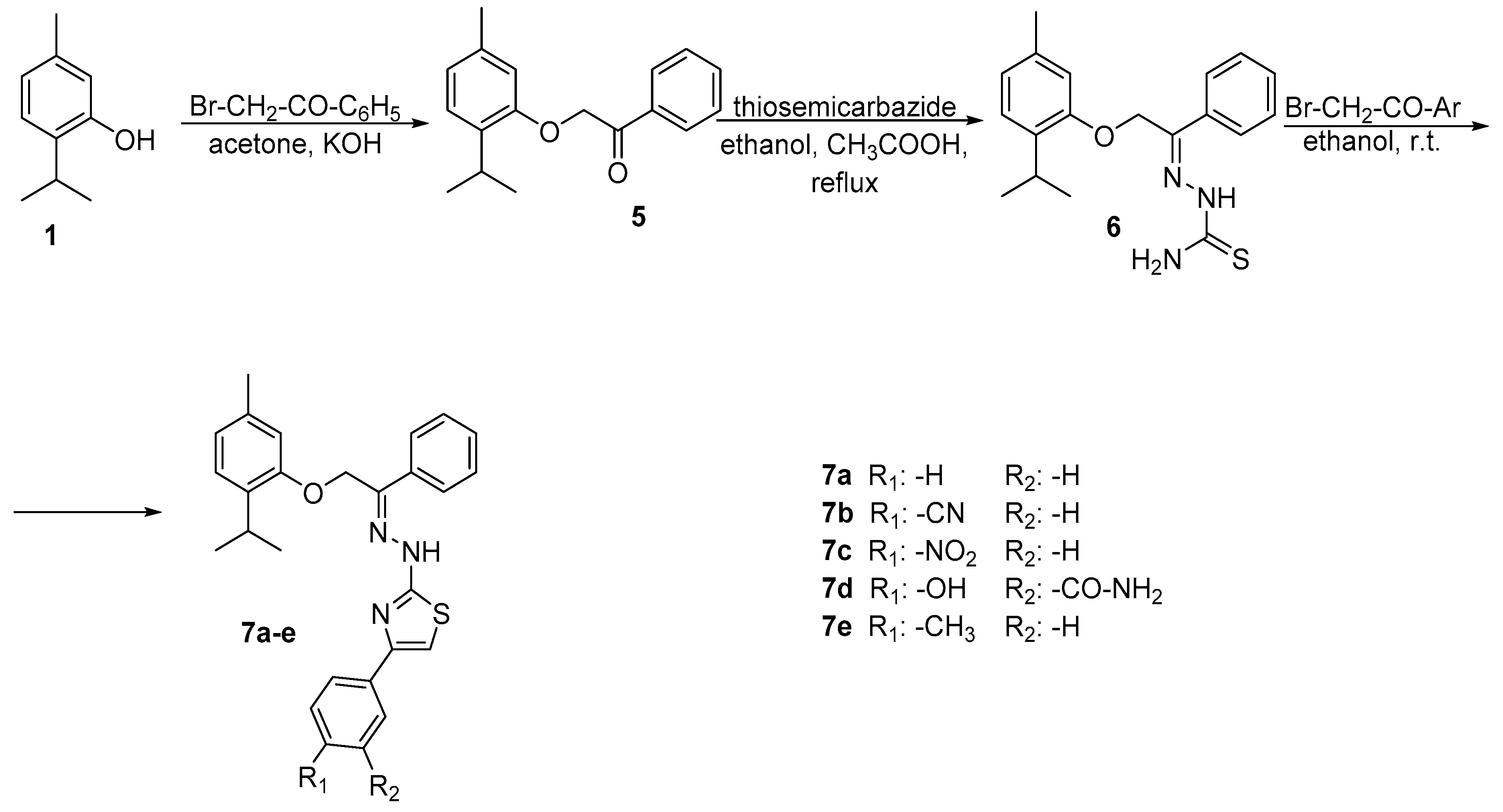
2.1. Chemistry
2.2. Anti-Candida Activity Assay
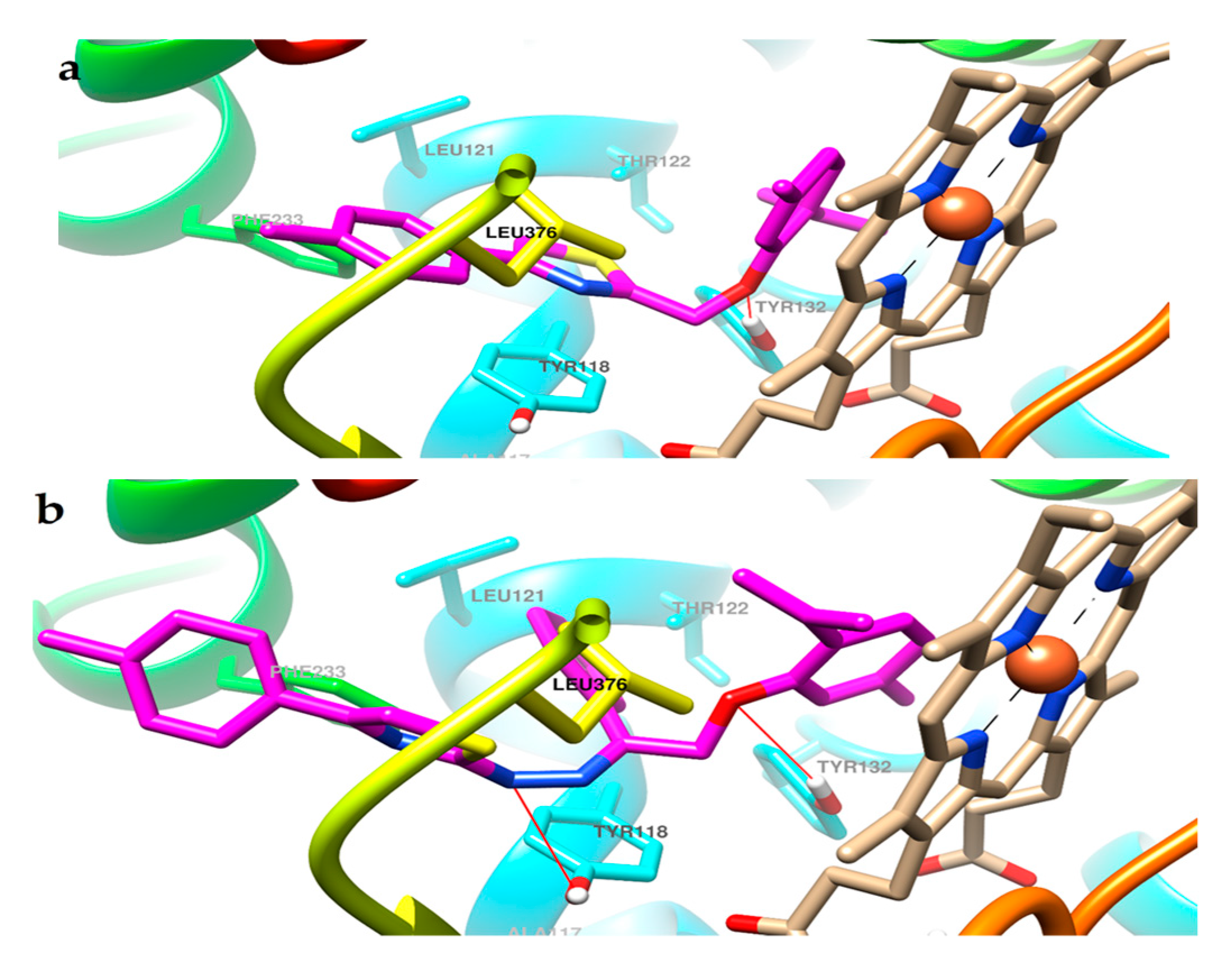
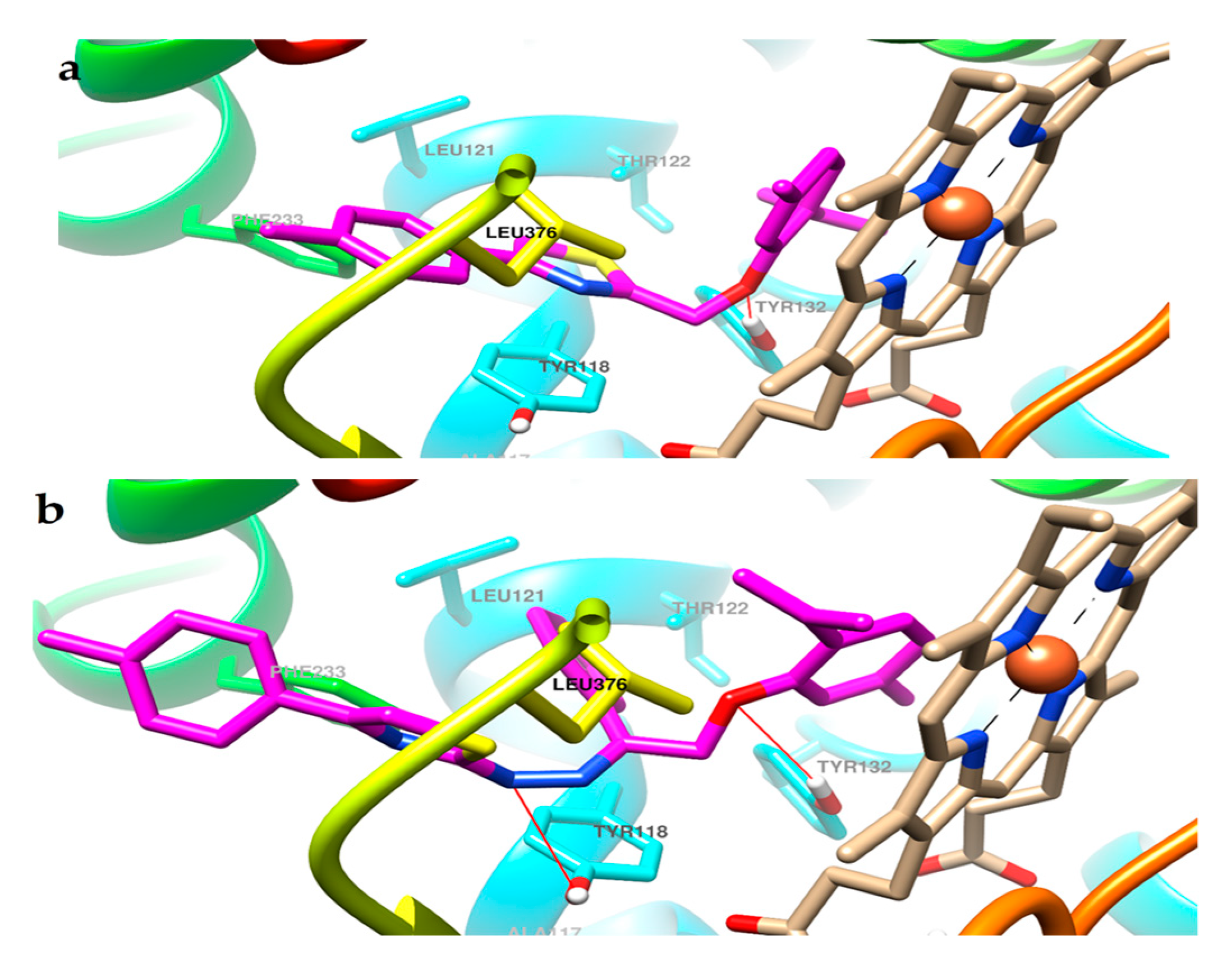
2.3. Molecular Docking
2.4. ADMET Profiling
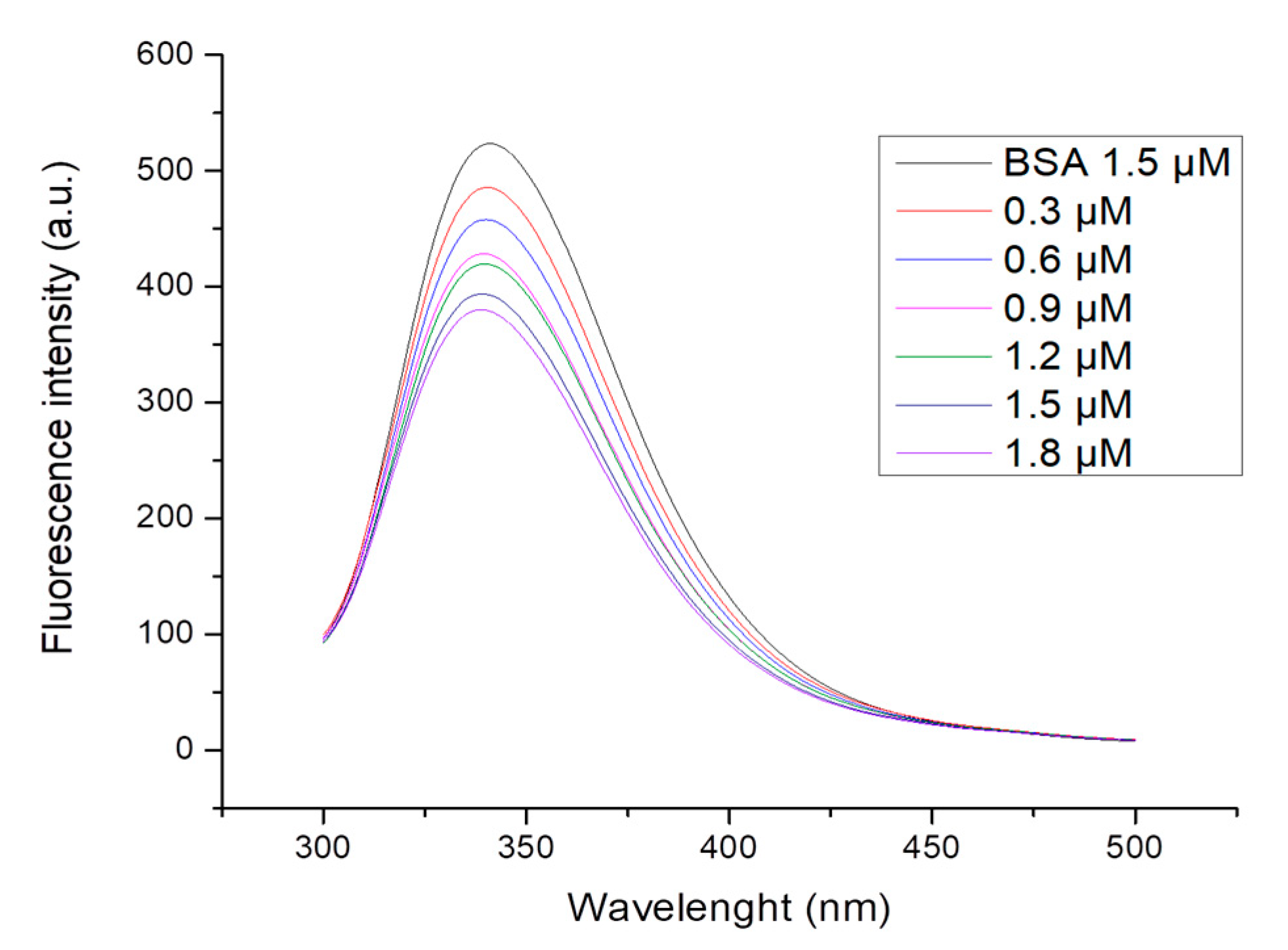
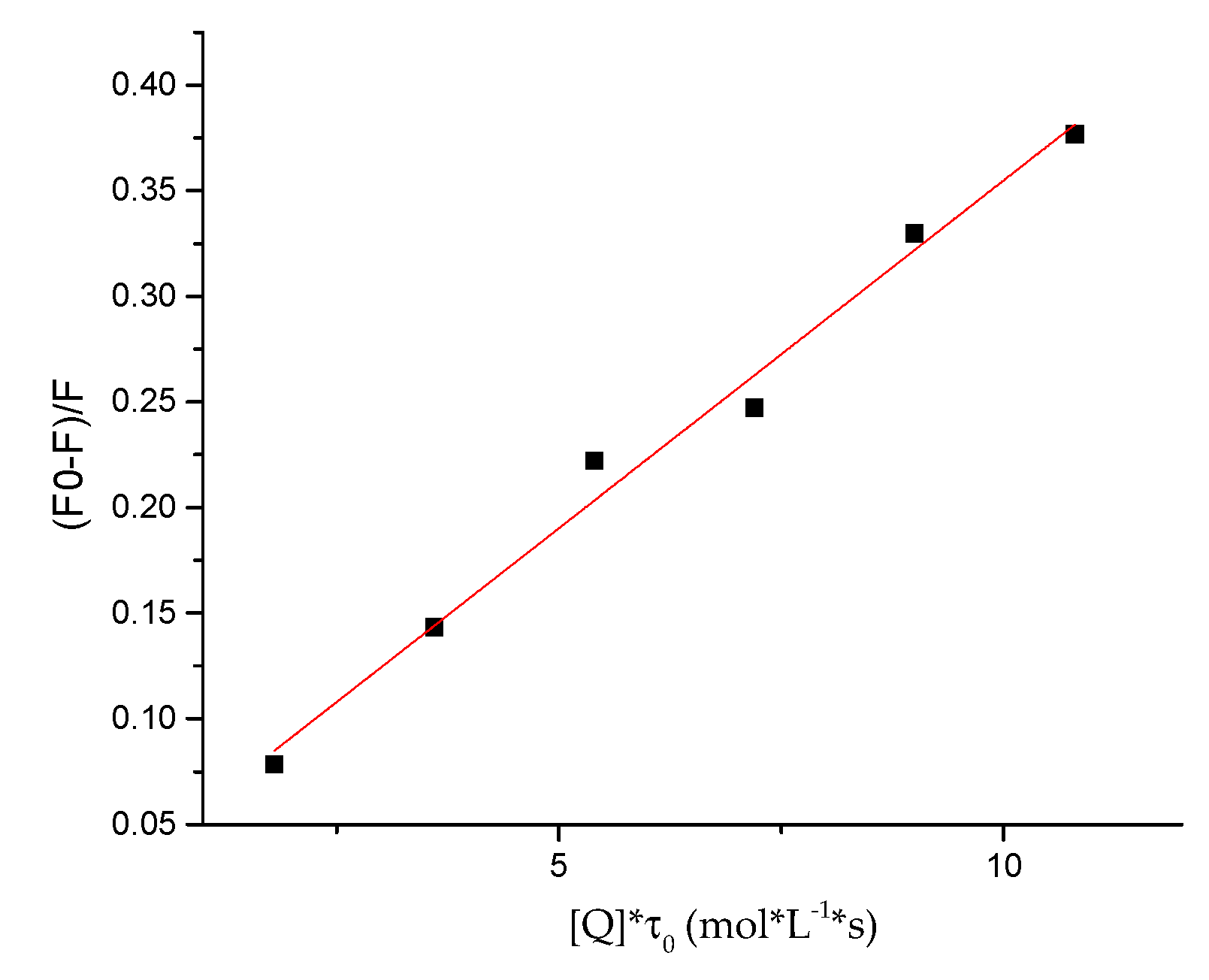
2.5. Protein-Binding Study
3. Materials and Methods
3.1. General Information
3.2. Chemistry
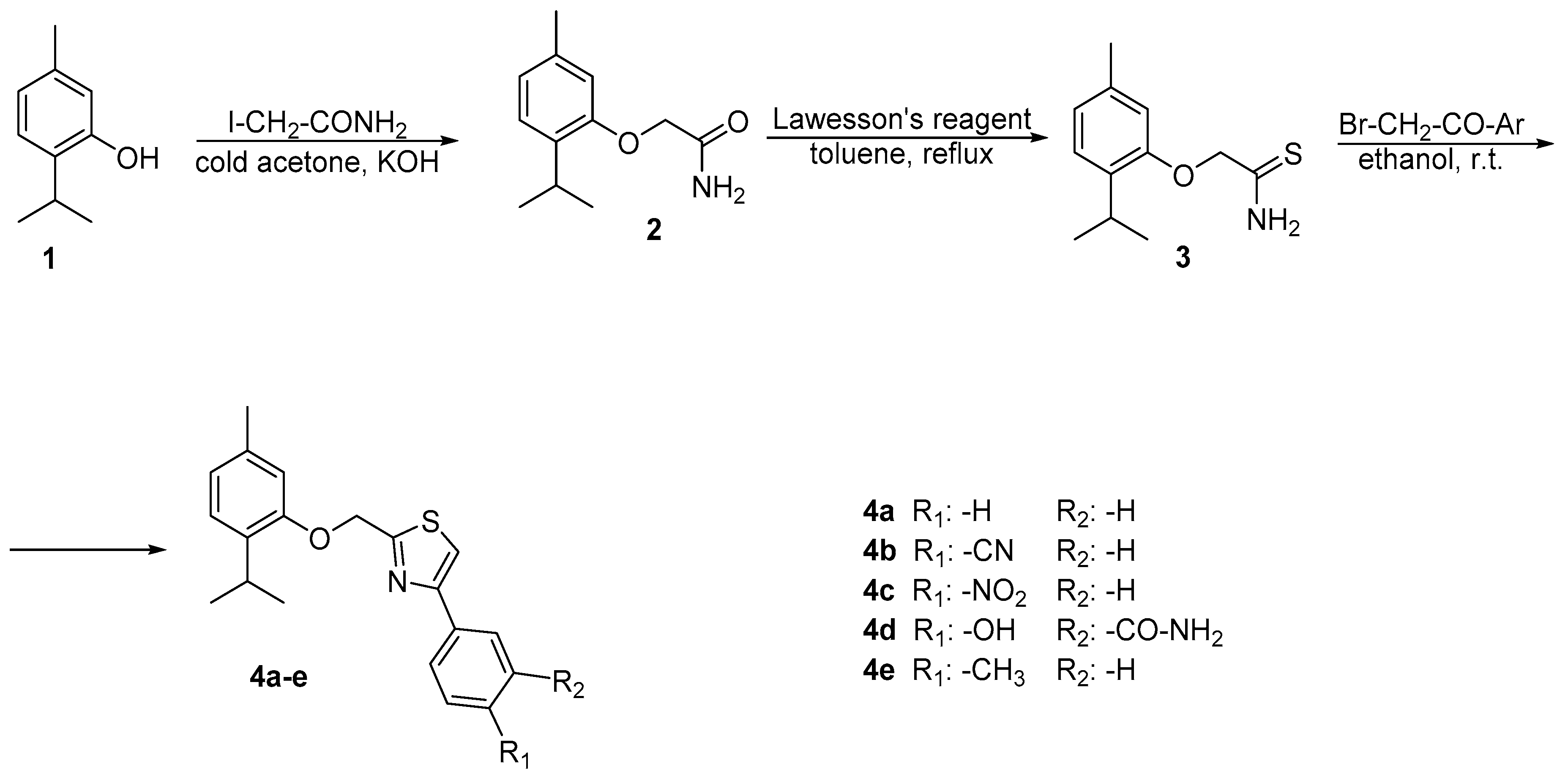
3.2.1. Synthesis of 2-(2-isopropyl-5-methylphenoxy)acetamide 2
3.2.2. Synthesis of 2-(2-isopropyl-5-methylphenoxy)ethanethioamide 3
3.2.3. General Procedure for the Synthesis of 2-((2-isopropyl-5-methylphenoxy)methyl)-4-phenyl thiazole Derivatives 4a–e
3.2.4. Synthesis of 2-(2-(2-isopropyl-5-methylphenoxy)-1-phenylethylidene)hydrazine-1-carbo-thioamide 6
3.2.5. General Procedure for the Synthesis of 2-(2-(2-(2-isopropyl-5-methylphenoxy)-1-phenyl ethylidene)hydrazineyl)-4-phenylthiazole Derivatives 7a–e
3.3. Anti-Candida Activity Assay
3.4. Molecular Docking
3.5. ADMET Predictions
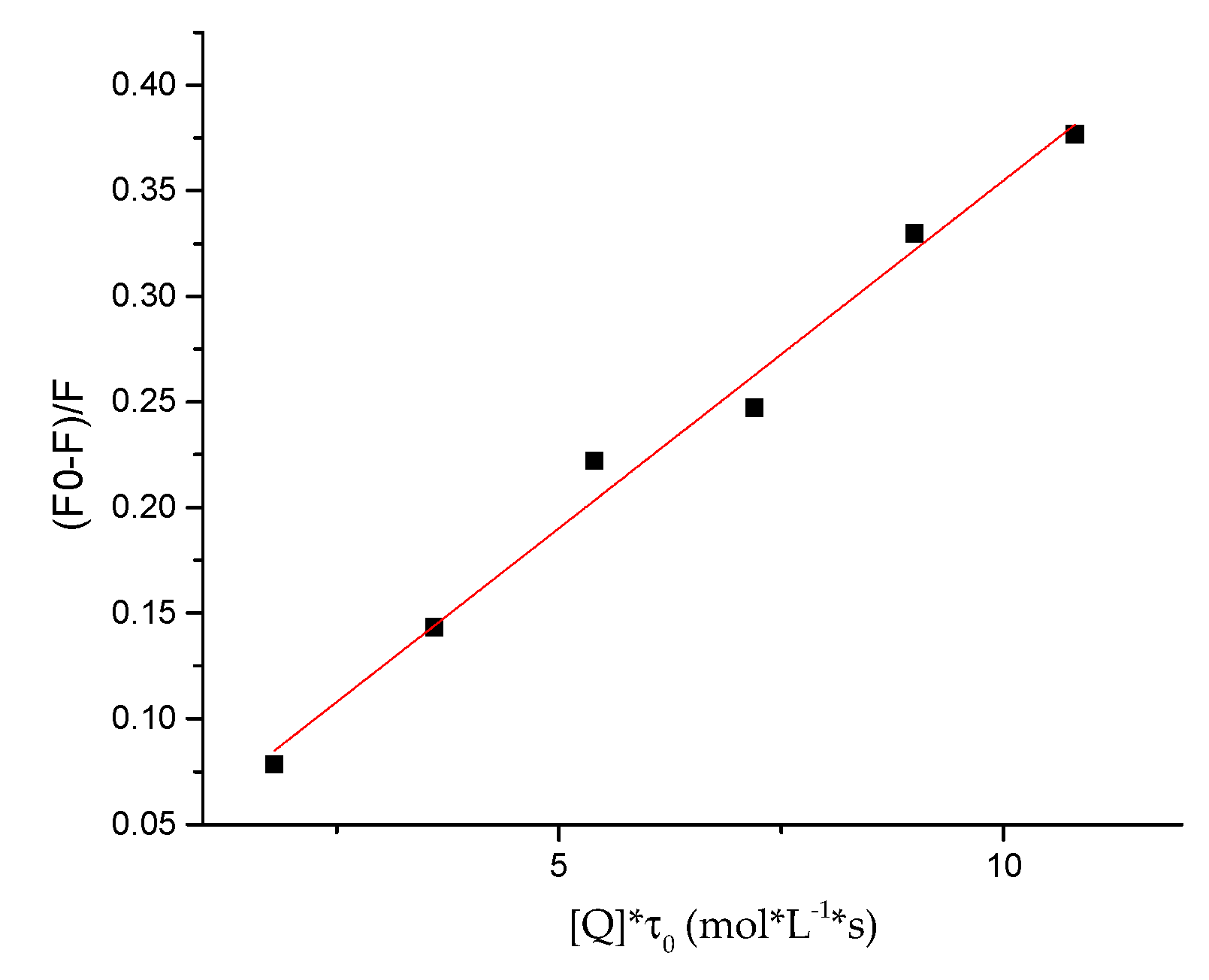
3.6. Protein-Binding Study
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pristov, K.E.; Ghannoum, M.A. Resistance of Candida to azoles and echinocandins worldwide. Clin. Microbiol. Infect. 2019, 25, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017, 16, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Revie, N.M.; Iyer, K.R.; Robbins, N.; Cowen, L.E. Antifungal drug resistance: Evolution, mechanisms and impact. Curr. Opin. Microbiol. 2018, 45, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Mazu, T.K.; Bricker, B.A.; Flores-Rozas, H.; Ablordeppey, S.Y. The Mechanistic Targets of Antifungal Agents: An Overview. Mini-Rev. Med. Chem. 2016, 16, 555–578. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.C.; Sagatova, A.A.; Hosseini, P.; Ruma, Y.N.; Wilson, R.K.; Keniya, M.V. Fungal Lanosterol 14α-demethylase: A target for next-generation antifungal design. Biochim. Biophys. Acta Proteins Proteom. 2019. [Google Scholar] [CrossRef]
- Singla, P.; Luxami, V.; Singh, R.; Tandon, V.; Paul, K. Novel pyrazolo [3,4-d]pyrimidine with 4-(1H-benzimidazol-2-yl)-phenylamine as broad spectrum anticancer agents: Synthesis, cell based assay, topoisomerase inhibition, DNA intercalation and bovine serum albumin studies. Eur. J. Med. Chem. 2017, 126, 24–35. [Google Scholar] [CrossRef]
- Bikobo, D.S.N.; Vodnar, D.C.; Stana, A.; Tiperciuc, B.; Nastasă, C.; Douchet, M.; Oniga, O. Synthesis of 2-phenylamino-thiazole derivatives as antimicrobial agents. J. Saudi Chem. Soc. 2017, 21, 861–868. [Google Scholar] [CrossRef]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef]
- Lino, C.I.; Gonçalves de Souza, I.; Borelli, B.M.; Silvério Matos, T.T.; Santos Teixeira, I.N.; Ramos, J.P.; Maria de Souza Fagundes, E.; de Oliveira Fernandes, P.; Maltarollo, V.G.; Johann, S.; et al. Synthesis, molecular modeling studies and evaluation of antifungal activity of a novel series of thiazole derivatives. Eur. J. Med. Chem. 2018, 151, 248–260. [Google Scholar] [CrossRef]
- Moraca, F.; De Vita, D.; Pandolfi, F.; Di Santo, R.; Costi, R.; Cirilli, R.; D’Auria, F.D.; Panella, S.; Palamara, A.T.; Simonetti, G.; et al. Synthesis, biological evaluation and structure-activity correlation study of a series of imidazol-based compounds as Candida albicans inhibitors. Eur. J. Med. Chem. 2014, 83, 665–673. [Google Scholar] [CrossRef]
- Carradori, S.; Secci, D.; Bolasco, A.; Rivanera, D.; Mari, E.; Zicari, A.; Lotti, L.V.; Bizzarri, B. Synthesis and cytotoxicity of novel (thiazol-2-yl)hydrazine derivatives as promising anti-Candida agents. Eur. J. Med. Chem. 2013, 65, 102–111. [Google Scholar] [CrossRef]
- Maillard, L.T.; Bertout, S.; Quinonéro, O.; Akalin, G.; Turan-Zitouni, G.; Fulcrand, P.; Demirci, F.; Martinez, J.; Masurier, N. Synthesis and anti-Candida activity of novel 2-hydrazino-1,3-thiazole derivatives. Bioorganic Med. Chem. Lett. 2013, 23, 1803–1807. [Google Scholar] [CrossRef]
- Hu, Y.J.; Liu, Y.; Shen, X.S.; Fang, X.Y.; Qu, S.S. Studies on the interaction between 1-hexylcarbamoyl-5-fluorouracil and bovine serum albumin. J. Mol. Struct. 2005, 738, 143–147. [Google Scholar] [CrossRef]
- Shi, J.H.; Chen, J.; Wang, J.; Zhu, Y.Y.; Wang, Q. Binding interaction of sorafenib with bovine serum albumin: Spectroscopic methodologies and molecular docking. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 149, 630–637. [Google Scholar] [CrossRef]
- De Oliveira Filho, G.B.; de Oliveira Cardoso, M.V.; Espíndola, J.W.P.; e Silva, D.A.O.; Ferreira, R.S.; Coelho, P.L.; dos Anjos, P.S.; de Souza Santos, E.; Meira, C.S.; Moreira, D.R.M.; et al. Structural design, synthesis and pharmacological evaluation of thiazoles against Trypanosoma cruzi. Eur. J. Med. Chem. 2017, 141, 346–361. [Google Scholar] [CrossRef]
- Stana, A.; Vodnar, D.C.; Tamaian, R.; Pîrnău, A.; Vlase, L.; Ionuț, I.; Oniga, O.; Tiperciuc, B. Design, synthesis and antifungal activity evaluation of new thiazolin-4-ones as potential lanosterol 14α-demethylase inhibitors. Int. J. Mol. Sci. 2017, 18, 177. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Borcea, A.M.; Marc, G.; Ionut, I.; Vodnar, D.C.; Vlase, L.; Gligor, F.; Pricopie, A.; Pîrnău, A.; Tiperciuc, B.; Oniga, O. A novel series of acylhydrazones as potential anti-Candida agents: Design, synthesis, biological evaluation and in silico studies. Molecules 2019, 24, 184. [Google Scholar] [CrossRef]
- Stana, A.; Enache, A.; Vodnar, D.C.; Nastasă, C.; Benedec, D.; Ionuț, I.; Login, C.; Marc, G.; Oniga, O.; Tiperciuc, B. New Thiazolyl-triazole Schiff Bases: Synthesis and Evaluation of the Anti-Candida Potential. Molecules 2016, 21, 1595. [Google Scholar] [CrossRef]
- Marc, G.; Stana, A.; Pîrnău, A.; Vlase, L.; Vodnar, D.C.; Duma, M.; Tiperciuc, B.; Oniga, O. 3,5-Disubstituted Thiazolidine-2,4-Diones: Design, Microwave-Assisted Synthesis, Antifungal Activity, and ADMET Screening. SLAS Discov. 2018, 23, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, B.; Abed, S.N.; Al-Attraqchi, O.; Kuche, K.; Tekade, R.K. Computer-Aided Prediction of Pharmacokinetic (ADMET) Properties; Tekade, R.K., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; ISBN 9780128144213. [Google Scholar]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Pereira, F.; Latino, D.A.R.S.; Gaudêncio, S.P. A chemoinformatics approach to the discovery of lead-like molecules from marine and microbial sources en route to antitumor and antibiotic drugs. Mar. Drugs 2014, 12, 757–778. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, J.; Yu, H.; Li, Y.; Hou, T. Recent Developments of In Silico Predictions of Oral Bioavailability. Comb. Chem. High Throughput Screen. 2011, 14, 362–374. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef]
- Artursson, P.; Bergstrom, C.A.S. Computational Approaches to Drug Absorption and Bioavailability Intestinal Absorption: The Role of Polar Surface Area; van de Waterbeemd, H., Lennernas, H., Artursson, P., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003; ISBN 352730438X. [Google Scholar]
- Molavipordanjani, S.; Emami, S.; Mardanshahi, A.; Talebpour Amiri, F.; Noaparast, Z.; Hosseinimehr, S.J. Novel 99m Tc-2-arylimidazo[2,1-b]benzothiazole derivatives as SPECT imaging agents for amyloid-β plaques. Eur. J. Med. Chem. 2019, 149–161. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Zhao, L.; Sun, N.; Tian, L.; Zhao, S.; Sun, B.; Sun, Y.; Zhao, D. Strategies for the development of highly selective cytochrome P450 inhibitors: Several CYP targets in current research. Bioorganic Med. Chem. Lett. 2019, 1–9. [Google Scholar] [CrossRef]
- Gardin, A.; Ufer, M.; Legangneux, E.; Rossato, G.; Jin, Y.; Su, Z.; Pal, P.; Li, W.; Shakeri-Nejad, K. Effect of Fluconazole Coadministration and CYP2C9 Genetic Polymorphism on Siponimod Pharmacokinetics in Healthy Subjects. Clin. Pharmacokinet. 2019, 58, 349–361. [Google Scholar] [CrossRef]
- Yap, C.W.; Chen, Y.Z. Prediction of cytochrome P450 3A4, 2D6, and 2C9 inhibitors and substrates by using support vector machines. J. Chem. Inf. Model. 2005, 45, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Singla, P.; Luxami, V.; Paul, K. Synthesis and in vitro evaluation of novel triazine analogues as anticancer agents and their interaction studies with bovine serum albumin. Eur. J. Med. Chem. 2016, 117, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Luan, F.; Chen, X. Binding analysis of glycyrrhetinic acid to human serum albumin: Fluorescence spectroscopy, FTIR, and molecular modeling. Bioorganic Med. Chem. 2006, 14, 3210–3217. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huang, C.R.; Jiang, M.; Zhu, Y.Y.; Wang, J.; Chen, J.; Shi, J.H. Binding interaction of atorvastatin with bovine serum albumin: Spectroscopic methods and molecular docking. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 156, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Alagesan, M.; Bhuvanesh, N.S.P.; Dharmaraj, N. Potentially cytotoxic new copper(ii) hydrazone complexes: Synthesis, crystal structure and biological properties. Dalt. Trans. 2013, 42, 7210–7223. [Google Scholar] [CrossRef]
- Suryawanshi, V.D.; Walekar, L.S.; Gore, A.H.; Anbhule, P.V.; Kolekar, G.B. Spectroscopic analysis on the binding interaction of biologically active pyrimidine derivative with bovine serum albumin. J. Pharm. Anal. 2016, 6, 56–63. [Google Scholar] [CrossRef]
- Xu, H.; Yao, N.; Xu, H.; Wang, T.; Li, G.; Li, Z. Characterization of the interaction between eupatorin and bovine serum albumin by spectroscopic and molecular modeling methods. Int. J. Mol. Sci. 2013, 14, 14185–14203. [Google Scholar] [CrossRef]
- Wani, T.A.; Bakheit, A.H.; Al-Majed, A.R.A.; Bhat, M.A.; Zargar, S. Study of the interactions of bovine serum albumin with the new anti-inflammatory agent 4-(1,3-dsioxo-1,3-dihydro-2H-isoindol-2-yl)-N-[(4-ethoxy-phenyl) methylidene]benzohydrazide using a multi-spectroscopic approach and molecular docking. Molecules 2017, 22, 1258. [Google Scholar] [CrossRef]
- Hudson, E.A.; de Paula, H.M.C.; Ferreira, G.M.D.; Ferreira, G.M.D.; Hespanhol, M. do C.; da Silva, L.H.M.; Pires, A.C. dos S. Thermodynamic and kinetic analyses of curcumin and bovine serum albumin binding. Food Chem. 2018, 242, 505–512. [Google Scholar] [CrossRef]
- Liu, J.; He, Y.; Liu, D.; He, Y.; Tang, Z.; Lou, H.; Huo, Y.; Cao, X. Characterizing the binding interaction of astilbin with bovine serum albumin: A spectroscopic study in combination with molecular docking technology. RSC Adv. 2018, 8, 7280–7286. [Google Scholar] [CrossRef]
- Dufour, C.; Dangles, O. Flavonoid-serum albumin complexation: Determination of binding constants and binding sites by fluorescence spectroscopy. Biochim. Biophys. Acta Gen. Subj. 2005, 1721, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Saunders, D.G.; Hoban, D.J.; Karlowsky, J.A. Influence of human serum on antifungal pharmacodynamics with Candida albicans. Antimicrob. Agents Chemother. 2001, 45, 2018–2022. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.D.; Procop, G.W.; Dufresne, P.; Fuller, J.; Ghannoum, M.A.; Hanson, K.E.; Holliday, D.; Holliday, N.M.; Kovanda, L.; Lockhart, S.R.; et al. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, 4th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2010, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Pricopie, A.-I.; Marc, G.; Nastasă, C.; Borcea, A.-M.; Ionuţ, I.; Tiperciuc, B.; Oniga, O.; Vlase, L.; Pîrnău, A.; Vodnar, D.C. Design and synthesis of some novel 1,2,4-triazole-3-yl-mercapto derivatives as potential anti-Candida agents. Farmacia 2018, 66, 948–958. [Google Scholar] [CrossRef]
- Borcea, A.-M.; Marc, G.; Pîrnău, A.; Vlase, L.; Ionuț, I.; Tiperciuc, B.; Oniga, O. Synthesis and molecular docking study of some new 1,4-phenylene-bisthiazoles as fungal lanosterol 14α-demethylase inhibitors. Farmacia 2017, 65, 683–689. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 4a–e and 7a–e are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | C. albicans ATCC 10231 | C. parapsilosis ATCC 22019 | C. zeylanoides ATCC 201082 |
|---|---|---|---|
| 4a | 62.5 | 62.5 | 125 |
| 4b | 62.5 | 62.5 | 125 |
| 4c | 62.5 | 62.5 | 125 |
| 4d | 62.5 | 62.5 | 125 |
| 4e | 62.5 | 62.5 | 125 |
| 7a | 7.81 | 15.62 | 62.5 |
| 7b | 62.5 | 62.5 | 62.5 |
| 7c | 62.5 | 62.5 | 125 |
| 7d | 62.5 | 62.5 | 125 |
| 7e | 3.9 | 15.62 | 15.62 |
| Fluconazole | 15.62 | 7.81 | 15.62 |
| Compound | C. albicans ATCC 10231 | C. parapsilosis ATCC 22019 | C. zeylanoides ATCC 201082 |
|---|---|---|---|
| 4a | 125 | 125 | 250 |
| 4b | 125 | 125 | 250 |
| 4c | 125 | 125 | 250 |
| 4d | 125 | 125 | 250 |
| 4e | 125 | 125 | 250 |
| 7a | 15.62 | 31.24 | 125 |
| 7b | 125 | 125 | 125 |
| 7c | 125 | 125 | 250 |
| 7d | 125 | 125 | 250 |
| 7e | 7.8 | 31.24 | 31.24 |
| Fluconazole | 31.24 | 15.62 | 31.24 |
| Compound | Best Binding Conformation | The 2 Å Cluster Containing the Top Binding Conformation | Number of Distinct Clusters | |||||
|---|---|---|---|---|---|---|---|---|
| ΔG (kcal/mol) | Ki (nM) | NoC 1 | Average ΔG (kcal/mol) | Standard Deviation | ||||
| ΔG (kcal/mol) | Cartesian Coordinates | Total | Multi-Member | |||||
| 4a | −10.51 | 19.77 | 19 | −9.89 | 0.54 | 0.73 | 26 | 15 |
| 4b | −11.49 | 3.78 | 41 | −10.60 | 0.46 | 0.36 | 21 | 15 |
| 4c | −10.93 | 9.73 | 32 | −9.95 | 0.56 | 0.51 | 23 | 12 |
| 4d | −10.83 | 11.52 | 6 | −10.52 | 0.27 | 0.76 | 38 | 19 |
| 4e | −11.00 | 8.65 | 27 | −10.63 | 0.51 | 0.47 | 23 | 16 |
| 7a | −12.52 | 0.66 | 3 | −11.35 | 1.20 | 1.04 | 50 | 18 |
| 7b | −13.21 | 0.21 | 14 | −12.51 | 0.74 | 0.50 | 42 | 16 |
| 7c | −12.20 | 1.14 | 1 | – | – | – | 44 | 18 |
| 7d | −12.39 | 0.83 | 1 | – | – | – | 64 | 17 |
| 7e | −12.71 | 0.48 | 10 | −12.63 | 0.70 | 0.70 | 48 | 19 |
| Compound | MW 1 (Da) | RoB 2 | HBA 3 | HBD 4 | tPSA 5 | mLogP 6 | Lipinski Violations | Veber Violations |
|---|---|---|---|---|---|---|---|---|
| 4a | 323.45 | 5 | 2 | 0 | 50.36 | 3.54 | 0 | 0 |
| 4b | 348.46 | 5 | 3 | 0 | 74.15 | 2.81 | 0 | 0 |
| 4c | 368.45 | 6 | 4 | 0 | 96.18 | 2.39 | 0 | 0 |
| 4d | 382.48 | 6 | 4 | 2 | 113.68 | 2.07 | 0 | 0 |
| 4e | 337.48 | 5 | 2 | 0 | 50.36 | 3.76 | 0 | 0 |
| 7a | 441.59 | 8 | 3 | 1 | 74.75 | 4.49 | 1 | 0 |
| 7b | 466.60 | 8 | 4 | 1 | 98.54 | 3.78 | 0 | 0 |
| 7c | 486.59 | 9 | 5 | 1 | 120.57 | 3.46 | 0 | 0 |
| 7d | 500.61 | 9 | 5 | 3 | 138.07 | 3.11 | 1 | 0 |
| 7e | 455.61 | 8 | 3 | 1 | 74.75 | 4.68 | 1 | 0 |
| Compound | %GI Abs 1 | BBBP 2 | PGP 3 Substrate | CYP450 Inhibition | |
|---|---|---|---|---|---|
| CYP3A4 | CYP2C9 | ||||
| 4a | 91.62 | Yes | Yes | Yes | Yes |
| 4b | 83.41 | No | Yes | Yes | Yes |
| 4c | 75.81 | No | No | Yes | Yes |
| 4d | 69.78 | No | No | Yes | Yes |
| 4e | 91.62 | No | Yes | Yes | Yes |
| 7a | 83.21 | No | Yes | Yes | No |
| 7b | 75.00 | No | Yes | Yes | Yes |
| 7c | 67.40 | No | No | Yes | No |
| 7d | 61.36 | No | No | No | No |
| 7e | 83.21 | No | Yes | Yes | No |
| Compound | Kq × 1013 (L/mol·s) | a KSV × 105 (L/mol) | b R2 |
|---|---|---|---|
| 7e | 3.29 ± 0.0017 | 1.97± 0.0124 | 0.9856 |
| Compound | n | logKb | Kb × 105 (M−1) | R2 |
|---|---|---|---|---|
| 7e | 0.873 ± 0.0355 | −0.6448 ± 0.0094 | 2.26 | 0.9917 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pricopie, A.-I.; Ionuț, I.; Marc, G.; Arseniu, A.-M.; Vlase, L.; Grozav, A.; Găină, L.I.; Vodnar, D.C.; Pîrnău, A.; Tiperciuc, B.; et al. Design and Synthesis of Novel 1,3-Thiazole and 2-Hydrazinyl-1,3-Thiazole Derivatives as Anti-Candida Agents: In Vitro Antifungal Screening, Molecular Docking Study, and Spectroscopic Investigation of their Binding Interaction with Bovine Serum Albumin. Molecules 2019, 24, 3435. https://doi.org/10.3390/molecules24193435
Pricopie A-I, Ionuț I, Marc G, Arseniu A-M, Vlase L, Grozav A, Găină LI, Vodnar DC, Pîrnău A, Tiperciuc B, et al. Design and Synthesis of Novel 1,3-Thiazole and 2-Hydrazinyl-1,3-Thiazole Derivatives as Anti-Candida Agents: In Vitro Antifungal Screening, Molecular Docking Study, and Spectroscopic Investigation of their Binding Interaction with Bovine Serum Albumin. Molecules. 2019; 24(19):3435. https://doi.org/10.3390/molecules24193435
Chicago/Turabian StylePricopie, Andreea-Iulia, Ioana Ionuț, Gabriel Marc, Anca-Maria Arseniu, Laurian Vlase, Adriana Grozav, Luiza Ioana Găină, Dan C. Vodnar, Adrian Pîrnău, Brîndușa Tiperciuc, and et al. 2019. "Design and Synthesis of Novel 1,3-Thiazole and 2-Hydrazinyl-1,3-Thiazole Derivatives as Anti-Candida Agents: In Vitro Antifungal Screening, Molecular Docking Study, and Spectroscopic Investigation of their Binding Interaction with Bovine Serum Albumin" Molecules 24, no. 19: 3435. https://doi.org/10.3390/molecules24193435