Cooperative Effects in Weak Interactions: Enhancement of Tetrel Bonds by Intramolecular Hydrogen Bonds





1
Trinity Biomedical Sciences Institute, School of Chemistry, The University of Dublin, Trinity College, Dublin 2, Ireland
2
Instituto de Química Médica, CSIC, Juan de la Cierva, 3, E-28006 Madrid, Spain
3
Irish Centre of High-End Computing, Grand Canal Quay, Dublin 2, Ireland
4
School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(2), 308; https://doi.org/10.3390/molecules24020308
Submission received: 3 December 2018
/
Revised: 8 January 2019
/
Accepted: 9 January 2019
/
Published: 16 January 2019
(This article belongs to the Special Issue Intramolecular Hydrogen Bonding 2018)
Abstract
:A series of silyl and germanium complexes containing halogen atoms (fluorine and chlorine atoms) and exhibiting tetrel bonds with Lewis bases were analyzed by means of Møller-Plesset computational theory. Binding energies of germanium derivatives were more negative than silicon ones. Amongst the different Lewis bases utilized, ammonia produced the strongest tetrel bonded complexes in both Ge and Si cases, and substitution of the F atom by Cl led to stronger complexes with an ethylene backbone. However, with phenyl backbones, the fluorosilyl complexes were shown to be less stable than the chlorosilyl ones, but the opposite occurred for halogermanium complexes. In all the cases studied, the presence of a hydroxyl group enhanced the tetrel bond. That effect becomes more remarkable when an intramolecular hydrogen bond between the halogen and the hydrogen atom of the hydroxyl group takes places.
1. Introduction
One of the major achievements in contemporary chemistry was the introduction by Jean-Marie Lehn of supramolecular chemistry [1]. According to Vögtle “In contrast to molecular chemistry, which is predominantly based upon the covalent bonding of atoms, supramolecular chemistry is based upon intermolecular interactions, i.e., on the association of two or more building blocks, which are held together by intermolecular bonds”. Today, these intermolecular bonds are called weak interactions regardless of whether they are intra or intermolecular. There are numerous weak interactions, also known as non-covalent interactions, and they have been shown to be of the upmost importance across different domains including biology, chemistry and material science [2]. These non-covalent interactions has been categorised based on the interacting atoms involved: hydrogen bonds [3,4], halogen bonds [5], hydride bonds [6,7], pnictogen bonds [8,9,10,11,12,13], chalcogen interactions [14,15,16,17,18] and tetrel bonds [19,20,21,22]. The latter, tetrel bonds, are defined, analogous to halogen bonds, as interactions between electron donors and tetrel atoms (C, Si and Ge), in which the tetrel atom acts as an electron acceptor, usually through an electron-deficient outer lobe of a p orbital, called a σ-hole [23] by Politzer and Murray [24,25,26], which is formed in the tetrel atom, especially when the atom bonded to the tetrel is highly electronegative. It has also been shown that interactions through σ-holes are mainly driven by the electrostatic interaction term [27,28,29,30,31,32,33,34,35,36,37,38].
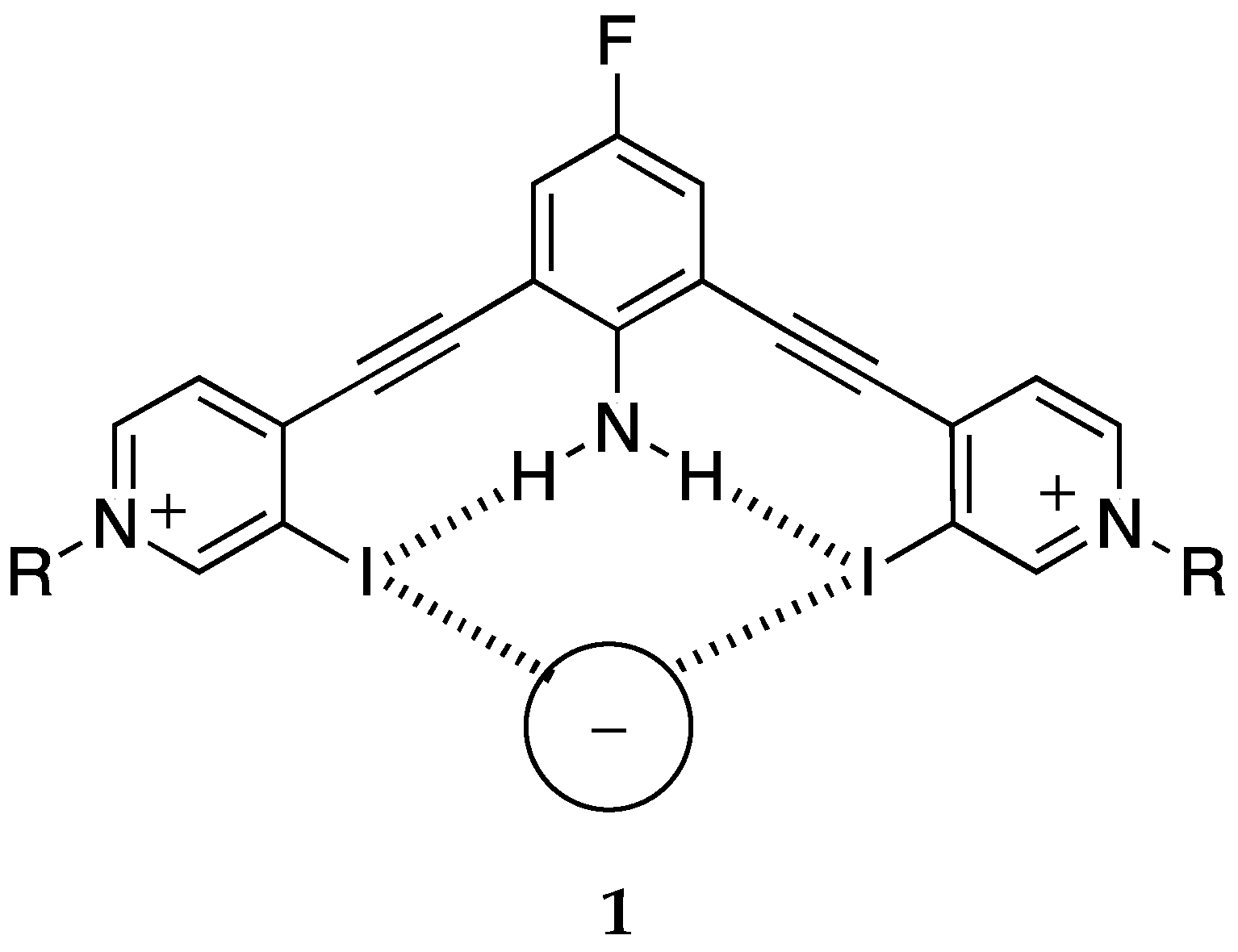
Related to this topic is cooperativity, usually intermolecular (allosteric), but also, although much less studied, intramolecular [39]. There are two relevant papers. Berryman et al. reported the synthesis and anion-binding properties of receptor 1 (2,6-bis(4-ethynylpyridinyl)-4-fluoroaniline) which can act as a halogen donor, trapping a wide variety of anions (Cl−, Br−, I−, SCN−, NO3−, HSO4−, H2PO4− and ReO4−) [40] (Scheme 1). The presence of intramolecular N–H···I hydrogen bonds (HBs) increases the strength of the I···anion halogen bonds (XBs).
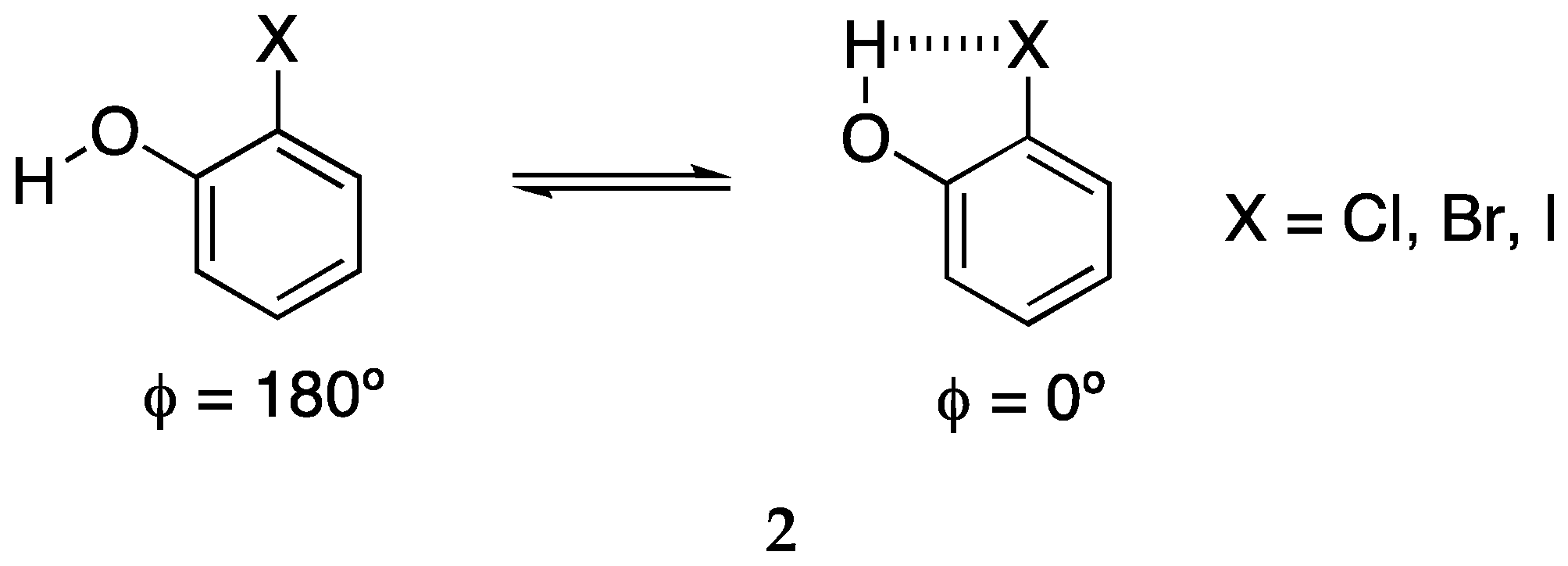
Ho and co-workers described how the σ-hole responsible for the XB is substantially increased by an intramolecular HB [41]. The model they selected was 2-halophenol (2) that can exist as two limit conformations (Scheme 2), the anti one without HB and the syn one with an intramolecular HB (IMHB).
For each X, they calculated the electrostatic potential map as a function of the torsion angle ϕ. They found that the positive zone increases considerably when the torsion angle approaches 0°, i.e., when there is an IMHB.
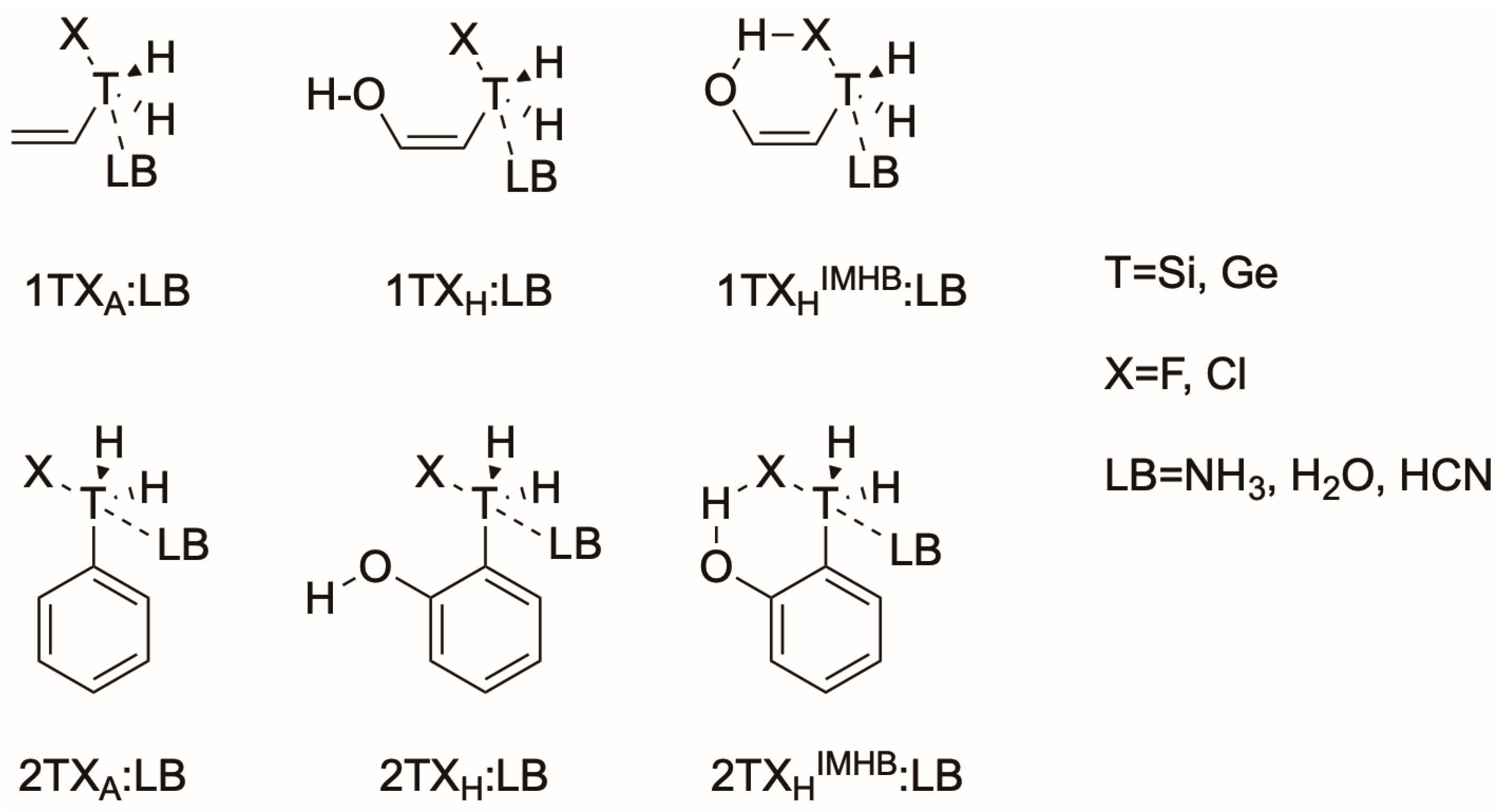
In the present article, the effect of the intramolecular hydrogen bond in fluorosilyl and fluorogermanium derivatives and their abilities as tetrel bond donors is studied (Scheme 3). The following notation will be used to label the complexes studied throughout the manuscript: nTXaZ:LB, where “n” stands for the two types of carbon backbone (1=allyl, 2=phenyl), “T” stands for the tetrel atom (Si or Ge), “Z” indicates whether the complex exhibits, or does not exhibit, an intramolecular hydrogen bond (IMHB or none), “a” indicates the presence of an OH group in the molecule (hydroxyl=H) or not (allyl=A). Finally, LB stands for the Lewis Base involved (NH3, H2O or HCN). For example, 1GeClAIMBH:NH3 corresponds to an allylchlorogermanium derivative interacting with ammonia in which the OH group is present and forming an intramolecular hydrogen bond. The short notation 1TX:LB will be used to refer the whole complex family.
2. Results
In order to evaluate the appropriate computational level for the present study, a benchmark across different basis sets has been carried out. For such purpose, 1SiF:NH3 complexes (1SiFA:NH3, 1SiFH:NH3, and 1SiFHIMHB:NH3) were optimized by means of Møller-Plesset (MP2) using different basis sets, i.e., aug-cc-pVDZ (avdz), aug-cc-pVTZ (avtz), aug-cc-pVQZ (avqz), and using Helgaker’s method to extrapolate to the complete basis set (CBS) with two pairs of basis: aug-cc-pVDZ and aug-cc-pVTZ (CBSDT) and aug-cc-pVTZ and aug-cc-pVQZ (CBSTQ). Using those levels, the binding energies (Eb), obtained as a difference of the energy of the complex minus the energies of the isolated monomers in their optimized structure, for these three complexes, were calculated and are summarized in Table 1. As observed, for the evolution of the energy values, avdz < avtz < avqz, it seems that there is a strong dependency of the binding energy with respect to the basis set size. When extrapolation to the complete basis set (CBS) is taken into account, it was also observed that extrapolations using two different pairs of basis set, CBSDT and CBSTQ, also provide different results, with the latter, in our opinion, more accurate than the former. Taken into account the outcome of the current benchmark and the computational feasibility of the calculations, the MP2/CBSTQ computational method was chosen to evaluate the rest of the complexes, and from hence forth, named MP2/CBS for simplicity.
2.1. Allylfluorotetrel Derivatives: Effect of the Lewis Bases (1TF:LB)
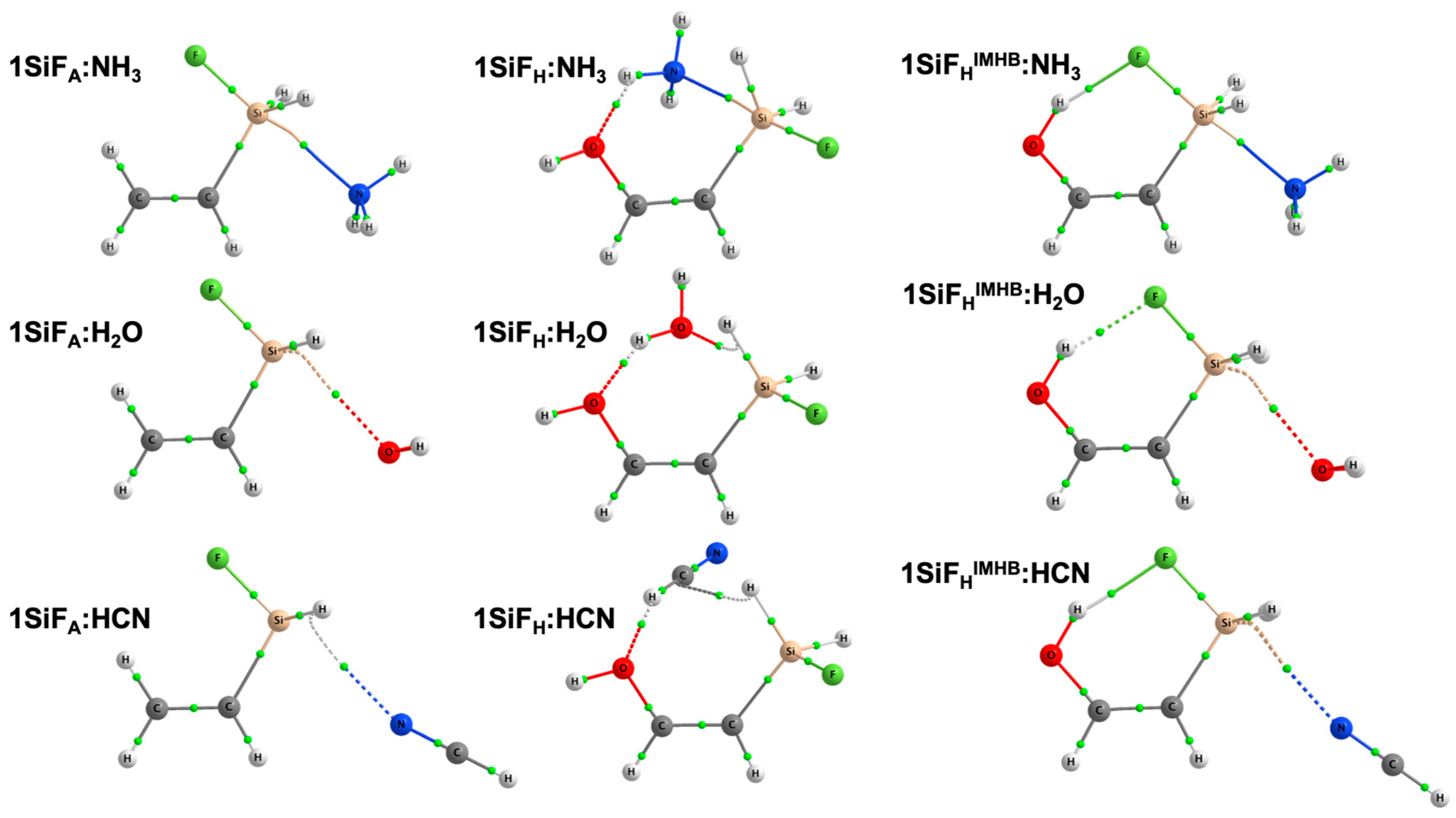
The effect of the Lewis bases upon complexation within 1TF:LB complexes (Figure 1) was evaluated using three different Lewis bases, i.e., NH3, H2O and HCN. The intermolecular T···Y (Y = N or O) distances at the MP2/aug-cc-pVTZ computational level are gathered in Table 2. Molecular graphs and Cartesian coordinates can be found in the electronic supplementary information (Table S1). The intermolecular Si···N distances found in 1SiF:NH3 range from 2.276 (1SiFHIMHB:NH3) to 2.518 Å (1SiFA:NH3), while in 1GeF:NH3, the Ge···Y distance ranges from 2.450 (1GeFHIMHB:NH3) to 2.661 Å (1GeFA:NH3). As observed, 1TFHIMHB:NH3 complexes present the shortest intermolecular T···Y distances (Table 2). Looking at the 1SiFA:NH3 complex, the intermolecular Si···N distance is 2.518 Å. When the 1SiFH:NH3 complex is taken into account, there is a shortening of 0.183 Å on the Si···N distance. This shortening can point to a decrease in the electron density on the Si atom due to the OH group and therefore an increase in the σ-hole depth, which eventually will enhance the tetrel bond. This shortening is even more visible in 1SiFHIMHB:NH3 complex (0.242 Å), which may indicate that the intramolecular hydrogen bond (IMHB) enhances the tetrel bond. In the case of 1GeF:LB germanium complexes, similar but more pronounced trends in the intermolecular Ge···N distances were found due to the IMHB and hydroxyl groups. Furthermore, the T-F distance was also analyzed to evaluate the polarity of the T-F bonds with the IMHB. T-F distance for the 1SiFA:NH3 complex is 1.643 Å, while in 1SiFH:NH3 and 1SiFHIMHB:NH3 complexes, they are 1.654 and 1.685 Å, respectively, indicating an increase in the charge transfer to the T-F σ*T-F antibonding orbital, particularly in the complexes exhibiting IMBH. The same is true for 1GeF:NH3 complexes (1.768, 1.776 and 1.815 Å for 1GeFA:NH3 complex, 1GeFH:NH3 and 1GeFHIMHB:NH3 complexes, respectively).
When different Lewis bases are considered (Table 2), it can be seen that complexes with ammonia have the shortest intermolecular T···Y distances. However, similar trends were observed for 1TF:H2O complexes compared with 1TF:NH3 complexes, where the T···Y distance evolves as follows: 1TFA:H2O > 1TFH:H2O > 1TFIMHBA:H2O. In the case of 1TF:HCN complexes, the 1TFH:HCN complex seems to deviate from this trend, exhibiting a lengthening in the intermolecular T···Y distance. The reason behind is that in those complexes (both for 1SiFH:HCN and 1GeFH:HCN), the O atom from the OH group acts as an electron donor and performs an intermolecular hydrogen bond with the Lewis base. In other words, the Lewis base acts as a hydrogen donor. That provokes a re-orientation of the Lewis base which lengthens the T···N distance. If the molecule is constrained to Cs symmetry, forcing those abovementioned to be co-planar, an imaginary frequency is found which, if followed, reverts into the actual rotated structure with C1 symmetry.
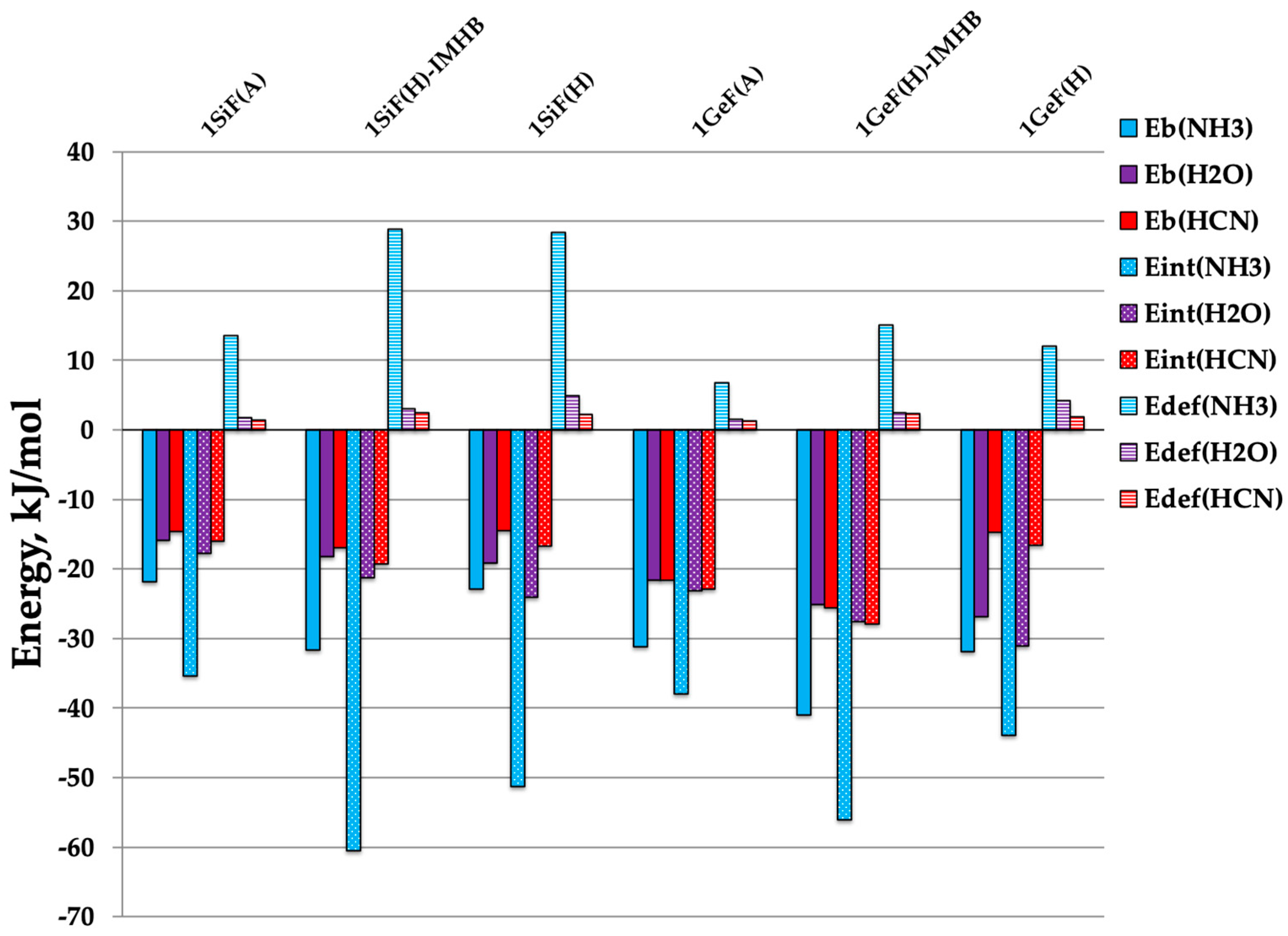
In order to evaluate the interaction energies between 1TF compounds and the Lewis base, binding energies (Eb), interaction energies (Eint) and deformation energies (Edef) were calculated at the MP2/CBS computational level and are summarized in Table 3 and with a histogram plotted in Figure 1. Binding energies (Eb) and interaction energies (Eint) were calculated as described in the “Materials and Methods” section. The deformation energy (Edef), also called re-organization energy, was calculated as the difference between Eb and Eint. Eb ranges from −21.8 to −41.0 kJ·mol−1 for 1TF:NH3 complexes. In 1TF:H2O, it ranges from −15.9 to −26.9 kJ·mol−1, and −14.5 to −25.1 kJ·mol−1 for 1TF:HCN ones. In all cases, 1TF:NH3 complexes show stronger interactions than 1TF:H2O or 1TF:HCN ones as per the Eb values. Focusing on the 1SiF:NH3 complexes, it was observed that the Eb value calculated for 1SiFA:NH3 is −21.8 kJ·mol−1, and when the hydroxyl groups are present, Eb becomes slightly more negative (−22.9 kJ·mol−1), which indicates a strengthening of the interaction. This strengthening is even larger in 1SiFHIMHB:NH3 (Eb = −31.7 kJ·mol−1), which shows that when the IMHB takes place, the interactions between the complex and the Lewis base are stronger. The same occurs for the 1GeF:NH3 complexes. In the case of water complexes, the Eb follows the same trend as in ammonia complexes. However, the 1TFH:H2O complex presents slightly more negative values of Eb than the 1TFHIMHB:H2O complex, which is likely due to a secondary intermolecular hydrogen bond in which water is acting as a hydrogen bond donor (Figure 1). This could be the reason behind the extra stabilization in the Eb. The opposite happens for the 1TF:HCN complexes in which 1TFH:HCN is less stable than its parental complex, 1TFA:HCN. As explained above, this is provoked by a re-orientation of the Lewis base in the 1TFH:HCN due to the presence of the hydroxyl group, which acts as an electron donor and forms an intermolecular hydrogen bond with the HCN (Figure 1). Poor correlations between the binding energy and the intermolecular distances were found, which may be due to the secondary hydrogen bonds in the 1TFH:LB complexes and electronic repulsions between atoms due to the electron lone pairs [42].
To evaluate the re-organization energy, i.e., the relaxation energy of the monomers and the energy penalty upon complexation, the interaction energies were evaluated. As observed (Figure 2), Eint values for all the complexes studied are more negative than the corresponding Eb values; this is particularly dramatic for the 1SiF:NH3 complexes in which the Eint values are the most negative of all complexes. When the difference of both quantities (Eb − Eint) is taken into account, the deformation energy, Edef, values (Table 3) indicate that 1TF:NH3 complexes suffer a larger penalty in binding energy due to re-organization than 1TF:H2O and 1TF:HCN complexes.
2.2. Allylhalotetrel Derivatives: Effect of the Halogen (1TX:NH3)
Once the effect of the Lewis base on the tetrel interaction and the relative energy between the three different complexes was determined, the effect of substituting the halogen atom (X) bonded to the tetrel atom (T=Si or Ge) was studied. For this purpose, and keeping the same Lewis base (NH3), fluorine atom was replaced by a chlorine atom within the 1TX:NH3 complexes. Intermolecular T···N distances are summarized in Table 4. The shortest T···N distance corresponds to the 1SiFHIMHB:NH3 complex (2.276 Å) while the largest is found in the 1GeClA:NH3 complex (2.767 Å). As observed in Table 4, 1TF:NH3 complexes exhibit shorter intermolecular T···Y distances than 1TCl:NH3 ones, with the exception of the 1SiFH:NH3 complex, which presents a slightly longer (0.044 Å) T···N distance than the 1SiClH:NH3 complex. Furthermore, the T-F distances found for 1TF:NH3 complexes suggest an increase in the polarity of the T-F bond, and a similar phenomenon is observed for 1TCl:NH3 (Table 4) in which the T-Cl bond distances exhibit a lengthening in 1TXH:NH3 and 1TXHIMHB:NH3 complexes with respect to those in 1TXA:NH3 complexes.
In terms of binding energies, 1TF:NH3 complexes show slightly more negative Eb values than 1TCl:NH3 complexes, which may be caused by an increase in the depth of the σ-hole on the tetrel atom (Table S2). Fluorine atoms withdraw more electron density, therefore the σ-hole on the tetrel atom is deeper, so 1TF:NH3 complexes will have more negative Eb values than 1TCl:NH3 complexes. Once again, the only exception corresponds to 1SiClH:NH3 with an Eb value of −24.5 kJ·mol−1, while the 1SiFH:NH3 Eb value is −22.9 kJ·mol−1. In both cases, the halogen atom does not form any IMHBs, which reinforces the idea that IMHBs are stabilizing the complex. Regarding the interaction energy, Eint is shown to be more negative than Eb, which is as expected because the Eb suffers from the re-organization penalty. In fact, Edef values indicate that this penalty is larger for silyl complexes than germanium ones, and within the same tetrel family (1TX:NH3), the deformation energy is much larger, up to three times larger in some cases, for complexes with hydroxyl groups (1TFH:NH3 and 1TXHIMHB:NH3) than in their parental complexes (1SiXA:NH3).
2.3. Phenyl Halogen Tetrel Derivatives: Effect of the Backbone (2TX:NH3)
Finally, different carbon backbones have been evaluated by means of replacing the allyl backbone with a phenyl ring focusing only on complexes with ammonia. The intermolecular T···N distances, Eb, Eint, Edef and T-X bond distances are summarized in Table 5. Intermolecular T···N distances in 2TX:NH3 complexes present similar values to those in 1TX:NH3 complexes, but with slight variations. 2TFA:NH3 complexes (both for Si and Ge) exhibit longer T···N distances (2.596 and 2.703 Å, respectively) than 1TFA:NH3 complexes (2.518 and 2.661 Å), and the same occurs for 2TXHIMHB:NH3 complexes, while the opposite is true for 2TFH:NH3 (Si = 2.330 and Ge = 2.553 Å) compared with 1TFH:NH3 (Si = 2.335 and Ge = 2.570 Å). As in 1TX:NH3 complexes, the ones with hydroxyl groups present, both 2TXH:NH3 and 2TXHIMHB:NH3 exhibit a shortening of the T···N distances. However, looking at the T-X bond distances, both for X=F and Cl, a lengthening is observed in 2TXH:NH3 and 2TXHIMHB:NH3 consistent with the results mentioned above and the increase in polarity of the T-X bond with the presence of hydroxyl groups and IMHBs.
Binding energies for 2TX:NH3 complexes are found to be within the same range as 1TX:NH3 complexes, with 2TXA:NH3 and 2TXHIMHB:NH3 complexes exhibiting larger Eb values than their corresponding allyl counterparts, while 2TXH:NH3 complexes present slightly smaller Eb values than 1TXH:NH3 complexes, both for T=Si, Ge and X=F, Cl. Eb values show that in all cases, 2SiCl:NH3 complexes have a stronger interaction with NH3 than 2SiF:NH3 complexes, while the opposite is true for the germanium derivatives. In terms of interaction energies, Eint, values in Table 5 reveal similar features to those found for the allyl complexes, i.e., large negative values of Eint, twice as much in some cases (for example: 2SiXH:NH3). This indicates a substantial re-organization energy, which was also confirmed when the Edef values were analyzed. Edef values are very large, in fact, silyl complexes with hydroxyl groups (2SXH:NH3) present the largest deformation energies, as occurred in 1SXH:NH3 complexes. This can be explained in terms of electronic repulsion between groups, as explained above [42].
If we compare the evolution of the binding energy across the 2TX:NH3 complexes, it is seen that the presence of hydroxyl (2TXH:NH3) enhances the interaction with the Lewis base in about 10.2% (2SiFH:NH3) and 10.7% (2GeFH:NH3). This enhancement is even larger within 2TCl:NH3 complexes (33.4% and 12.4% in 2SiClH:NH3 and 2GeClH:NH3, respectively). Besides, the enhancement of the binding energy reaches the maximum value when the IMHB takes place in the complex, reaching up to 38.2% of enhancement on the Eb with respect to the 2TXA:NH3 complex.
2.4. Electron Density Properties
The electron density properties of the intermolecular tetrel interactions were studied by means of atoms in molecules (AIM) theory. In most of the cases, the tetrel bond is characterized by the existence of a bond critical point (BCP) between the tetrel atom and the electron donor on the Lewis base moiety (N or O) (Table S1). As it is denoted in the literature, this should be taken carefully, since the opposite is not true, i.e., the absence of a BCP between two interacting atoms does not necessary imply the absence of interactions [43]. It has been found that in some cases dominant weak interactions exhibit neither BCP nor bond paths [44]. However, while the BCPs should be taken cautiously and there is a huge debate about the interpretation of the bond paths and their relationship to the chemical bond, the existence of BCPs and their associated properties has been proven to be a useful tool to identify non-covalent interactions across the different varieties: hydrogen [45,46], halogen [47], pnicogen [48], chalcogen [14] and tetrel bonds [49].
The electron density parameters obtained are summarized in Table S3. Values of the electron density at the bond critical (ρBCP) for 1SiF:NH3 and 1GeF:NH3 complexes range from 0.0270 to 0.045 a.u. and 0.0241 to 0.0366 a.u., respectively. Values for 1SiF:HCN and 1GeF:HCN complexes, which show similar T···N interactions, are smaller at 0.0101–0.0129 a.u. and 0.0132–0.0158 a.u., respectively. This is aligned with the interaction and binding energy values found and the intermolecular T···N distance evolution. Similar comparison trends are found for the Laplacian (∇2ρ BCP) values and total electron energy density (HBCP) values between those complexes.
1TCl:NH3 complexes show very similar ρBCP values to 1TF:NH3, concomitantly with similar Laplacian and HBCP values, with slight differences. Cremer et al. demonstrated that the sign of the total electron energy density (HBCP), defined as the sum of GBCP + VBCP, could be used to indicate the degree of covalency for chemical interactions [50,51,52]. HBCP values have been found to be negative for interactions that significantly share electrons. In fact, in other interactions, such as pnicogen bonds, the electron density increment within intermonomeric regions has been postulated as a stabilizing factor [53]. As observed for 1TX:NH3 complexes (Table S3), negative HBCP values are found and they may indicate a certain covalent character of the tetrel interaction. Furthermore, 1TXHIMHB:NH3 complexes present the most negative HBCP values, which reinforces the idea that hydroxyl groups performing IMHB enhance tetrel bonds.
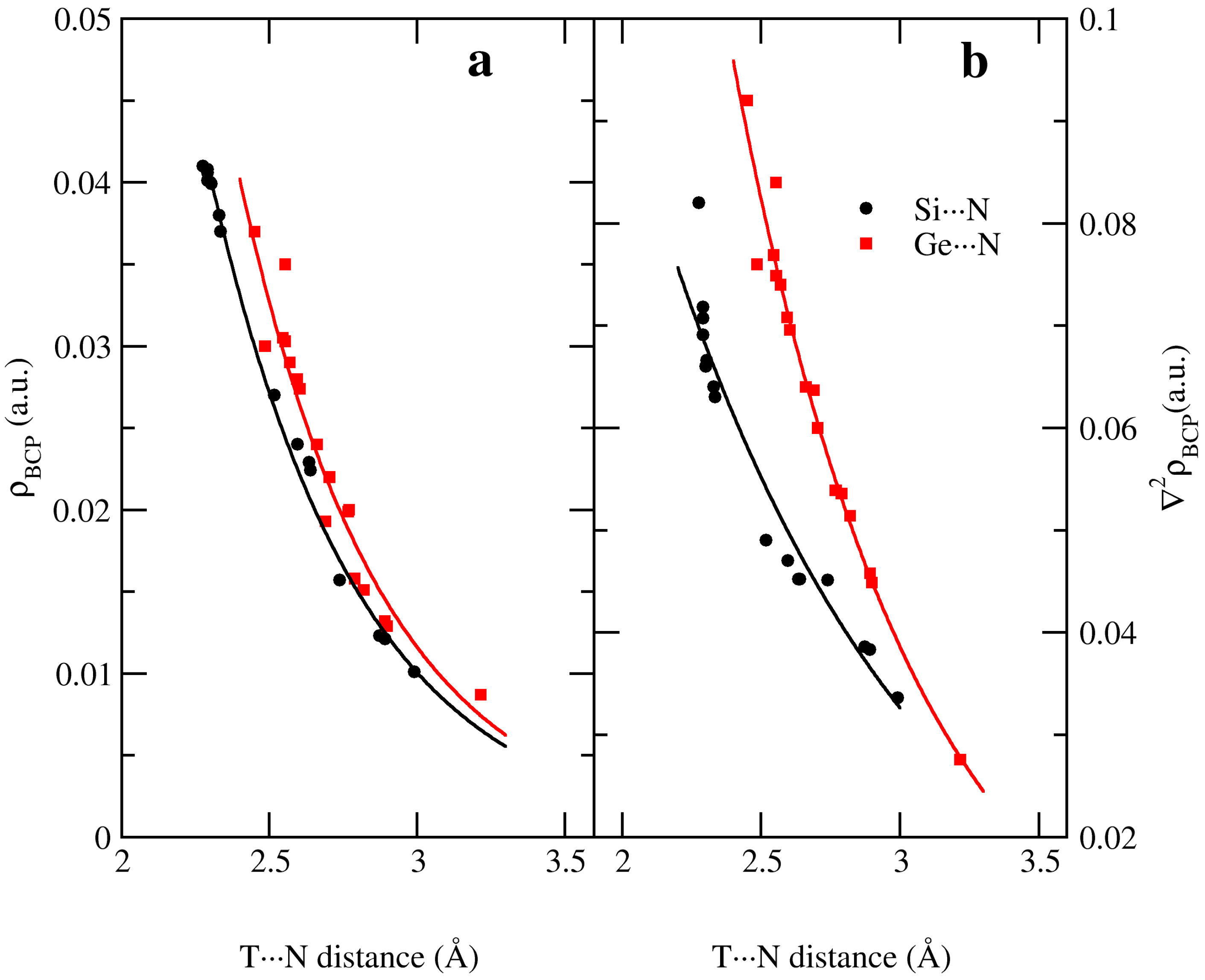
Exponential relationships have been found between the electron density at the BCP and the intermolecular T···N distance; Si···N distances show better correlations (ρ = 3.8959 e−1.9863 d(T···N), R2 = 0.991) than Ge···N (ρ = 5.7894 e−2.0706 d(T···N), R2 = 0.943) interactions as observed in Figure 3a. Similar relationships have also been found between the Laplacian and T···N distance (∇2ρ = 0.76454 e−1.0514 d(T···N), R2 = 0.947 and ∇2ρ = 3.6611 e−1.5177 d(T···N), R2 = 0.982 for T= Si and Ge complexes, respectively) as depicted in Figure 3b.
In order to provide further insight into the electron density changes upon complexation, electron density shift maps (EDS) at the ±0.002 a.u. isosurfaces are plotted in Figure 4. Blue areas correspond to those regions with a decrease in the electron density upon complexation, while yellow areas indicate those regions in which an increase in the electron density occurs. As observed, the 1SiFA:NH3 complex shows an increase (positive yellow area) in electron density between the N and Si atoms, corresponding to the donation of electron density from the Lewis base into the Si σ-hole. This pattern of depletion–increase in electron density has been observed for a wide range of inter/intramolecular interactions [14,54,55,56,57,58]. When 1SiFH:NH3 and 1SiFHIMHB:NH3 complexes are considered, the changes in electron density upon complexation become even more evident: blue (negative) and yellow (positive) areas are larger for 1SiFH:NH3 and 1SiFHIMHB:NH3 complexes than for 1SiFA:NH3 complexes, particularly in the areas surrounding the ammonia in which a drastic decrease in electron density is observed. This decrease is due to the larger electron density donation from the Lewis base in complexes with hydroxyl groups (1SiFH:NH3 and 1SiFHIMHB:NH3) than in the 1SiFA:NH3 one. When the phenyl backboned complexes are taken into account, the electron density shift maps show very similar increase/decrease patterns compared to the allyl one. Furthermore, there are no appreciable changes in the shape or size of the blue and yellow areas in comparison with the 1SiF:NH3 complexes, which is in consonance with the binding/interaction energies and the evolution of intermolecular T···N distances.
3. Materials and Methods
The structures of the complexes were optimized at the Møller-Plesset (MP2) [59]/aug-cc-pVTZ [60,61] computational level. Harmonic vibrational frequencies were computed at the same level used for the geometry optimizations in order to confirm that the stationary points are local minima. Calculations were performed using the Gaussian09 program [62]. Binding energies (Eb) were calculated as a difference in the energy of the complex minus the energy of each monomer in their optimized geometry. Interaction energies (Eint) were calculated as a difference in the energy of the complex minus the energy of each monomer, keeping the monomer geometry fixed in the complex optimized geometry. Finally, the deformation energy (Edef), also called the re-organization energy, was calculated as the difference between Eb and Eint. In order to provide more accurate energies the interaction energies were also estimated at the MP2/CBS (complete basis set) limit using the method of Helgaker et al. [63,64] from the calculated interaction energies with the aug-cc-pVTZ and aug-cc-pVQZ basis sets:
where EX and ECBS are the energies for the basis set with the largest angular momentum X and for the complete basis set, respectively.
The atoms in molecules (AIM) methodology [65,66] was used to analyze the electron density of the systems with the AIMAll program [67].
The intermolecular electron density shift (EDS) [58] was calculated using Equation (3):
where ρ(nTX:NH3), ρ(nTX) and ρ(NH3) stand for the electron density of the complex and both fragments in the geometry of the complex, respectively.
EDS = ρ(nTX:NH3) − ρ(nTX) − ρ(NH3)
4. Conclusions
The complexation of a series of silyl (nSiX:LB) and germanium (nGeX:LB) complexes, with two different halogen atoms (X=F and Cl) was studied. Three different Lewis bases (NH3, H2O and HCN) were considered to form the complexes and the effect of the existence or absence of intramolecular hydrogen bonds on the intermolecular tetrel bond were analyzed.
Calculated geometrical parameters and binding and interaction energies suggested that the presence of hydroxyl groups enhances the intermolecular tetrel bond. In general, nTXH:LB and nTXHIMHB:LB complexes presented shorter intermolecular T···Y distances than their parental nTXA:LB complexes. This fact, concomitantly with the Eb values found: Eb(nTXHIMHB:LB) < Eb(nTXA:LB), indicates a strengthening of the tetrel bond with the presence of hydroxyl groups, whether it is forming an IMHB or not. However, the data analyzed reveal that when the IMHB takes place, the enhancement is even larger than without it.
Regarding the Lewis base, complexes with NH3 showed the most negative binding energies, followed by H2O and HCN.
The substitution of the fluorine atom (X=F) by a chlorine atom, in general, weakens the tetrel bond in allyl complexes (1TCl:NH3), which show Eb and Eint values larger than its fluorine counterparts.
Finally, the effect of the carbon backbone was evaluated using two different backbones, allyl and phenyl. In spite of some differences, the intermolecular T···N distances and binding energies remain within the same range values.
Supplementary Materials
The supplementary materials are available online.
Author Contributions
Investigation, I.A., C.T., J.E., G.S.-S.; data curation, I.A., C.T., G.S.-S.; writing—original draft preparation, I.A., C.T., J.E., G.S.-S.; writing—review and editing, I.A., C.T., J.E., G.S.-S.; funding acquisition, I.A.
Funding
We thank the Ministerio de Ciencia e Innovación (Project No. CTQ2015-63997-C2-2-P) and the Comunidad Autónoma de Madrid (Project FOTOCARBON, ref S2013/MIT-2841) for continuous support.
Acknowledgments
Thanks are given to the CTI (CSIC), and to the Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lehn, J.M. Supramolecular Chemistry Concepts and Perspectives; Wiley-VCH: Weinheim, Germany, 1995. [Google Scholar]
- Müller-Dethlefs, K.; Hobza, P. Noncovalent Interactions: A Challenge for Experiment and Theory. Chem. Rev. 2000, 100, 143–168. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Sensitivity of noncovalent bonds to intermolecular separation: Hydrogen, halogen, chalcogen, and pnicogen bonds. CrystEngComm 2013, 15, 3119–3124. [Google Scholar] [CrossRef]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G.; Pilati, T.; Biella, S. Halogen Bonding: Fundamentals and Applications; Springer: Berlin, Germany, 2008. [Google Scholar]
- Rozas, I.; Alkorta, I.; Elguero, J. Field effects on dihydrogen bonded systems. Chem. Phys. Lett. 1997, 275, 423–428. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Inverse Hydrogen-Bonded Complexes. J. Phys. Chem. A 1997, 101, 4236–4244. [Google Scholar] [CrossRef]
- Scheiner, S. Effects of substituents upon the P···N noncovalent interaction: The limits of its strength. J. Phys. Chem. A 2011, 115, 11202–11209. [Google Scholar] [CrossRef] [PubMed]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen bonds: A new molecular linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef]
- Sundberg, M.R.; Uggla, R.; Viñas, C.; Teixidor, F.; Paavola, S.; Kivekäs, R. Nature of intramolecular interactions in hypercoordinate C-substituted 1,2-dicarba-closo-dodecaboranes with short P⋯P distances. Inorg. Chem. Commun. 2007, 10, 713–716. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J.; Sánchez-Sanz, G. Lone-pair hole on P: P···N pnicogen bonds assisted by halogen bonds. J. Phys. Chem. A 2017, 121, 1362–1370. [Google Scholar] [CrossRef]
- Sanchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Modulating intramolecular P···N pnictogen interactions. Phys. Chem. Chem. Phys. 2016, 18, 9148–9160. [Google Scholar] [CrossRef]
- Trujillo, C.; Sanchez-Sanz, G.; Alkorta, I.; Elguero, J. Halogen, chalcogen and pnictogen interactions in (XNO2)2 homodimers (X = F, Cl, Br, I). New J. Chem. 2015, 39, 6791–6802. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Intermolecular weak interactions in HTeXH dimers (X=O, S, Se, Te): Hydrogen bonds, chalcogen–chalcogen contacts and chiral discrimination. Chem. Phys. Chem. 2012, 13, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Alkorta, I.; Elguero, J. Theoretical study of the HXYH dimers (X, Y = O, S, Se). Hydrogen bonding and chalcogen–chalcogen interactions. Mol. Phys. 2011, 109, 2543–2552. [Google Scholar] [CrossRef] [Green Version]
- Azofra, L.; Alkorta, I.; Scheiner, S. Noncovalent interactions in dimers and trimers of SO3 and CO. Theor. Chem. Acc. 2014, 133, 1–8. [Google Scholar] [CrossRef]
- Sanz, P.; Yáñez, M.; Mó, O. Resonance-assisted intramolecular chalcogen–chalcogen interactions? Chem. Eur. J. 2003, 9, 4548–4555. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C. Improvement of anion transport systems by modulation of chalcogen interactions: The influence of solvent. J. Phys. Chem. A 2018, 122, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-bonding interaction: Rediscovered supramolecular force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.-Y. Significant evidence of C⋯O and C⋯C long-range contacts in several heterodimeric complexes of CO with CH3–X, should one refer to them as carbon and dicarbon bonds! Phys. Chem. Chem. Phys. 2014, 16, 17238–17252. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular complexes between silicon derivatives and electron-rich groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. Chem. Phys. Chem. 2015, 16, 2496–2517. [Google Scholar]
- Murray, J.S.; Lane, P.; Politzer, P. A predicted new type of directional noncovalent interaction. Int. J. Quantum Chem. 2007, 107, 2286–2292. [Google Scholar] [CrossRef]
- Murray, J.; Concha, M.; Lane, P.; Hobza, P.; Politzer, P. Blue shifts vs. red shifts in σ-hole bonding. J. Mol. Model. 2008, 14, 699–704. [Google Scholar] [CrossRef]
- Mohajeri, A.; Pakiari, A.H.; Bagheri, N. Theoretical studies on the nature of bonding in σ-hole complexes. Chem. Phys. Lett. 2009, 467, 393–397. [Google Scholar] [CrossRef]
- Buckingham, A.D.; Fowler, P.W. A model for the geometries of Van der Waals complexes. Can. J. Chem. 1985, 63, 2018–2025. [Google Scholar] [CrossRef] [Green Version]
- Legon, A.C.; Millen, D.J. Angular geometries and other properties of hydrogen-bonded dimers: A simple electrostatic interpretation of the success of the electron-pair model. Chem. Soc. Rev. 1987, 16, 467–498. [Google Scholar] [CrossRef]
- Stone, A.J.; Price, S.L. Some new ideas in the theory of intermolecular forces: Anisotropic atom-atom potentials. J. Phys. Chem. 1988, 92, 3325–3335. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface electrostatic potentials of halogenated methanes as indicators of directional intermolecular interactions. Int. J. Quantum Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Burling, F.T.; Goldstein, B.M. Computational studies of nonbonded sulfur-oxygen and selenium-oxygen interactions in the thiazole and selenazole nucleosides. J. Am. Chem. Soc. 1992, 114, 2313–2320. [Google Scholar] [CrossRef]
- Price, S.L. Applications of realistic electrostatic modelling to molecules in complexes, solids and proteins. J. Chem. Soc. Faraday Trans. 1996, 92, 2997–3008. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Awwadi, F.F.; Willett, R.D.; Peterson, K.A.; Twamley, B. The nature of Halogen⋅⋅⋅Halogen synthons: Crystallographic and theoretical studies. Chem. Eur. J. 2006, 12, 8952–8960. [Google Scholar] [CrossRef]
- Politzer, P.; Riley, K.E.; Bulat, F.A.; Murray, J.S. Perspectives on halogen bonding and other σ-hole interactions: Lex parsimoniae (Occam’s Razor). Comput. Theor. Chem. 2012, 998, 2–8. [Google Scholar] [CrossRef]
- Hennemann, M.; Murray, J.; Politzer, P.; Riley, K.; Clark, T. Polarization-induced σ-holes and hydrogen bonding. J. Mol. Model. 2012, 18, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Clark, T. σ-Holes. WIREs Comput. Mol. Sci. 2013, 3, 13–20. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Halogen bonding: An interim discussion. Chem. Phys. Chem. 2013, 14, 278–294. [Google Scholar] [CrossRef]
- Hunter, C.A.; Anderson, H.L. What is cooperativity? Angew. Chem. Int. Ed. 2009, 48, 7488–7499. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Riel, A.M.S.; Berryman, O.B. Solvatochromism and fluorescence response of a halogen bonding anion receptor. New J. Chem. 2018, 42, 10489–10492. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.-C.C.; Scholfield, M.R.; Rowe, R.K.; Ford, M.C.; Alexander, A.T.; Mehl, R.A.; Ho, P.S. Increasing enzyme stability and activity through hydrogen bond-enhanced halogen bonds. Biochemistry 2018, 57, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Ośmiałowski, B.; Kolehmainen, E.; Gawinecki, R.; Kauppinen, R.; Koivukorpi, J.; Valkonen, A. NMR and quantum chemical studies on association of 2,6-bis(acylamino)pyridines with selected imides and 2,2′-dipyridylamine. Struct. Chem. 2010, 21, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Lane, J.R.; Contreras-García, J.; Piquemal, J.-P.; Miller, B.J.; Kjaergaard, H.G. Are bond critical points really critical for hydrogen bonding? J. Chem. Theor. Comput. 2013, 9, 3263–3266. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M. Bond paths between distant atoms do not necessarily indicate dominant interactions. J. Comput. Chem. 2018, 39, 2183–2195. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H···F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Universal features of the electron density distribution in hydrogen-bonding regions: A comprehensive study involving H···X (X=H, C, N, O, F, S, Cl, π) interactions. Chem. Eur. J. 2010, 16, 2442–2452. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Charge-transfer complexes between dihalogen compounds and electron donors. J. Phys. Chem. A 1998, 102, 9278–9285. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. Structures, energies, bonding, and NMR properties of pnicogen complexes H2XP:NXH2 (X = H, CH3, NH2, OH, F, Cl). J. Phys. Chem. A 2011, 115, 13724–13731. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Blanco, F.; Elguero, J.; Dobado, J.A.; Ferrer, S.M.; Vidal, I. Carbon···Carbon weak interactions. J. Phys. Chem. A 2009, 113, 8387–8393. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croatica Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Jenkins, S.; Morrison, I. The chemical character of the intermolecular bonds of seven phases of ice as revealed by ab initio calculation of electron densities. Chem. Phys. Lett. 2000, 317, 97–102. [Google Scholar] [CrossRef]
- Aronld, W.D.; Oldfield, E. The chemical nature of hydrogen bonding in proteins via NMR: J-couplings, chemical shifts, and AIM theory. J. Am. Chem. Soc. 2000, 122, 12835–12841. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Description of pnicogen bonding with the help of vibrational spectroscopy—The missing link between theory and experiment. Chem. Phys. Lett. 2014, 614, 136–142. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Theoretical study of cyanophosphines: Pnicogen vs. dipole-dipole interactions. Comput. Theor. Chem. 2015, 1053, 305–314. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Intramolecular pnicogen interactions in phosphorus and arsenic analogues of proton sponges. Phys. Chem. Chem. Phys. 2014, 16, 15900–15909. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Alkorta, I.; Trujillo, C.; Elguero, J. Intramolecular pnicogen interactions in PHF-(CH2)n-PHF (n = 2–6) systems. Chem. Phys. Chem. 2013, 14, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Weak interactions between hypohalous acids and dimethylchalcogens. Phys. Chem. Chem. Phys. 2012, 14, 9880–9889. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Electron density shift description of non-bonding intramolecular interactions. Comput. Theor. Chem. 2012, 991, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian-Basis Sets for Use in Correlated Molecular Calculations 1. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Halkier, A.; Helgaker, T.; Jørgensen, P.; Klopper, W.; Olsen, J. Basis-set convergence of the energy in molecular Hartree–Fock calculations. Chem. Phys. Lett. 1999, 302, 437–446. [Google Scholar] [CrossRef]
- Halkier, A.; Klopper, W.; Helgaker, T.; Jørgensen, P.; Taylor, P.R. Basis set convergence of the interaction energy of hydrogen-bonded complexes. J. Chem. Phys. 1999, 111, 9157–9167. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Claredon Press: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms in Molecules. An Introduction; Prentice-Hall: Manchester, UK, 2000. [Google Scholar]
- Keith, T.A. AIMAll (Version 15.05.18); TK Gristmill Software: Overland Park, KS, USA, 2015. [Google Scholar]
Scheme 1.
Schematic description of 2,6-bis(4-ethynylpyridinyl)-4-fluoroaniline interacting with anions.
Scheme 1.
Schematic description of 2,6-bis(4-ethynylpyridinyl)-4-fluoroaniline interacting with anions.

Scheme 2.
The two conformers of 2-halophenol.

Scheme 3.
Schematic representation of the nTX:LB complexes subject to study.

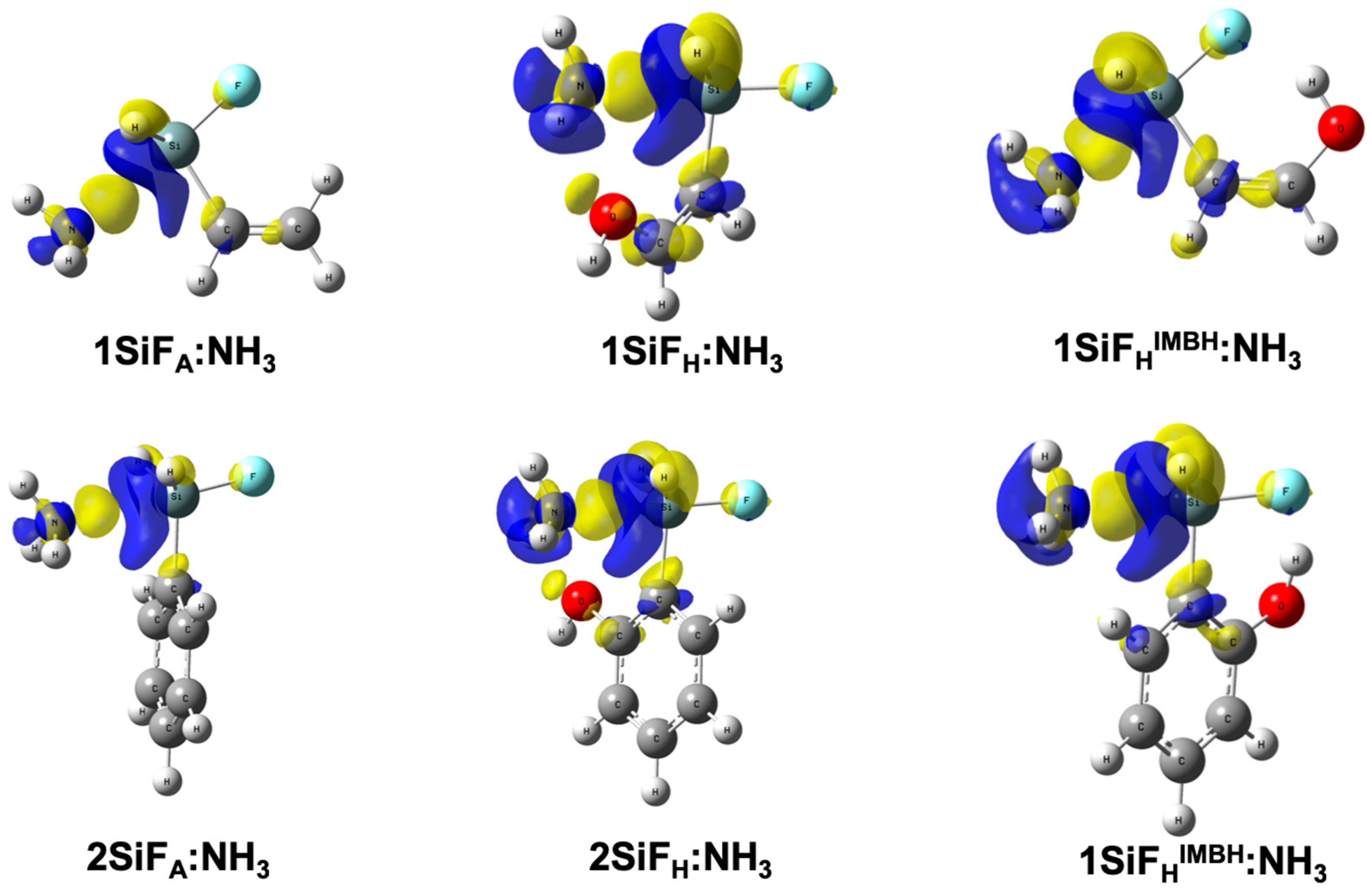
Figure 1.
Molecular graphs for 1SiF:LB complexes at the MP2/aug-cc-pVTZ computational level. Green dots correspond to bond critical points.
Figure 1.
Molecular graphs for 1SiF:LB complexes at the MP2/aug-cc-pVTZ computational level. Green dots correspond to bond critical points.

Figure 2.
Histogram for the different energies: Eb (solid), Eint (dotted) and Edef (stripped) for each Lewis base considered: NH3 (blue), H2O (purple) and HCN (red).
Figure 2.
Histogram for the different energies: Eb (solid), Eint (dotted) and Edef (stripped) for each Lewis base considered: NH3 (blue), H2O (purple) and HCN (red).

Figure 3.
Exponential relationships between (a) electron density at the bond critical point and (b) Laplacian at the bond critical point with the intermolecular T···N distance at the MP2/aug-cc-pVTZ computational level.
Figure 3.
Exponential relationships between (a) electron density at the bond critical point and (b) Laplacian at the bond critical point with the intermolecular T···N distance at the MP2/aug-cc-pVTZ computational level.

Figure 4.
Electron density shift maps for the nSiF:NH3 (n = 1, 2) complexes on the ±0.002 a.u. electron density isosurfaces at the MP2/aug-cc-pVTZ computational level. Blue and yellow areas correspond to regions with a decrease and increase in electron density, respectively.
Figure 4.
Electron density shift maps for the nSiF:NH3 (n = 1, 2) complexes on the ±0.002 a.u. electron density isosurfaces at the MP2/aug-cc-pVTZ computational level. Blue and yellow areas correspond to regions with a decrease and increase in electron density, respectively.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Binding energies (Eb), in kJ·mol−1, of 1SiF:NH3 complexes at the MP2 computational level with different basis sets including extrapolation to the complete basis set limit (CBS) with two different basis set pairs.
Table 1.
Binding energies (Eb), in kJ·mol−1, of 1SiF:NH3 complexes at the MP2 computational level with different basis sets including extrapolation to the complete basis set limit (CBS) with two different basis set pairs.
| avdz a | avtz a | avqz a | CBSDT b | CBSTQ b | |
|---|---|---|---|---|---|
| 1SiFA:NH3 | −29.3 | −25.4 | −23.8 | −24.5 | −21.8 |
| 1SiFH:NH3 | −32.3 | −28.2 | −25.9 | −27.4 | −22.9 |
| 1SiFHIMHB:NH3 | −40.5 | −36.3 | −34.4 | −35.4 | −31.7 |
a avdz, avtz and avqz stand for aug-cc-pVDZ, aug-cc-pVTZ and aug-cc-pVQZ respectively. b CBS stands for extrapolation to the complete basis set limit, and DT and TQ refer to the pair of basis set used to extrapolate: avdz-avtz and avtz-avqz, respectively.
Table 2.
Intermolecular distances, T···N(O) (T=Si, Ge) in Å for 1TF:LB complexes at the MP2/aug-cc-pVTZ computational level.
Table 2.
Intermolecular distances, T···N(O) (T=Si, Ge) in Å for 1TF:LB complexes at the MP2/aug-cc-pVTZ computational level.
| Comp. | NH3 | H2O | HCN |
|---|---|---|---|
| 1SiFA:LB | 2.518 | 2.885 | 2.991 |
| 1SiFH:LB | 2.335 | 2.883 | 4.033 |
| 1SiFHIMHB:LB | 2.276 | 2.765 | 2.873 |
| 1GeFA:LB | 2.661 | 2.829 | 2.898 |
| 1GeFH:LB | 2.570 | 2.782 | 2.891 |
| 1GeFHIMHB:LB | 2.450 | 2.720 | 2.789 |
Table 3.
Binding energies (Eb), interaction energies (Eint) and deformation energies (Edef) for the 1TF:LB complexes at the MP2/CBS computational level, in kJ·mol−1.
Table 3.
Binding energies (Eb), interaction energies (Eint) and deformation energies (Edef) for the 1TF:LB complexes at the MP2/CBS computational level, in kJ·mol−1.
| Complex | Eb | Eint | Edef | ||||||
|---|---|---|---|---|---|---|---|---|---|
| NH3 | H2O | HCN | NH3 | H2O | HCN | NH3 | H2O | HCN | |
| 1SiFA:NH3 | −21.8 | −15.9 | −14.6 | −35.2 | −17.7 | −16.1 | 13.6 | 1.8 | 1.4 |
| 1SiFH:NH3 | −22.9 | −19.2 | −14.5 | −51.3 | −24.1 | −16.7 | 28.4 | 4.9 | 2.2 |
| 1SiFHIMHB:NH3 | −31.7 | −18.2 | −16.9 | −60.6 | −21.2 | −19.2 | 28.8 | 3.0 | 2.4 |
| 1GeFA:NH3 | −31.2 | −21.6 | −21.6 | −38.0 | −23.1 | −22.9 | 6.8 | 1.5 | 1.3 |
| 1GeFH:NH3 | −31.9 | −26.9 | −14.7 | −43.9 | −31.1 | −16.6 | 12.0 | 4.2 | 1.9 |
| 1GeFHIMHB:NH3 | −41.0 | −25.1 | −25.6 | −56.1 | −27.6 | −27.9 | 15.1 | 2.4 | 2.3 |
Table 4.
Binding energies (Eb), interaction energies (Eint) and deformation energies (Edef), at the MP2/CBS computational level, in kJ·mol−1, intermolecular T···N distances and T-X bond distances (T=Si, Ge and X=F, Cl), in Å for 1TX:NH3 complexes at the MP2/aug-cc-pVTZ computational level.
Table 4.
Binding energies (Eb), interaction energies (Eint) and deformation energies (Edef), at the MP2/CBS computational level, in kJ·mol−1, intermolecular T···N distances and T-X bond distances (T=Si, Ge and X=F, Cl), in Å for 1TX:NH3 complexes at the MP2/aug-cc-pVTZ computational level.
| Eb | Eint | Edef | T···N | T-X | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl |
| 1SiXA:NH3 | −21.8 | −20.1 | −35.4 | −30.5 | 13.6 | 10.4 | 2.518 | 2.640 | 1.643 | 2.110 |
| 1SiXH:NH3 | −22.9 | −24.5 | −51.3 | −58.5 | 28.4 | 34.1 | 2.335 | 2.291 | 1.654 | 2.156 |
| 1SiXHIMHB:NH3 | −31.7 | −27.5 | −60.6 | −58.9 | 28.8 | 31.4 | 2.276 | 2.291 | 1.685 | 2.185 |
| 1GeXA:NH3 | −31.2 | −28.2 | −38.0 | −32.7 | 6.8 | 4.6 | 2.661 | 2.767 | 1.768 | 2.193 |
| 1GeXH:NH3 | −31.9 | −31.6 | −43.9 | −42.3 | 12.0 | 10.7 | 2.570 | 2.603 | 1.776 | 2.217 |
| 1GeXHIMHB:NH3 | −41.0 | −34.8 | −56.1 | −46.6 | 15.1 | 11.8 | 2.450 | 2.545 | 1.815 | 2.251 |
Table 5.
Binding energies (Eb), interaction energies (Eint) and deformation energies (Edef), at the MP2/CBS computational level, in kJ·mol−1, intermolecular T···N distances and T-X bond distances (T=Si, Ge and X=F, Cl), in Å for 2TX:NH3 complexes at the MP2/aug-cc-pVTZ computational level.
Table 5.
Binding energies (Eb), interaction energies (Eint) and deformation energies (Edef), at the MP2/CBS computational level, in kJ·mol−1, intermolecular T···N distances and T-X bond distances (T=Si, Ge and X=F, Cl), in Å for 2TX:NH3 complexes at the MP2/aug-cc-pVTZ computational level.
| Eb | Eint | Edef | T···N | T-X | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl | X=F | X=Cl |
| 2SiXA:NH3 | −21.5 | −21.5 | −30.99 | −30.78 | 9.5 | 9.2 | 2.596 | 2.635 | 1.638 | 2.111 |
| 2SiXH:NH3 | −23.7 | −28.7 | −59.13 | −61.07 | 33.9 | 33.2 | 2.330 | 2.305 | 1.652 | 2.150 |
| 2SiXHIMHB:NH3 | −25.2 | −27.9 | −55.27 | −59.79 | 31.6 | 31.1 | 2.301 | 2.291 | 1.677 | 2.176 |
| 2GeXA:NH3 | −29.0 | −27.8 | −34.45 | −31.73 | 5.4 | 3.9 | 2.703 | 2.771 | 1.766 | 2.194 |
| 2GeXH:NH3 | −32.1 | −31.3 | −55.44 | −48.18 | 15.3 | 13.3 | 2.553 | 2.594 | 1.776 | 2.214 |
| 2GeXHIMHB:NH3 | −40.1 | −34.9 | −47.60 | −44.99 | 15.5 | 13.7 | 2.485 | 2.554 | 1.809 | 2.244 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Trujillo, C.; Alkorta, I.; Elguero, J.; Sánchez-Sanz, G. Cooperative Effects in Weak Interactions: Enhancement of Tetrel Bonds by Intramolecular Hydrogen Bonds. Molecules 2019, 24, 308. https://doi.org/10.3390/molecules24020308
AMA Style
Trujillo C, Alkorta I, Elguero J, Sánchez-Sanz G. Cooperative Effects in Weak Interactions: Enhancement of Tetrel Bonds by Intramolecular Hydrogen Bonds. Molecules. 2019; 24(2):308. https://doi.org/10.3390/molecules24020308
Chicago/Turabian StyleTrujillo, Cristina, Ibon Alkorta, José Elguero, and Goar Sánchez-Sanz. 2019. "Cooperative Effects in Weak Interactions: Enhancement of Tetrel Bonds by Intramolecular Hydrogen Bonds" Molecules 24, no. 2: 308. https://doi.org/10.3390/molecules24020308