
Monitoring of Nonadiabatic Effects in Individual Chromophores by Femtosecond Double-Pump Single-Molecule Spectroscopy: A Model Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theoretical Framework
2.1. Hamiltonian, Master Equation and the SM Signal
2.2. Computational Details
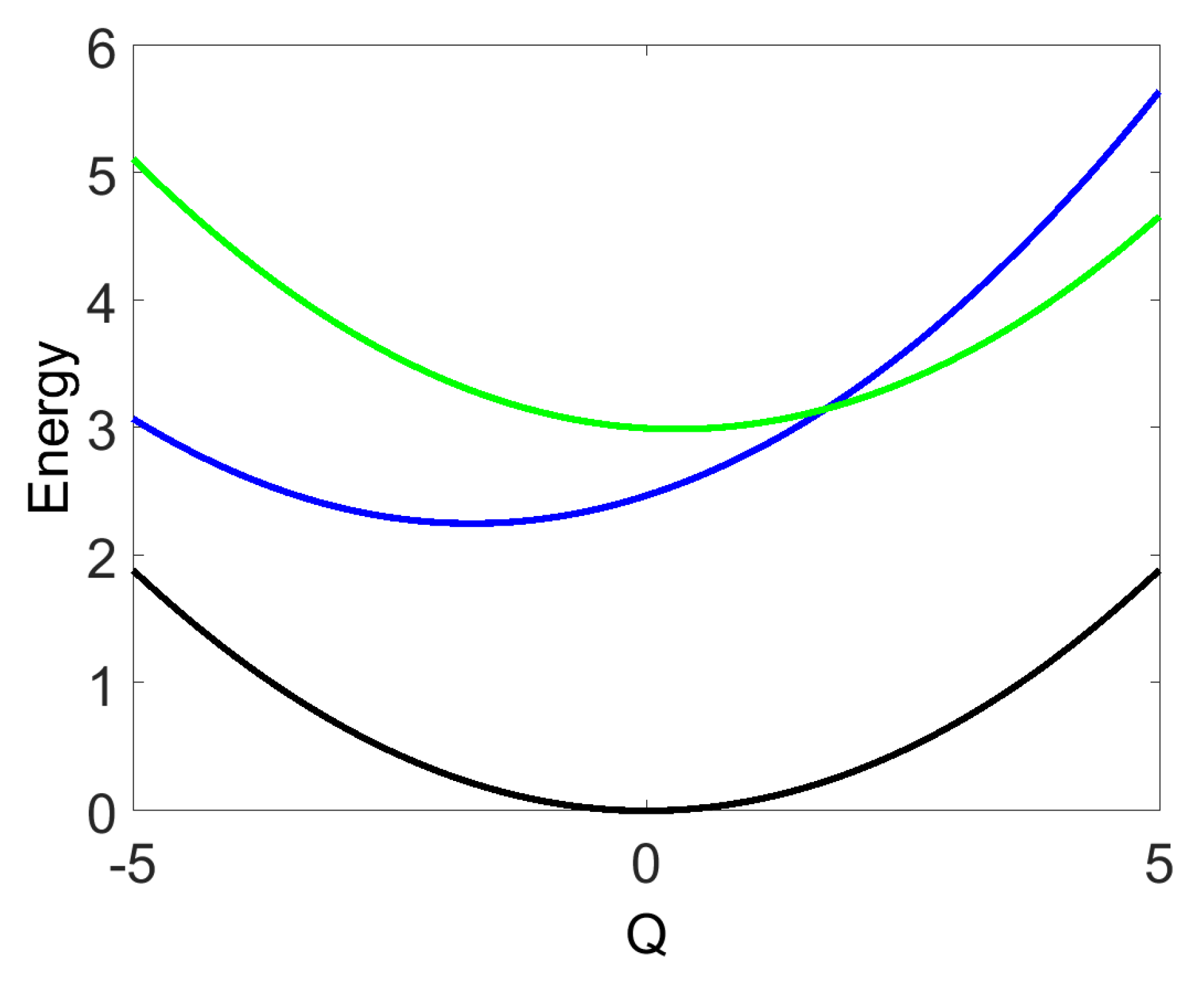
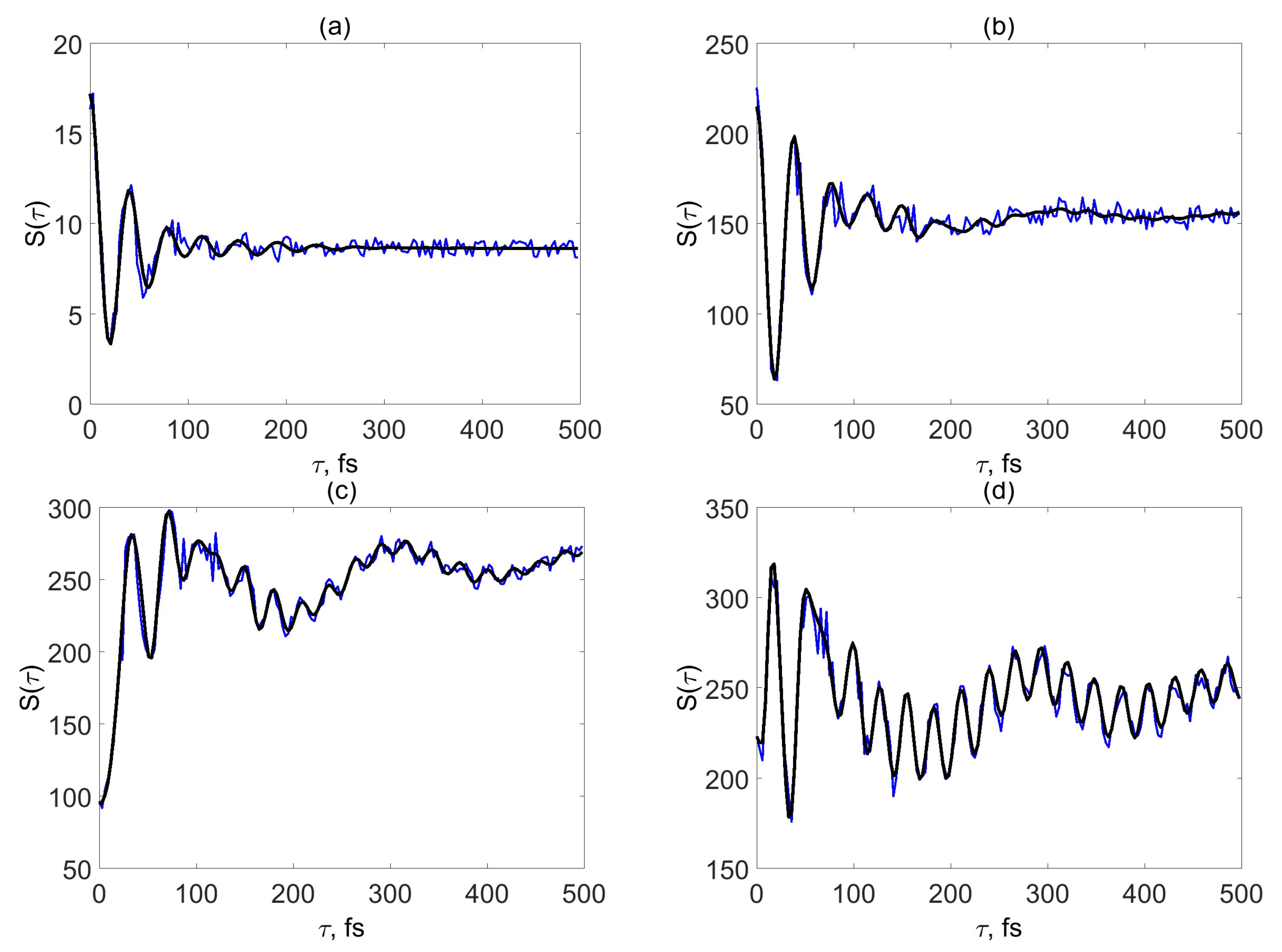
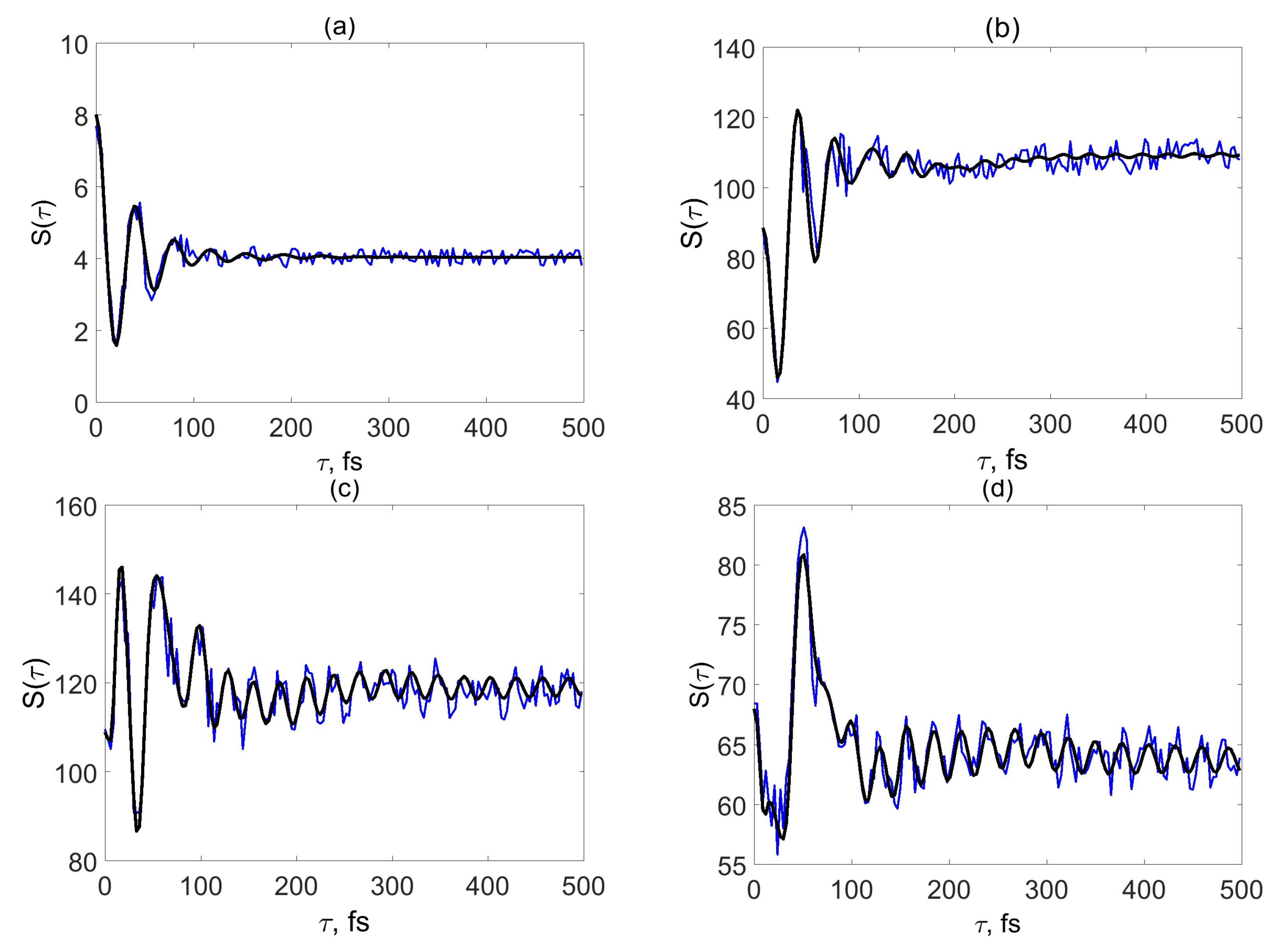
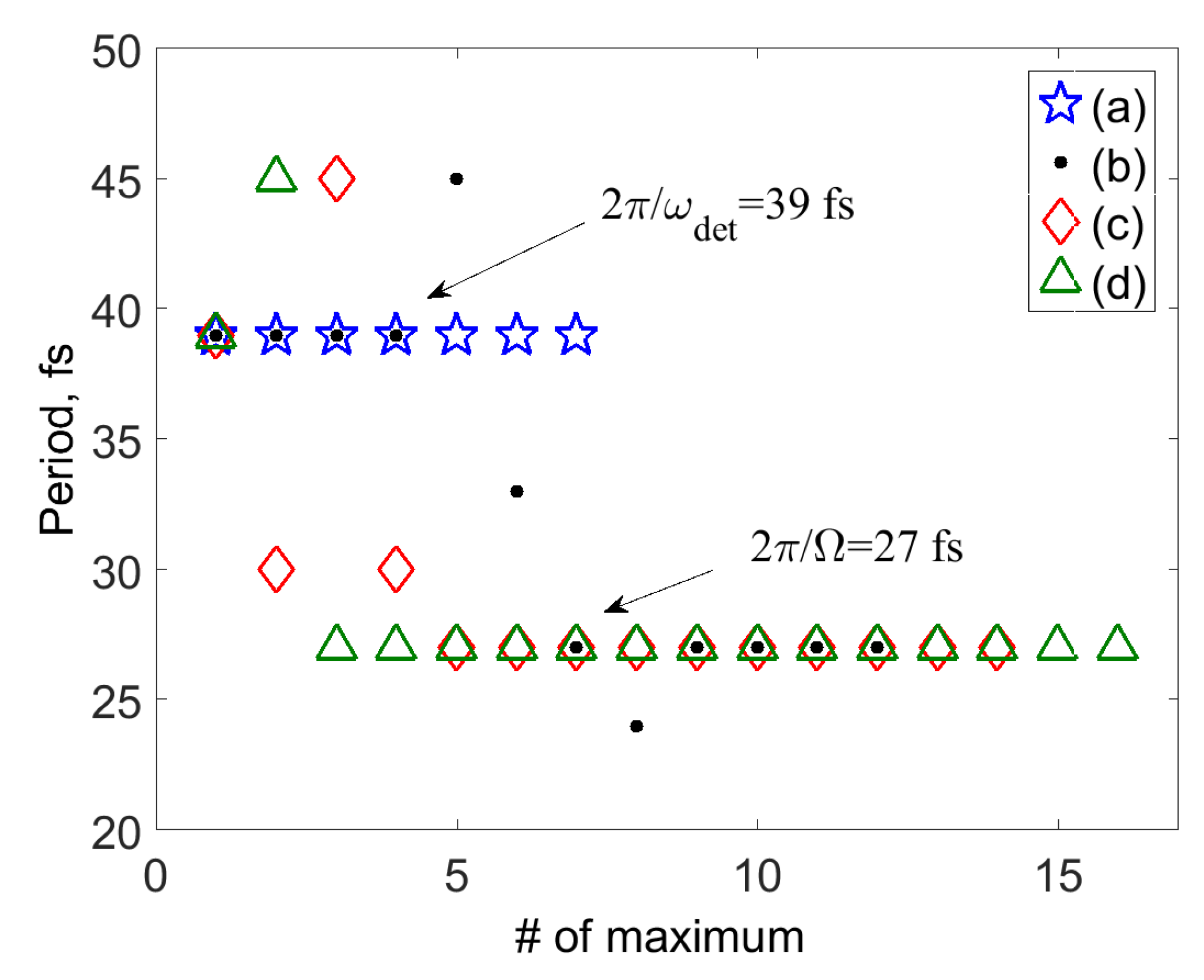
3. SM Signals
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zewail, A.H. Femtochemistry: Atomic-Scale Dynamics of the Chemical Bond. J. Phys. Chem. A 2000, 104, 5660–5694. [Google Scholar] [CrossRef]
- Brinks, D.; Hildner, R.; van Dijk, E.M.H.P.; Stefani, F.D.; Nieder, J.B.; Hernando, J.; van Hulst, N.F. Ultrafast dynamics of single molecules. Chem. Soc. Rev. 2014, 43, 2476–2491. [Google Scholar] [CrossRef] [PubMed]
- Piatkowski, L.; Accanto, N.; van Hulst, N.F. Ultrafast meets ultrasmall: controlling nanoantennas and molecules. ACS Photonics 2016, 3, 1401–1414. [Google Scholar] [CrossRef]
- Orrit, M.; Bernard, J. Single pentacene molecules detected by fluorescence excitation in a p-terphenyl crystal. Phys. Rev. Lett. 1990, 65, 2716. [Google Scholar] [CrossRef] [PubMed]
- Scherer, N.F.; Carlson, R.J.; Matro, A.; Du, M.; Ruggiero, A.J.; Romero-Rochin, V.; Cina, J.A.; Fleming, G.R.; Rice, S.A. Fluorescence-detected wave packet interferometry: Time resolved molecular spectroscopy with sequences of femtosecond phase-locked pulses. J. Chem. Phys. 1991, 95, 1487–1511. [Google Scholar] [CrossRef]
- Brinks, D.; Stefani, F.D.; Kulzer, F.; Hildner, R.; Haminiau, T.H.; Avlasevich, Y.; van Hulst, N.F. Visualizing and controlling vibrational wave packets of single molecules. Nature 2010, 465, 905–906. [Google Scholar] [CrossRef] [PubMed]
- Hildner, R.; Brinks, D.; Stefani, F.D.; van Hulst, N.F. Electronic coherences and vibrational wave-packets in single molecules studied with femtosecond phase-controlled spectroscopy. Phys. Chem. Chem. Phys. 2011, 13, 1888–1894. [Google Scholar] [CrossRef] [PubMed]
- Hildner, R.; Brinks, D.; van Hulst, N.F. Femtosecond coherence and quantum control of single molecules at room temperature. Nat. Phys. 2011, 7, 172–177. [Google Scholar] [CrossRef]
- Piatkowski, L.; Gellings, E.; van Hulst, N.F. Broadband single-molecule excitation spectroscopy. Nat. Commun. 2015, 7, 104111–104112. [Google Scholar] [CrossRef]
- Weigel, A.; Sebesta, A.; Kukura, P. Shaped and Feedback-Controlled Excitation of Single Molecules in the Weak-Field Limit. J. Phys. Chem. Lett. 2015, 6, 4032–4037. [Google Scholar] [CrossRef]
- Palacino-González, E.; Gelin, M.F.; Domcke, W. Theoretical aspects of femtosecond double-pump single-molecule spectroscopy. I. Weak-field regime. Phys. Chem. Chem. Phys. 2017, 19, 32296–32306. [Google Scholar] [CrossRef]
- Palacino-González, E.; Gelin, M.F.; Domcke, W. Theoretical aspects of femtosecond double-pump single-molecule spectroscopy. II. Strong-field regime. Phys. Chem. Chem. Phys. 2017, 19, 32307–32319. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.; Troiani, F.; Corni, S. Probing quantum coherence in ultrafast molecular processes: An ab initio approach to open quantum systems. J. Chem. Phys. 2018, 148, 204112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildner, R.; Brinks, D.; Nieder, J.B.; Cogdell, R.J.; van Hulst, N.F. Quantum coherent energy transfer over varying pathways in single light-harvesting complexes. Science 2013, 340, 1448–1451. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gelin, M.F.; Domcke, W.; Zhao, Y. Theory of femtosecond coherent double-pump single-molecule spectroscopy: Application to light harvesting complexes. J. Chem. Phys. 2015, 142, 164106. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gelin, M.F.; Domcke, W.; Zhao, Y. Simulation of Femtosecond Phase-Locked Double-Pump Signals of Individual Light-Harvesting Complexes LH2. J. Phys. Chem. Lett. 2018, 9, 4488–4494. [Google Scholar] [CrossRef] [PubMed]
- Caycedo-Soler, F.; Lim, J.; Oviedo-Casado, S.; van Hulst, N.F.; Huelga, S.F.; Plenio, M.B. Theory of Excitonic Delocalization for Robust Vibronic Dynamics in LH2. J. Phys. Chem. Lett. 2018, 9, 3446–3453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bixon, M.; Jortner, J. Vibrational coherence in nonadiabatic dynamics. J. Chem. Phys. 1997, 107, 1470–1482. [Google Scholar] [CrossRef]
- Kovalenko, S.A.; Dobryakov, A.L.; Farztdinov, V. Detecting Electronic Coherence in Excited-State Electron Transfer in Fluorinated Benzenes. Phys. Rev. Lett. 2006, 96, 068301. [Google Scholar] [CrossRef]
- Hüter, O.; Sala, M.; Neumann, H.; Zhang, S.; Studzinski, H.; Egorova, D.; Temps, F. Long-lived coherence in pentafluorobenzene as a probe of ππ*–πσ* vibronic coupling. J. Chem. Phys. 2016, 145, 014302. [Google Scholar] [CrossRef]
- Rubtsov, I.V.; Yoshihara, K. Vibrational Coherence in Electron Donor–Acceptor Complexes: Assignment of the Oscillatory Mode. In Femtochemistry, 1st ed.; De Schryver, F.C., De Feyter, S., Gerd Schweitzer, G., Eds.; Wiley-VCH: Weinheim, Germany, 2001; pp. 367–380. [Google Scholar]
- Domcke, W.; Stock, G. Theory of Ultrafast Nonadiabatic Excited-State Processes and their Spectroscopic Detection in Real Time. Adv. Chem. Phys. 1997, 100, 1–169. [Google Scholar]
- May, V.; Kühn, O. Charge and Energy Transfer Dynamics in Molecular Systems; Wiley-VCH: Berlin, Germany, 2004. [Google Scholar]
- Di Maiolo, F.; Painelli, A. Intermolecular Energy Transfer in Real Time. J. Chem. Theory Comput. 2018, 14, 5339–5349. [Google Scholar] [CrossRef] [PubMed]
- Redfield, A.G. The Theory of Relaxation Processes. Adv. Magn. Reson. 1965, 1, 1–32. [Google Scholar]
- Egorova, D.; Thoss, M.; Domcke, W.; Wang, H. Modeling of ultrafast electron-transfer processes: Validity of multilevel Redfield theory. J. Chem. Phys. 2003, 119, 2761–2773. [Google Scholar] [CrossRef]
- Gelin, M.F.; Tanimura, Y.; Domcke, W. Simulation of femtosecond “double-slit” experiments for a chromophore in a dissipative environment. J. Chem. Phys. 2013, 139, 214302. [Google Scholar] [CrossRef] [PubMed]
- Yampolsky, S.; Fishman, D.A.; Dey, S.; Hulkko, E.; Banik, M.; Potma, E.O.; Apkarian, V.A. Seeing a single molecule vibrate through time-resolved coherent anti-Stokes Raman scattering. Nat. Photonics 2014, 8, 650–656. [Google Scholar] [CrossRef]
- Gelin, M.F.; Egorova, D.; Domcke, W. Time-resolved spontaneous emission beyond the doorway-window approximation. Chem. Phys. 2004, 301, 129–139. [Google Scholar] [CrossRef]
- Li, X.; Gurzadyan, G.G.; Gelin, M.F.; Domcke, W.; Gong, C.; Liu, J.; Sun, L. Enhanced S2 Fluorescence from a Free-Base Tetraphenylporphyrin Surface-Mounted Metal Organic Framework. J. Phys. Chem. C 2018, 122, 23321–23328. [Google Scholar] [CrossRef]
- Egorova, D.; Gelin, M.F.; Thoss, M.; Domcke, W.; Wang, H. Effects of intense femtosecond pumping on ultrafast electronic-vibrational dynamics in molecular systems with relaxation. J. Chem. Phys. 2008, 129, 214303. [Google Scholar] [CrossRef]
- Haase, M.; Hübner, C.G.; Nolde, F.; Müllen, K.; Basché, T. Photoblinking and photobleaching of rylene diimide dyes. Phys. Chem. Chem. Phys. 2011, 13, 1776–1785. [Google Scholar] [CrossRef]
- Mitsui, M.; Fukui, H.; Takahashi, R.; Takakura, Y.; Mizukami, T. Single-Molecule Fluorescence Spectroscopy of Perylene Diimide Dyes in a γ-Cyclodextrin Film: Manifestation of Photoinduced H-Atom Transfer via Higher Triplet (n,n*) Excited States. J. Phys. Chem. A 2017, 121, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Gelin, M.F.; Rao, B.J.; Nest, M.; Domcke, W. Domain of validity of the perturbative approach to femtosecond optical spectroscopy. J. Chem. Phys. 2013, 139, 224107. [Google Scholar] [CrossRef] [PubMed]
- Pisliakov, A.V.; Gelin, M.F.; Domcke, W. Detection of Electronic and Vibrational Coherence Effects in Electron-Transfer Systems by Femtosecond Time-Resolved Fluorescence Spectroscopy: Theoretical Aspects. J. Phys. Chem. A 2003, 107, 2657–2666. [Google Scholar] [CrossRef]
- Egorova, D.; Gelin, M.F.; Domcke, W. Time- and frequency-resolved fluorescence spectra of nonadiabatic dissipative systems: What photons can tell us. J. Chem. Phys. 2005, 122, 134504. [Google Scholar] [CrossRef] [PubMed]
- Mukamel, S. Principles of Nonlinear Optical Spectroscopy; Oxford University Press: New York, NY, USA, 1995. [Google Scholar]
- Leibel, M.; Toninelli, C.; van Hulst, N.F. Room-temperature ultrafast nonlinear spectroscopy of a single molecule. Nat. Photonics 2018, 12, 45–49. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gelin, M.F.; Palacino-González, E.; Chen, L.; Domcke, W. Monitoring of Nonadiabatic Effects in Individual Chromophores by Femtosecond Double-Pump Single-Molecule Spectroscopy: A Model Study. Molecules 2019, 24, 231. https://doi.org/10.3390/molecules24020231
Gelin MF, Palacino-González E, Chen L, Domcke W. Monitoring of Nonadiabatic Effects in Individual Chromophores by Femtosecond Double-Pump Single-Molecule Spectroscopy: A Model Study. Molecules. 2019; 24(2):231. https://doi.org/10.3390/molecules24020231
Chicago/Turabian StyleGelin, Maxim F., Elisa Palacino-González, Lipeng Chen, and Wolfgang Domcke. 2019. "Monitoring of Nonadiabatic Effects in Individual Chromophores by Femtosecond Double-Pump Single-Molecule Spectroscopy: A Model Study" Molecules 24, no. 2: 231. https://doi.org/10.3390/molecules24020231