
Synthesis, Structural Confirmation, and Biosynthesis of 22-OH-PD1n-3 DPA †

 ,
, 

Abstract
:
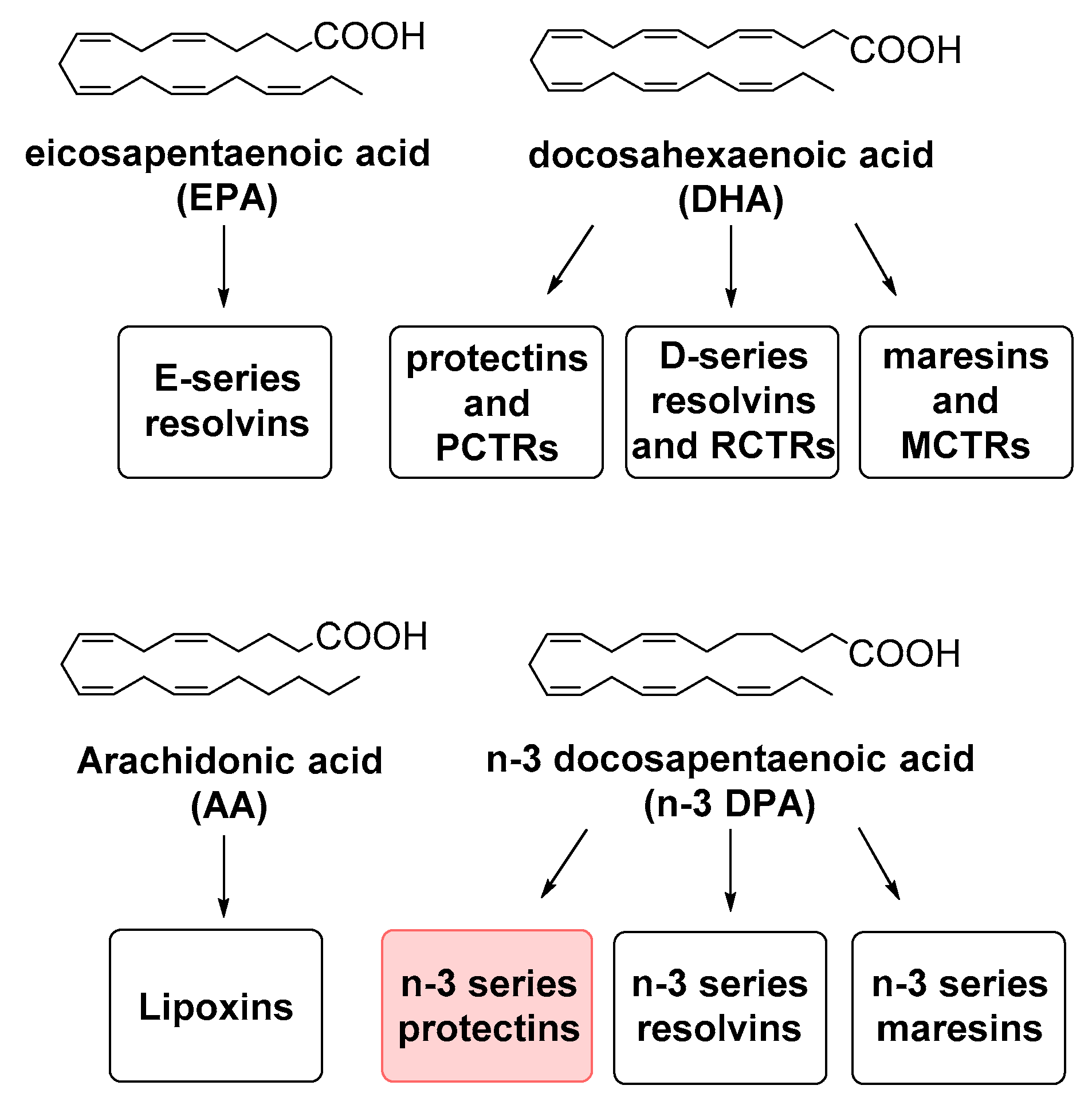
1. Introduction
2. Results and Discussion
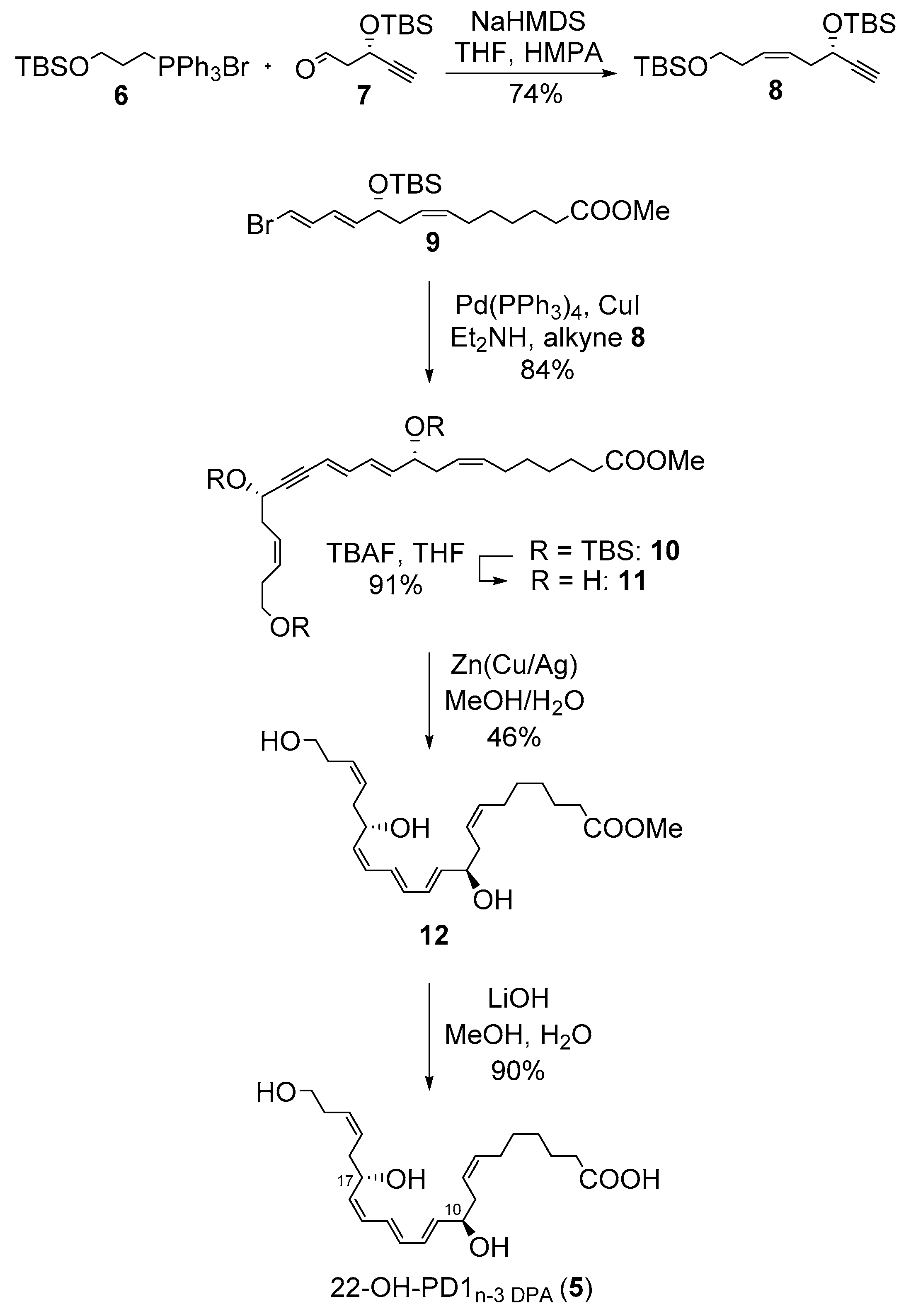
2.1. Synthesis of 22-OH-PD1n-3 DPA
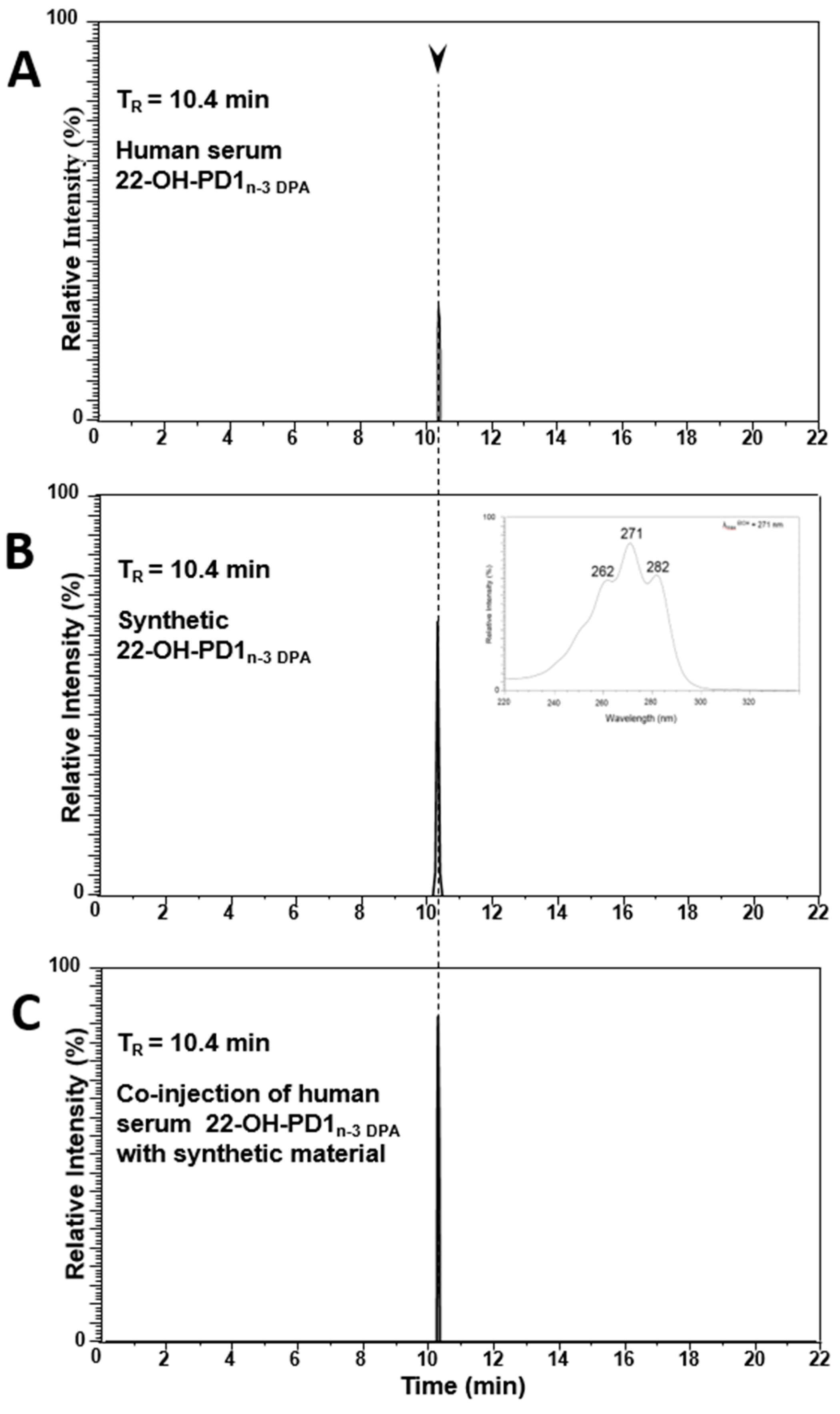
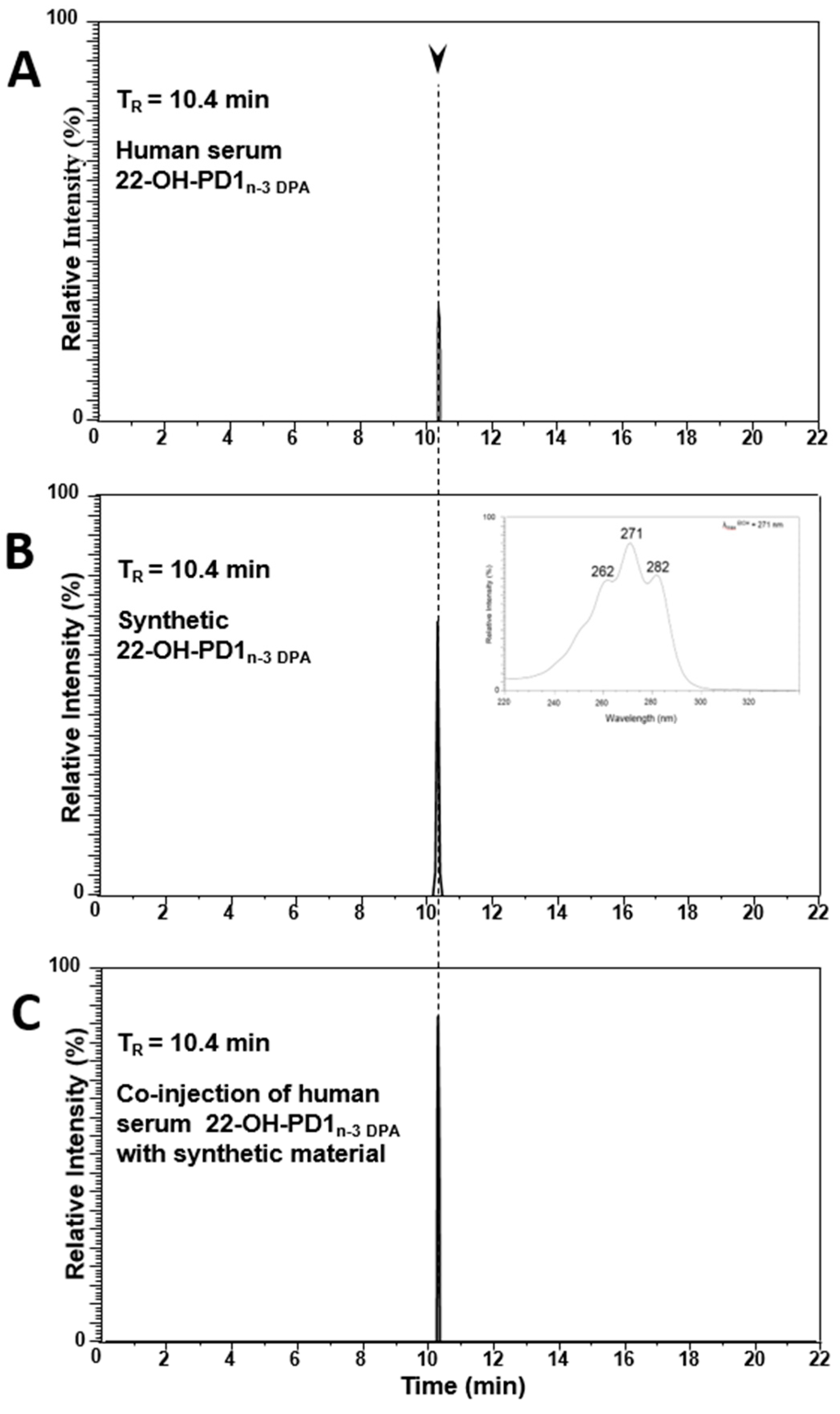
2.2. Matching of Synthetic 22-OH-PD1n-3 DPA with Material Formed in Human Serum
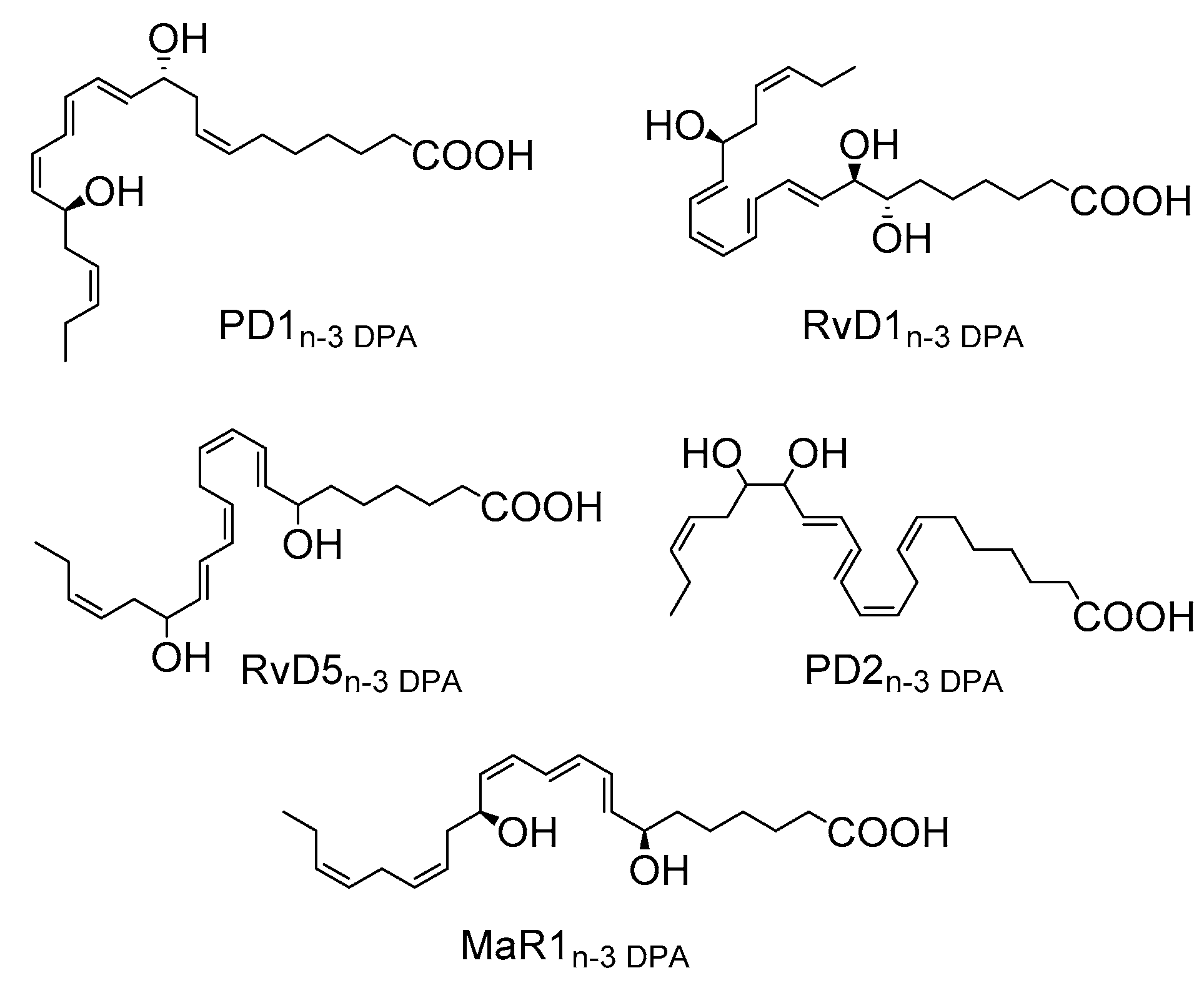
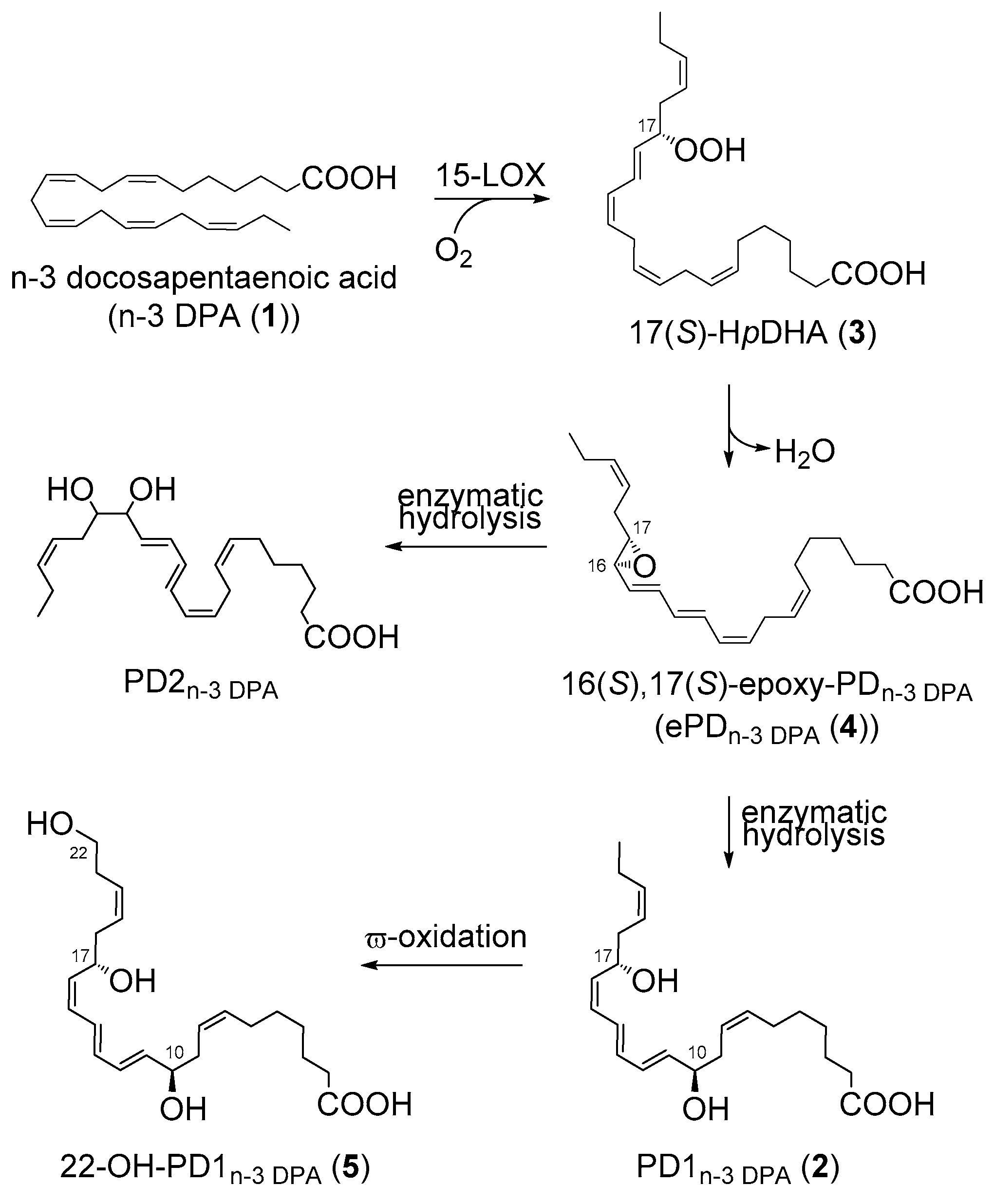
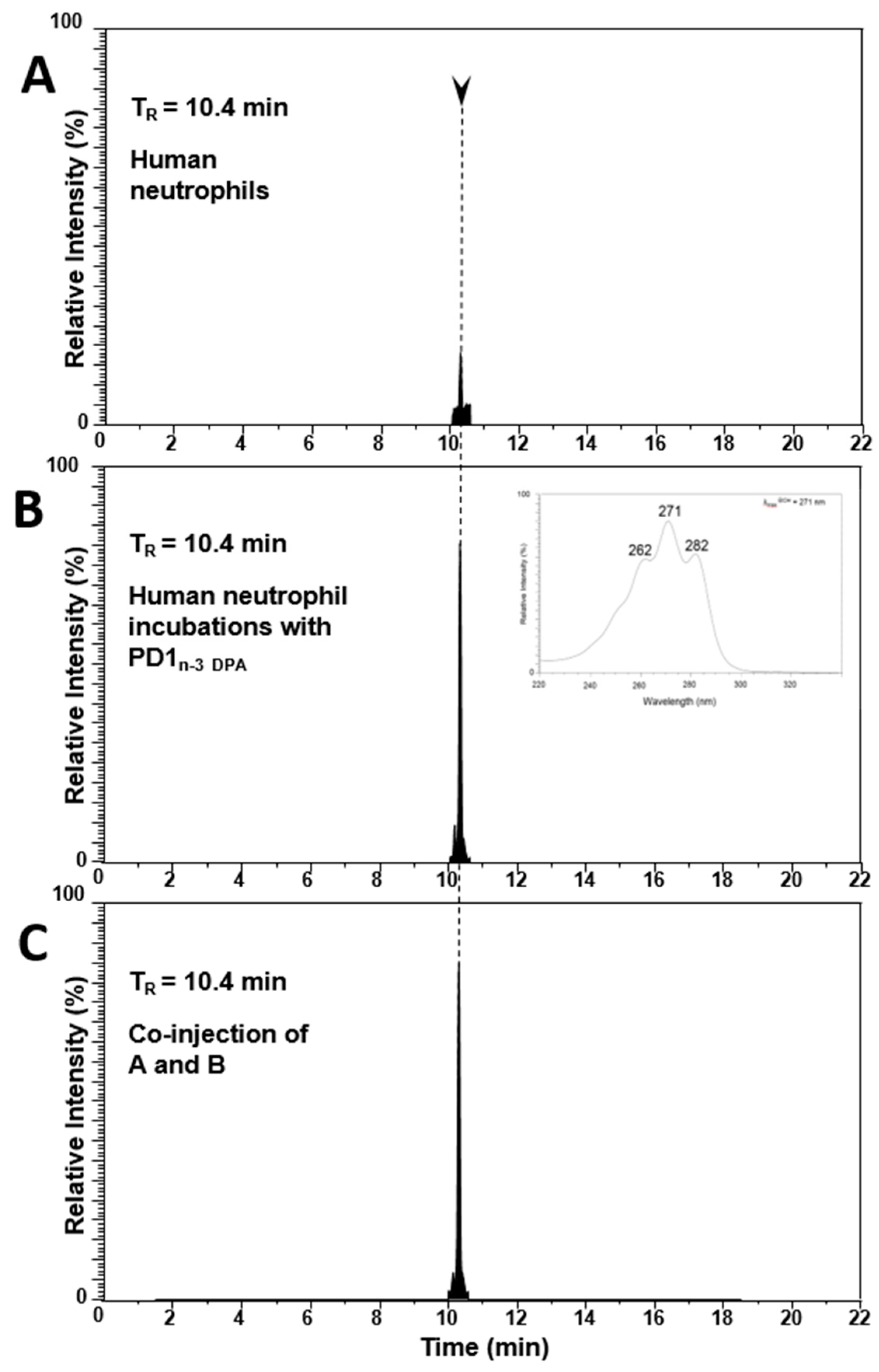
2.3. PD1n-3 DPA is A Precursor in the Biosynthesis of 22-OH-PD1n-3 DPA in Human Neutrophils
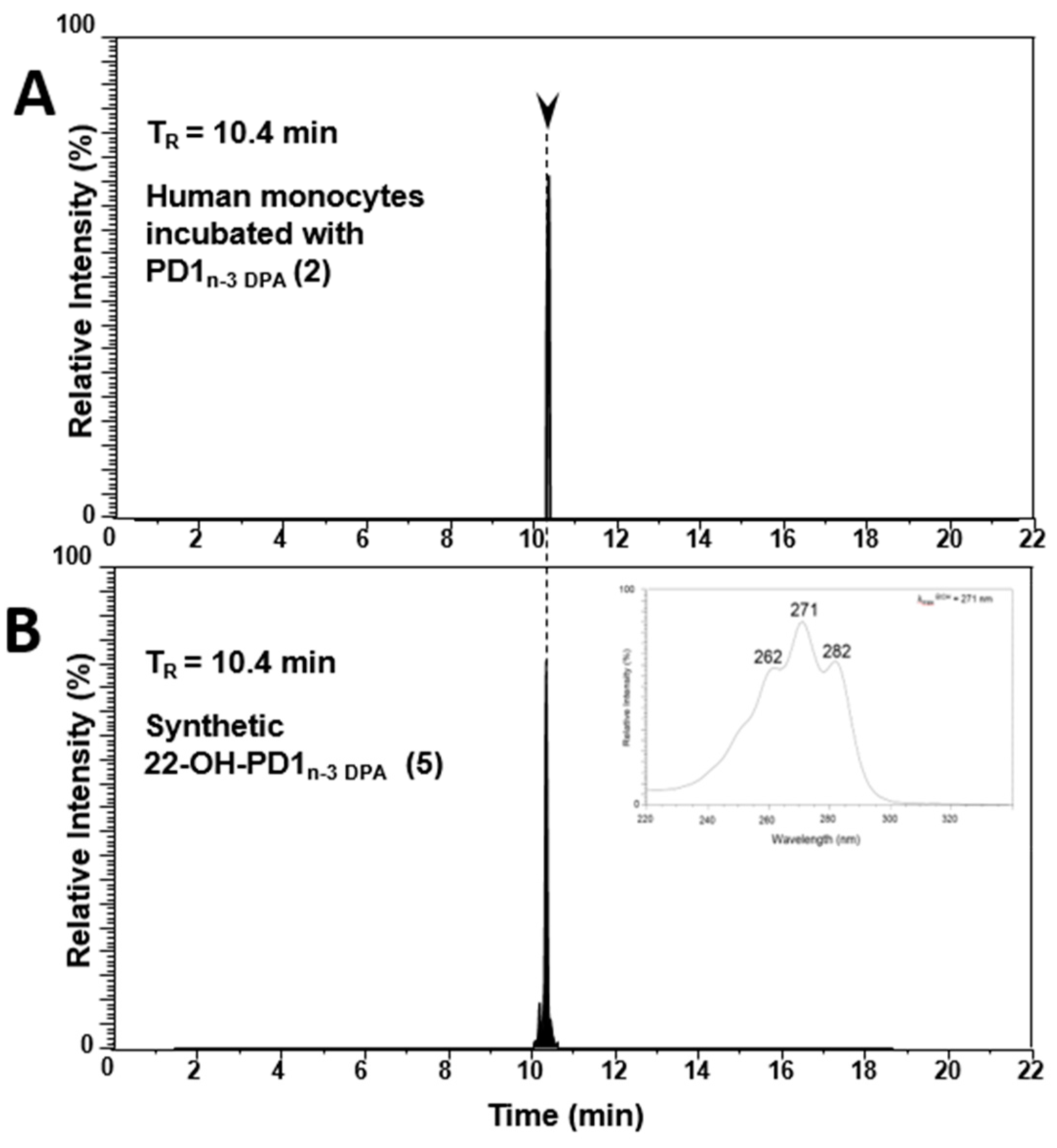
2.4. Biosynthesis of 22-OH-PD1n-3 DPA from PD1n-3 DPA in Human Monocytes
3. Materials and Methods
3.1. Synthesis of Compounds
3.1.1. Bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenyl-λl5-phosphane (6)
3.1.2. (S,Z)-5-Ethynyl-2,2,3,3,12,12,13,13-octamethyl-4,11-dioxa-3,12-disilatetradec-7-ene (8)
3.1.3. Methyl (7Z,10R,11E,13E,17S,19Z)-10,17,22-tris((tert-butyldimethylsilyl)oxy) docosa-7,11,13,19-tetraen-15-ynoate (10)
3.1.4. Methyl (7Z,10R,11E,13E,17S,19Z)-10,17,22-trihydroxydocosa-7,11,13,19-tetraen-15-ynoate (11)
3.1.5. 22-OH-PD1n-3 DPA Methyl Ester (12)
3.1.6. 22-OH-PD1n-3 DPA (5)
3.2. Lipid Mediator Profiling
3.3. Human Neutrophil Incubations
3.4. Human Serum Incubations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rather, L.J. Disturbance of function (function laesa): The legendary fifth cardinal sign of inflammation, added by Galen to the four cardinal signs of Celsus. Bull. N. Y. Acad. Med. 1971, 47, 303–322. [Google Scholar] [PubMed]
- Libby, P. Inflammatory mechanisms: The molecular basis of inflammation and disease. Nutr. Rev. 2007, 65, 140–146. [Google Scholar] [CrossRef]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Glass, C.K. Anti-inflammatory therapy in chronic disease: Challenges and opportunities. Science 2013, 339, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Petasis, N.A. Resolvins and protectins in inflammation resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, T.E.; Serhan, C.N. Resolution of inflammation: A new paradigm for the pathogenesis of periodontal disease. J. Dent. Res. 2003, 82, 82–90. [Google Scholar] [CrossRef]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N. Resolution phase lipid mediators of inflammation: Agonists of resolution. Curr. Opin. Pharmacol. 2013, 13, 632–640. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef]
- Dalli, J.; Colas, R.A.; Serhan, C.N. Novel n-3 immunoresolvents: Structures and actions. Sci. Rep. 2013, 3, 1940. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Chiang, N.; Serhan, C.N. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat. Med. 2015, 21, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Cameron-Smith, D.; Garg, M.; Sinclair, A.J. Docosapentaenoic acid (22:5n-3): A review of its biological effects. Prog. Lipid Res. 2011, 50, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aursnes, M.; Tungen, J.E.; Vik, A.; Colas, R.; Cheng, C.-Y.C.; Dalli, J.; Serhan, C.N.; Hansen, T.V. Total synthesis of the lipid mediator PD1n-3 DPA: Configural assignments and anti-inflammatory and pro-resolving actions. J. Nat. Prod. 2014, 77, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.V.; Vik, A.; Serhan, C.N. The protectin family of specialized pro-resolving mediators: Potent immunoresolvents enabling innovative approaches to target obesity and diabetes. Front. Pharmacol. 2019, 9, 1582–1598. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.V.; Dalli, J.; Serhan, C.N. The novel lipid mediator PD1n-3 DPA: An overview of the structural elucidation, synthesis, biosynthesis and bioactions. Prostaglandins Other Lipid Mediat. 2017, 133, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Vik, A.; Dalli, J.; Hansen, T.V. Recent advances in the chemistry and biology of anti-inflammatory and specialized pro-resolving mediators biosynthesized from n-3 docosapentaenoic acid. Bioorg. Med. Chem. Lett. 2017, 27, 2259–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobbetti, T.; Dalli, J.; Colas, R.A.; Canova, D.F.; Aursnes, M.; Bonnet, D.; Alric, L.; Vergnolle, N.; Deraison, C.; Hansen, T.V.; et al. Protectin D1n-3 DPA and resolvin D5n-3 DPA are novel effectors of intestinal protection. Proc. Natl. Acad. Sci. USA 2017, 114, 3963–3968. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, F.; Pasqualini, G.; Craparotta, I.; Marchini, S.; Van Vliet, E.A.; Foerch, P.; Vandenplas, C.; Leclercq, K.; Aronica, E.; Porcu, L.; et al. n-3 Docosapentaenoic acid-derived protectin D1 promotes resolution of neuroinflammation and arrests epileptogenesis. Brain 2018, 141, 3130–3143. [Google Scholar] [CrossRef] [PubMed]
- Pistorius, K.; Souza, P.R.; De Matteis, R.; Austin-Williams, S.; Primdahl, K.G.; Vik, A.; Mazzacuva, F.; Colas, R.A.; Marques, R.M.; Hansen, T.V.; et al. PDn-3 DPA pathway regulates human monocyte differentiation and macrophage function. Cell Chem. Biol. 2018, 25, 749–760. [Google Scholar] [CrossRef]
- Primdahl, K.G.; Tungen, J.E.; De Souza, P.R.S.; Colas, R.A.; Dalli, J.; Hansen, T.V.; Vik, A. Stereocontrolled synthesis and investigation of the biosynthetic transformations of 16(S), 17(S)-epoxy-PDn-3 DPA. Org. Biomol. Chem. 2017, 15, 8606–8613. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H.; Minakami, S. Oxidation of 20- hydroxyleukotriene B4 by human neutrophil microsomes. Role of aldehyde dehydrogenase and leukotriene B4 ω-hydroxylase (cytochrome P-450LTB4) in leukotriene B4 ω-oxidation. J. Biol. Chem. 1990, 265, 4348–4353. [Google Scholar] [PubMed]
- Divanovic, S.; Dalli, J.; Jorge-Nebert, L.F.; Flick, L.M.; Gálvez-Peralta, M.; Boespflug, N.D.; Stankiewicz, T.E.; Fitzgerald, J.M.; Somarathna, M.; Karp, C.L.; et al. Contributions of the three CYP1 monooxygenases to pro-inflammatory and inflammation-resolution lipid mediator pathways. J. Immunol. 2013, 191, 3347–3357. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.-L. Resolvins. A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Tungen, J.E.; Aursnes, M.; Vik, A.; Colas, R.A.; Dalli, J.; Serhan, C.N.; Hansen, T.V. Synthesis, anti-inflammatory and pro-resolving activities of 22-OH-PD1, a mono-hydroxylated metabolite of protectin D1. J. Nat. Prod. 2014, 77, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Tungen, J.E.; Aursnes, M.; Ramon, S.; Colas, R.A.; Serhan, C.N.; Olberg, D.E.; Nuruddin, S.; Willoch, F.; Hansen, T.V. Synthesis of protectin D1 analogs: Novel pro-resolution and radiotracer agents. Org. Biomol. Chem. 2018, 16, 6818–6823. [Google Scholar] [CrossRef]
- Tungen, J.E.; Aursnes, M.; Dalli, J.; Arnardottir, H.; Serhan, C.N.; Hansen, T.V. Total synthesis of the anti-inflammatory and pro-resolving lipid mediator MaRn-3 DPA. Chem. Eur. J. 2014, 20, 14575–14578. [Google Scholar] [CrossRef]
- Ramon, S.; Dalli, J.; Sanger, J.M.; Winkler, J.W.; Aursnes, M.; Tungen, J.E.; Hansen, T.V.; Serhan, C.N. The protectin PCTR1 is produced by human M2 macrophages and enhances resolution of infectious inflammation. Am. J. Phatol. 2016, 186, 962–973. [Google Scholar] [CrossRef]
- Primdahl, K.G.; Aursnes, M.; Tungen, J.E.; Hansen, T.V.; Vik, A. An efficient synthesis of leukotriene B4. Org. Biomol. Chem. 2015, 13, 5412–5417. [Google Scholar] [CrossRef]
- Tungen, J.E.; Aursnes, M.; Hansen, T.V. Stereoselective synthesis of maresin 1. Tetrahedron Lett. 2015, 56, 1843–1846. [Google Scholar] [CrossRef]
- Nolsøe, J.M.; Aursnes, M.; Tungen, J.E.; Hansen, T.V. Dienals derived from pyridinium salts and their subsequent application in natural product synthesis. J. Org. Chem. 2015, 80, 5377–5385. [Google Scholar] [CrossRef] [PubMed]
- Aursnes, M.; Tungen, J.E.; Vik, A.; Dalli, J.; Hansen, T.V. Stereoselective synthesis of protectin D1: A potent anti-inflammatory and pro-resolving lipid mediator. Org. Biomol. Chem. 2014, 12, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Primdahl, K.G.; Aursnes, M.; Walker, M.E.; Colas, R.A.; Serhan, C.N.; Dalli, J.; Hansen, T.V.; Vik, A. Synthesis of 13(R)-Hydroxy-7Z,10Z,13R,14E,16Z,19Z docosapentaenoic acid (13R-HDPA) and its biosynthetic conversion to the 13-series resolvins. J. Nat. Prod. 2016, 79, 2693–2702. [Google Scholar] [CrossRef] [PubMed]
- Tungen, J.E.; Gerstmann, L.; Vik, A.; De Mateis, R.; Colas, R.A.; Dalli, J.; Chiang, N.; Serhan, C.N.; Kalesse, M.; Hansen, T.V. Resolving inflammation: synthesis, configurational assignment, and biological evaluations of RvD1n− 3 DPA. Chem. Eur. J. 2019, 25, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Boland, W.; Schroer, C.N.; Sieler, C.M.; Feigel, M. Stereospecific syntheses and spectroscopic properties of isomeric 2,4,6,8-undecatetraenes. New hydrocarbons from the marine brown alga Giffordia mitchellae. Part IV. Helv. Chim. Acta. 1987, 70, 1025–1040. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C.N. Specific lipid mediator signatures of human phagocytes: Microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 2012, 120, e60–e72. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J. Does promoting resolution instead of inhibiting inflammation represent the new paradigm in treating infections? Mol. Aspects Med. 2017, 58, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, F.; Simone, J.-M.; Carcache, D.; Bobal, P.; Neier, R. Radical couplings as key steps for the preparation of derivatives of nonactic acid. Montash. Chem. 2007, 138, 121–129. [Google Scholar] [CrossRef]
- Dalli, J.; Colas, R.A.; Walker, M.E.; Serhan, C.N. Lipid mediator metabolomics via LC-MS/MS profiling and analysis. Methods Mol. Biol. 2018, 1730, 59–72. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds 6, 8, 12 and 5 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesman, J.I.; Gangestad Primdahl, K.; Tungen, J.E.; Palmas, F.; Dalli, J.; Hansen, T.V. Synthesis, Structural Confirmation, and Biosynthesis of 22-OH-PD1n-3 DPA. Molecules 2019, 24, 3228. https://doi.org/10.3390/molecules24183228
Nesman JI, Gangestad Primdahl K, Tungen JE, Palmas F, Dalli J, Hansen TV. Synthesis, Structural Confirmation, and Biosynthesis of 22-OH-PD1n-3 DPA. Molecules. 2019; 24(18):3228. https://doi.org/10.3390/molecules24183228
Chicago/Turabian StyleNesman, Jannicke Irina, Karoline Gangestad Primdahl, Jørn Eivind Tungen, Fransesco Palmas, Jesmond Dalli, and Trond Vidar Hansen. 2019. "Synthesis, Structural Confirmation, and Biosynthesis of 22-OH-PD1n-3 DPA" Molecules 24, no. 18: 3228. https://doi.org/10.3390/molecules24183228
APA StyleNesman, J. I., Gangestad Primdahl, K., Tungen, J. E., Palmas, F., Dalli, J., & Hansen, T. V. (2019). Synthesis, Structural Confirmation, and Biosynthesis of 22-OH-PD1n-3 DPA. Molecules, 24(18), 3228. https://doi.org/10.3390/molecules24183228