All chemical reagents and solvents were obtained from commercial sources and used without additional purification. Thin-layer chromatography (TLC) was performed on silica gel plates (250 μm, Sorbent Technologies, Atlanta, GA, USA) and the plates were visualized under UV at 254 nm. Standard grade silica gel (230–400 mesh, Sorbent Technologies, Atlanta, GA, USA) was used for flash column chromatography. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III HD 600 MHz NMR or JEOL Model DELTA-270 (270 MHz) NMR spectrometer. Chemical shifts are reported in parts per million (δ) from an internal standard of residual DMSO-d6 (2.50 or 39.5 ppm) or CDCl3 (7.27 or 77.2 ppm). Data are reported as follows: chemical shift (δ), multiplicity (s, singlet; d, doublet; dd, doublet of doublet; t, triplet; dt, doublet of triplet, q, quartet; sep, septet; br s, broad singlet; m, multiplet), coupling constant (J) in Hertz (Hz), integration. High resolution mass spectrometry (HRMS) data were obtained on an Applied Biosystem 4000 Q TRAP® LC/MS/MS system using electrospray ionization (ESI). Low-resolution mass spectra (LRMS) were recorded on a Shimadzu AXIMA Confidence MALDI-TOF mass spectrometer (nitrogen UV laser, 50 Hz, 337 nm) by using α-cyano-4-hydroxycinnamic acid (CHCA) as a matrix. Melting points were determined with a Mel-Temp® capillary apparatus (Cole-Parmer, Staffordshire, UK) and are uncorrected. High performance liquid chromatography (HPLC) analyses were carried out on Agilent 1100 series HPLC system (Foster City, CA) equipped with a diode-array UV detector and a C18-bounded HPLC column (Vydac 218TP104, 4.6 × 250 mm, 10 μm) by using a 40 min-gradient elution from 10% to 90% acetonitrile in water (0.1% TFA) and a flow rate of 1.0 mL/min. Eluents were monitored at 280 nm. Solid-phase reactions were carried out in 12 mL polypropylene cartridges with 20 μ PE frit (Applied Separations, Allentown, PA) and a labquake tube shaker (Fisher Scientific, Pittsburgh, PA) was used for mixing.
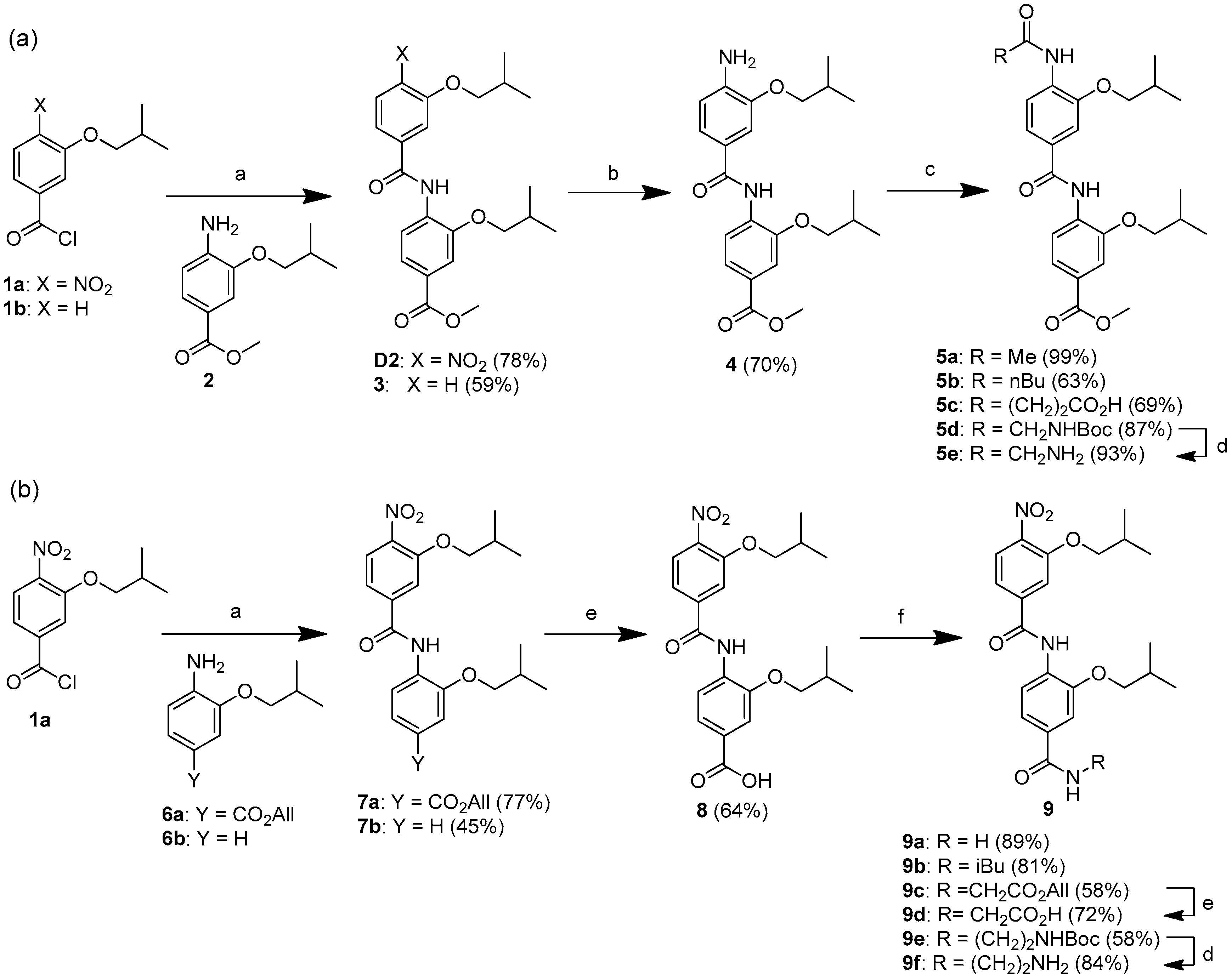
4.1. Synthesis of Bis-Benzamides
Methyl 3-isobutoxy-4-[(3-isobutoxybenzoyl)amino]benzoate (3): Oxalyl chloride (0.26 mL, 3.0 mmol) was slowly added to a solution of 3-isobutoxybenzoic acid (300 mg, 1.5 mmol) in DCM (20 mL). The resulting mixture was stirred at room temperature for 2 h. The solvent and excess oxalyl chloride were then removed under reduced pressure, and the residue was dissolved in DCM (5 mL). The resulting solution was then slowly added to a solution of methyl 4-amino-3-isobutoxybenzoate (226 mg, 1.0 mmol) and DIEA (0.52 mL, 3.0 mmol) in DCM (20 mL). After stirring at room temperature for 12 h, the resulting solution was concentrated under reduced pressure, and diluted with EtOAc (20 mL) and 1N HCl (20 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (20 mL). The organic layers were combined, washed with saturated NaHCO3 and brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resulting solid was washed with EtOAc and dried in vacuo to afford the compound 3 as a white solid (240 mg, 59%). 1H NMR (DMSO-d6, 600 MHz): δ 9.44 (br s, 1 H), 8.12 (d, J = 8.3 Hz, 1 H), 7.63 (dd, J = 8.3, 1.7 Hz, 1 H), 7.56 (d, J = 1.7 Hz, 1 H), 7.49 (d, J = 7.7 Hz, 1 H), 7.46 (d, J = 7.7 Hz, 1 H), 7.45 (d, J = 7.7 Hz, 1 H), 7.19–7.17 (m, 1 H), 3.92 (d, J = 6.2 Hz, 2 H), 3.86 (s, 3 H), 3.83 (d, J = 6.6 Hz, 2 H), 2.14–2.01 (m, 2 H), 1.02 (d, J = 6.6 Hz, 6 H), 1.00 (d, J = 6.7 Hz, 6 H). 13C NMR (DMSO-d6, 150 MHz): δ 165.8, 164.6, 158.9, 149.5, 135.7, 131.8, 129.9, 125.9, 122.0, 121.9, 119.4, 118.5, 112.8, 112.0, 74.5, 73.9, 52.1, 27.8, 27.7, 18.99, 18.97. MALDI-TOF (m/z): [M+H]+ calcd for C23H30NO5: 400.21, found 400.81.
Methyl 4-[(4-amino-3-isobutoxybenzoyl)amino]-3-isobutoxybenzoate (
4): The title compound was synthesized as previously described [
10].
Methyl 4-[(4-acetamido-3-isobutoxybenzoyl)amino]-3-isobutoxybenzoate (5a): Acetyl chloride (0.015 mL, 0.21 mmol) was slowly added to a solution of compound 4 (60 mg, 0.14 mmol) and DIEA (0.097 mL, 0.56 mmol) in EtOAc (10 mL). After stirring at room temperature for 6 h, the resulting solution was concentrated under reduced pressure, and diluted with EtOAc (20 mL) and 1N HCl (10 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (20 mL). The organic layers were combined, washed with saturated NaHCO3 and brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (hexanes/DCM 1:1) to afford the compound 5a as a light yellow solid (63 mg, 99%). Rf = 0.16 (hexanes/EtOAc 4:1). 1H NMR (DMSO-d6, 600 MHz): δ 9.37 (br s, 1 H), 9.10 (br s, 1 H), 8.15 (d, J = 8.4 Hz, 1 H), 8.12 (d, J = 8.3 Hz, 1 H), 7.63 (dd, J = 8.3, 1.5 Hz, 1 H), 7.56 (d, J = 1.5 Hz, 1 H), 7.54 (d, J = 1.5 Hz, 1 H), 7.51 (dd, J = 8.3, 1.5 Hz, 1 H), 3.92 (d, J = 6.6 Hz, 2 H), 3.89 (d, J = 6.6 Hz, 2 H), 3.86 (s, 3 H), 2.19–2.11 (m, 2 H), 2.15 (s, 3 H), 1.03 (d, J = 7.0 Hz, 12 H). 13C NMR (DMSO-d6, 150 MHz): δ 168.8, 165.8, 164.2, 149.3, 148.4, 131.9, 131.1, 129.5, 125.6, 122.1, 121.6, 121.1, 119.8, 112.0, 110.8, 74.8, −74.6, 52.1, 27.8, 27.5, 24.0, 19.10, 19.05. MALDI-TOF (m/z): [M + Na]+ calcd for C25H32N2NaO6: 479.52, found 479.79.
Methyl 3-isobutoxy-4-[[3-isobutoxy-4-(pentanoylamino)benzoyl]amino]benzoate (5b): The title compound was prepared following the same procedure as for compound 5a, using compound 4 (250 mg, 0.60 mmol) and valeroyl chloride (0.15 mL, 1.21 mmol). The crude product was purified by flash column chromatography (hexanes/EtOAc 2:1) to afford the compound 5b as a white solid (189 mg, 63%). Rf = 0.49 (hexanes/EtOAc 2:1). 1H NMR (DMSO-d6, 600 MHz): δ 9.37 (br s, 1 H), 9.02 (br s, 1 H), 8.15 (d, J = 8.4 Hz, 1 H), 8.08 (d, J = 8.4 Hz, 1 H), 7.64 (dd, J = 8.3, 1.7 Hz, 1 H), 7.56 (d, J = 1.8 Hz, 1 H), 7.54 (d, J = 1.8 Hz, 1 H), 7.51 (dd, J = 8.4, 1.8 Hz, 1 H), 3.92 (d, J = 6.2 Hz, 2 H), 3.89 (d, J = 6.6 Hz, 2 H), 3.86 (s, 3 H), 2.44 (t, J = 7.3 Hz, 2 H), 2.17–2.09 (m, 2 H), 1.61–1.56 (m, 2 H), 1.37–1.31 (m, 2 H), 1.03 (d, J = 6.6 Hz, 12 H), 0.90 (t, J = 7.3 Hz, 3 H). 13C NMR (DMSO-d6, 150 MHz): δ 171.6, 165.8, 164.2, 149.3, 148.6, 131.9, 131.0, 129.6, 125.6, 122.1, 121.5, 121.3, 119.8, 111.9, 110.7, 74.7, 74.5, 52.1, 36.0, 27.8, 27.6, 27.3, 21.7, 19.1, 13.7. MALDI-TOF (m/z): [M + H]+ calcd for C28H39N2O6: 499.28, found 499.82.
4-[2-Isobutoxy-4-[(2-isobutoxy-4-methoxycarbonylphenyl)carbamoyl]anilino]-4-oxobutanoic acid (5c): Succinic anhydride (3.4 g, 33.8 mmol) was added to a solution of compound 4 (2.8 g, 6.8 mmol) in DCM (300 mL). After stirring at room temperature for 12 h, the resulting precipitate was collected by vacuum filtration, washed with DCM, and dried in vacuo to afford compound 5c as a white solid (2.4 g, 69%). 1H NMR (DMSO-d6, 600 MHz): δ 12.14 (br s, 1 H), 9.37 (br s, 1 H), 9.12 (br s, 1 H), 8.15 (d, J = 8.1 Hz, 1 H), 8.13 (d, J = 7.4 Hz, 1 H), 7.63 (dd, J = 8.3, 1.7 Hz, 1 H), 7.56 (d, J = 1.7 Hz, 1 H), 7.54 (d, J = 1.7 Hz, 1 H), 7.51 (dd, J = 8.3, 1.7 Hz, 1 H), 3.92 (d, J = 6.2 Hz, 2 H), 3.89 (d, J = 6.6 Hz, 2 H), 3.86 (s, 3 H), 2.69 (t, J = 6.7 Hz, 2 H), 2.53 (t, J = 6.7 Hz, 2 H), 2.19–2.10 (m, 2 H), 1.04 (d, J = 6.7 Hz, 6 H), 1.03 (d, J = 6.7 Hz, 6 H). 13C NMR (DMSO-d6, 150 MHz): δ 173.8, 170.6, 165.8, 164.2, 149.2, 148.3, 131.9, 131.1, 129.4, 125.6, 122.1, 121.5, 120.7, 119.9, 111.9, 110.7, 74.8, 74.5, 52.1, 31.3, 28.9, 27.8, 27.6, 19.12, 19.05. MALDI-TOF (m/z): [M + H]+ calcd for C27H35N2O8: 515.24, found 515.69.
[2-[2-Isobutoxy-4-[(2-isobutoxy-4-methoxycarbonylphenyl)carbamoyl]anilino]-2-oxoethyl]ammonium trifluoroacetate (
5e): Compound
5d was synthesized as previously described [
10]. Then, compound
5d (1.8 g, 3.2 mmol) was dissolved in 50% TFA in DCM (40 mL) and the mixture was stirred at room temperature for 1 h. The reaction mixture was concentrated under reduced pressure. The product was precipitated by adding cold diethyl ether, washed with diethyl ether, and dried in vacuo. The TFA salt of compound
5e was obtained as a white solid (1.7 g, 93%).
1H NMR (DMSO-
d6, 600 MHz):
δ 9.73 (br s, 1 H), 9.43 (br s, 1 H), 8.15 (d,
J = 8.1 Hz, 1 H), 8.13 (d,
J = 8.4 Hz, 1 H), 8.00 (br s, 3 H), 7.64 (dd,
J = 8.4, 1.5 Hz, 1 H), 7.60 (d,
J = 1.5 Hz, 1 H), 7.57–7.56 (m, 2 H), 3.92 (d,
J = 5.5 Hz, 4 H), 3.91 (s, 2 H), 3.86 (s, 3 H), 1.04 (d,
J = 5.8 Hz, 6 H), 1.03 (d,
J = 6.2 Hz, 6 H).
13C NMR (DMSO-
d6, 150 MHz):
δ 165.8, 164.2, 158.4, 158.2, 149.4, 148.7, 131.9, 130.4, 129.9, 125.8, 122.1, 121.8, 121.3, 120.0, 112.0, 111.0, 75.0, 74.6, 52.1, 41.4, 27.8, 27.5, 19.13, 19.06. MALDI-TOF (
m/
z): [M + H]
+ calcd for C
25H
34N
3O
6: 472.24, found 472.67.
3-Isobutoxy-N-(2-isobutoxyphenyl)-4-nitrobenzamide (7b): The title compound was prepared following the same procedure as for compound 3, using 3-isobutoxy-4-nitrobenzoic acid (431 mg, 0.18 mmol) and 2-isobutoxyaniline (6b) (200 mg, 1.2 mmol). Recrystallization from EtOAc/hexanes afforded compound 7b as a light yellow solid (210 mg, 45%). 1H NMR (DMSO-d6, 600 MHz): δ 9.70 (br s, 1 H), 8.01 (d, J = 8.1 Hz, 1 H), 7.80 (s, 1 H), 7.66 (d, J = 8.1 Hz, 1 H), 7.60 (d, J = 8.4 Hz, 1 H), 7.23–7.20 (m, 1 H), 7.10–7.09 (m, 1 H), 6.99–6.97 (m, 1 H), 4.02 (d, J = 6.2 Hz, 2 H), 3.80 (d, J = 6.2 Hz, 2 H), 2.10–2.00 (m, 2 H), 0.99 (d, J = 6.6 Hz, 6 H), 0.97 (d, J = 6.6 Hz, 6 H). 13C NMR (DMSO-d6, 150 MHz): δ 163.2, 151.5, 150.9, 140.8, 139.5, 126.4, 126.2, 125.0, 124.9, 120.0, 119.3, 113.6, 112.4, 75.1, 75.0, 74.1, 27.7, 27.5, 18.9, 18.6. MALDI-TOF (m/z): [M + H]+ calcd for C21H27N2O5: 387.19, found 387.67.
3-Isobutoxy-4-[(3-isobutoxy-4-nitrobenzoyl)amino]benzoic acid (
8): Compound
7a was synthesized as previously described [
10]. Then, Pd(PPh
3)
4 (198 mg, 0.17 mmol) and PhSiH
3 (0.43 mL, 3.4 mmol) were added to a solution of compound
7a (800 mg, 1.7 mmol) in DCM (30 mL). After stirring at room temperature for 1 h, the reaction mixture was concentrated under reduced pressure. The product was precipitated by adding diethyl ether, washed with diethyl ether, and dried in vacuo. Compound
8 was obtained as a white solid (465 mg, 64%), m.p. 242–245 °C.
1H NMR (DMSO-
d6, 600 MHz):
δ 12.98 (br s, 1 H), 9.78 (br s, 1 H), 8.03 (d,
J = 8.4 Hz, 1 H), 7.96 (d,
J = 8.4 Hz, 1 H), 7.80 (d,
J = 1.4 Hz, 1 H), 7.62–7.60 (m, 2 H), 7.57 (d,
J = 1.5 Hz, 1 H), 4.03 (d,
J = 6.2 Hz, 2 H), 3.89 (d,
J = 6.6 Hz, 2 H), 2.11–2.05 (m, 2 H), 1.005 (d,
J = 6.4 Hz, 6 H), 0.995 (d,
J = 6.3 Hz, 6 H).
13C NMR (DMSO-
d6, 150 MHz):
δ 167.4, 164.0, 151.6, 150.8, 141.7, 139.8, 131.3, 128.6, 125.6, 123.6, 122.4, 120.0, 114.3, 113.0, 75.8, 75.0, 40.2, 28.3, 28.1, 19.5, 19.2. MALDI-TOF (
m/
z): [M + Na]
+ calcd for C
22H
26N
2NaO
7: 453.16, found 453.67.
3-Isobutoxy-4-[(3-isobutoxy-4-nitrobenzoyl)amino]benzamide (9a): DIEA (0.16 mL, 0.92 mmol) was added to a solution of compound 8 (200 mg, 0.46 mmol) and PyBOP (263 mg, 0.51 mmol) in DMF (10 mL). After stirring at room temperature for 1 h, ammonium chloride (246 mg, 4.6 mmol) and additional DIEA (0.80 mL, 4.6 mmol) were added. The resulting mixture was stirred at room temperature for 12 h and diluted with EtOAc (50 mL) and 1N HCl (30 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (30 mL). The organic layers were combined, washed with saturated NaHCO3, and concentrated under reduced pressure. The resulting solid was washed with EtOAc and dried in vacuo to afford the compound 9a as a white solid (175 mg, 89%), m.p. 249–251 °C. 1H NMR (DMSO-d6, 270 MHz): δ 9.76 (br s, 1 H), 8.02 (d, J = 8.4 Hz, 1 H), 8.00 (br s, 1 H), 7.84 (d, J = 8.2 Hz, 1 H), 7.79 (d, J = 1.2 Hz, 1 H), 7.60 (dd, J = 8.4, 1.2 Hz, 1 H), 7.57 (d, J = 1.5 Hz, 1 H), 7.53 (dd, J = 8.2, 1.5 Hz, 1 H), 7.37 (br s, 1 H), 4.02 (d, J = 6.7 Hz, 2 H), 3.87 (d, J = 6.4 Hz, 2 H), 2.12–2.00 (m, 2 H), 0.994 (d, J = 6.7 Hz, 6 H), 0.992 (d, J = 6.7 Hz, 6 H). 13C NMR (DMSO-d6, 150 MHz): δ 167.2, 163.5, 151.0, 150.6, 141.1, 139.4, 131.9, 129.2, 125.1, 123.5, 119.8, 119.5, 113.8, 111.4, 75.3, 74.5, 27.8, 27.6, 19.1, 18.7. MALDI-TOF (m/z): [M + Na]+ calcd for C22H27N3NaO6: 452.18, found 452.54.
3-Isobutoxy-4-[(3-isobutoxy-4-nitrobenzoyl)amino]-N-isobutylbenzamide (9b): DIEA (0.084 mL 0.48 mmol) was added to a mixture of compound 8 (50 mg, 0.12 mmol) and PyBOP (73 mg, 0.14 mmol) in DMF (5 mL), followed by isobutylamine (0.024 mL, 0.24 mmol). After stirring at room temperature for 12 h, the resulting solution was diluted with EtOAc (20 mL) and 1N HCl (20 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (20 mL). The organic layers were combined, washed with saturated NaHCO3 and brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (hexanes/EtOAc 1:1) to afford the compound 9b as a light yellow solid (47 mg, 81%). Rf = 0.41 (hexanes/EtOAc 1:1). 1H NMR (DMSO-d6, 600 MHz): δ 9.77 (br s, 1 H), 8.47 (d, J = 5.7 Hz, 1 H), 8.02 (d, J = 8.2 Hz, 1 H), 7.84 (d, J = 8.2 Hz, 1 H), 7.80 (br s, 1 H), 7.61 (dd, J = 8.2, 1.4 Hz, 1 H), 7.54 (br s, 1 H), 7.51 (d, J = 8.2 Hz, 1 H), 4.03 (d, J = 6.2 Hz, 2 H), 3.88 (d, J = 6.2 Hz, 2 H), 3.09 (t, J = 6.4 Hz, 2 H), 2.11–2.04 (m, 2 H), 1.90–1.82 (m, 1 H), 1.00 (d, J = 5.7 Hz, 6 H), 0.996 (d, J = 5.7 Hz, 6 H), 0.90 (d, J = 6.6 Hz, 6 H). 13C NMR (DMSO-d6, 150 MHz): δ 165.5, 163.4, 151.0, 150.7, 141.1, 139.4, 132.4, 128.9, 125.1, 123.7, 119.5, 119.4, 113.8, 111.1, 75.2, 74.5, 46.8, 28.1, 27.8, 27.6, 20.2, 19.1, 18.7. MALDI-TOF (m/z): [M + H]+ calcd for C26H36N3O6: 486.26, found 486.76.
2-[[3-Isobutoxy-4-[(3-isobutoxy-4-nitrobenzoyl)amino]benzoyl]amino]acetic acid (9d): DIEA (0.35 mL, 2.0 mmol) was added to a solution of compound 8 (430 mg, 1.0 mmol) and PyBOP (624 mg, 1.2 mmol) in DMF (20 mL). After stirring at room temperature for 1 h, glycine allyl ester trifluoroacetate (344 mg, 1.5 mmol) and additional DIEA (0.35 mL, 2.0 mmol) were added. The resulting mixture was stirred at room temperature for 12 h and diluted with EtOAc (50 mL) and 1N HCl (30 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (20 mL). The organic layers were combined, washed with saturated NaHCO3 and brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. Recrystallization from EtOAc/hexanes afforded compound 9c as a white solid (308 mg, 58%). Then, Pd(PPh3)4 (54 mg, 0.047 mmol) and PhSiH3 (0.12mL, 0.94 mmol) were added to a solution of compound 9c (250 mg, 0.47 mmol) in THF (20 mL). After stirring at room temperature for 1 h, the reaction mixture was concentrated under reduced pressure. The product was precipitated by adding diethyl ether, washed with diethyl ether, and dried in vacuo. Compound 9d was obtained as a white solid (165 mg, 72%). 1H NMR (DMSO-d6, 600 MHz): δ 12.6 (br s, 1 H), 9.79 (br s, 1 H), 8.86 (t, J = 5.7 Hz, 1 H), 8.02 (d, J = 8.4 Hz, 1 H), 7.88 (d, J = 8.1 Hz, 1 H), 7.81 (br s, 1 H), 7.62 (d, J = 8.4 Hz, 1 H), 7.58 (br s, 1 H), 7.54 (d, J = 8.2 Hz, 1 H), 4.03 (d, J = 6.6 Hz, 2 H), 3.94 (d, J = 5.9 Hz, 2 H), 3.89 (d, J = 6.2 Hz, 2 H), 2.12–2.05 (m, 2 H), 1.01 (d, J = 6.6 Hz, 6 H), 1.00 (d, J = 6.7 Hz, 6 H). 13C NMR (DMSO-d6, 150 MHz): δ 171.4, 165.8, 163.5, 151.1, 150.7, 141.1, 139.4, 131.4, 129.4, 125.1, 123.6, 119.7, 119.5, 113.8, 111.1, 75.3, 74.6, 41.3, 27.8, 27.7, 19.1, 18.7. MALDI-TOF (m/z): [M + Na]+ calcd for C24H29N3NaO8: 510.19, found 510.68.
2-[[3-Isobutoxy-4-[(3-isobutoxy-4-nitrobenzoyl)amino]benzoyl]amino]ethylammonium trifluoroacetate (
9f): The title compound was synthesized as previously described [
10].
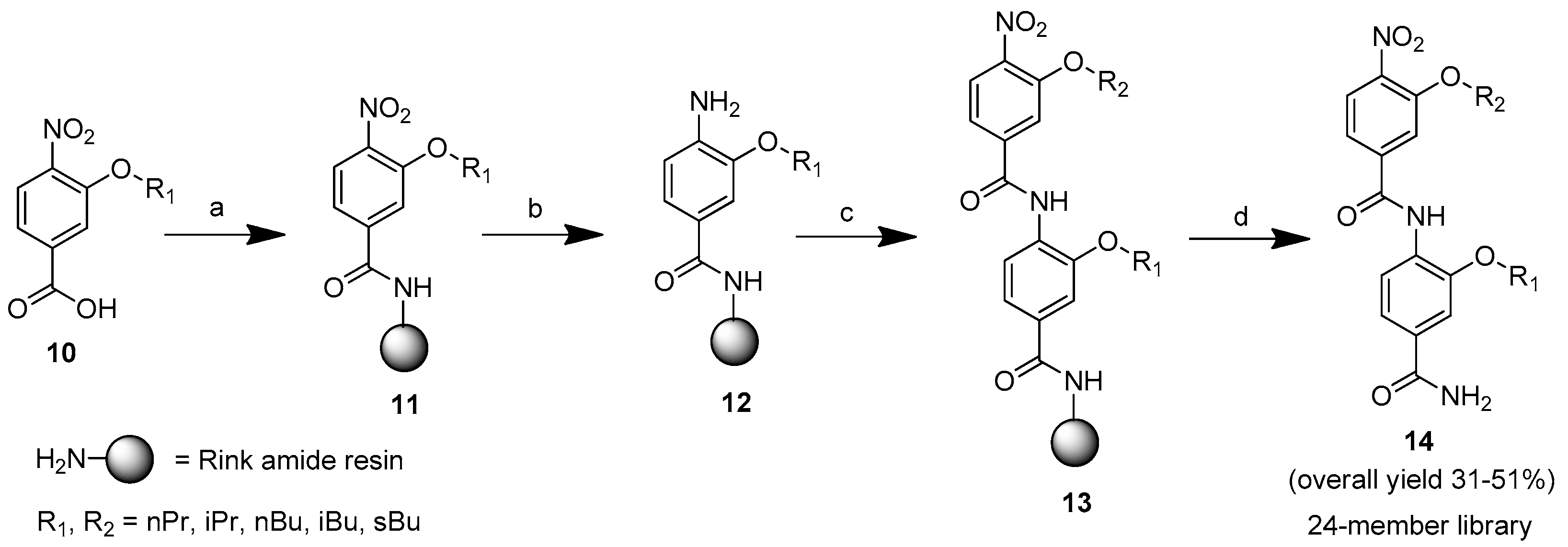
4.2. Synthesis of Bis-Benzamides Library 14
These compounds were synthesized as previously described [
21]. We briefly describe their syntheses. Fmoc-Rink amide MBHA resin (0.50 mmol/g, 300 mg, 0.15 mmol) was swollen in DMF for 2 h and washed with DMF (×3). The Fmoc protecting group was removed by treating with piperidine (20% in DMF, 5 × 30 min), and washed with DMF (×3). Then, 3-alkoxy-4-nitrobenzoic acid
10 was introduced by using a preactivated ester which was prepared by mixing 3-alkoxy-4-nitrobenzoic acid
10 (4.0 equiv.), PyBOP (4.0 equiv), and DIEA (4.0 equiv) in DMF (8 mL) for 5 min. The solution was added to the resin and shaken at room temperature for 24 h. The resin was then filtered and washed with DMF (×3) affording compound
11. The nitro group of the compound
11 was swollen in AcOH (50% in H
2O)/HCl (0.5 N in H
2O)/THF (1:1:6, 8 mL) for 20 min and treated with SnCl
2·2H
2O (5.0 equiv.). The reaction mixture was shaken at room temperature for 24 h. The resin was filtered, and washed with HCl (0.5 N in H
2O)/DMF (1:6) (×3), H
2O/DMF (1:6) (×3) and DMF (×3) affording compound
12. Then 3-alkoxy-4-nitrobenzoic acid
10 was introduced by using a preactivated HOAt ester which was prepared by mixing 3-alkoxy-4-nitrobenzoic acid
10 (4.0 equiv.), HATU (4.0 equiv), and DIEA (4.0 equiv) in DMF (8 mL) for 1 h. The solution was added to the resin and shaken at room temperature for 24 h. The resin was then filtered and washed with DMF (×3) affording the resin-bound bis-benzamide
13. The bis-benzamide
13 was washed with DCM (×3) and dried in vacuo. The dried resin was treated with a cleavage mixture of TFA/H
2O (95:5, 6 mL) for 90 min. The TFA solution was then filtered, and the resin was washed with TFA (2 mL). The combined TFA solution was concentrated to a volume of approximately 0.5 mL with a gentle stream of nitrogen. The product was precipitated by adding cold diethyl ether, washed with diethyl ether, and dried in vacuo affording compound
14.
4-[(4-Nitro-3-propoxybenzoyl)amino]-3-propoxybenzamide (14a): Light yellow solid, 22 mg, 47% overall yield, 95% purity by HPLC. 1H NMR (DMSO-d6, 270 MHz): δ 9.76 (br s, 1 H), 8.01 (d, J = 8.4 Hz, 1 H), 7.99 (br s, 1 H), 7.85 (d, J = 8.2 Hz, 1 H), 7.80 (d, J = 1.4 Hz, 1 H), 7.61 (dd, J = 8.4, 1.5 Hz, 1 H), 7.58 (d, J = 1.6 Hz, 1 H), 7.53 (dd, J = 8.0, 1.6 Hz, 1 H), 7.37 (br s, 1 H), 4.22 (t, J = 6.4 Hz, 2 H), 4.05 (t, J = 6.4 Hz, 2 H), 1.82–1.73 (m, 4 H), 0.988 (t, J = 7.2 Hz, 3 H), 0.986 (t, J = 7.2 Hz, 3 H). 13C NMR (DMSO-d6, 150 MHz): δ 167.2, 163.5, 151.0, 150.4, 141.2, 139.4, 131.8, 129.2, 125.0, 123.4, 119.7, 119.5, 114.0, 111.4, 70.8, 69.9, 22.0, 21.8, 10.4, 10.2. MALDI-TOF (m/z): [M + Na]+ calcd for C20H23N3NaO6: 424.15, found 424.87.
4-[(3-Isopropoxy-4-nitrobenzoyl)amino]-3-propoxybenzamide (14b): Light yellow solid, 22 mg, 37% overall yield, 97% purity by HPLC. 1H NMR (DMSO-d6, 270 MHz): δ 9.74 (br s, 1 H), 7.99 (br s, 1 H), 7.97 (d, J = 8.4 Hz, 1 H), 7.86 (d, J = 8.2 Hz, 1 H), 7.85 (br s, 1 H), 7.81 (d, J = 8.3, 1 H), 7.57 (br s, 1 H), 7.53 (d, J = 8.4, 1 H), 7.37 (br s, 1 H), 4.94 (sep, J = 6.2, 1 H), 4.05 (t, J = 6.3 Hz, 2 H), 1.82–1.71 (m, 2 H), 1.33 (d, J = 6.2, 6 H), 0.99 (t, J = 7.3 Hz, 3 H). 13C NMR (DMSO-d6, 150 MHz): δ 167.2, 163.6, 150.4, 149.7, 142.3, 139.2, 131.7, 129.2, 124.9, 123.3, 119.7, 119.6, 115.1, 111.4, 72.5, 69.9, 22.0, 21.6, 10.4. MALDI-TOF (m/z): [M + Na]+ calcd for C20H23N3NaO6: 424.15, found 424.97.
4-[(3-Butoxy-4-nitrobenzoyl)amino]-3-propoxybenzamide (14c): Light yellow solid, 30 mg, 48% overall yield based on the loading of Fmoc-Rink amide resin, 95% purity by HPLC, m.p. 247–248 °C. 1H NMR (DMSO-d6, 270 MHz): δ 9.76 (br s, 1 H), 8.00 (d, J = 8.2 Hz, 1 H), 7.99 (br s, 1 H), 7.85 (d, J = 8.2 Hz, 1 H), 7.81 (d, J = 1.5 Hz, 1 H), 7.60 (dd, J = 8.4, 1.5 Hz, 1 H), 7.58 (d, J = 1.7 Hz, 1 H), 7.53 (dd, J = 8.2, 1.7 Hz, 1 H), 7.37 (br s, 1 H), 4.26 (t, J = 6.3 Hz, 2 H), 4.05 (t, J = 6.3 Hz, 2 H), 1.85–1.69 (m, 4 H), 1.51–1.38 (m, 2 H), 0.99 (t, J = 7.4 Hz, 3 H), 0.94 (t, J = 7.7 Hz, 3 H). 13C NMR (DMSO-d6, 68 MHz): δ 167.8, 164.1, 151.5, 150.9, 141.8, 140.0, 132.3, 129.8, 125.6, 123.9, 120.3, 120.0, 114.5, 112.0, 70.5, 70.0, 30.9, 22.6, 19.1, 14.1, 11.0. HRMS-ESI (m/z): [M + H]+ calcd for C21H26N3O6: 416.1822, found 416.1815.
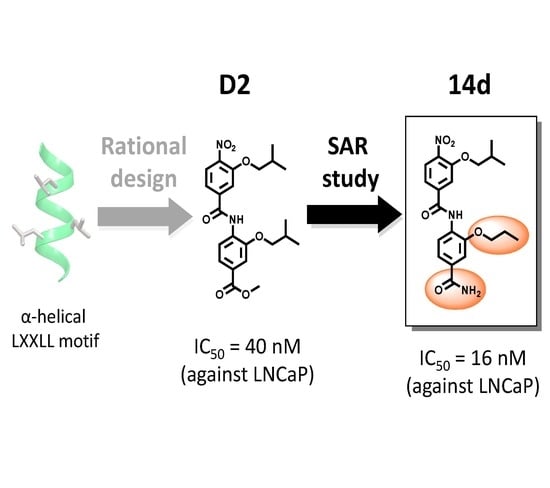
4-[(3-Isobutoxy-4-nitrobenzoyl)amino]-3-propoxybenzamide (14d): Light yellow solid, 32 mg, 51% overall yield based on the loading of Fmoc-Rink amide resin, 93% purity by HPLC, m.p. 255–257 °C. 1H NMR (DMSO-d6, 270 MHz): δ 9.76 (br s, 1 H), 8.02 (d, J = 8.2 Hz, 1 H), 7.99 (br s, 1 H), 7.86 (d, J = 8.2 Hz, 1 H), 7.79 (d, J = 1.6 Hz, 1 H), 7.61 (dd, J = 8.3, 1.6 Hz, 1 H), 7.58 (d, J = 1.7 Hz, 1 H), 7.53 (dd, J = 8.2, 1.7 Hz, 1 H), 7.37 (br s, 1 H), 4.05 (t, J = 6.4 Hz, 2 H), 4.03 (d, J = 6.5 Hz, 2 H), 2.12–2.02 (m, 1 H), 1.85–1.72 (m, 2 H), 0.994 (t, J = 7.2 Hz, 3 H), 0.992 (d, J = 6.7 Hz, 6 H). 13C NMR (DMSO-d6, 68 MHz): δ 167.8, 164.1, 151.6, 151.0, 141.7, 140.0, 132.3, 129.8, 125.6, 123.9, 120.3, 120.1, 114.5, 112.0, 75.8, 70.5, 28.2, 22.6, 19.3, 11.2. HRMS-ESI (m/z): [M + H]+ calcd for C21H26N3O6: 416.1816, found 416.1822.
3-Isopropoxy-4-[(4-nitro-3-propoxy-benzoyl)amino]benzamide (14f): Light yellow solid, 24 mg, 40% overall yield, 95% purity by HPLC. 1H NMR (DMSO-d6, 270 MHz): δ 9.66 (br s, 1 H), 8.01(d, J = 8.4 Hz, 1 H), 7.98 (br s, 1 H), 7.91 (d, J = 8.2 Hz, 1 H), 7.78 (d, J = 1.4 Hz, 1 H), 7.60 (dd, J = 8.2, 1.4 Hz, 1 H), 7.58 (d, J = 1.5 Hz, 1 H), 7.52 (dd, J = 8.4, 1.6 Hz, 1 H), 7.37 (br s, 1 H), 4.70 (sep, J = 6.2 Hz, 1 H), 4.23 (t, J = 6.4 Hz, 2 H), 1.83–1.71 (m, 2 H), 1.32 (d, J = 5.7 Hz, 6 H), 0.99 (t, J = 7.4 Hz, 3 H). 13C NMR (DMSO-d6, 150 MHz): δ 167.2, 163.6, 151.0, 148.9, 141.2, 139.5, 131.5, 130.2, 125.1, 123.1, 119.9, 119.4, 114.1, 113.1, 71.2, 70.8, 21.8, 21.7, 10.2. MALDI-TOF (m/z): [M + Na]+ calcd for C20H23N3NaO6: 424.15, found 424.62.
4-[(3-Butoxy-4-nitrobenzoyl)amino]-3-isopropoxybenzamide (14h): Light yellow solid, 28 mg, 45% overall yield based on the loading of Fmoc-Rink amide resin, 96% purity by HPLC, m.p. 213–214 °C. 1H NMR (DMSO-d6, 270 MHz): δ 9.65 (br s, 1 H), 8.01 (d, J = 8.4 Hz, 1 H), 7.99 (br s, 1 H), 7.92 (d, J = 8.4 Hz, 1 H), 7.79 (br s, 1 H), 7.59 (d, J = 8.3 Hz, 1 H), 7.58 (br s, 1 H), 7.52 (d, J = 8.4 Hz, 1 H), 7.37 (br s, 1 H), 4.70 (sep, J = 5.9 Hz, 1 H), 4.27 (t, J = 6.4 Hz, 2 H), 1.79–1.68 (m, 2 H), 1.51–1.37 (m, 2 H), 1.32 (d, J = 5.9 Hz, 6 H), 0.93 (t, J = 7.4 Hz, 3 H). 13C NMR (DMSO-d6, 150 MHz): δ 167.2, 163.5, 150.9, 148.9, 141.2, 139.4, 131.5, 130.2, 125.0, 122.9, 119.9, 119.4, 114.0, 113.0, 71.2, 69.1, 30.3, 21.8, 18.5, 13.5. MALDI-TOF (m/z): [M + Na]+ calcd for C21H25N3NaO6: 438.16, found 438.51.
4-[(3-Isobutoxy-4-nitrobenzoyl)amino]-3-isopropoxybenzamide (14i): Light yellow solid, 31 mg, 48% overall yield, 93% purity by HPLC. 1H NMR (DMSO-d6, 270 MHz): δ 9.66 (br s, 1 H), 8.02 (d, J = 8.4 Hz, 1 H), 7.99 (br s, 1 H), 7.92 (d, J = 8.2 Hz, 1 H), 7.77 (br s, 1 H), 7.59 (d, J = 8.3 Hz, 1 H), 7.58 (br s, 1 H), 7.52 (d, J = 8.4 Hz, 1 H), 7.36 (br s, 1 H), 4.70 (sep, J = 5.9 Hz, 1 H), 4.04 (d, J = 6.4 Hz, 2 H), 2.11–2.02 (m, 1 H), 1.32 (d, J = 5.7 Hz, 6 H), 0.99 (d, J = 6.7 Hz, 6 H). 13C NMR (DMSO-d6, 150 MHz): δ 167.2, 163.5, 151.0, 148.9, 141.1, 139.5, 131.5, 130.2, 125.1, 123.0, 119.9, 119.4, 114.0, 113.0, 75.2, 71.2, 27.6, 21.8, 18.7. MALDI-TOF (m/z): [M + Na]+ calcd for C21H25N3NaO6: 438.16, found 438.95.
3-Butoxy-4-[(4-nitro-3-propoxybenzoyl)amino]benzamide (14k): Light yellow solid, 23 mg, 37% overall yield, >99% purity by HPLC. 1H NMR (DMSO-d6, 270 MHz): δ 9.75 (br s, 1 H), 8.01 (d, J = 8.4 Hz, 1 H), 7.99 (br s, 1 H), 7.85 (d, J = 8.2 Hz, 1 H), 7.80 (d, J = 1.2 Hz, 1 H), 7.60 (dd, J = 8.4, 1.4 Hz, 1 H), 7.58 (d, J = 1.4 Hz, 1 H), 7.52 (dd, J = 8.4, 1.5 Hz, 1 H), 7.37 (br s, 1 H), 4.22 (t, J = 6.4 Hz, 2 H), 4.09 (t, J = 6.4 Hz, 2 H), 1.84–1.70 (m, 4 H), 1.52–1.38 (m, 2 H), 0.99 (t, J = 7.4 Hz, 3 H), 0.91 (t, J = 7.4 Hz, 3 H). 13C NMR (DMSO-d6, 150 MHz): δ 167.2, 163.5, 150.9, 150.4, 141.2, 139.4, 131.8, 129.2, 125.0, 123.3, 119.8, 119.5, 114.0, 111.4, 70.8, 68.1, 30.7, 21.8, 18.7, 13.7, 10.2. MALDI-TOF (m/z): [M + Na]+ calcd for C21H25N3NaO6: 438.16, found 438.75.
3-Butoxy-4-[(3-isopropoxy-4-nitrobenzoyl)amino]benzamide (14l): Light yellow solid, 25 mg, 40% overall yield based on the loading of Fmoc-Rink amide resin, >99% purity by HPLC, m.p. 233–235 °C. 1H NMR (DMSO-d6, 270 MHz): δ 9.73 (br s, 1 H), 7.99 (br s, 1 H), 7.97 (d, J = 8.4 Hz, 1 H), 7.86 (d, J = 8.2 Hz, 1 H), 7.81 (d, J = 1.2 Hz, 1 H), 7.59 (dd, J = 8.2, 1.2 Hz, 1 H), 7.58 (d, J = 1.4 Hz, 1 H), 7.52 (dd, J = 8.3, 1.4 Hz, 1 H), 7.37 (br s, 1 H), 4.94 (sep, J = 5.9 Hz, 1 H), 4.09 (t, J = 6.2 Hz, 2 H), 1.80–1.70 (m, 2 H), 1.52–1.38 (m, 2 H), 1.33 (d, J = 6.2 Hz, 6 H), 0.91 (t, J = 7.4 Hz, 3 H). 13C NMR (DMSO-d6, 150 MHz): δ 167.2, 163.5, 150.4, 149.7, 142.3, 139.2, 131.7, 129.2, 124.9, 123.3, 119.7, 119.6, 115.1, 111.4, 72.5, 68.2, 30.7, 21.5, 18.7, 13.7. MALDI-TOF (m/z): [M + H]+ calcd for C21H26N3O6: 416.18, found 416.56.
3-Butoxy-4-[(3-butoxy-4-nitrobenzoyl)amino]benzamide (14m): Light yellow solid, 20 mg, 31% overall yield based on the loading of Fmoc-Rink amide resin, 92% purity by HPLC, m.p. 249–251 °C. 1H NMR (DMSO-d6, 270 MHz): δ 9.74 (br s, 1 H), 8.00 (d, J = 8.4 Hz, 1 H), 7.99 (br s, 1 H), 7.86 (d, J = 8.4 Hz, 1 H), 7.80 (d, J = 1.2 Hz, 1 H), 7.60 (dd, J = 8.4, 1.4 Hz, 1 H), 7.58 (br s, 1 H), 7.52 (dd, J = 8.3, 1.4 Hz, 1 H), 7.37 (br s, 1 H), 4.26 (t, J = 6.2 Hz, 2 H), 4.09 (t, J = 6.2 Hz, 2 H), 1.80–1.69 (m, 4 H), 1.52–1.37 (m, 4 H), 0.94 (t, J = 7.2 Hz, 3 H), 0.91 (t, J = 7.3 Hz, 3 H). 13C NMR (DMSO-d6, 68 MHz): δ 167.8, 164.1, 151.5, 151.0, 141.8, 140.0, 132.3, 129.8, 125.6, 123.9, 120.3, 120.1, 114.5, 112.0, 69.7, 68.7, 31.2, 30.9, 19.3, 19.1, 14.3, 14.1. MALDI-TOF (m/z): [M + Na]+ calcd for C22H27N3NaO6: 452.18, found 452.56.
3-Butoxy-4-[(3-sec-butoxy-4-nitrobenzoyl)amino]benzamide (14o): Light yellow solid, 20 mg, 31% overall yield based on the loading of Fmoc-Rink amide resin, 94% purity by HPLC, m.p. 218–220 °C. 1H NMR (DMSO-d6, 270 MHz): δ 9.73 (br s, 1 H), 7.99 (br s, 1 H), 7.97 (d, J = 8.4 Hz, 1 H), 7.87 (d, J = 8.2 Hz, 1 H), 7.80 (d, J = 1.2 Hz, 1 H), 7.580 (d, J = 1.4 Hz, 1 H), 7.578 (dd, J = 8.2, 1.5 Hz, 1 H), 7.52 (dd, J = 8.3, 1.6 Hz, 1 H), 7.37 (br s, 1 H), 4.81–4.70 (m, 1 H), 4.09 (d, J = 6.4 Hz, 2 H), 1.80–1.63 (m, 4 H), 1.52–1.38 (m, 2 H), 1.30 (d, J = 5.9 Hz, 3 H), 0.93 (t, J = 7.4 Hz, 3 H), 0.90 (t, J = 7.4 Hz, 3 H). 13C NMR (DMSO-d6, 150 MHz): δ 167.2, 163.6, 150.4, 150.0, 142.2, 139.2, 131.7, 129.2, 124.9, 123.3, 119.8, 119.5, 114.8, 111.4, 77.0, 68.2, 30.7, 28.4, 18.7, 18.7, 13.7, 9.2. MALDI-TOF (m/z): [M + Na]+ calcd for C22H27N3NaO6: 452.18, found 452.60.
4-[(3-Butoxy-4-nitrobenzoyl)amino]-3-isobutoxybenzamide (14r): Light yellow solid, 20 mg, 31% overall yield based on the loading of Fmoc-Rink amide resin, 92% purity by HPLC, m.p. 239–240 °C. 1H NMR (DMSO-d6, 270 MHz): δ 9.76 (br s, 1 H), 8.01 (d, J = 8.2 Hz, 1 H), 7.99 (br s, 1 H), 7.83 (d, J = 8.4 Hz, 1 H)), 7.81 (br s, 1 H), 7.60 (dd, J = 8.3, 1.4 Hz, 1 H), 7.57 (br s, 1 H), 7.52 (dd, J = 8.2, 1.5 Hz, 1 H), 7.37 (br s, 1 H), 4.25 (t, J = 6.2 Hz, 2 H), 3.87 (d, J = 6.4 Hz, 2 H), 2.14–2.00 (m, 1 H), 1.79–1.68 (m, 2 H), 1.51–1.40 (m, 2 H), 0.99 (d, J = 6.7 Hz, 6 H), 0.93 (t, J = 7.4 Hz, 3 H). 13C NMR (DMSO-d6, 68 MHz): δ 167.8, 164.1, 151.5, 151.2, 141.8, 140.0, 132.5, 129.7, 125.6, 124.1, 120.3, 120.1, 114.5, 112.0, 75.1, 69.7. 30.9, 28.4, 19.6, 19.1, 14.1. HRMS-ESI (m/z): [M + Na]+ calcd for C22H27N3NaO6: 452.1798, found 452.1792.
4-[(3-sec-Butoxy-4-nitrobenzoyl)amino]-3-isobutoxybenzamide (14s): Light yellow solid, 26 mg, 40% overall yield based on the loading of Fmoc-Rink amide resin, >99% purity by HPLC, m.p. 216–218 °C. 1H NMR (DMSO-d6, 270 MHz): δ 9.75 (br s, 1 H), 7.99 (br s, 1 H), 7.97 (d, J = 8.4 Hz, 1 H), 7.83 (d, J = 8.2 Hz, 1 H), 7.81 (br s, 1 H), 7.59 (d, J = 8.2 Hz, 1 H), 7.59 (br s, 1 H), 7.53 (d, J = 8.4 Hz, 1 H), 7.37 (br s, 1 H), 4.78–4.71 (m, 1 H), 3.86 (d, J = 6.4 Hz, 2 H), 2.11–2.02 (m, 1 H), 1.73–1.62 (m, 2 H), 1.29 (d, J = 6.2 Hz, 3 H), 0.98 (d, J = 6.7 Hz, 6 H), 0.93 (t, J = 7.4 Hz, 3 H). 13C NMR (DMSO-d6, 68 MHz): δ 167.8, 164.1, 151.3, 150.6, 142.8, 139.8, 132.5, 129.8, 125.5, 124.3, 120.3, 120.0, 115.3, 112.0, 77.6, 75.1, 29.0, 28.4, 19.6, 19.3, 9.8. HRMS-ESI (m/z): [M + Na]+ calcd for C22H27N3NaO6: 452.1798, found 452.1798.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}