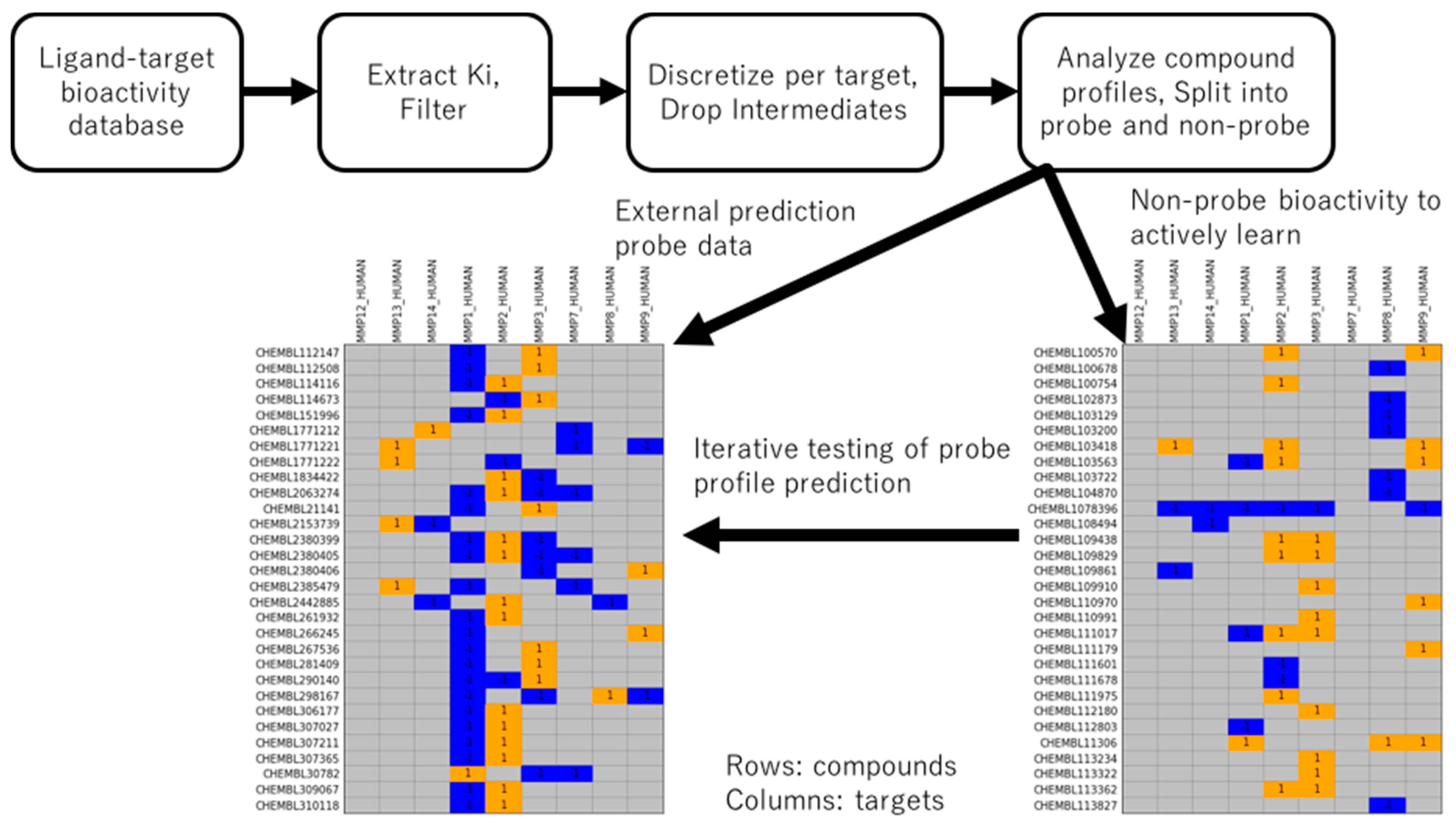
2.1. Generation of Probe and Non-Probe Datasets
The process of data retrieval, discretization, and split into non-probe training and probe external prediction datasets is given in
Figure 1. Inhibitory activities against the matrix metalloproteinase (MMP) family, a collection of more than 20 enzymes with both tumor suppression and tumor progression roles [
21], were retrieved from ChEMBL [
22]. MMP inhibitors were once expected to be a central strategy for inhibition of cancer growth, yet promising preclinical developments in small molecules did not translate to successful clinical trials [
23]. Despite the setback, there is renewed interest in development of small molecules for MMPs due to their roles aside from oncology [
24], and antibodies for several MMPs are commercially available.
MMP targets with sufficient
Ki measurements were used to derive active and inactive annotations, with ligands subsequently categorized into probe and non-probe statuses (see Methods). In short, probes had strong potency for exactly one MMP target and non-potency for at least one other MMP. The non-probe dataset was made available to the active learning method for training, and the external probe dataset was tested for predictability at each iteration of the fit-predict-pick active learning cycle. This choice of train-test data represents a chemical-centric domain of applicability challenge different from a prior target-centric study [
16].
Table 1 shows the statistics of the resulting dataset. Nine MMP targets had sufficient ligand bioactivity data to be included in the study. In total, there were, respectively, 1181 and 72 unique compounds among 2397 and 165 bioactivities in the training and external datasets. 750 compounds in the non-probe training data contained at least one active annotation; among them, 708 (94%) had exclusively active annotations. 473 compounds contained at least one inactive annotation; among them, 431 (91%) had exclusively inactive annotations. 42 compounds were present with both inactive and active annotations. For the external set of probe bioactivities, in accordance with definition, all 72 compounds have one active annotation and at least one inactive annotation (c.f.,
Figure 1).
No compound satisfying the requirements to be included as a probe contains annotations against MMP12; all ligands with MMP12 annotation are present only in the non-probe training bioactivity data. The percentages of actives per target in the training data ranged from 19% (MMP7) to 88% (MMP12). With respect to fraction of active annotations per target between the training and test sets, the overall trend was positive (Pearson R 0.73); MMP1 did not follow the trend, with 54% (200) active annotations in the training data but only 4% (2) active annotations in the test data.
2.2. Longitudinal Performance of Active Learning Strategies and Descriptor Impact
The first probe profile prediction challenge executed was a control experiment for which subsequent experiments could be gauged for significance. We chose simple physicochemical descriptions of compounds (molecular weight, total polarizable surface area, estimated LogP, etc.) and combined them with a dummy identity descriptor for each target (see Methods). That is, each MMP target has a unique descriptor profile with a single value of unity in one position and a value of zero in all other positions. Combining all identity descriptor profiles results in an identity matrix. By using this descriptor and then comparing results to those obtained from a more biologically relevant descriptor, the contribution of a biological descriptor can be assessed more objectively.
Using the control descriptor, an initial active learning of and prediction on the training (non-probe) data, which can be construed as retrospective active learning, was done with evaluation of each of the random, greedy, and curiosity strategies (see Introduction). The F1 measure, a metric combining correct active predictions, type-I error, and type-II error, as well as the Matthews Correlation Coefficient, a metric incorporating all types of prediction results with multiplicative penalty for mistakes and thus reflects accounting for specificity, were chosen as evaluation metrics (see Methods for formulas). Compared to the well-known Accuracy metric (fraction of correct predictions) or recently proposed Power Metric [
25], these two metrics are less subject to over-estimating model performance [
26].
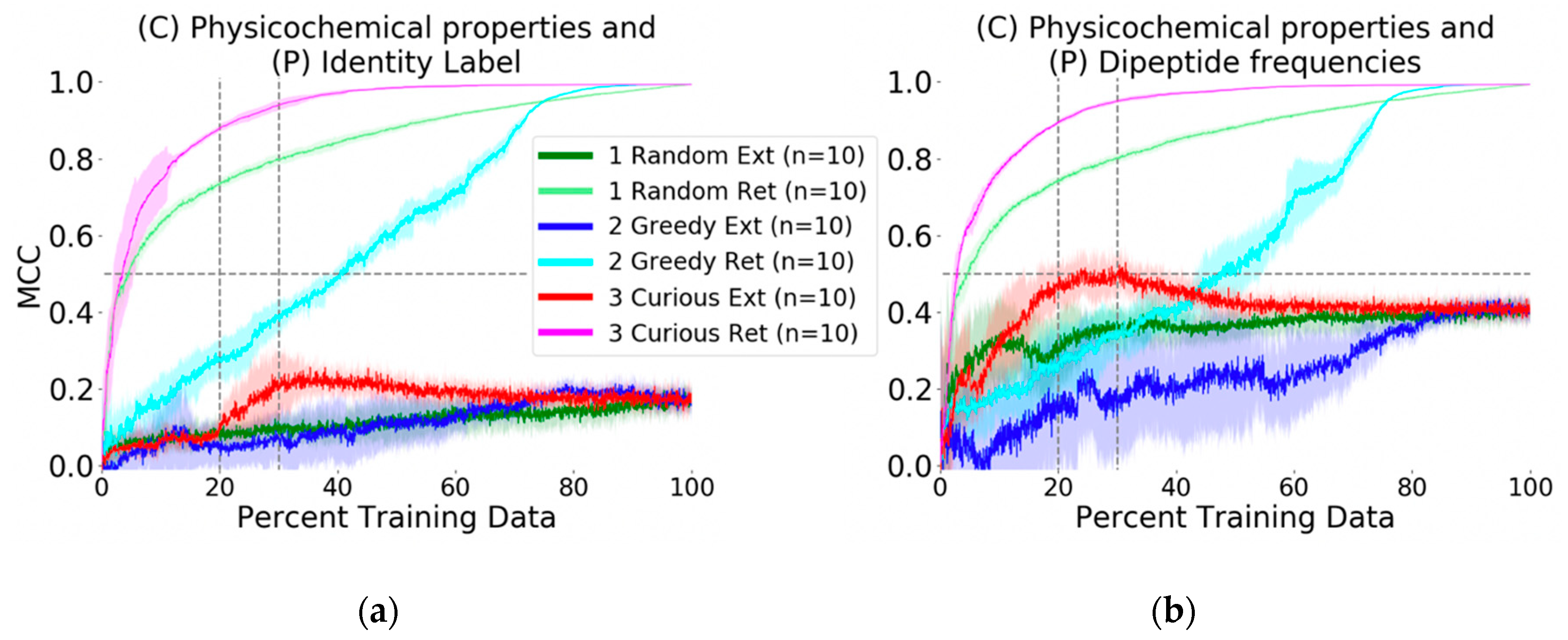
The baseline evaluation with random picking resulted in an average (
n = 10) Matthews Correlation Coefficient (MCC) value of 0.70 (see Methods for metric formulas) at the point by which 20% of the available training data had been selected. At the same data volume, the greedy picker had an MCC of 0.30, whereas the curiosity picker had an MCC of 0.90 (
Figure 2a). At 80% of the training data, all picking methods yielded models with MCC values greater than 0.90, meaning that the structure-activity relationships (SAR) of the remaining 20% of data could be inferred from the 80% used by any method.
The more crucial question and second experiment, prospective active prediction of the external probe dataset, was performed. Whereas in retrospective or recall-type active learning with the physicochemical-identity descriptor the curiosity-picked model achieved MCC of 0.90 with 20% of the training data, the same model was not predictive of the probe compound bioactivity profiles. In fact, none of the physicochemical-identity SARs developed by the picking methods were predictive against the challenge (MCC ~0.10,
Figure 2a). At best, the curious picker at 35% of the data reached a peak of MCC = 0.20 and then dropped to MCC = 0.18 after the addition of more SAR training data. The other picking strategies arrived at the same predictive performance at 80% of the training data.
We proceeded to execute the same challenge but to describe the MMP targets in a more biologically relevant way and ask if SAR modeling would be improved. Identity description of MMPs was replaced with their dipeptide frequencies as protein descriptors (see Methods for calculation details). This did not make any mentionable changes in the retrospective model evaluation; prediction performance for each selection strategy was similar to that achieved using the identity descriptor. However, for the external dataset, improvement was marked. The curious picker reached MCC of 0.46 with 20% of the training data, which further improved to MCC = 0.50 at 23% (
Figure 2b). Interestingly, performance decayed after including more than 30% of the training data, with stable convergence to MCC = 0.45 at 45% of the training data. The random and greedy pickers obtained respective MCC values of 0.30 and 0.10 at the benchmark 20% training data point, and eventually converged to the same MCC = 0.45 at later stages.
Intrigued by the prediction improvement on the probe data via the inclusion of dipeptide descriptors, we sought to clarify the contribution of the protein sequence information. Experiments were repeated with single amino acid frequency and tripeptide frequency descriptors, keeping the compound physicochemical description constant. Separately, we wanted to know the extent by which alternative chemical descriptions would affect prediction performance, and if the improvement over the identity target descriptor could be reproduced with a different chemical perception. In total, eleven additional experiments analogous to
Figure 2 were executed, with all 13 experiments summarized in
Table 2 and discussed next.
Exchanging the control target protein identity descriptor by amino acid, dipeptide, and tripeptide frequencies led to a continuous improvement in probe profile MCC performance, with respective MCCs of 0.35, 0.46, and 0.48 at 20% of non-probe training data (all experiments curiosity picking). Despite the fact that sequentially neighboring residues are not necessarily spatially near, there is a clear difference that addition of amino acid and peptide frequencies boosted the prediction performance of the model on the external dataset. As the underlying estimator algorithm (random forest) is not designed with biological or chemical context in mind, the best we could initially reason is that the extra protein description yielded increased matching correlation with distributions in the physicochemical description, that decision trees subsequently could identify these differential distributions, and that rules based on such differential distributions were transferrable to the external probe SAR data in many cases.
Dipeptide and tripeptide peak performance on the probe dataset was achieved at approximately 32% of training data, with MCC values above 0.52 (
Table 2). In these experiments as well, curious active learning outperformed random and greedy strategies (
Figure S1a). The results clearly re-iterate that the assumption of more data yielding better models is invalid.
Next, we address the latter of two questions posed above by changing the perception of chemical space from physicochemical properties to structural fingerprints. The first descriptor change was to use atomic neighborhood fingerprints (ECFPs). A clear improvement in prospective prediction performance was again observed when replacing identity target descriptors with dipeptide descriptors (see
Figure S1b and
Table 2). In the case of ECFPr2-1024 (maximum neighborhood radius of 2 from a central atom, with fingerprints hashed into 1024 bits), the change to a biological descriptor improved average performance on the external dataset (at 20% training) from MCC of 0.23 to 0.59. Additionally, the peak of mean MCC of the same system was 0.67 at 57% of training data.
Given these results from separate physicochemical and structural perceptions of compounds, we sought to know whether combining the two different types of descriptors could improve predictive performance. Upon combining the pChem and ECFPr2-1024 descriptors with dipeptide description, the curiousity picker scored a mean MCC of 0.57 at 20% of the training data, with a surprising peak mean MCC of 0.62 at 21% of the data (
Table 2 and
Figure S1a). In this situation there was thus no gain from joining complementary chemical space descriptions.
To address the question if compression to more or less hash bits in the ECFP representation of compounds had any effect on external performance, additional experiments were undertaken. Quite unexpectedly, a change from 1024 bits to either 512 or 4096 bits resulted in improvement (
Table 2 and
Figure S1c). While the expansion to more bits to reduce hashing collisions and empower the decision trees to find additional separation criteria appears rational, the details of how fewer bits could yield improved average performance is unclear and will require further investigation. The 4096-bit representation was notable in that prospective active learning performance was generally stable after achieving peak performance, unlike most other representations.
Finally, we additionally considered a pharmacophoric representation of compounds. The combination of CATS2D chemical descriptors (see Methods) and dipeptide frequency also resulted in improvement over the physicochemical description, and once again there was marked improvement when replacing the target identity descriptor with the dipeptide descriptor. Extension to tripeptide sequences led to a minor (+ ~0.03 MCC) yet improvement. Further testing with tetrapeptide subsequences showed no major change in long-term performance though marginal gains were made in early (5–15%) stages.
One remarkable feature for all external predictions using the curious picking strategy was that model performance peaked after learning from 20–25% of the training data. Further learning events yielded performances that were either unchanged or degraded. While performance in prior experiments [
15,
16,
27,
28] suggested that prediction performance monotonically increases with more data, those results were all in retrospective contexts; indeed, Rakers and colleagues [
16] found that in biological contexts of simulated de-orphanization, performance could recede with increases in training data. Here, this finding has been replicated in a chemical context.
In addition to the MCC, it is equally prudent to consider the F1 score, which places emphasis on the ability to detect active (inhibiting) annotations. Values in
Table 2, notably peak F1 scores, suggest that the majority of probe compounds could have their inhibiting annotation detected; corresponding MCC values were a bit lower, with the difference in metric values signaling that specificity (True Negative) prediction was the challenging aspect.
2.3. Deconstructing Behavior Dynamics by Active Projection
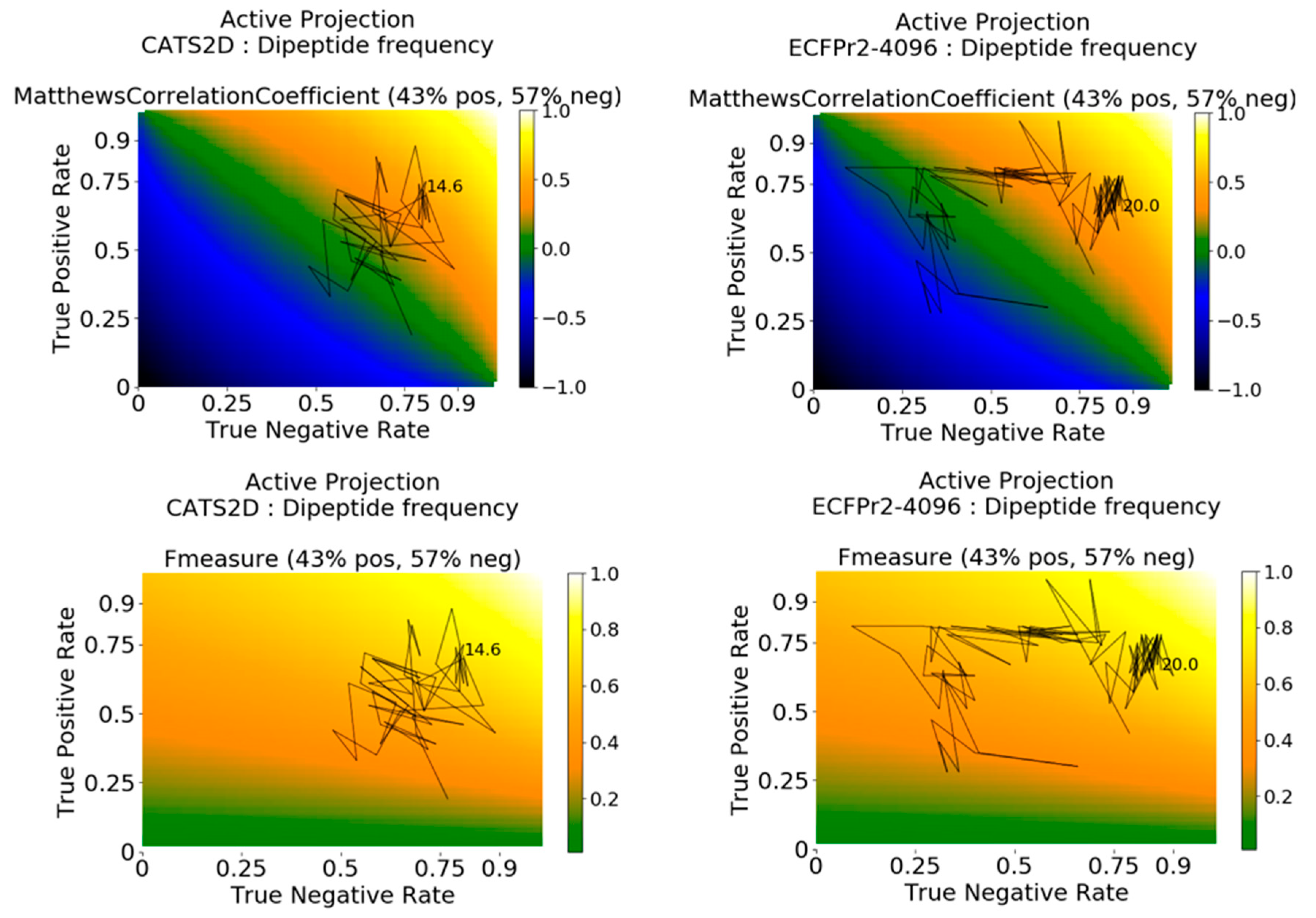
We sought to know further details about how the performance metrics evolve with iteration. To address this question, we applied the Active Projection method recently proposed [
29] (see Methods). This enabled us to track a model’s dynamic balance between its True Positive Rate (TPR, also called sensitivity) and True Negative Rate (TNR, also called specificity). Notably, the method visually clarifies why TPR or TNR can be high and yet MCC or F1 can be low for a model.
Active projections on the external probe dataset using curiosity-picked CATS2D-dipeptide and ECFP-dipeptide models are shown in
Figure 3. They clarify that predictions for initial iterations of the CATS2D-dipeptide model were heavily biased toward inactive predictions, as the TNR was close to 0.75 but the TPR rate was less than 0.25; the resulting MCC was approximately 0 (
Figure 2, left panels). However, the curiosity-picked examples iteratively contributed to a more balanced model, which can be seen from the active projection. Where the CATS2D-dipeptide model reaches its approximate peak of MCC = 0.50, the TPR and TNR were 0.75 and 0.80, respectively.
Interestingly, the corresponding ECFP-dipeptide model followed a different trajectory, with lower initial TNR, higher initial TPR, and an initial evolution that better predicted the inhibiting ligand-target pairs. At approximately 4% of the training data, the model has uncovered the patterns in the bioactive non-probe data with matching correspondence in the probe dataset, and further data selection from the non-probe data enhances selectivity prediction in the probe dataset. The model finally arrives in a stable zone around TNR 0.85, TPR 0.70 and MCC of 0.50 at 20% of training data (
Figure 3, right panels).
Analyses with pChem-dipeptide and pChem-tripeptide displayed jittering behavior in early iterations of learning. For instance, by use of active projection, we observed the dipeptide-based model had a high TNR before the model shifted focus to actives, and subsequently developed predictive ability of both actives and inactives (
Figure S2). For the tripeptide-based model, TPR was high early on with subsequent shift to inactives. Eventually both models stabilized at TPR of more than 0.80 and TNR of more than 0.65 with MCCs near 0.50. The dynamic behaviors of active learning were clearly deconvoluted through the use of active projection.
2.4. Feature Analysis and Sequential Decision Making from Features
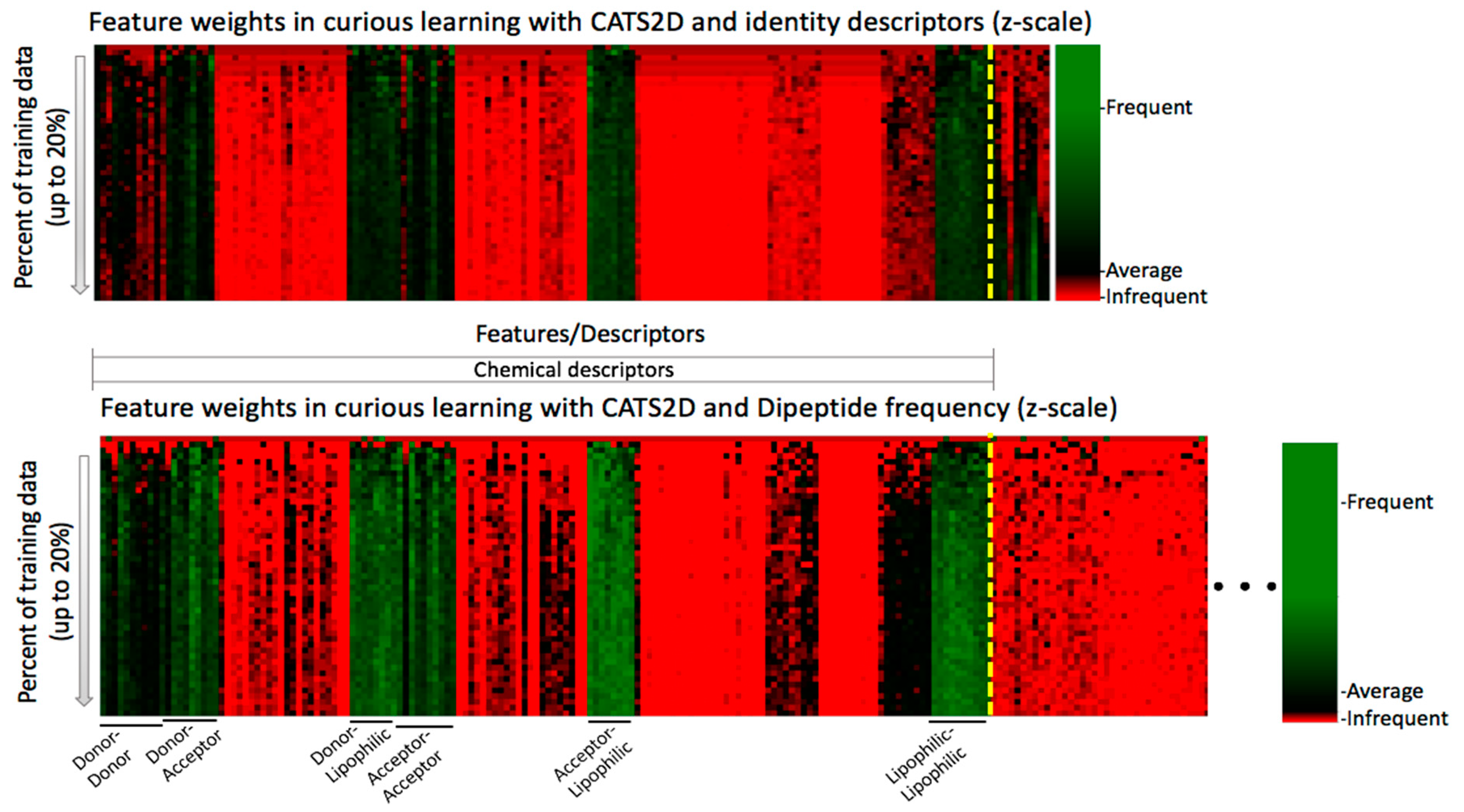
In chemogenomic active learning, the model is always trying to establish statistical patterns correlating chemical-protein descriptors to the endpoint (active or inactive in this case). Having identified that target descriptors did contribute to probe compound profile prediction, and having deconstructed the evolution of model performance by active projection, we also wanted to understand which of the descriptors contributed to this pair of results. To find out the answer, the time series evolution of feature weights was calculated and visualized by heatmap representation (see Methods).
Feature weights for the CATS2D-identity and CATS2D-dipeptide experiments, for up to 20% of training on the non-probe data, are shown in
Figure 3. It is essential to note that because weights differ in the experiments and the relative scalings of feature weights are different, only intra-feature comparison in a single experimental setting (i.e., fixed descriptors) is appropriate. For intra-experiment comparison, we can only compare the values of weights relative to the average weight per iteration. The feature weight heatmaps of CATS2D-identity and CATS2D-dipeptide experiments show that some of the features from CATS2D descriptors are highly weighted from early iterations (e.g., hydrogen bond donor atom pairs or donor-lipophilic atom pairs). Some of the CATS2D descriptors are almost never considered (e.g., positively/negatively-charged atom pair), either by coincidence due to random feature selection for evaluation, or due to lack of discriminative power. Other descriptors are initially uninformative or not considered yet are factored into models at later iterations. This pattern was present in both identity- and dipeptide-described experiments (
Figure 4). As a result of scaling, analysis of the CATS2D-dipeptide feature weights demonstrates that target descriptors were indeed contributing to decisions made by the random forest, albeit with lower frequency than the compound descriptors (
Figure 4).
Examination of the target descriptor weights in the CATS2D-identity experiment reveals that one of the identity descriptors had unusually high weighting. This would suggest that chemogenomic active learning is building per-target SAR models within the structure of the decision trees developed by placing target identification decision nodes at early decision branches, a hypothesis considered previously in the context of a much larger ligand-target dataset [
15]. This hypothesis is explored in further detail below.
A similar analysis was performed for the pChem-identity and pChem-dipeptide models (
Figure S3b). Most of the highly weighted features belonged to the physicochemical descriptors. Among the chemical descriptors, some of the features are always weighted low, i.e., the number of sp hybridized carbons, total charge, the numbers of 11- and 12-membered rings, and the distance/detour ring indices of order 6, 11, and 12. On the other hand, some of the features including the percentages of hydrogen, carbon, nitrogen and oxygen, total polarizable surface area, logP estimates, compound surface areas (total, acceptor, or donor), hydrophilic factor, Ghose-Crippen molecular refractivity, McGowan volume, and van der Waals volume from McGowan volume, are often weighed highly (
Figure S3b). Compound feature weightings were similar for the identity- and dipeptide-type models. Via feature weight analysis, it is clear that dipeptide frequencies were also factors in model behavior, though iteration-dependent feature weight evolution knowledge was difficult to extract.
Considering reproducible improvement from target descriptors (
Figure 2 and
Table 2) and that target feature values were part of decision tree formulation (
Figure 4), we wanted to even further clarify the behavior of the system. Toward this goal, we considered deciphering the decision making process through visualization of decision trees.
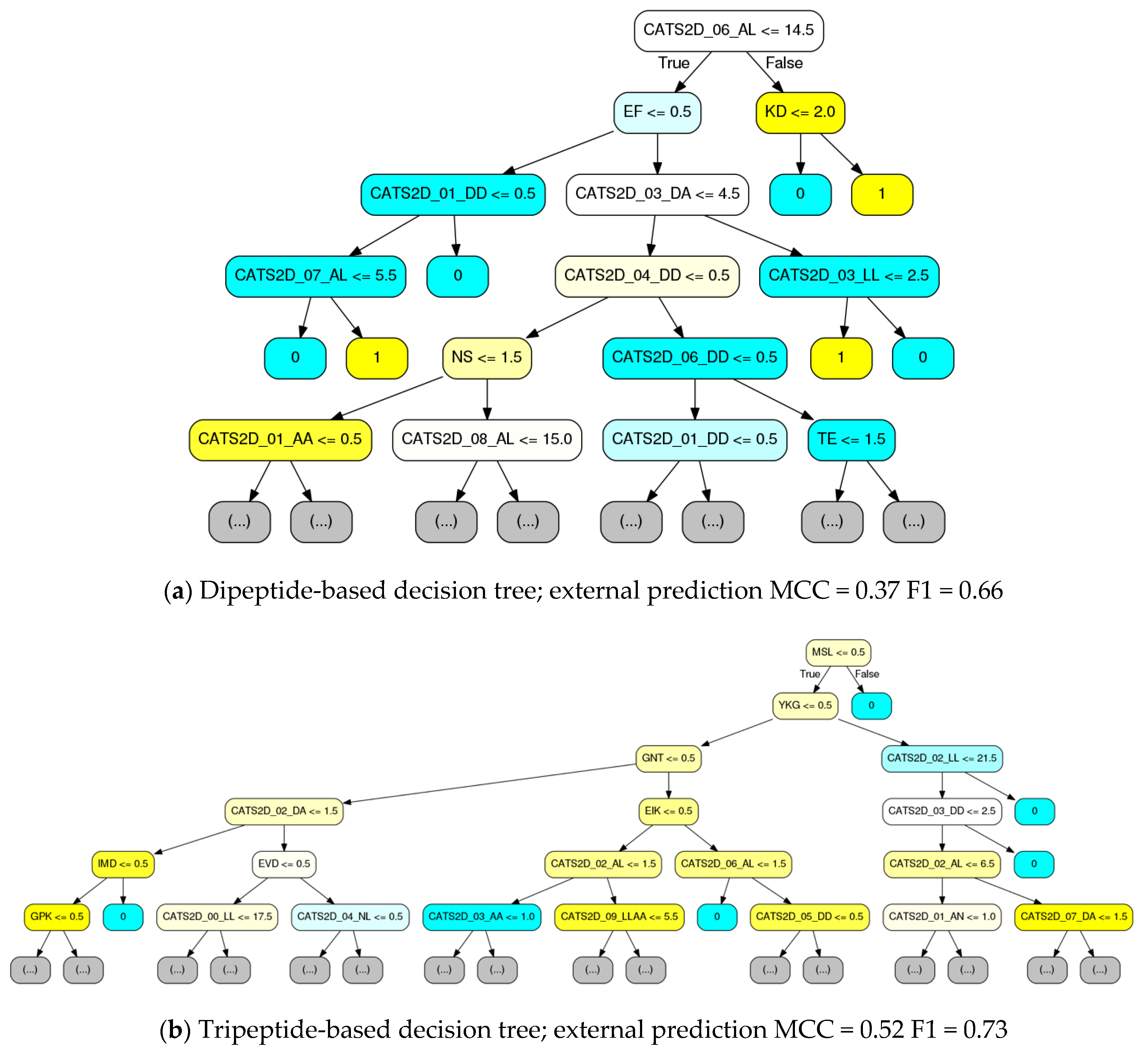
Representative decision trees from the chemogenomic random forest, where decision trees were fit at the 10% data level with curiosity picking, were rendered (
Figure 5). For comparison to
Figure 3 and
Figure 4, the CATS2D-dipeptide descriptor was again used. Complementary to
Figure 4, the order in which the features are considered is made unambiguous. To our knowledge, this is the first report of a chemogenomic model that unambiguously details the decision making process by a statistical pattern recognition (machine learning) estimator.
Consistent with
Figure 4, it is evident from this visualization that the majority of the decision nodes use the chemical descriptors. We see from
Figure 5 that pharmacophoric features, notably those groups labeled in
Figure 4, are highly present in early decision making nodes. While CATS2D considers all pairs of atom types separated by 1–10 bonds (0–9 intermediate atoms), and we see high weighting on all path lengths for some of the pharmacophoric groups (
Figure 4), short-distance atom pairs appear more frequent in early decision nodes. Nonetheless, it is clear that peptide subsequence information leads to both local SAR development as well as to final active/inactive decisions. By comparing with
Table 2, it is notable that the individual trees in
Figure 5 have performance approximate to or only marginally less than the forests that they belong to.
2.5. Consequences of Subsequence Absence-Presence Tests; Local SAR Model Development
Decision tree visualization contributed to a clarification that peptide sequences were in fact a part of the active/inactive decision process; comparing them to trees with identity descriptors confirmed their higher relative weights (frequency of use in decision making, data not shown). Yet, it is not intuitive from a biological standpoint how a single dipeptide or tripeptide without any other structural or biophysical context could explain activity. Spurred by a desire to unambiguously explain machine learning, an additional in-depth analysis was executed.
First, a multiple sequence alignment of the MMPs was performed using the Clustal Omega service [
30]. Decision tree branches were visually inspected for dipeptide and tripeptide sequences, then compared to the multiple sequence alignment. This revealed that some tripeptide sequences were unique to a single MMP (e.g., the tripeptide “MSL” was only present in MMP9, see
Figure 5). In some cases, (in)active decision was an immediate consequence of the subsequence presence test. In other cases, a collection of chemical descriptors was cascaded below the tripeptide until a final decision was made. This suggests that the tripeptide sequences are simply markers that provide fast access to the identification of a target and its SAR. Based on manual analysis of rendered trees, we have compiled a rough sketch of tripeptide-based decisions possible, available as
Figure S4a.
Reducing the resolution to dipeptides also demonstrated this effect, but decision nodes tended to use dipeptide count thresholds higher than one. Nonetheless, cascades of dipeptide decision nodes or alternating compound-protein nodes made clear that target identification with subsequent SAR generation was the dominant process for final classification of ligand-target interaction.
It is also worth considering that there were loose correlations in training data between MMP targets and their fraction of active data points. For example, the Pearson correlation between the fraction of actives per target and asparagine-valine (NV) dipeptide frequency was 0.43. Hence, despite lacking biophysical context, such a dipeptide fills the role of a first-line inquiry (
Figure S4b). After substantial manual inspection and discovery of such correlations, we proceeded to systematically quantify the percentages of decision trees that had target descriptors as their root decision node. Compared to the frequency of dipeptide descriptors as root decision nodes (95% confidence interval: 20–28% of decision trees per n = 5 random forests), tripeptides were significantly more present as roots (95% CI: 38–54%).
As mentioned above, all we could do after initial experiments (
Figure 2) was to speculate about correlations between descriptors and active/inactive relationships. However, combining those results with the feature weighting and detailed tree rendering (
Figure 5) has yielded a far clearer picture about how ligand-target interaction decisions are made, and how we should design computational experiments for interaction modeling.
2.6. Yoked Prediction by SVM and ANNs
In experiments so far, we have only trained random forest models. We also wanted to expand prospective evaluation to other machine learning algorithms such as SVMs and ANNs by feeding them with data points selected by active learning. At least for ANNs, the idea to simply swap out one algorithm with a replacement ANN is also referred to as “shallow deep learning” [
31]. In experiments with CATS2D-dipeptide descriptors, SVMs and ANNs were able to achieve relatively high maximum MCC values between 0.60 to 0.70 in several experimental settings. At first glance, these algorithms can be said to outperform the random forest models. Yet the metrics were generally unstable and small changes to the model configuration such as the number of hidden layers or regularization coefficient easily resulted in a model with much lower MCC of around 0.40, or a dysfunctional model which classifies all data points into one class and does not have measurable MCC (data not shown).
By comparing ANN models with a different number of layers, it was evident that adding more layers to ANN models did not significantly improve the MCC. Although the highest MCC of 0.72 was achieved with an ANN having 10 layers, MCC of approximately 0.70 could also be achieved by an ANN having six or seven layers. Even smaller, we found that an ANN with two hidden layers could achieve performance of MCC = 0.71 on the external set (75% of training data, regularization alpha = 0.1), yet the very same architecture and regularization could only achieve MCC = 0.46 when initialized using a different seed value. SVM models yielded identical prediction performance across multiple trials with different seeds, as the SVM algorithm is guaranteed to converge on a unique, optimum solution.
We should also note that some individual decision trees trained on 75% of the training data could achieve MCC of 0.72. In both RF and ANN, random number generator seed values affect the unitary decision elements of the estimators (respectively, decision trees and non-input units of ANNs). While the standard RF algorithm does not feature a feedback correction mechanism similar to backpropagation in ANNs, it would appear that weight initialization in ANNs is sensitive to the point that even application of backpropagation cannot adjust faulty-initialized decision criteria in network units enough to recover the network to a state of predictive utility. Since there is no prediction difference in an optimal decision tree and a full ANN model, we would also be led to believe that ANNs, as constructed here using the multi-layer perception implementation, are similarly performing the target identification and local SAR modeling steps by adjustment of (linear) perceptrons that are cascaded together for final decision making.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}