
Inhibition of the Self-Assembly of Aβ and of Tau by Polyphenols: Mechanistic Studies


Abstract
:1. Introduction
2. Chemical Properties of Polyphenols of Relevance to This Review
3. Polyphenols Inhibit Aβ Self-Assembly
3.1. Modulation of Aβ Production
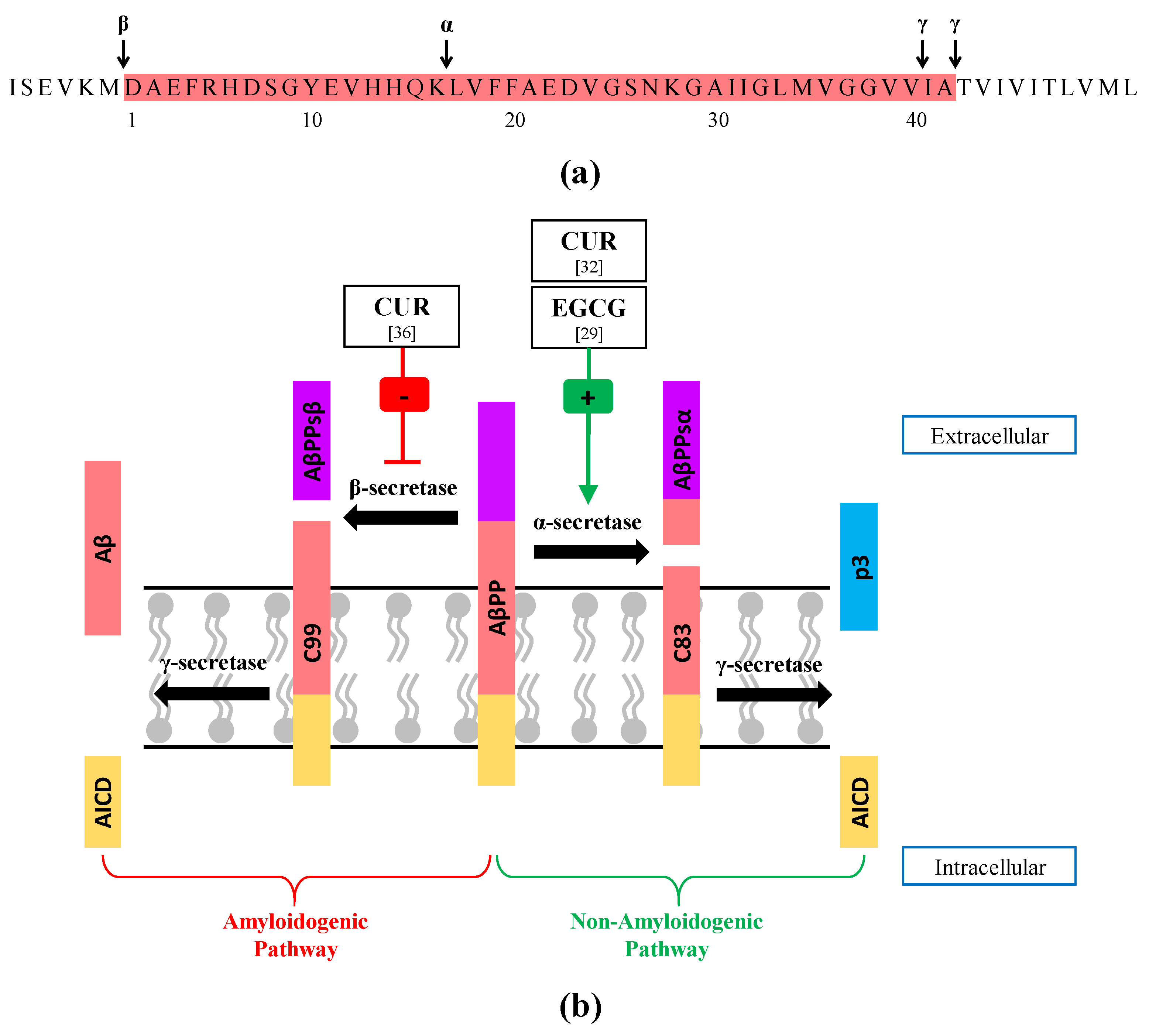
3.1.1. Enhancement of α-Secretase Activity
3.1.2. Inhibition of β-Secretase
3.2. Polyphenols Inhibit Toxic Aβ Oligomerization
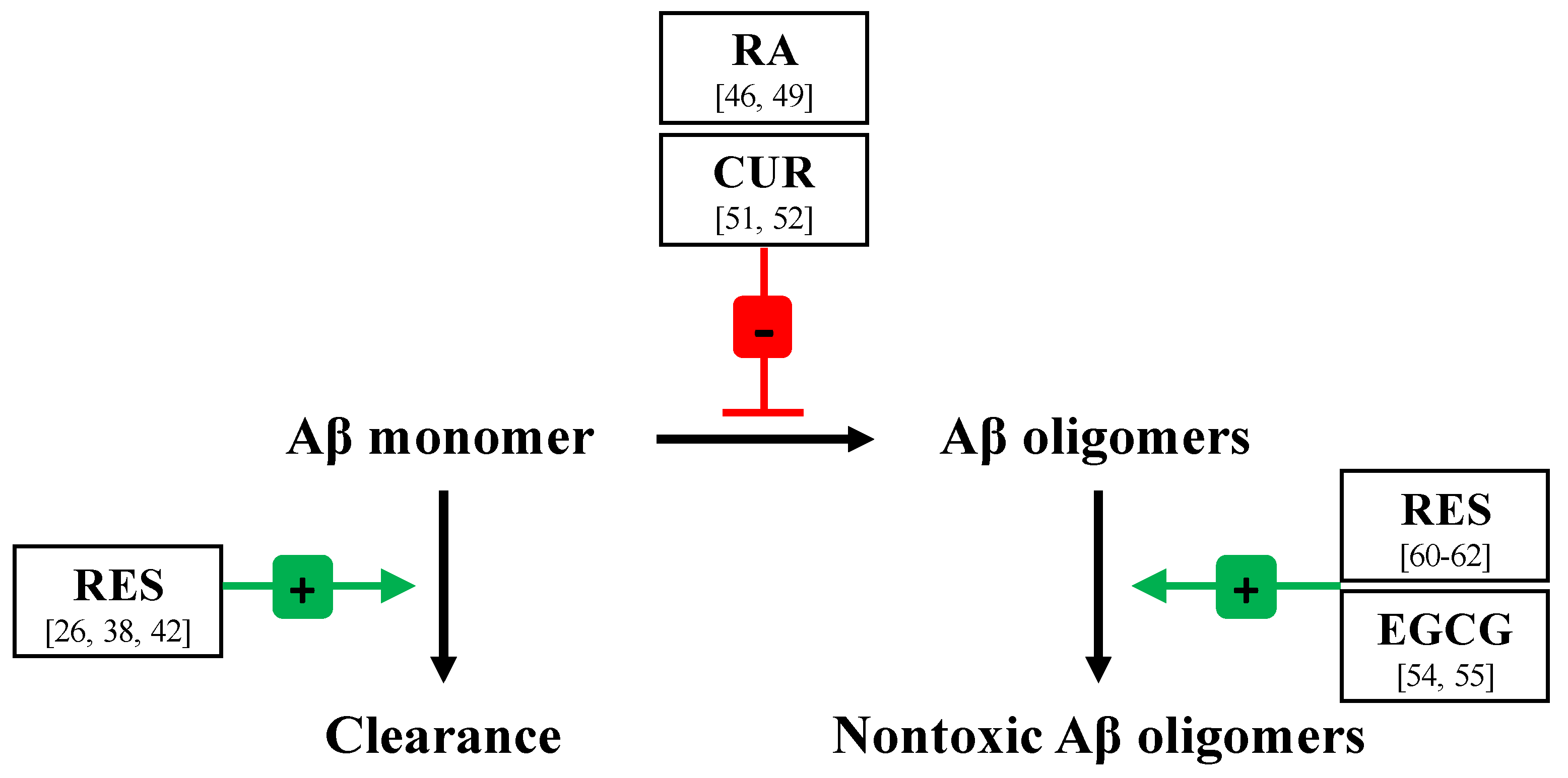
3.2.1. Enhancement of Aβ Monomer Clearance
3.2.2. Modulation of Aβ Monomer–Aβ Monomer Contacts
3.2.3. Remodeling of Aβ Oligomers to Nontoxic Forms
4. Polyphenols Inhibit Tau Self-Assembly
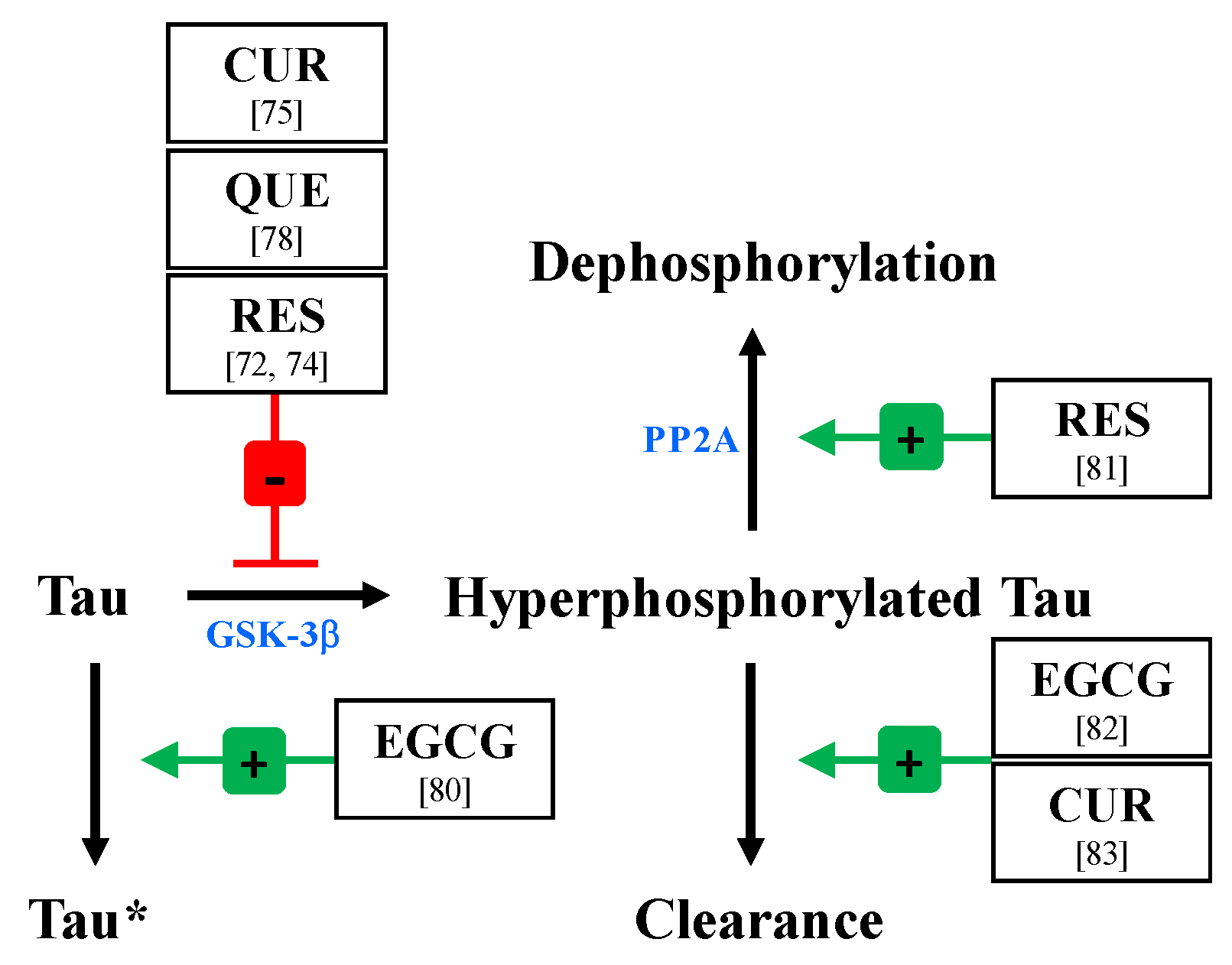
4.1. Polyphenols Modulate Tau Hyperphosphorylation
4.1.1. Inhibition of GSK-3β and Other Kinases
4.1.2. Remodeling of Tau to Tau*
4.1.3. Enhancement of PP2A Activity
4.1.4. Increased Clearance of Phosphorylated Tau
4.2. Polyphenols Inhibit Tau β-Sheet Formation
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef] [PubMed]
- Lazo, N.D.; Maji, S.K.; Fradinger, E.A.; Bitan, G.; Teplow, D.B. The amyloid-β protein. In Amyloid Proteins-the Beta Sheet Conformation and Disease; Sipe, J.D., Ed.; Wiley-VCH: Weinheim, Germany, 2005; pp. 385–491. [Google Scholar]
- Hayden, E.Y.; Teplow, D.B. Amyloid β-protein oligomers and Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The amyloid-β oligomer hypothesis: Beginning of the third decade. J. Alzheimers Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- Brglez Mojzer, E.; Knez Hrncic, M.; Skerget, M.; Knez, Z.; Bren, U. Polyphenols: Extraction methods, antioxidative action, bioavailability and anticarcinogenic effects. Molecules 2016, 21, 901. [Google Scholar] [CrossRef]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef]
- Stivala, L.A.; Savio, M.; Carafoli, F.; Perucca, P.; Bianchi, L.; Maga, G.; Forti, L.; Pagnoni, U.M.; Albini, A.; Prosperi, E.; et al. Specific structural determinants are responsible for the antioxidant activity and the cell cycle effects of resveratrol. J. Biol. Chem. 2001, 276, 22586–22594. [Google Scholar] [CrossRef]
- Bastianetto, S.; Zheng, W.H.; Quirion, R. Neuroprotective abilities of resveratrol and other red wine constituents against nitric oxide-related toxicity in cultured hippocampal neurons. Br. J. Pharmacol. 2000, 131, 711–720. [Google Scholar] [CrossRef] [Green Version]
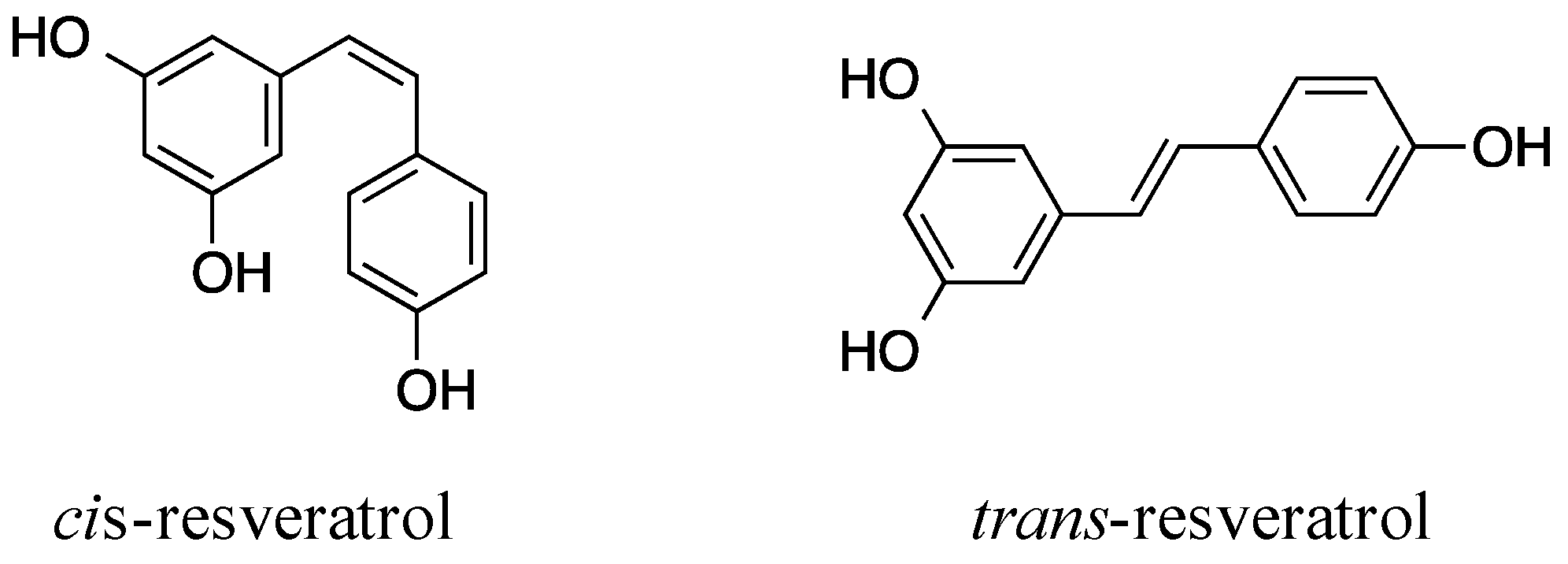
- Orallo, F. Comparative studies of the antioxidant effects of cis- and trans-resveratrol. Curr. Med. Chem. 2006, 13, 87–98. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Pasinetti, G.M.; Wang, J.; Ho, L.; Zhao, W.; Dubner, L. Roles of resveratrol and other grape-derived polyphenols in Alzheimer’s disease prevention and treatment. Biochim. Biophys. Acta 2015, 1852, 1202–1208. [Google Scholar] [CrossRef]
- Habtemariam, S. Molecular pharmacology of rosmarinic and salvianolic acids: Potential seeds for Alzheimer’s and vascular dementia drugs. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Lazo, N.D. Mechanistic studies of the inhibition of insulin fibril formation by rosmarinic acid. J. Phys. Chem. B 2018, 122, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Zhang, H.; Qi, R.; Tsao, R.; Mine, Y. Recent advances in the understanding of the health benefits and molecular mechanisms associated with green tea polyphenols. J. Agric. Food Chem. 2019, 67, 1029–1043. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Bharali, D.J.; Adhami, V.M.; Siddiqui, I.A.; Cui, H.; Shabana, S.M.; Mousa, S.A.; Mukhtar, H. Oral administration of naturally occurring chitosan-based nanoformulated green tea polyphenol EGCG effectively inhibits prostate cancer cell growth in a xenograft model. Carcinogenesis 2014, 35, 415–423. [Google Scholar] [CrossRef] [PubMed]
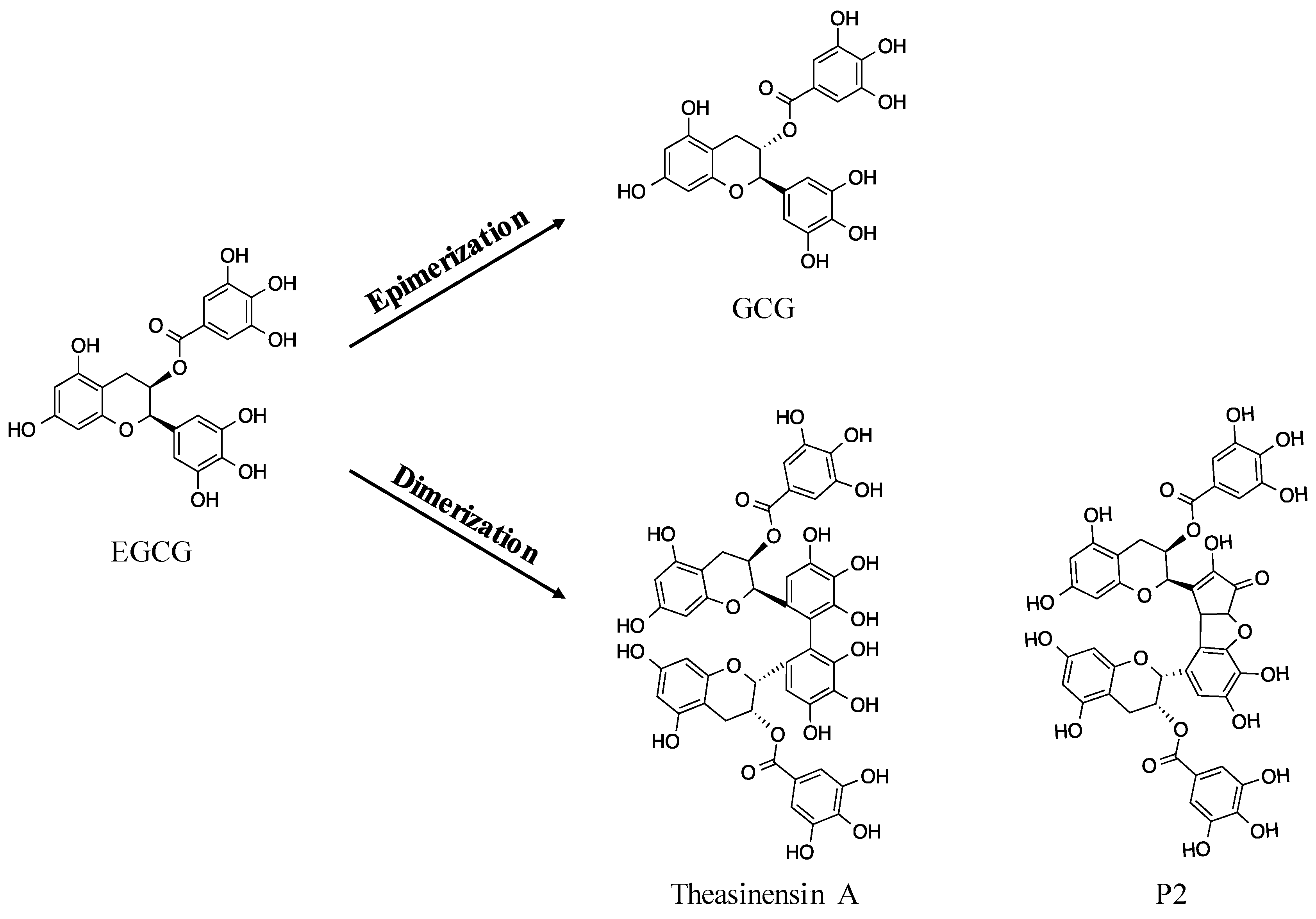
- Sang, S.; Lee, M.J.; Hou, Z.; Ho, C.T.; Yang, C.S. Stability of tea polyphenol (-)-epigallocatechin-3-gallate and formation of dimers and epimers under common experimental conditions. J. Agric. Food Chem. 2005, 53, 9478–9484. [Google Scholar] [CrossRef] [PubMed]
- Esatbeyoglu, T.; Huebbe, P.; Ernst, I.M.; Chin, D.; Wagner, A.E.; Rimbach, G. Curcumin—From molecule to biological function. Angew. Chem. Int. Ed. Engl. 2012, 51, 5308–5332. [Google Scholar] [CrossRef]
- Begum, A.N.; Jones, M.R.; Lim, G.P.; Morihara, T.; Kim, P.; Heath, D.D.; Rock, C.L.; Pruitt, M.A.; Yang, F.; Hudspeth, B.; et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2008, 326, 196–208. [Google Scholar] [CrossRef]
- Wang, Y.J.; Pan, M.H.; Cheng, A.L.; Lin, L.I.; Ho, Y.S.; Hsieh, C.Y.; Lin, J.K. Stability of curcumin in buffer solutions and characterization of its degradation products. J. Pharm. Biomed. Anal. 1997, 15, 1867–1876. [Google Scholar] [CrossRef]
- Verma, S.; Singh, A.; Mishra, A. Gallic acid: Molecular rival of cancer. Environ. Toxicol. Pharmacol. 2013, 35, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Cartron, E.; Fouret, G.; Carbonneau, M.A.; Lauret, C.; Michel, F.; Monnier, L.; Descomps, B.; Leger, C.L. Red-wine beneficial long-term effect on lipids but not on antioxidant characteristics in plasma in a study comparing three types of wine--description of two o-methylated derivatives of gallic acid in humans. Free Radic. Res. 2003, 37, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.V.; Badia, E.; Carbonneau, M.A.; Grimaldi, P.; Fouret, G.; Lauret, C.; Leger, C.L. Potential anti-atherogenic cell action of the naturally occurring 4-o-methyl derivative of gallic acid on ANG II-treated macrophages. FEBS Lett. 2004, 577, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.F.; Borge, G.I.A.; Piskula, M.; Tudose, A.; Tudoreanu, L.; Valentova, K.; Williamson, G.; Santos, C.N. Bioavailability of quercetin in humans with a focus on interindividual variation. Compr. Rev. Food Sci. Food Saf. 2018, 17, 714–731. [Google Scholar] [CrossRef]
- Krasinski, C.A.; Ivancic, V.A.; Zheng, Q.; Spratt, D.E.; Lazo, N.D. Resveratrol sustains insulin-degrading enzyme activity toward Aβ42. ACS Omega 2018, 3, 13275–13282. [Google Scholar] [CrossRef] [PubMed]
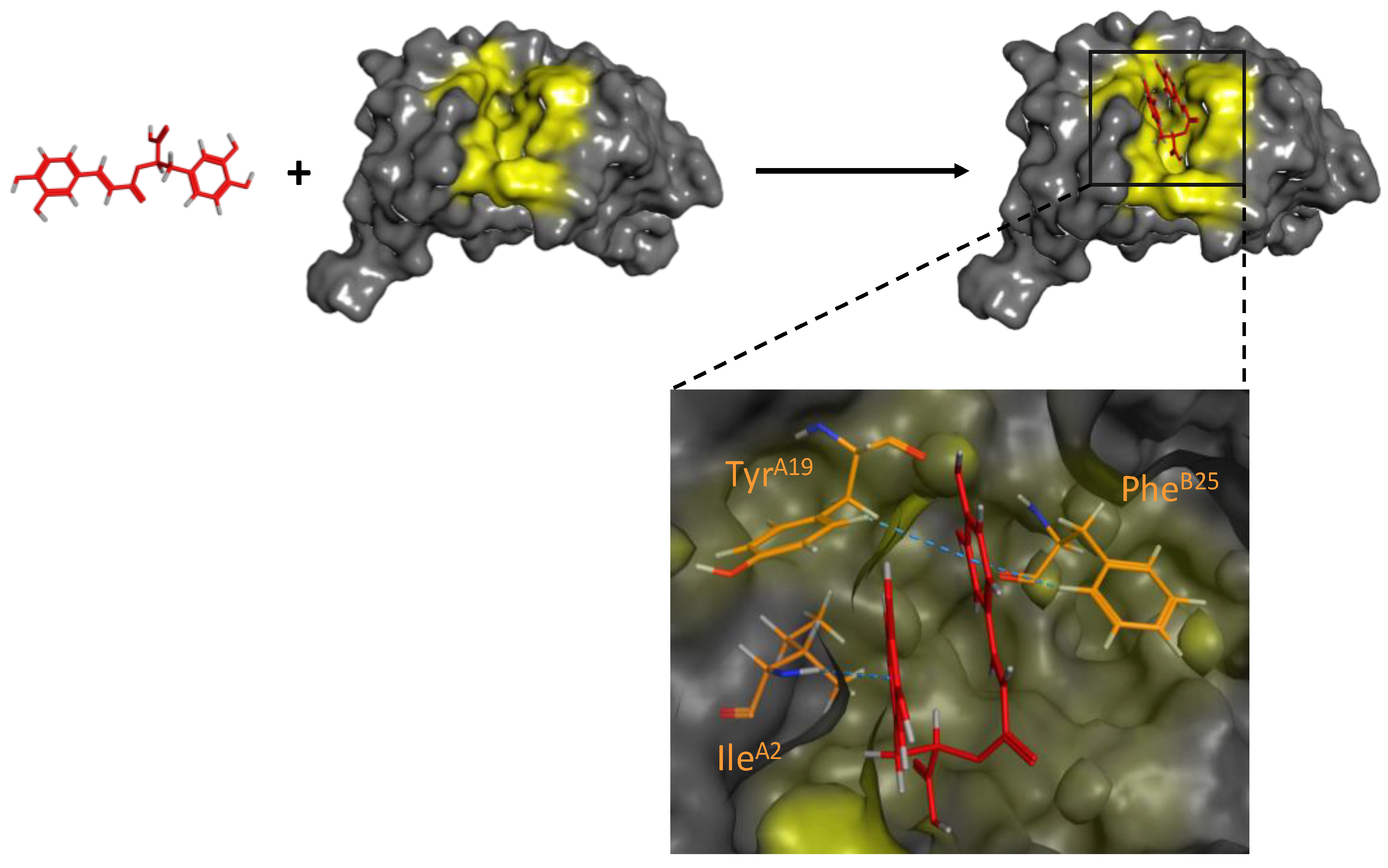
- Liu, G.; Gaines, J.C.; Robbins, K.J.; Lazo, N.D. Kinetic profile of amyloid formation in the presence of an aromatic inhibitor by nuclear magnetic resonance. ACS Med. Chem. Lett. 2012, 10, 856–859. [Google Scholar] [CrossRef]
- Sparks, S.; Liu, G.; Robbins, K.J.; Lazo, N.D. Curcumin modulates the self-assembly of the islet amyloid polypeptide by disassembling α-helix. Biochem. Biophys. Res. Commun. 2012, 422, 551–555. [Google Scholar] [CrossRef]
- Rezai-Zadeh, K.; Shytle, D.; Sun, N.; Mori, T.; Hou, H.; Jeanniton, D.; Ehrhart, J.; Townsend, K.; Zeng, J.; Morgan, D.; et al. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J. Neurosci. 2005, 25, 8807–8814. [Google Scholar] [CrossRef]
- Obregon, D.F.; Rezai-Zadeh, K.; Bai, Y.; Sun, N.; Hou, H.; Ehrhart, J.; Zeng, J.; Mori, T.; Arendash, G.W.; Shytle, D.; et al. ADAM10 activation is required for green tea (-)-epigallocatechin-3-gallate-induced α-secretase cleavage of amyloid precursor protein. J. Biol. Chem. 2006, 281, 16419–16427. [Google Scholar] [CrossRef]
- Wetzel, S.; Seipold, L.; Saftig, P. The metalloproteinase ADAM10: A useful therapeutic target? Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2071–2081. [Google Scholar] [CrossRef]
- Narasingappa, R.B.; Javagal, M.R.; Pullabhatla, S.; Htoo, H.H.; Rao, J.K.; Hernandez, J.F.; Govitrapong, P.; Vincent, B. Activation of α-secretase by curcumin-aminoacid conjugates. Biochem. Biophys. Res. Commun. 2012, 424, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Neumann, U.; Rueeger, H.; Machauer, R.; Veenstra, S.J.; Lueoend, R.M.; Tintelnot-Blomley, M.; Laue, G.; Beltz, K.; Vogg, B.; Schmid, P.; et al. A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-β and neuroinflammation in APP transgenic mice. Mol. Neurodegener. 2015, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Keskin, A.D.; Kekus, M.; Adelsberger, H.; Neumann, U.; Shimshek, D.R.; Song, B.; Zott, B.; Peng, T.; Forstl, H.; Staufenbiel, M.; et al. BACE inhibition-dependent repair of Alzheimer’s pathophysiology. PNAS 2017, 114, 8631–8636. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized trial of verubecestat for prodromal Alzheimer’s disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kim, J.R.; Lee, S.B.; Kim, Y.J.; Jung, M.Y.; Kwon, H.W.; Ahn, Y.J. Effects of curcuminoids identified in rhizomes of curcuma longa on BACE-1 inhibitory and behavioral activity and lifespan of Alzheimer’s disease drosophila models. BMC Complement. Altern. Med. 2014, 14, 88. [Google Scholar] [CrossRef]
- Cheng, X.R.; Zhou, J.W.; Zhou, Y.; Cheng, J.P.; Yang, R.F.; Zhou, W.X.; Zhang, Y.X.; Yun, L.H. The green tea polyphenol (2)-epigallocatechin-3-gallate (EGCG) is not a β-secretase inhibitor. Bioorg. Med. Chem. Lett. 2012, 22, 1408–1414. [Google Scholar] [CrossRef]
- Marambaud, P.; Zhao, H.; Davies, P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-β peptides. J. Biol. Chem. 2005, 280, 37377–37382. [Google Scholar] [CrossRef]
- Mori, T.; Koyama, N.; Guillot-Sestier, M.V.; Tan, J.; Town, T. Ferulic acid is a nutraceutical β-secretase modulator that improves behavioral impairment and Alzheimer-like pathology in transgenic mice. PLoS ONE 2013, 8, e55774. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jaggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef]
- Mori, T.; Koyama, N.; Tan, J.; Segawa, T.; Maeda, M.; Town, T. Combined treatment with the phenolics (-)-epigallocatechin-3-gallate and ferulic acid improves cognition and reduces Alzheimer-like pathology in mice. J. Biol. Chem. 2019, 294, 2714–2731. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-β peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef] [PubMed]
- Rege, S.D.; Geetha, T.; Broderick, T.L.; Babu, J.R. Resveratrol protects β amyloid-induced oxidative damage and memory associated proteins in H19-7 hippocampal neuronal cells. Curr. Alzheimer Res. 2015, 12, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Lazo, N.D.; Grant, M.A.; Condron, M.C.; Rigby, A.C.; Teplow, D.B. On the nucleation of amyloid β-protein monomer folding. Protein Sci. 2005, 14, 1581–1596. [Google Scholar] [CrossRef] [PubMed]
- Walti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Bockmann, A.; Guntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. PNAS 2016, 113, E4976–E4984. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-β aggregation pathway. Am. J. Pathol. 2009, 175, 2557–2565. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the β-amyloid precursor protein in familial Alzheimer’s disease increases β-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef]
- Glabe, C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008, 283, 29639–29643. [Google Scholar] [CrossRef]
- Ono, K.; Li, L.; Takamura, Y.; Yoshiike, Y.; Zhu, L.; Han, F.; Mao, X.; Ikeda, T.; Takasaki, J.; Nishijo, H.; et al. Phenolic compounds prevent amyloid β-protein oligomerization and synaptic dysfunction by site-specific binding. J. Biol. Chem. 2012, 287, 14631–14643. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef]
- Reinke, A.A.; Gestwicki, J.E. Structure-activity relationships of amyloid beta-aggregation inhibitors based on curcumin: Influence of linker length and flexibility. Chem. Biol. Drug Des. 2007, 70, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. Egcg remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. PNAS 2010, 107, 7710–7715. [Google Scholar] [CrossRef]
- Lopez del Amo, J.M.; Fink, U.; Dasari, M.; Grelle, G.; Wanker, E.E.; Bieschke, J.; Reif, B. Structural properties of EGCG-induced, nontoxic Alzheimer’s disease Aβ oligomers. J. Mol. Biol. 2012, 421, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; VanSchouwen, B.; Jafari, N.; Ni, X.; Ortega, J.; Melacini, G. Molecular mechanism for the (-)-epigallocatechin gallate-induced toxic to nontoxic remodeling of Aβ oligomers. J. Am. Chem. Soc. 2017, 139, 13720–13734. [Google Scholar] [CrossRef]
- Nguyen, P.; Derreumaux, P. Understanding amyloid fibril nucleation and Aβ oligomer/drug interactions from computer simulations. Acc. Chem. Res. 2014, 47, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, J.; Derreumaux, P.; Mu, Y. Molecular mechanism of the inhibition of EGCG on the Alzheimer Aβ(1-42) dimer. J. Phys. Chem. B 2013, 117, 3993–4002. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, X.P.; Yang, S.G.; Wang, Y.J.; Zhang, X.; Du, X.T.; Sun, X.X.; Zhao, M.; Huang, L.; Liu, R.T. Resveratrol inhibits β-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. Neurotoxicology 2009, 30, 986–995. [Google Scholar] [CrossRef]
- Ladiwala, A.R.; Lin, J.C.; Bale, S.S.; Marcelino-Cruz, A.M.; Bhattacharya, M.; Dordick, J.S.; Tessier, P.M. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Aβ into off-pathway conformers. J. Biol. Chem. 2010, 285, 24228–24237. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Aucoin, D.; Ahmed, M.; Ziliox, M.; Van Nostrand, W.E.; Smith, S.O. Capping of Aβ42 oligomers by small molecule inhibitors. Biochemistry 2014, 53, 7893–7903. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.D.; Grundkeiqbal, I.; Iqbal, K.; Koudinov, A.R.; Koudinova, N.V.; Kumar, A.; Beavis, R.C.; Ghiso, J. Alzheimers disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules biochemical characterization of Alzheimers soluble amyloid β protein in human cerebrospinal fluid-association with high density lipoproteins. Nat. Med. 1996, 2, 783–787. [Google Scholar] [PubMed]
- Alonso, A.D.; Zaidi, T.; Novak, M.; Barra, H.S.; Grundke-Iqbal, I.; Iqbal, K. Interaction of tau isoforms with Alzheimer’s disease abnormally hyperphosphorylated tau and in vitro phosphorylation into the disease-like protein. J. Biol. Chem. 2001, 276, 37967–37973. [Google Scholar] [PubMed]
- Jeganathan, S.; von Bergen, M.; Mandelkow, E.M.; Mandelkow, E. The natively unfolded character of tau and its aggregation to Alzheimer-like paired helical filaments. Biochemistry 2008, 47, 10526–10539. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef] [PubMed]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef]
- Guillozet-Bongaarts, A.L.; Cahill, M.E.; Cryns, V.L.; Reynolds, M.R.; Berry, R.W.; Binder, L.I. Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: In vitro evidence and implications for tangle formation in vivo. J. Neurochem. 2006, 97, 1005–1014. [Google Scholar] [CrossRef]
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927. [Google Scholar] [CrossRef]
- Cavallini, A.; Brewerton, S.; Bell, A.; Sargent, S.; Glover, S.; Hardy, C.; Moore, R.; Calley, J.; Ramachandran, D.; Poidinger, M.; et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J. Biol. Chem. 2013, 288, 23331–23347. [Google Scholar] [CrossRef]
- Medina, M.; Garrido, J.J.; Wandosell, F.G. Modulation of GSK-3 as a therapeutic strategy on tau pathologies. Front. Mol. Neurosci. 2011, 4, 24. [Google Scholar] [CrossRef]
- He, X.; Li, Z.; Rizak, J.D.; Wu, S.; Wang, Z.; He, R.; Su, M.; Qin, D.; Wang, J.; Hu, X. Resveratrol attenuates formaldehyde induced hyperphosphorylation of tau protein and cytotoxicity in N2A cells. Front. Neurosci. 2016, 10, 598. [Google Scholar] [CrossRef] [PubMed]
- Akiguchi, I.; Pallas, M.; Budka, H.; Akiyama, H.; Ueno, M.; Han, J.; Yagi, H.; Nishikawa, T.; Chiba, Y.; Sugiyama, H.; et al. SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio takeda’s legacy and future directions. Neuropathology 2017, 37, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Porquet, D.; Casadesus, G.; Bayod, S.; Vicente, A.; Canudas, A.M.; Vilaplana, J.; Pelegri, C.; Sanfeliu, C.; Camins, A.; Pallas, M.; et al. Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age (Dordr) 2013, 35, 1851–1865. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sui, H.; Zheng, Y.; Jiang, Y.; Shi, Y.; Liang, J.; Zhao, L. Curcumin-primed exosomes potently ameliorate cognitive function in AD mice by inhibiting hyperphosphorylation of the tau protein through the AKT/GSK-3β pathway. Nanoscale 2019, 11, 7481–7496. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, E.R.; Kalinine, E.; Haas, C.B.; Torrez, V.R.; Souza, D.O.; Muller, A.P.; Portela, L.V. Pretreatment with memantine prevents Alzheimer-like alterations induced by intrahippocampal okadaic acid administration in rats. Curr. Alzheimer Res. 2012, 9, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Broetto, N.; Hansen, F.; Brolese, G.; Batassini, C.; Lirio, F.; Galland, F.; Dos Santos, J.P.; Dutra, M.F.; Goncalves, C.A. Intracerebroventricular administration of okadaic acid induces hippocampal glucose uptake dysfunction and tau phosphorylation. Brain Res. Bull. 2016, 124, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Luo, T.; Li, S.; Zhou, Y.; Shen, X.Y.; He, F.; Xu, J.; Wang, H.Q. Quercetin protects against okadaic acid-induced injury via MAPK and PI3K/AKT/GSK3β signaling pathways in HT22 hippocampal neurons. PLoS ONE 2016, 11, e0152371. [Google Scholar]
- Guo, Y.; Zhao, Y.; Nan, Y.; Wang, X.; Chen, Y.; Wang, S. (−)-epigallocatechin-3-gallate ameliorates memory impairment and rescues the abnormal synaptic protein levels in the frontal cortex and hippocampus in a mouse model of Alzheimer’s disease. Neuroreport 2017, 28, 590–597. [Google Scholar] [CrossRef]
- Gueroux, M.; Fleau, C.; Slozeck, M.; Laguerre, M.; Pianet, I. Epigallocatechin 3-gallate as an inhibitor of tau phosphorylation and aggregation: A molecular and structural insight. J. Prev. Alzheimer Dis. 2017, 4, 218–225. [Google Scholar]
- Schweiger, S.; Matthes, F.; Posey, K.; Kickstein, E.; Weber, S.; Hettich, M.M.; Pfurtscheller, S.; Ehninger, D.; Schneider, R.; Krauss, S. Resveratrol induces dephosphorylation of tau by interfering with the MID1-PP2A complex. Sci. Rep. 2017, 7, 13753. [Google Scholar] [CrossRef]
- Chesser, A.S.; Ganeshan, V.; Yang, J.; Johnson, G.V. Epigallocatechin-3-gallate enhances clearance of phosphorylated tau in primary neurons. Nutr. Neurosci. 2016, 19, 21–31. [Google Scholar] [CrossRef] [PubMed]
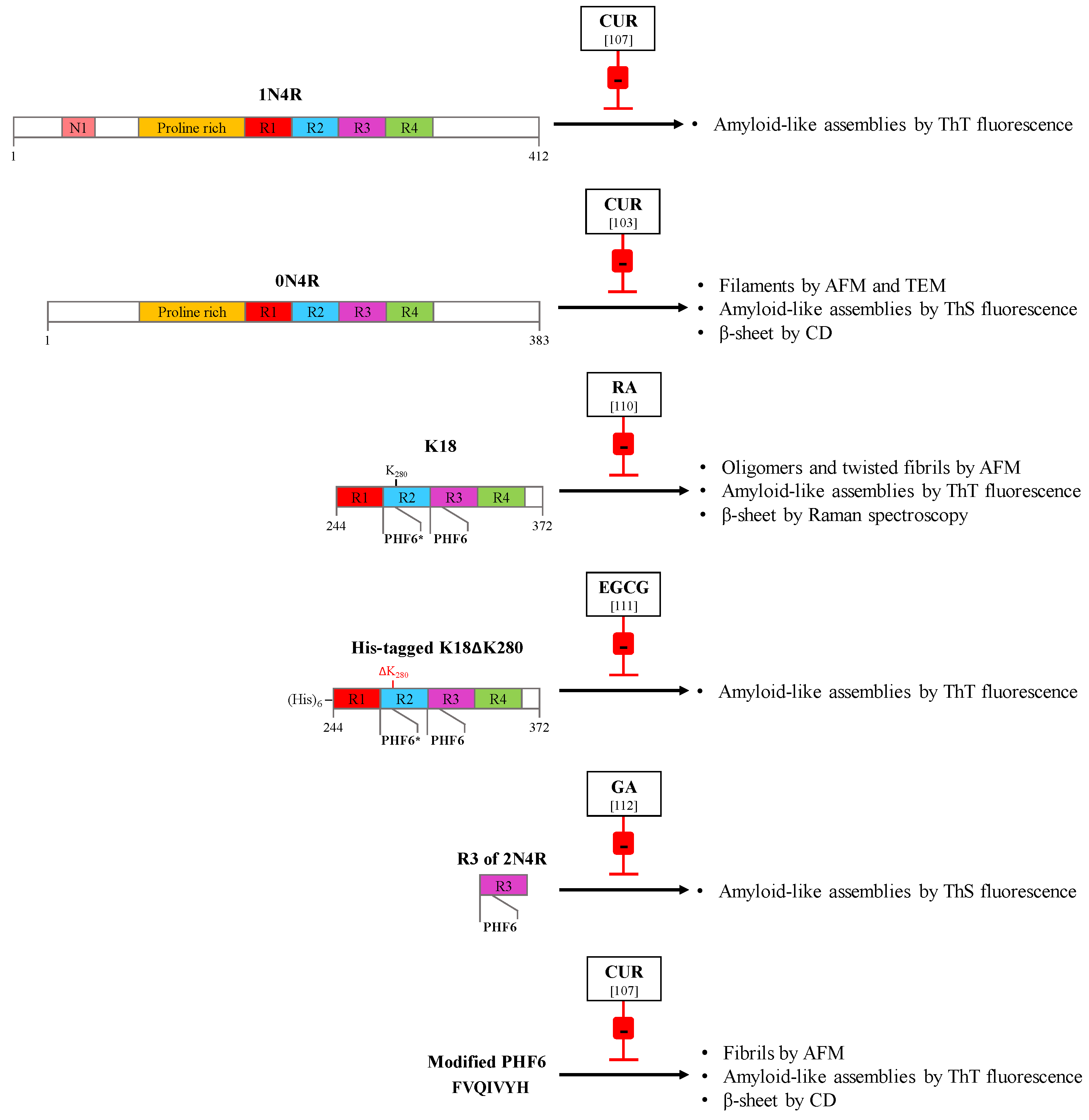
- Ma, Q.L.; Zuo, X.; Yang, F.; Ubeda, O.J.; Gant, D.J.; Alaverdyan, M.; Teng, E.; Hu, S.; Chen, P.P.; Maiti, P.; et al. Curcumin suppresses soluble tau dimers and corrects molecular chaperone, synaptic, and behavioral deficits in aged human tau transgenic mice. J. Biol. Chem. 2013, 288, 4056–4065. [Google Scholar] [CrossRef] [PubMed]
- Polydoro, M.; Acker, C.M.; Duff, K.; Castillo, P.E.; Davies, P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J. Neurosci. 2009, 29, 10741–10749. [Google Scholar] [CrossRef] [PubMed]
- Gota, V.S.; Maru, G.B.; Soni, T.G.; Gandhi, T.R.; Kochar, N.; Agarwal, M.G. Safety and pharmacokinetics of a solid lipid curcumin particle formulation in osteosarcoma patients and healthy volunteers. J. Agric. Food Chem. 2010, 58, 2095–2099. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J. Alzheimer Dis. 2008, 14, 393–399. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Biernat, J.; von Bergen, M.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Sites of tau important for aggregation populate β-structure and bind to microtubules and polyanions. J. Biol. Chem. 2005, 280, 24978–24986. [Google Scholar] [CrossRef] [PubMed]
- Barghorn, S.; Zheng-Fischhofer, Q.; Ackmann, M.; Biernat, J.; von Bergen, M.; Mandelkow, E.M.; Mandelkow, E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 2000, 39, 11714–11721. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Crowther, R.A. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999, 450, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.M. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef]
- Wilson, D.M.; Binder, L.I. Free fatty acids stimulate the polymerization of tau and amyloid β peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am. J. Pathol. 1997, 150, 2181–2195. [Google Scholar] [PubMed]
- Berriman, J.; Serpell, L.C.; Oberg, K.A.; Fink, A.L.; Goedert, M.; Crowther, R.A. Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-β structure. PNAS 2003, 100, 9034–9038. [Google Scholar] [CrossRef] [PubMed]
- Daebel, V.; Chinnathambi, S.; Biernat, J.; Schwalbe, M.; Habenstein, B.; Loquet, A.; Akoury, E.; Tepper, K.; Muller, H.; Baldus, M.; et al. β-sheet core of tau paired helical filaments revealed by solid-state NMR. J. Am. Chem. Soc. 2012, 134, 13982–13989. [Google Scholar] [CrossRef] [PubMed]
- von Bergen, M.; Barghorn, S.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Tau aggregation is driven by a transition from random coil to β sheet structure. Biochim. Biophys. Acta 2005, 1739, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Novak, M.; Thogersen, H.C.; Edwards, P.C.; Runswick, M.J.; Jakes, R.; Walker, J.E.; Milstein, C.; Rother, M.; Klug, A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer’s disease. PNAS 1988, 85, 4506–4510. [Google Scholar] [CrossRef] [PubMed]
- Barghorn, S.; Davies, P.; Mandelkow, E. Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro are based on β-structure in the core domain. Biochemistry 2004, 43, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Binder, L.I.; Guillozet-Bongaarts, A.L.; Garcia-Sierra, F.; Berry, R.W. Tau, tangles, and Alzheimer’s disease. Biochim. Biophys. Acta 2005, 1739, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Jung, Y.J.; Chinnathambi, S.; Mandelkow, E.M.; Mandelkow, E.; Muller, D.J. Human tau isoforms assemble into ribbon-like fibrils that display polymorphic structure and stability. J. Biol. Chem. 2010, 285, 27302–27313. [Google Scholar] [CrossRef] [PubMed]
- Santa-Maria, I.; Diaz-Ruiz, C.; Ksiezak-Reding, H.; Chen, A.; Ho, L.; Wang, J.; Pasinetti, G.M. GSPE interferes with tau aggregation in vivo: Implication for treating tauopathy. Neurobiol. Aging 2012, 33, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Murphy, M.P.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef]
- Wang, J.; Bi, W.; Cheng, A.; Freire, D.; Vempati, P.; Zhao, W.; Gong, B.; Janle, E.M.; Chen, T.Y.; Ferruzzi, M.G.; et al. Targeting multiple pathogenic mechanisms with polyphenols for the treatment of Alzheimer’s disease-experimental approach and therapeutic implications. Front. Aging Neurosci. 2014, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.S.; Bhaumik, P.; Panda, D. Curcumin inhibits tau aggregation and disintegrates preformed tau filaments in vitro. J. Alzheimer Dis. 2017, 60, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Robbins, K.J.; Liu, G.; Lin, G.; Lazo, N.D. Detection of strongly bound thioflavin T species in amyloid fibrils by ligand-detected 1H NMR. J. Phys. Chem. Lett. 2011, 2, 735–740. [Google Scholar] [CrossRef]
- Robbins, K.J.; Liu, G.; Selmani, V.; Lazo, N.D. Conformational analysis of thioflavin T bound to the surface of amyloid fibrils. Langmuir 2012, 28, 16490–16495. [Google Scholar] [CrossRef] [PubMed]
- Ivancic, V.A.; Ekanayake, O.; Lazo, N.D. Binding modes of thioflavin T on the surface of amyloid fibrils studied by NMR. Chemphyschem 2016, 17, 2461–2464. [Google Scholar] [CrossRef] [PubMed]
- Bijari, N.; Balalaie, S.; Akbari, V.; Golmohammadi, F.; Moradi, S.; Adibi, H.; Khodarahmi, R. Effective suppression of the modified PHF6 peptide/1N4R tau amyloid aggregation by intact curcumin, not its degradation products: Another evidence for the pigment as preventive/therapeutic “functional food”. Int. J.Biol. Macromol. 2018, 120, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. PNAS USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005, 280, 7614–7623. [Google Scholar] [CrossRef] [PubMed]
- Cornejo, A.; Aguilar Sandoval, F.; Caballero, L.; Machuca, L.; Munoz, P.; Caballero, J.; Perry, G.; Ardiles, A.; Areche, C.; Melo, F. Rosmarinic acid prevents fibrillization and diminishes vibrational modes associated to β sheet in tau protein linked to Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2017, 32, 945–953. [Google Scholar] [CrossRef]
- Wobst, H.J.; Sharma, A.; Diamond, M.I.; Wanker, E.E.; Bieschke, J. The green tea polyphenol (-)-epigallocatechin gallate prevents the aggregation of tau protein into toxic oligomers at substoichiometric ratios. FEBS Lett. 2015, 589, 77–83. [Google Scholar] [CrossRef]
- Yao, J.; Gao, X.; Sun, W.; Yao, T.; Shi, S.; Ji, L. Molecular hairpin: A possible model for inhibition of tau aggregation by tannic acid. Biochemistry 2013, 52, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Prusiner, S.B. β-amyloid prions and the pathobiology of Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 2018, 8, a023507. [Google Scholar] [CrossRef] [PubMed]
- Mudher, A.; Colin, M.; Dujardin, S.; Medina, M.; Dewachter, I.; Alavi Naini, S.M.; Mandelkow, E.M.; Mandelkow, E.; Buee, L.; Goedert, M.; et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol. Commun. 2017, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, B.; Stancu, I.C.; Buist, A.; Bird, M.; Wang, P.; Vanoosthuyse, A.; Van Kolen, K.; Verheyen, A.; Kienlen-Campard, P.; Octave, J.N.; et al. Heterotypic seeding of tau fibrillization by pre-aggregated Aβ provides potent seeds for prion-like seeding and propagation of tau-pathology in vivo. Acta Neuropathol. 2016, 131, 549–569. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.E.; DeVos, S.L.; Dujardin, S.; Corjuc, B.; Gor, R.; Gonzalez, J.; Roe, A.D.; Frosch, M.P.; Pitstick, R.; Carlson, G.A.; et al. Enhanced tau aggregation in the presence of amyloid β. Am. J. Pathol. 2017, 187, 1601–1612. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
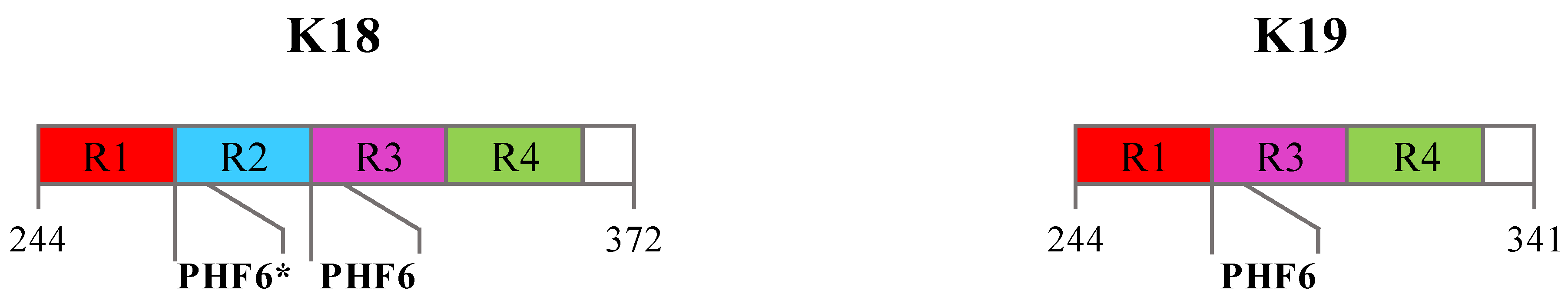{kind=link}
{kind=link}
| Molecule Common Source/s Solubility in H2O | Chemical Structure |
|---|---|
| Resveratrol |  |
| red wine & red grapes | C14H12O3 |
| sparingly soluble | 5-[(E)-2-(4-hydroxyphenyl)ethenyl]benzene-1,3-diol |
| Rosmarinic acid |  |
| rosemary, basil & sage | C18H16O8 |
| soluble | (2R)-3-(3,4-dihydroxyphenyl)-2-[(E)-3-(3,4-dihydroxyphenyl)prop-2-enoyl]oxypropanoic acid |
| Epigallocatechin-3-gallate |  |
| green tea | C22H18O11 |
| slightly soluble | [(2R,3R)-5,7-dihydroxy-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-chromen-3-yl] 3,4,5-trihydroxybenzoate |
| Curcumin |  |
| turmeric | C21H20O6 |
| sparingly soluble | (1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione |
| Gallic acid |  |
| black tea, fruits & nuts | C7H6O5 |
| sparingly soluble | 3,4,5-trihydroxybenzoic acid |
| Quercetin |  |
| onions, spinach & apples | C15H10O7 |
| sparingly soluble | 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxychromen-4-one |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Q.; Kebede, M.T.; Kemeh, M.M.; Islam, S.; Lee, B.; Bleck, S.D.; Wurfl, L.A.; Lazo, N.D. Inhibition of the Self-Assembly of Aβ and of Tau by Polyphenols: Mechanistic Studies. Molecules 2019, 24, 2316. https://doi.org/10.3390/molecules24122316
Zheng Q, Kebede MT, Kemeh MM, Islam S, Lee B, Bleck SD, Wurfl LA, Lazo ND. Inhibition of the Self-Assembly of Aβ and of Tau by Polyphenols: Mechanistic Studies. Molecules. 2019; 24(12):2316. https://doi.org/10.3390/molecules24122316
Chicago/Turabian StyleZheng, Qiuchen, Micheal T. Kebede, Merc M. Kemeh, Saadman Islam, Bethany Lee, Stuart D. Bleck, Liliana A. Wurfl, and Noel D. Lazo. 2019. "Inhibition of the Self-Assembly of Aβ and of Tau by Polyphenols: Mechanistic Studies" Molecules 24, no. 12: 2316. https://doi.org/10.3390/molecules24122316