The Amino Acid Composition of Quadruplex Binding Proteins Reveals a Shared Motif and Predicts New Potential Quadruplex Interactors

 ,
, 
 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
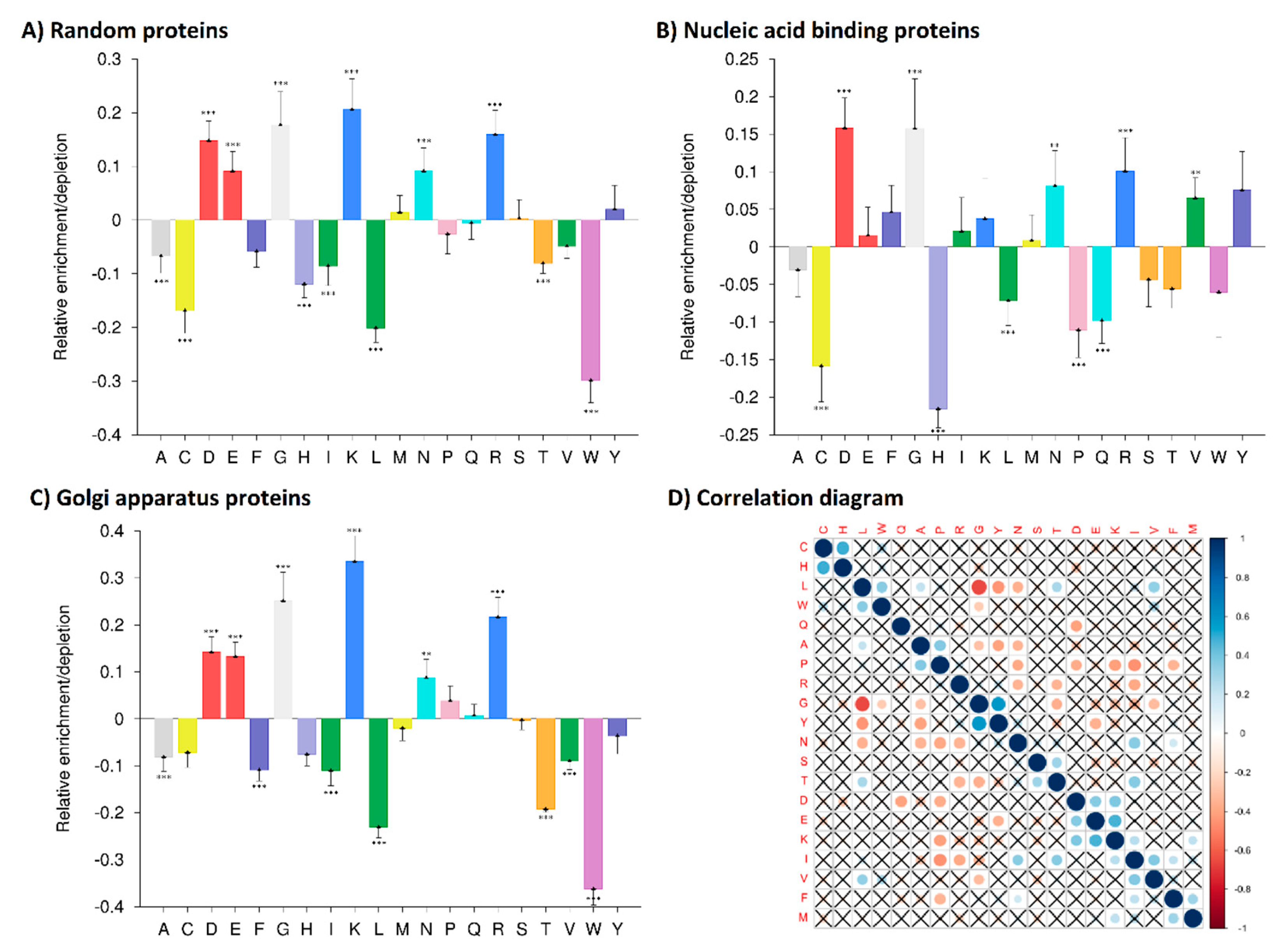
2.1. Amino Acid Composition Analyses
2.2. Correlation Analysis of Quadruplex Binding Proteins Amino Acid Composition
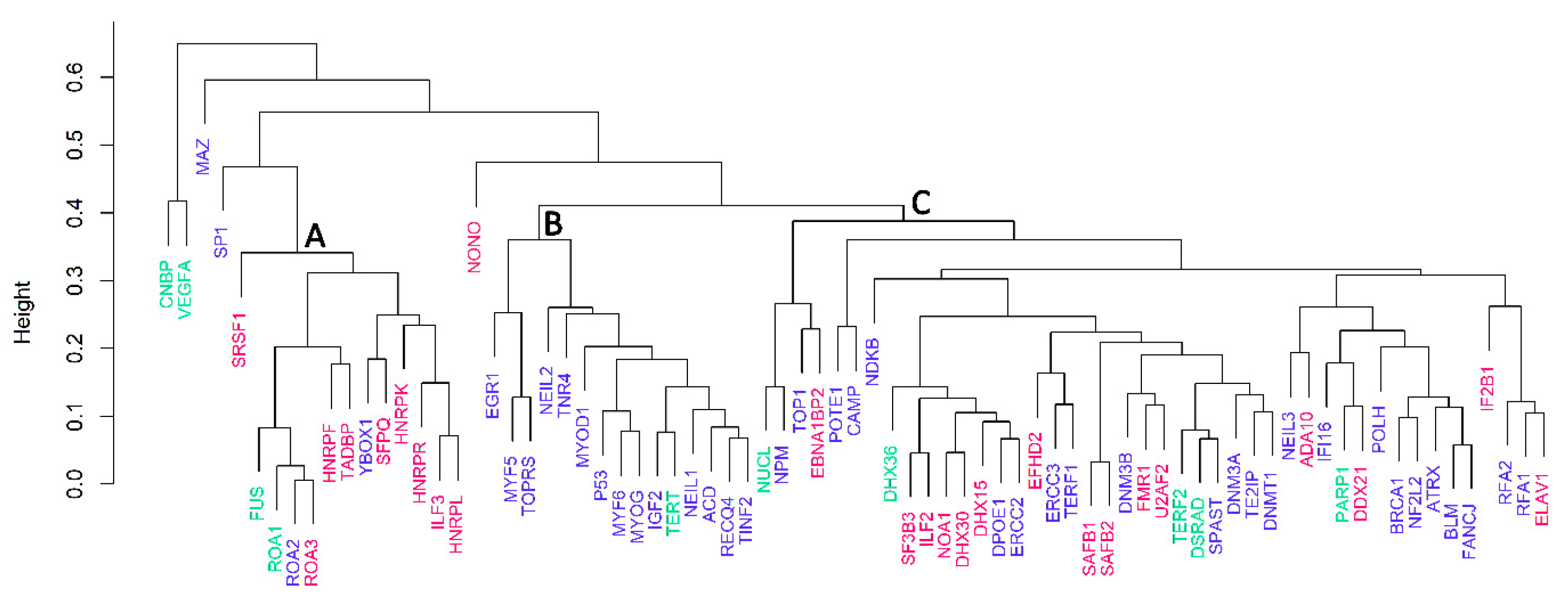
2.3. Cluster Dendrogram Analysis Based on Amino Acid Composition Matrix
2.4. Cluster Dendrogram Analysis Based on CLAP Approach
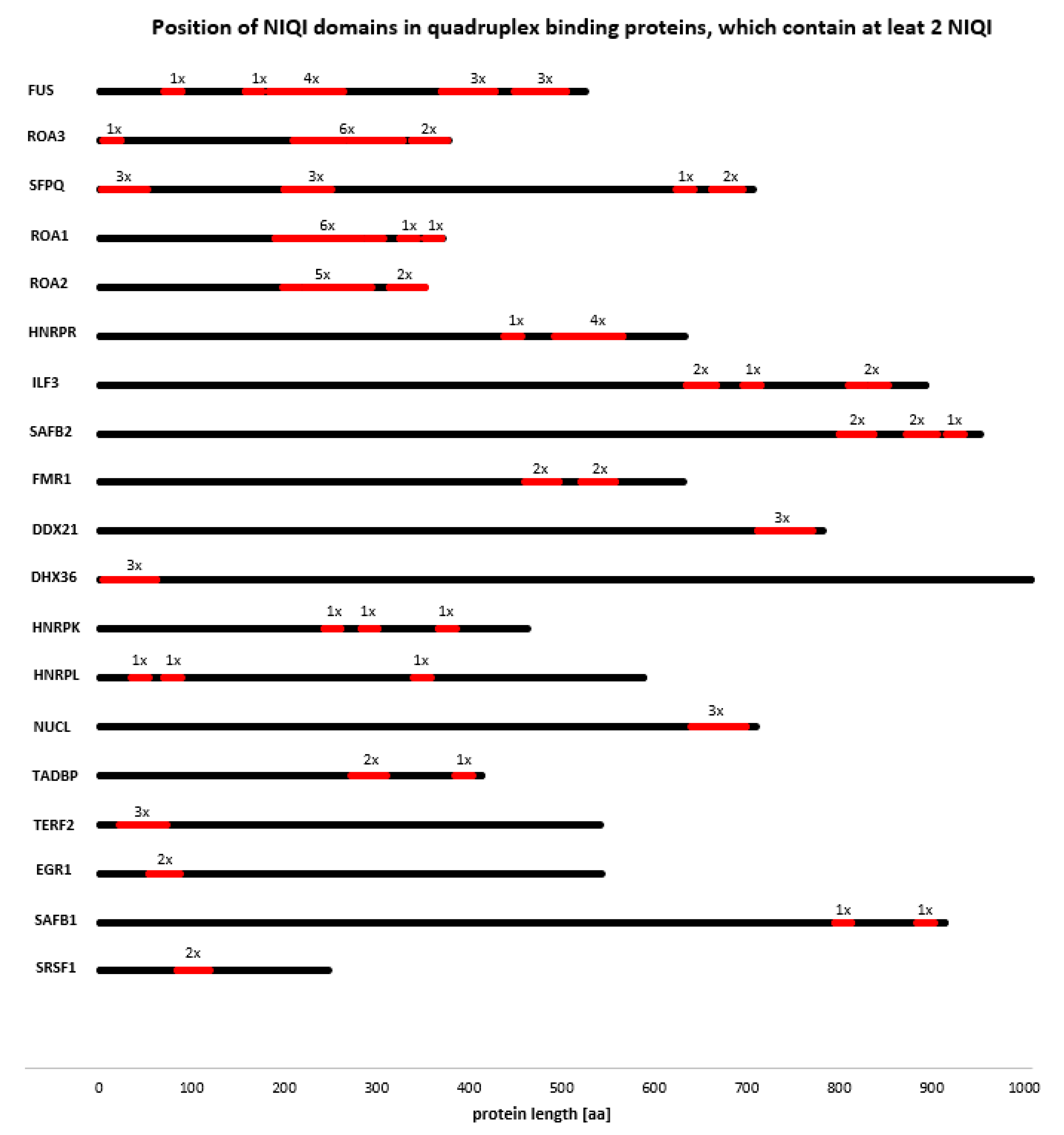
2.5. Motif Scanning of Known Quadruplex Interaction Sequence from FMR1
2.6. De novo Sequence Logo Generation
2.7. Motif Alignments
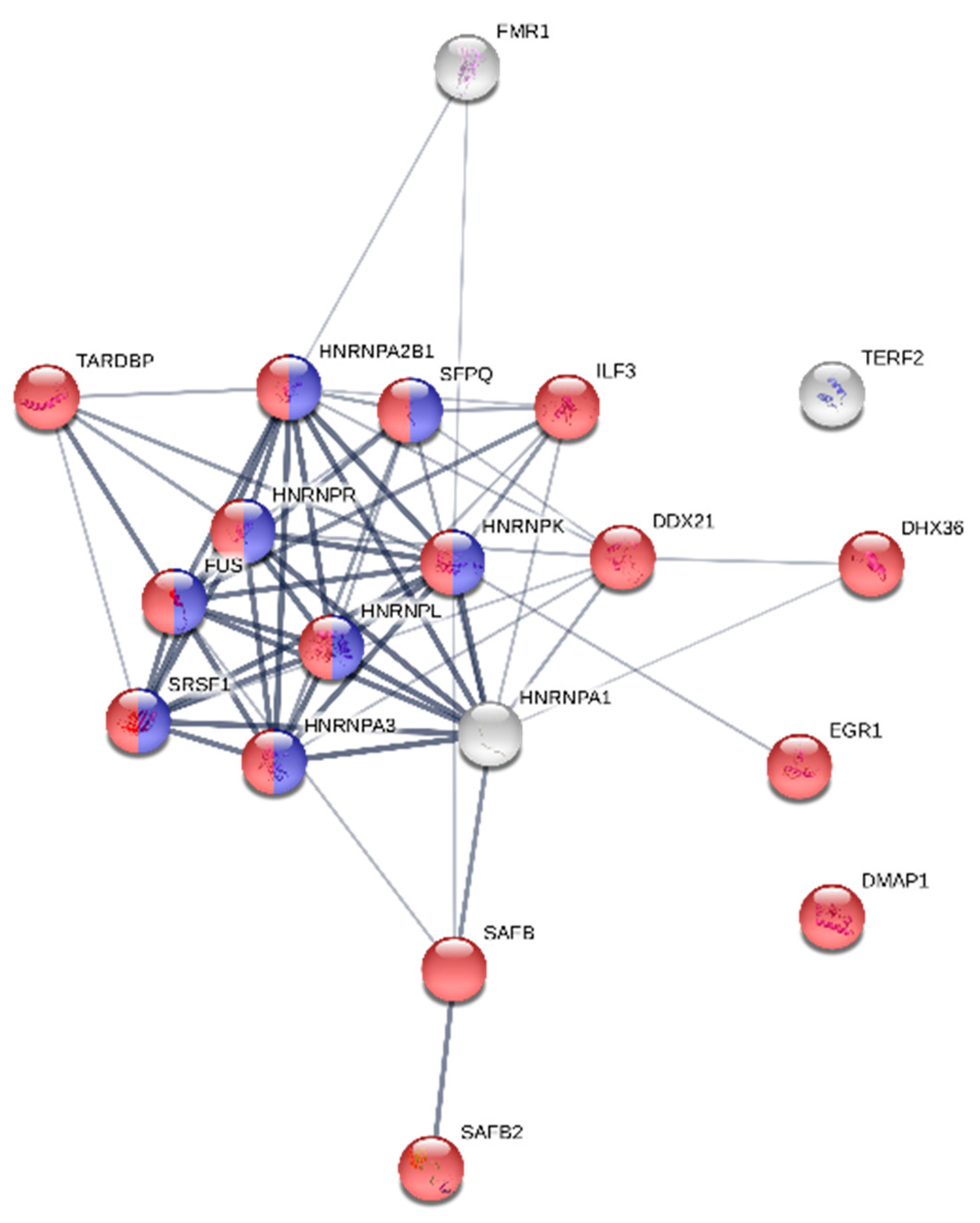
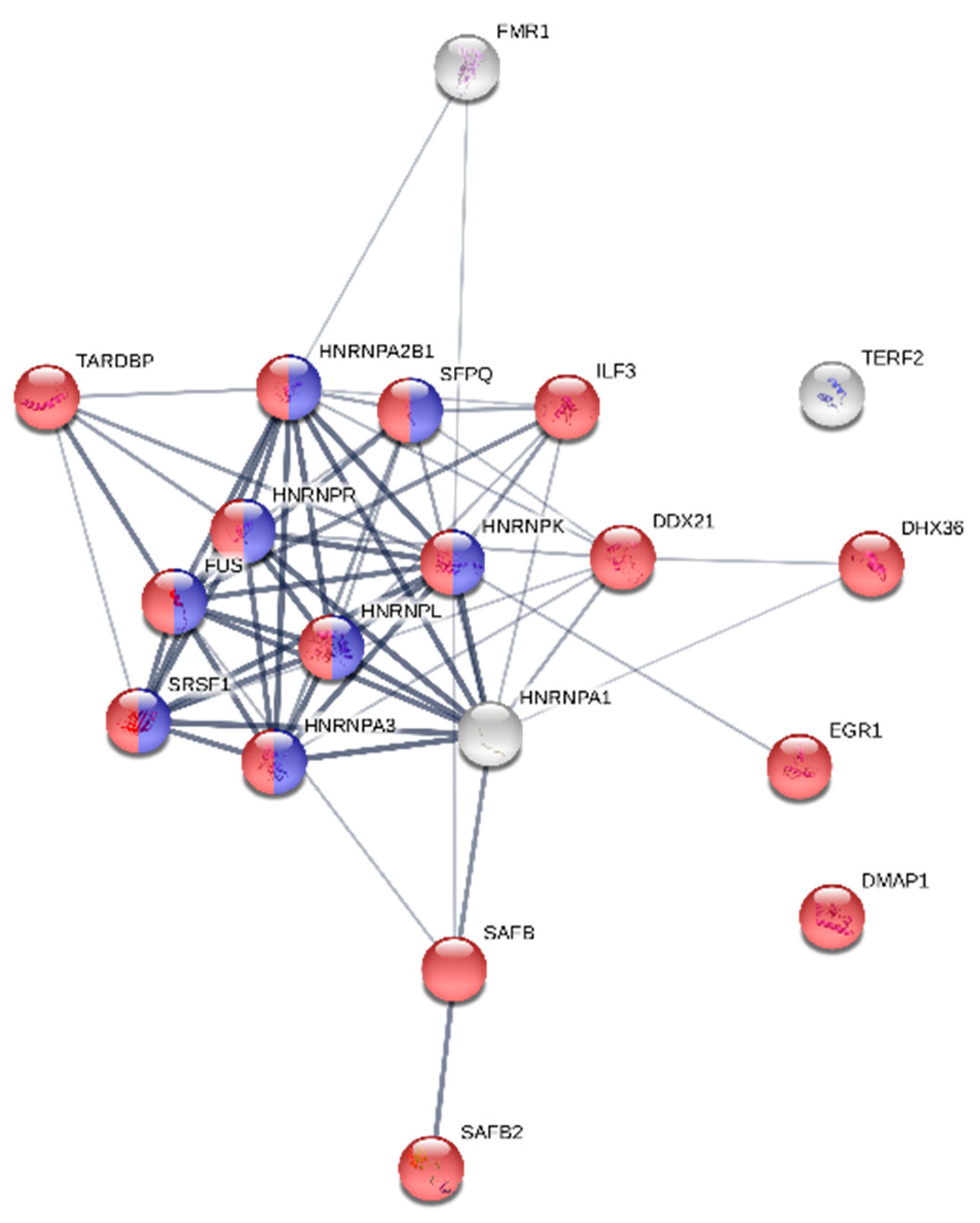
2.8. Protein Functional Network Analysis
2.9. Prediction of New Quadruplex Binding Proteins
3. Results
3.1. Amino Acid Residue Composition Analyses—Identifications of Distinct Enrichments and Depletions in Human Quadruplex Binding Proteins
3.2. Correlation Analysis of Human CBPs Amino Acid Composition
3.3. Cluster Analyses
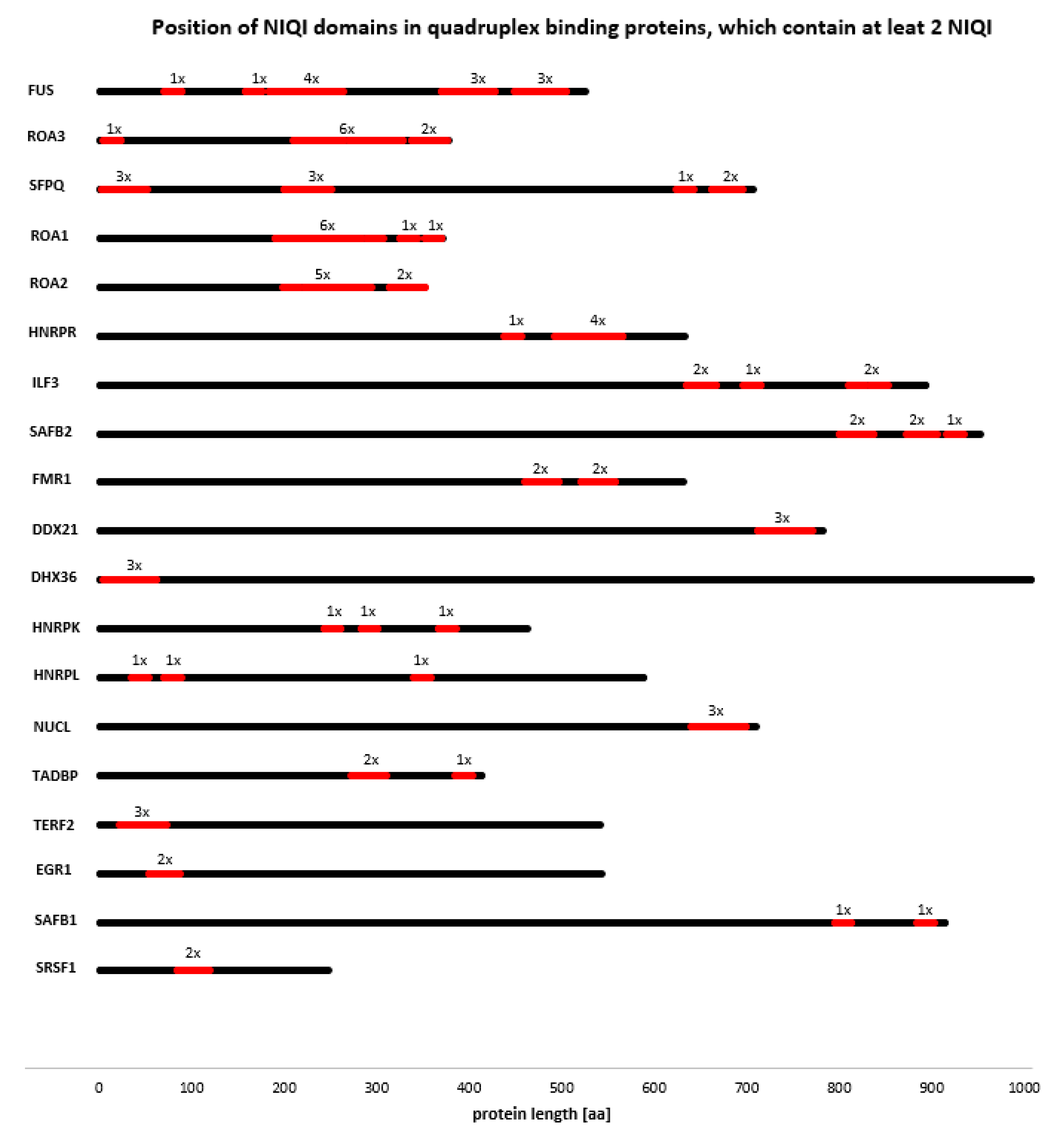
3.4. Novel Interesting Quadruplex Interaction Motif (NIQI)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic Acids: A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Palecek, E. Local supercoil-stabilized DNA structures. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 151–226. [Google Scholar] [CrossRef] [PubMed]
- Van Holde, K.; Zlatanova, J. Unusual DNA structures, chromatin and transcription. Bioessays 1994, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.D. Non-B DNA conformations, mutagenesis and disease. Trends Biochem. Sci. 2007, 32, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Majima, T. Conformational changes of non-B DNA. Chem. Soc. Rev. 2011, 40, 5893–5909. [Google Scholar] [CrossRef] [PubMed]
- Chasovskikh, S.; Dimtchev, A.; Smulson, M.; Dritschilo, A. DNA transitions induced by binding of PARP-1 to cruciform structures in supercoiled plasmids. Cytometry A 2005, 68, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cer, R.Z.; Bruce, K.H.; Donohue, D.E.; Temiz, N.A.; Mudunuri, U.S.; Yi, M.; Volfovsky, N.; Bacolla, A.; Luke, B.T.; Collins, J.R.; et al. Searching for non-B DNA-forming motifs using nBMST (non-B DNA motif search tool). In Current Protocols in Human Genetics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 1–22. [Google Scholar]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, G.N.; Lee, M.P.; Neidle, S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S.; Parkinson, G.N. Quadruplex DNA crystal structures and drug design. Biochimie 2008, 90, 1184–1196. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Todd, A.K.; Zloh, M.; Gunaratnam, M.; Parkinson, G.N.; Neidle, S. Crystal structure of a promoter sequence in the B-raf gene reveals an intertwined dimer quadruplex. J. Am. Chem. Soc. 2013, 135, 19319–19329. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef] [PubMed]
- Scaria, V.; Hariharan, M.; Arora, A.; Maiti, S. Quadfinder: Server for identification and analysis of quadruplex-forming motifs in nucleotide sequences. Nucleic Acids Res. 2006, 34, W683–W685. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedrat, A.; Lacroix, L.; Mergny, J.-L. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016, 44, 1746–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, C.R.; Singh, S.; Hambarde, S.; Griffin, W.C.; Gao, J.; Chib, S.; Yu, Y.; Ira, G.; Raney, K.D.; Kim, N. Yeast Sub1 and human PC4 are G-quadruplex binding proteins that suppress genome instability at co-transcriptionally formed G4 DNA. Nucleic Acids Res. 2017, 45, 5850–5862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mergny, J.-L.; Hélène, C. G-quadruplex DNA: A target for drug design. Nat. Med. 1998, 4, 1366–1367. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S.; Parkinson, G. Telomere maintenance as a target for anticancer drug discovery. Nat. Rev. Drug Discov. 2002, 1, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Waller, Z.A.; Sewitz, S.A.; Hsu, S.-T.D.; Balasubramanian, S. A small molecule that disrupts G-quadruplex DNA structure and enhances gene expression. J. Am. Chem. Soc. 2009, 131, 12628–12633. [Google Scholar] [CrossRef] [PubMed]
- Lyonnais, S.; Tarrés-Solé, A.; Rubio-Cosials, A.; Cuppari, A.; Brito, R.; Jaumot, J.; Gargallo, R.; Vilaseca, M.; Silva, C.; Granzhan, A.; et al. The human mitochondrial transcription factor A is a versatile G-quadruplex binding protein. Sci. Rep. 2017, 7, 43992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Bugaut, A.; Huppert, J.L.; Balasubramanian, S. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nat. Chem. Biol. 2007, 3, 218–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffitzel, C.; Berger, I.; Postberg, J.; Hanes, J.; Lipps, H.J.; Pluckthun, A. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc. Natl. Acad. Sci. USA 2001, 98, 8572–8577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Xiang, J.; Yang, S.; Zhou, Q.; Li, Q.; Tang, Y.; Xu, G. Verification of specific G-quadruplex structure by using a novel cyanine dye supramolecular assembly: I. recognizing mixed G-quadruplex in human telomeres. Chem. Commun. 2009, 9, 1103–1105. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.W.; Bolderson, E.; Cubeddu, L.; O’Byrne, K.J.; Richard, D.J. Human single-stranded DNA binding proteins are essential for maintaining genomic stability. BMC Mol. Biol. 2013, 14, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brázda, V.; Laister, R.C.; Jagelská, E.B.; Arrowsmith, C. Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 2011, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Brázda, V.; Hároníková, L.; Liao, J.C.; Fojta, M. DNA and RNA quadruplex-binding proteins. Int. J. Mol. Sci. 2014, 15, 17493–17517. [Google Scholar] [CrossRef] [PubMed]
- Jagelská, E.B.; Pivoňková, H.; Fojta, M.; Brázda, V. The potential of the cruciform structure formation as an important factor influencing p53 sequence-specific binding to natural DNA targets. Biochem. Biophys. Res. Commun. 2010, 391, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Coufal, J.; Jagelská, E.B.; Liao, J.C.; Brázda, V. Preferential binding of p53 tumor suppressor to p21 promoter sites that contain inverted repeats capable of forming cruciform structure. Biochem. Biophys. Res. Commun. 2013, 441, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Brázda, V.; Coufal, J. Recognition of local DNA structures by p53 protein. Int. J. Mol. Sci. 2017, 18, 375. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Tawani, A.; Mishra, A.; Kumar, A. G4IPDB: A database for G-quadruplex structure forming nucleic acid interacting proteins. Sci. Rep. 2016, 6, 38144. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.; Li, L.; Dong, X.; Wang, Y. Identification of SLIRP as a G Quadruplex-Binding Protein. J. Am. Chem. Soc. 2017, 139, 12426–12429. [Google Scholar] [CrossRef] [PubMed]
- Lago, S.; Tosoni, E.; Nadai, M.; Palumbo, M.; Richter, S.N. The cellular protein nucleolin preferentially binds long-looped G-quadruplex nucleic acids. BBA-Gen. Subj. 2017, 1861, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K.; Yoshizawa-Sugata, N.; Masai, H. Oligomer formation and G-quadruplex binding by purified murine Rif1 protein, a key organizer of higher-order chromatin architecture. J. Biol. Chem. 2018, 293, 3607–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pane, K.; Durante, L.; Crescenzi, O.; Cafaro, V.; Pizzo, E.; Varcamonti, M.; Zanfardino, A.; Izzo, V.; Di Donato, A.; Notomista, E. Antimicrobial potency of cationic antimicrobial peptides can be predicted from their amino acid composition: Application to the detection of “cryptic” antimicrobial peptides. J. Theor. Biol. 2017, 419, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Settanni, G.; Zhou, J.; Suo, T.; Schöttler, S.; Landfester, K.; Schmid, F.; Mailänder, V. Protein corona composition of poly (ethylene glycol)-and poly (phosphoester)-coated nanoparticles correlates strongly with the amino acid composition of the protein surface. Nanoscale 2017, 9, 2138–2144. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.D.; Ben-Hur, A. Amino acid composition predicts prion activity. PLoS Comput. Biol. 2017, 13, e1005465. [Google Scholar]
- Bartas, M.; Bažantová, P.; Brázda, V.; Liao, J.C.; Červeň, J.; Pečinka, P. Identification of distinct amino acid composition of human cruciform binding proteins. Mol. Biol. 2019, 53. in press. [Google Scholar]
- Wu, Y.; Shin-ya, K.; Brosh, R.M. FANCJ Helicase Defective in Fanconia Anemia and Breast Cancer Unwinds G-Quadruplex DNA To Defend Genomic Stability. Mol. Cell. Biol. 2008, 28, 4116–4128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkies, P.; Murat, P.; Phillips, L.G.; Patel, K.J.; Balasubramanian, S.; Sale, J.E. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res. 2012, 40, 1485–1498. [Google Scholar] [CrossRef] [PubMed]
- Quante, T.; Otto, B.; Brázdová, M.; Kejnovská, I.; Deppert, W.; Tolstonog, G.V. Mutant p53 is a transcriptional co-factor that binds to G-rich regulatory regions of active genes and generates transcriptional plasticity. Cell Cycle 2012, 11, 3290–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Su, L.; Qiu, J.; Xiao, N.; Lin, J.; Tan, J.; Ou, T.; Gu, L.; Huang, Z.; Li, D. Mechanistic studies for the role of cellular nucleic-acid-binding protein (CNBP) in regulation of c-myc transcription. BBA-Gen. Subj. 2013, 1830, 4769–4777. [Google Scholar] [CrossRef] [PubMed]
- König, S.L.B.; Huppert, J.L.; Sigel, R.K.O.; Evans, A.C. Distance-dependent duplex DNA destabilization proximal to G-quadruplex/i-motif sequences. Nucleic Acids Res. 2013, 41, 7453–7461. [Google Scholar] [CrossRef] [PubMed]
- Bosch, P.C.; Segura-Bayona, S.; Koole, W.; van Heteren, J.T.; Dewar, J.M.; Tijsterman, M.; Knipscheer, P. FANCJ promotes DNA synthesis through G-quadruplex structures. EMBO J. 2014, e201488663. [Google Scholar] [CrossRef]
- Saito, T.; Yoshida, W.; Yokoyama, T.; Abe, K.; Ikebukuro, K. Identification of RNA Oligonucleotides Binding to Several Proteins from Potential G-Quadruplex Forming Regions in Transcribed Pre-mRNA. Molecules 2015, 20, 20832–20840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hároníková, L.; Coufal, J.; Kejnovská, I.; Jagelská, E.B.; Fojta, M.; Dvořáková, P.; Muller, P.; Vojtesek, B.; Brázda, V. IFI16 Preferentially Binds to DNA with Quadruplex Structure and Enhances DNA Quadruplex Formation. PLoS ONE 2016, 11, e0157156. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: New York, NY, USA, 2005; pp. 571–607. [Google Scholar]
- Kozlowski, L.P. Proteome-pI: Proteome isoelectric point database. Nucleic Acids Res. 2017, 45, D1112–D1116. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Uversky, V.N.; Dunker, A.K.; Lonardi, S. Composition Profiler: A tool for discovery and visualization of amino acid composition differences. BMC Bioinform. 2007, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Kasprzyk, A. BioMart: Driving a paradigm change in biological data management. Database (Oxford) 2011, 2011, bar049. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Cao, D.-S.; Zhu, M.-F.; Xu, Q.-S. protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics 2015, 31, 1857–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobanov, M.Y.; Sokolovskiy, I.V.; Galzitskaya, O.V. HRaP: Database of occurrence of HomoRepeats and patterns in proteomes. Nucleic Acids Res. 2014, 42, D273–D278. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Simko, V. R package “corrplot”: Visualization of a Correlation Matrix (Version 0.84). Available online: https://github.com/taiyun/corrplot (accessed on 1 August 2017).
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Anamika, K.; Srinivasan, N. Classification of protein kinases on the basis of both kinase and non-kinase regions. PLoS ONE 2010, 5, e12460. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, R.M.; Mehrotra, P.; Rakshambikai, R.; Gnanavel, M.; Martin, J.; Srinivasan, N. The relationship between classification of multi-domain proteins using an alignment-free approach and their functions: A case study with immunoglobulins. Mol. Biosyst. 2014, 10, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Gnanavel, M.; Mehrotra, P.; Rakshambikai, R.; Martin, J.; Srinivasan, N.; Bhaskara, R.M. CLAP: A web-server for automatic classification of proteins with special reference to multi-domain proteins. BMC Bioinform. 2014, 15, 343. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]
- Frith, M.C.; Saunders, N.F.; Kobe, B.; Bailey, T.L. Discovering sequence motifs with arbitrary insertions and deletions. PLoS Comput. Biol. 2008, 4, e1000071. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Team, U. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Vasilyev, N.; Polonskaia, A.; Darnell, J.C.; Darnell, R.B.; Patel, D.J.; Serganov, A. Crystal structure reveals specific recognition of a G-quadruplex RNA by a β-turn in the RGG motif of FMRP. Proc. Natl. Acad. Sci. USA 2015, 112, E5391–E5400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, R.; Miyazaki, T.; Oyoshi, T. G-quadruplex binding ability of TLS/FUS depends on the β-spiral structure of the RGG domain. Nucleic Acids Res. 2018, 46, 5894–5901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gaetano, C.M.; Williams, K.R.; Bassell, G.J.; Mihailescu, M.R. FMRP interacts with G-quadruplex structures in the 3′-UTR of its dendritic target Shank1 mRNA. RNA Biol. 2014, 11, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- McRae, E.K.; Booy, E.P.; Padilla-Meier, G.P.; McKenna, S.A. On Characterizing the Interactions between Proteins and Guanine Quadruplex Structures of Nucleic Acids. J. Nucleic Acids 2017, 2017, 9675348. [Google Scholar] [CrossRef] [PubMed]
- Thandapani, P.; O’Connor, T.R.; Bailey, T.L.; Richard, S. Defining the RGG/RG motif. Mol. Cell 2013, 50, 613–623. [Google Scholar] [CrossRef] [PubMed]
- DeForte, S.; Uversky, V.; DeForte, S.; Uversky, V.N. Order, Disorder, and Everything in Between. Molecules 2016, 21, 1090. [Google Scholar] [CrossRef] [PubMed]
- Bartas, M.; Brázda, V.; Karlický, V.; Červeň, J.; Pečinka, P. Bioinformatics analyses and in vitro evidence for five and six stacked G-quadruplex forming sequences. Biochimie 2018, 150, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, W.; Saito, T.; Yokoyama, T.; Ferri, S.; Ikebukuro, K. Aptamer selection based on G4-forming promoter region. PLoS ONE 2013, 8, e65497. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Shchekotikhin, A.E.; Xodo, L.E. HRAS is silenced by two neighboring G-quadruplexes and activated by MAZ, a zinc-finger transcription factor with DNA unfolding property. Nucleic Acids Res. 2014, 42, 8379–8388. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänsel-Hertsch, R.; Spiegel, J.; Marsico, G.; Tannahill, D.; Balasubramanian, S. Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2018, 13, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Takahama, K.; Miyawaki, A.; Shitara, T.; Mitsuya, K.; Morikawa, M.; Hagihara, M.; Kino, K.; Yamamoto, A.; Oyoshi, T. G-quadruplex DNA-and RNA-specific-binding proteins engineered from the RGG domain of TLS/FUS. ACS Chem. Biol. 2015, 10, 2564–2569. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brázda, V.; Červeň, J.; Bartas, M.; Mikysková, N.; Coufal, J.; Pečinka, P. The Amino Acid Composition of Quadruplex Binding Proteins Reveals a Shared Motif and Predicts New Potential Quadruplex Interactors. Molecules 2018, 23, 2341. https://doi.org/10.3390/molecules23092341
Brázda V, Červeň J, Bartas M, Mikysková N, Coufal J, Pečinka P. The Amino Acid Composition of Quadruplex Binding Proteins Reveals a Shared Motif and Predicts New Potential Quadruplex Interactors. Molecules. 2018; 23(9):2341. https://doi.org/10.3390/molecules23092341
Chicago/Turabian StyleBrázda, Václav, Jiří Červeň, Martin Bartas, Nikol Mikysková, Jan Coufal, and Petr Pečinka. 2018. "The Amino Acid Composition of Quadruplex Binding Proteins Reveals a Shared Motif and Predicts New Potential Quadruplex Interactors" Molecules 23, no. 9: 2341. https://doi.org/10.3390/molecules23092341
APA StyleBrázda, V., Červeň, J., Bartas, M., Mikysková, N., Coufal, J., & Pečinka, P. (2018). The Amino Acid Composition of Quadruplex Binding Proteins Reveals a Shared Motif and Predicts New Potential Quadruplex Interactors. Molecules, 23(9), 2341. https://doi.org/10.3390/molecules23092341