Current Concepts and Treatments of Schizophrenia


Abstract
:
1. Introduction
2. Schizophrenia as a Complex Disease
2.1. Dopaminergic Hypothesis
2.2. Glutamatergic Hypothesis
2.3. Serotoninergic Hypothesis of Schizophrenia
2.4. Other Aminergic GPCRs in Schizophrenia
2.5. GABAergic Hypothesis of Schizophrenia
2.6. Nicotinic Receptors in Schizophrenia
2.7. The Endocannabinoid System in Schizophrenia
2.8. Role of Inflammation and Oxidative Stress in the Pathomechanism of Schizophrenia
3. Classical Approaches to Treat Schizophrenia
- -
- (1) Some patients lack response to drug treatment. Clozapine is recommended in patients resistant to other neuroleptics. The 30% of patients that do not respond are classified as “treatment resistant” and represent a major problem regarding treatment. It is still unknown what underlies the difference between responsive and unresponsive patients, although there are some presumptions that polymorphisms within the dopamine and serotonin receptors family may be involved.
- -
- (2) They are effective in relieving the positive symptoms (delusions, hallucinations, thought disorders, etc.) but most of them lack effectiveness in controlling the negative symptoms (social isolation, emotional flattening) and cognitive dysfunctions.
- -
- (3) They may result in a wide range of side effects including extrapyramidal, sedative and endocrine effects that can limit patient compliance.
- -
- (4) They may decline survival through pro-arrhythmic effects.
3.1. First-Generation Antipsychotics
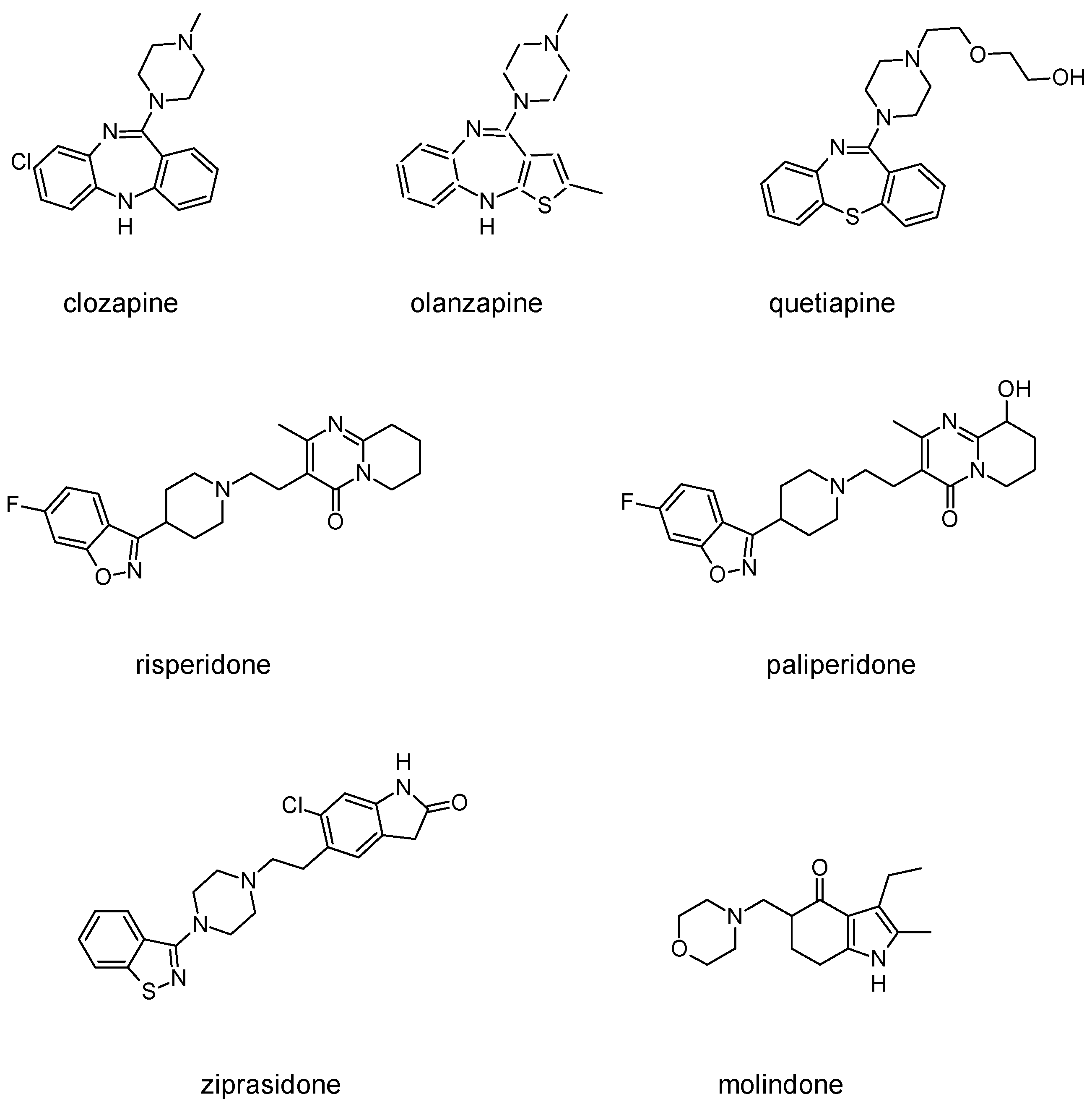
3.2. Second-Generation Antipsychotics
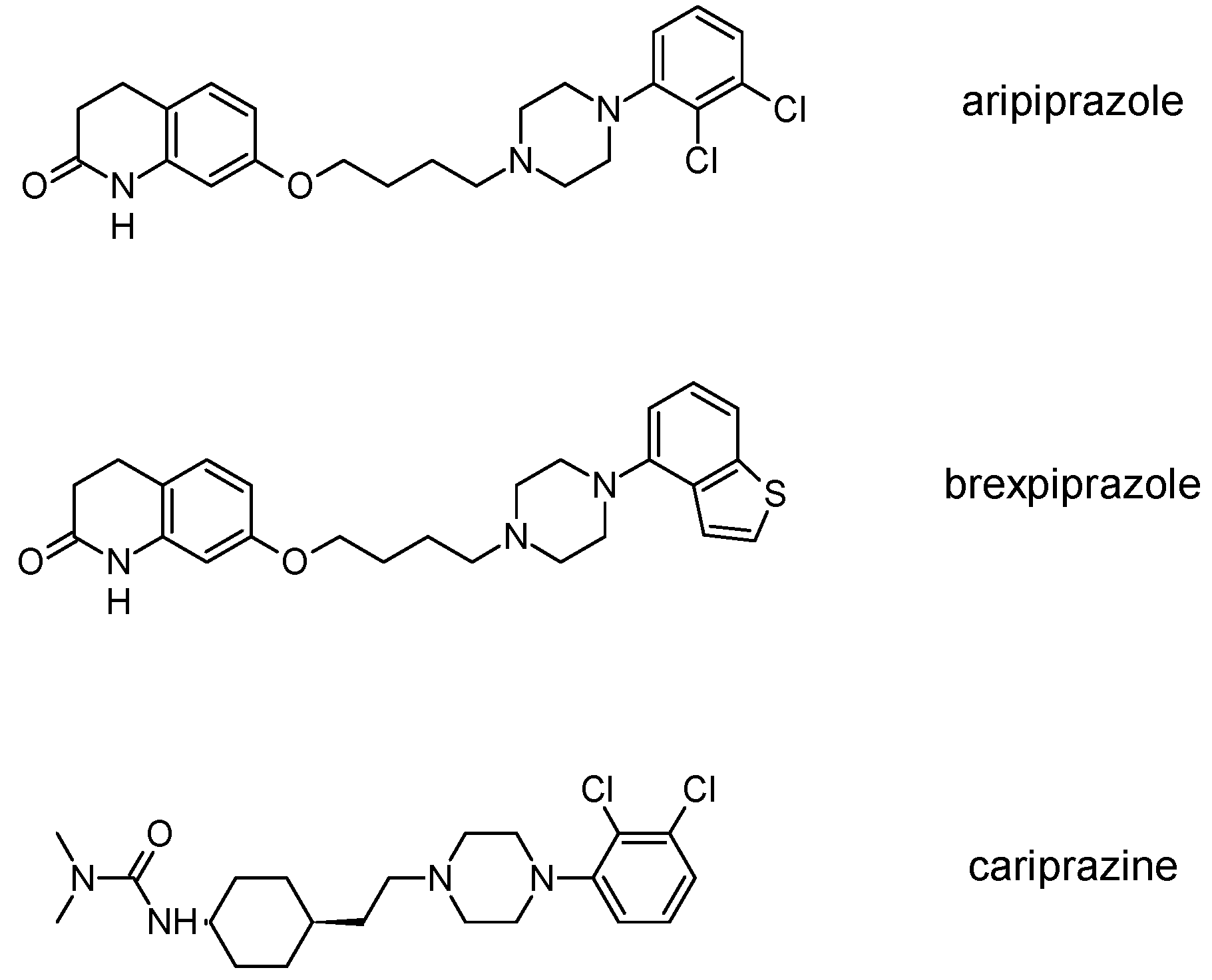
3.3. Third-Generation Antipsychotics
4. Targeting Novel GPCR Signaling Mechanisms in Schizophrenia
4.1. GPCRs and Their Novel Signaling Mechanisms as Drug Targets
4.2. Targeting GPCR Signaling Complexity in Schizophrenia
5. Other Non-Classical Approaches for the Treatment of Schizophrenia
6. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Andlin-Sobocki, P.; Jönsson, B.; Wittchen, H.-U.; Olesen, J. Cost of disorders of the brain in Europe. Eur. J. Neurol. 2005, 12 (Suppl. 1), 1–27. [Google Scholar] [CrossRef] [Green Version]
- Stroup, T.S.; Lieberman, J.A.; Swartz, M.S.; McEvoy, J.P. Comparative effectiveness of antipsychotic drugs in schizophrenia. Dialogues Clin. Neurosci. 2000, 2, 373–379. [Google Scholar] [PubMed]
- Carbon, M.; Correll, C.U. Thinking and acting beyond the positive: The role of the cognitive and negative symptoms in schizophrenia. CNS Spectr. 2014, 19 (Suppl. 1), 35–53. [Google Scholar] [CrossRef] [PubMed]
- De Berardis, D.; Rapini, G.; Olivieri, L.; Di Nicola, D.; Tomasetti, C.; Valchera, A.; Fornaro, M.; Di Fabio, F.; Perna, G.; Di Nicola, M.; et al. Safety of antipsychotics for the treatment of schizophrenia: A focus on the adverse effects of clozapine. Ther. Adv. Drug Saf. 2018, 9, 237–256. [Google Scholar] [CrossRef] [PubMed]
- Laruelle, M. Schizophrenia: From dopaminergic to glutamatergic interventions. Curr. Opin. Pharmacol. 2014, 14, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and dopamine in schizophrenia: An update for the 21st century. J. Psychopharmacol. Oxf. Engl. 2015, 29, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Yost, J.M.; Setola, V.; Chen, X.; Sassano, M.F.; Chen, M.; Peterson, S.; Yadav, P.N.; Huang, X.; Feng, B.; et al. Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. USA 2011, 108, 18488–18493. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Roth, B.L. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 117–144. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.F.; Tarazi, F.I.; Shahid, M. The effectiveness of multi-target agents in schizophrenia and mood disorders: Relevance of receptor signature to clinical action. Pharmacol. Ther. 2010, 126, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.D.; Clarke, W.P.; von Zastrow, M.; Nichols, D.E.; Kobilka, B.; Weinstein, H.; Javitch, J.A.; Roth, B.L.; Christopoulos, A.; Sexton, P.M.; et al. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 2007, 320, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Stott, L.A.; Hall, D.A.; Holliday, N.D. Unravelling intrinsic efficacy and ligand bias at G protein coupled receptors: A practical guide to assessing functional data. Biochem. Pharmacol. 2016, 101, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Weïwer, M.; Xu, Q.; Gale, J.P.; Lewis, M.; Campbell, A.J.; Schroeder, F.A.; Van de Bittner, G.C.; Walk, M.; Amaya, A.; Su, P.; et al. Functionally Biased D2R Antagonists: Targeting the β-Arrestin Pathway to Improve Antipsychotic Treatment. ACS Chem. Biol. 2018, 13, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Opportunities and Challenges in the Discovery of Allosteric Modulators of GPCRs. Methods Mol. Biol. Clifton NJ 2018, 1705, 297–319. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Signaling within Allosteric Machines: Signal Transmission Pathways Inside G Protein-Coupled Receptors. Molecules 2017, 22, 1188. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.J.; Lindsley, C.W.; Meiler, J.; Niswender, C.M. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat. Rev. Drug Discov. 2014, 13, 692–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
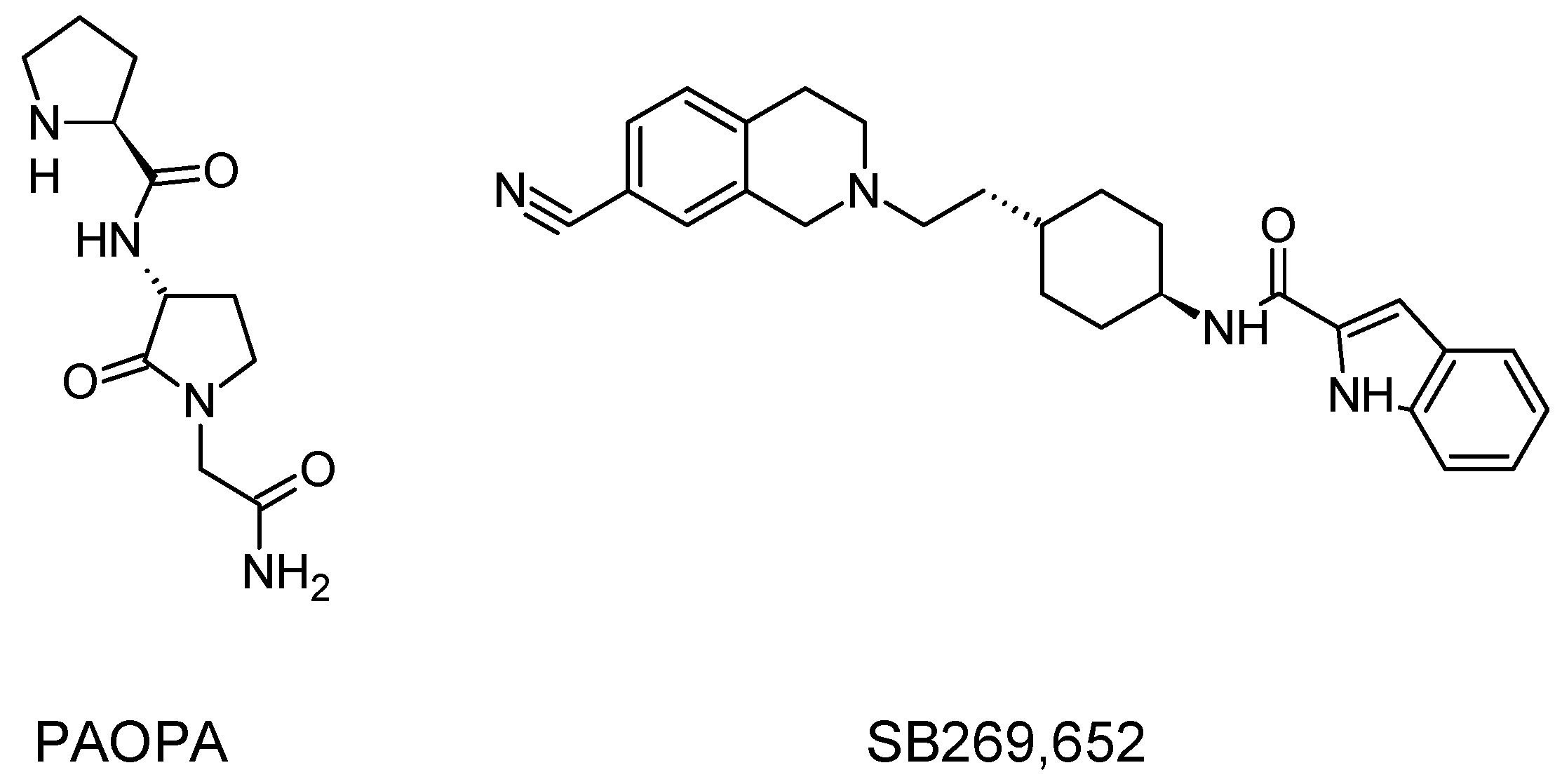
- Beyaert, M.G.R.; Daya, R.P.; Dyck, B.A.; Johnson, R.L.; Mishra, R.K. PAOPA, a potent dopamine D2 receptor allosteric modulator, prevents and reverses behavioral and biochemical abnormalities in an amphetamine-sensitized preclinical animal model of schizophrenia. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2013, 23, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Fasciani, I.; Marampon, F.; Maggio, R.; Scarselli, M. The First Negative Allosteric Modulator for Dopamine D2 and D3 Receptors, SB269652 May Lead to a New Generation of Antipsychotic Drugs. Mol. Pharmacol. 2017, 91, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Agnati, L.F.; Ferré, S.; Genedani, S.; Leo, G.; Guidolin, D.; Filaferro, M.; Carriba, P.; Casadó, V.; Lluis, C.; Franco, R.; et al. Allosteric modulation of dopamine D2 receptors by homocysteine. J. Proteome Res. 2006, 5, 3077–3083. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Selent, J. Oligomerization of G protein-coupled receptors: Biochemical and biophysical methods. Curr. Med. Chem. 2011, 18, 4606–4634. [Google Scholar] [CrossRef] [PubMed]
- Selent, J.; Kaczor, A.A. Oligomerization of G protein-coupled receptors: Computational methods. Curr. Med. Chem. 2011, 18, 4588–4605. [Google Scholar] [CrossRef] [PubMed]
- Ng, G.Y.; O’Dowd, B.F.; Lee, S.P.; Chung, H.T.; Brann, M.R.; Seeman, P.; George, S.R. Dopamine D2 receptor dimers and receptor-blocking peptides. Biochem. Biophys. Res. Commun. 1996, 227, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.-I.; Wang, H.-C.; Hsu, J.-L.; Liu, M.-E. Does the dopamine hypothesis explain schizophrenia? Rev. Neurosci. 2013, 24, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.; Lindqvist, M. Effect of chlorpromazine or haloperidol on formation of 3methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol. Toxicol. (Copenh.) 1963, 20, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.L.; Kahn, R.S.; Ko, G.; Davidson, M. Dopamine in schizophrenia: A review and reconceptualization. Am. J. Psychiatry 1991, 148, 1474–1486. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Kapur, S. The Dopamine Hypothesis of Schizophrenia: Version III–The Final Common Pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Berridge, K.C.; Robinson, T.E. What is the role of dopamine in reward: Hedonic impact, reward learning, or incentive salience? Brain Res. Brain Res. Rev. 1998, 28, 309–369. [Google Scholar] [CrossRef]
- Kapur, S. Psychosis as a state of aberrant salience: A framework linking biology, phenomenology, and pharmacology in schizophrenia. Am. J. Psychiatry 2003, 160, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Luedtke, R.R.; Rangel-Barajas, C.; Malik, M.; Reichert, D.E.; Mach, R.H. Bitropic D3 Dopamine Receptor Selective Compounds as Potential Antipsychotics. Curr. Pharm. Des. 2015, 21, 3700–3724. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, W.; Zhang, X.; Yin, L.; Chen, B.; Song, J. Synthesis and pharmacological evaluation of piperidine (piperazine)-substituted benzoxazole derivatives as multi-target antipsychotics. Bioorg. Med. Chem. Lett. 2015, 25, 5299–5305. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Wicke, K.; Drescher, K.U. Dopamine D₃ receptor antagonism--still a therapeutic option for the treatment of schizophrenia. Naunyn. Schmiedebergs Arch. Pharmacol. 2013, 386, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Castner, S.A.; Williams, G.V.; Goldman-Rakic, P.S. Reversal of antipsychotic-induced working memory deficits by short-term dopamine D1 receptor stimulation. Science 2000, 287, 2020–2022. [Google Scholar] [CrossRef] [PubMed]
- Natesan, S.; Reckless, G.E.; Barlow, K.B.L.; Odontiadis, J.; Nobrega, J.N.; Baker, G.B.; George, S.R.; Mamo, D.; Kapur, S. The antipsychotic potential of l-stepholidine–A naturally occurring dopamine receptor D1 agonist and D2 antagonist. Psychopharmacology (Berl.) 2008, 199, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, B.; Javitt, D. From Revolution to Evolution: The Glutamate Hypothesis of Schizophrenia and its Implication for Treatment. Neuropsychopharmacology 2012, 37, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Tsai, S.-J. New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C.; Coyle, J.T. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am. J. Psychiatry 2001, 158, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J.; Weinberger, D.R. Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol. Psychiatry 2005, 10, 40–68. [Google Scholar] [CrossRef] [PubMed]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Paoletti, P.; Neyton, J. NMDA receptor subunits: Function and pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Farber, N.B. The NMDA receptor hypofunction model of psychosis. Ann. N. Y. Acad. Sci. 2003, 1003, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C. Glutamate receptors in schizophrenia and antipsychotic drugs. In Neurotransmitter Receptors in Actions of Antipsychotic Medications; Lidow, M.S., Ed.; CRC Press: New York, NY, USA, 2000; pp. 126–141. [Google Scholar]
- Meador-Woodruff, J.H.; Healy, D.J. Glutamate receptor expression in schizophrenic brain. Brain Res. Brain Res. Rev. 2000, 31, 288–294. [Google Scholar] [CrossRef]
- Gao, X.M.; Sakai, K.; Roberts, R.C.; Conley, R.R.; Dean, B.; Tamminga, C.A. Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: Effects of schizophrenia. Am. J. Psychiatry 2000, 157, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.M.; Hogg, A.J.; Healy, D.J.; Haroutunian, V.; Davis, K.L.; Meador-Woodruff, J.H. Ionotropic glutamate receptor binding and subunit mRNA expression in thalamic nuclei in schizophrenia. Am. J. Psychiatry 2000, 157, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Heckers, S.; Goff, D.; Schacter, D.L.; Savage, C.R.; Fischman, A.J.; Alpert, N.M.; Rauch, S.L. Functional imaging of memory retrieval in deficit vs nondeficit schizophrenia. Arch. Gen. Psychiatry 1999, 56, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Kondziella, D.; Brenner, E.; Eyjolfsson, E.M.; Sonnewald, U. How do glial–neuronal interactions fit into current neurotransmitter hypotheses of schizophrenia? Neurochem. Int. 2007, 50, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.M.; Morrison, P.D.; Pilowsky, L.S. Glutamate and dopamine dysregulation in schizophrenia–A synthesis and selective review. J. Psychopharmacol. Oxf. Engl. 2007, 21, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, L.G.; Schuff, N.; Cashdollar, N.; Weiner, M.W. Age-related glutamate and glutamine concentration changes in normal human brain: 1H MR spectroscopy study at 4 T. Neurobiol. Aging 2005, 26, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leveque, J.C.; Macías, W.; Rajadhyaksha, A.; Carlson, R.R.; Barczak, A.; Kang, S.; Li, X.M.; Coyle, J.T.; Huganir, R.L.; Heckers, S.; et al. Intracellular modulation of NMDA receptor function by antipsychotic drugs. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 4011–4020. [Google Scholar] [CrossRef]
- Arvanov, V.L.; Liang, X.; Schwartz, J.; Grossman, S.; Wang, R.Y. Clozapine and haloperidol modulate N-methyl-D-aspartate- and non-N-methyl-D-aspartate receptor-mediated neurotransmission in rat prefrontal cortical neurons in vitro. J. Pharmacol. Exp. Ther. 1997, 283, 226–234. [Google Scholar] [PubMed]
- Aringhieri, S.; Carli, M.; Kolachalam, S.; Verdesca, V.; Cini, E.; Rossi, M.; McCormick, P.J.; Corsini, G.U.; Maggio, R.; Scarselli, M. Molecular targets of atypical antipsychotics: From mechanism of action to clinical differences. Pharmacol. Ther. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Menniti, F.S.; Lindsley, C.W.; Conn, P.J.; Pandit, J.; Zagouras, P.; Volkmann, R.A. Allosteric modulators for the treatment of schizophrenia: Targeting glutamatergic networks. Curr. Top. Med. Chem. 2013, 13, 26–54. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.E.; Pennicott, L.E.; Beswick, P. AMPA receptor-positive allosteric modulators for the treatment of schizophrenia: An overview of recent patent applications. Future Med. Chem. 2015, 7, 473–491. [Google Scholar] [CrossRef] [PubMed]
- Herman, E.J.; Bubser, M.; Conn, P.J.; Jones, C.K. Metabotropic glutamate receptors for new treatments in schizophrenia. Handb. Exp. Pharmacol. 2012, 297–365. [Google Scholar] [CrossRef]
- Aghajanian, G. Serotonin model of schizophrenia: Emerging role of glutamate mechanisms. Brain Res. Rev. 2000, 31, 302–312. [Google Scholar] [CrossRef]
- Kapur, S.; Remington, G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am. J. Psychiatry 1996, 153, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Eggers, A.E. Extending David Horrobin’s membrane phospholipid theory of schizophrenia: Overactivity of cytosolic phospholipase A2 in the brain is caused by overdrive of coupled serotonergic 5HT2A/2C receptors in response to stress. Med. Hypotheses 2012, 79, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Eggers, A.E. A serotonin hypothesis of schizophrenia. Med. Hypotheses 2013, 80, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A. Alterations of serotonin transmission in schizophrenia. Int. Rev. Neurobiol. 2007, 78, 133–164. [Google Scholar] [CrossRef] [PubMed]
- Nikiforuk, A.; Hołuj, M.; Kos, T.; Popik, P. The effects of a 5-HT5A receptor antagonist in a ketamine-based rat model of cognitive dysfunction and the negative symptoms of schizophrenia. Neuropharmacology 2016, 105, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; Li, Z.; Kaneda, Y.; Ichikawa, J. Serotonin receptors: Their key role in drugs to treat schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; Matsubara, S.; Lee, J.C. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J. Pharmacol. Exp. Ther. 1989, 251, 238–246. [Google Scholar] [PubMed]
- Kusumi, I.; Boku, S.; Takahashi, Y. Psychopharmacology of atypical antipsychotic drugs: From the receptor binding profile to neuroprotection and neurogenesis. Psychiatry Clin. Neurosci. 2015, 69, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Mombereau, C.; Arnt, J.; Mørk, A. Involvement of presynaptic 5-HT1A receptors in the low propensity of brexpiprazole to induce extrapyramidal side effects in rats. Pharmacol. Biochem. Behav. 2017, 153, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Godlewska, B.R.; Olajossy-Hilkesberger, L.; Ciwoniuk, M.; Olajossy, M.; Marmurowska-Michałowska, H.; Limon, J.; Landowski, J. Olanzapine-induced weight gain is associated with the -759C/T and -697G/C polymorphisms of the HTR2C gene. Pharmacogenomics J. 2009, 9, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Gressier, F.; Porcelli, S.; Calati, R.; Serretti, A. Pharmacogenetics of clozapine response and induced weight gain: A comprehensive review and meta-analysis. Eur. Neuropsychopharmacol. 2016, 26, 163–185. [Google Scholar] [CrossRef] [PubMed]
- Hołuj, M.; Popik, P.; Nikiforuk, A. Improvement of ketamine-induced social withdrawal in rats: The role of 5-HT7 receptors. Behav. Pharmacol. 2015, 26, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Nikiforuk, A.; Popik, P. Amisulpride promotes cognitive flexibility in rats: The role of 5-HT7 receptors. Behav. Brain Res. 2013, 248, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Ellenbroek, B.A.; Ghiabi, B. Do Histamine receptor 3 antagonists have a place in the therapy for schizophrenia? Curr. Pharm. Des. 2015, 21, 3760–3770. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, A.; Rook, J.M.; Dickerson, J.W.; Roop, G.N.; Morrison, R.D.; Jalan-Sakrikar, N.; Lamsal, A.; Noetzel, M.J.; Poslusney, M.S.; Wood, M.R.; et al. Potentiation of M1 Muscarinic Receptor Reverses Plasticity Deficits and Negative and Cognitive Symptoms in a Schizophrenia Mouse Model. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2016, 41, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Maletic, V.; Eramo, A.; Gwin, K.; Offord, S.J.; Duffy, R.A. The Role of Norepinephrine and Its α-Adrenergic Receptors in the Pathophysiology and Treatment of Major Depressive Disorder and Schizophrenia: A Systematic Review. Front. Psychiatry 2017, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M. The GABA system in schizophrenia: Cells, molecules and microcircuitry. Schizophr. Res. 2015, 167, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Tso, I.F.; Fang, Y.; Phan, K.L.; Welsh, R.C.; Taylor, S.F. Abnormal GABAergic function and face processing in schizophrenia: A pharmacologic-fMRI study. Schizophr. Res. 2015, 168, 338–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benes, F.M.; Berretta, S. GABAergic interneurons: Implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2001, 25, 1–27. [Google Scholar] [CrossRef]
- Guidotti, A.; Auta, J.; Davis, J.M.; Dong, E.; Grayson, D.R.; Veldic, M.; Zhang, X.; Costa, E. GABAergic dysfunction in schizophrenia: New treatment strategies on the horizon. Psychopharmacology (Berl.) 2005, 180, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Garbutt, J.C.; van Kammen, D.P. The interaction between GABA and dopamine: Implications for schizophrenia. Schizophr. Bull. 1983, 9, 336–353. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Lim, B.; Matzilevich, D.; Walsh, J.P.; Subburaju, S.; Minns, M. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc. Natl. Acad. Sci. USA 2007, 104, 10164–10169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlhaas, P.J.; Singer, W. Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci. 2010, 11, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Cardin, J.A.; Carlén, M.; Meletis, K.; Knoblich, U.; Zhang, F.; Deisseroth, K.; Tsai, L.-H.; Moore, C.I. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 2009, 459, 663–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassef, A.; Baker, J.; Kochan, L.D. GABA and schizophrenia: A review of basic science and clinical studies. J. Clin. Psychopharmacol. 2003, 23, 601–640. [Google Scholar] [CrossRef] [PubMed]
- Manzella, F. Smoking in schizophrenic patients: A critique of the self-medication hypothesis. World J. Psychiatry 2015, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Raedler, T.J.; Bymaster, F.P.; Tandon, R.; Copolov, D.; Dean, B. Towards a muscarinic hypothesis of schizophrenia. Mol. Psychiatry 2007, 12, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Brunzell, D.H.; McIntosh, J.M. Alpha7 Nicotinic Acetylcholine Receptors Modulate Motivation to Self-Administer Nicotine: Implications for Smoking and Schizophrenia. Neuropsychopharmacology 2012, 37, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Forchuk, C.; Norman, R.; Malla, A.; Martin, M.-L.; McLean, T.; Cheng, S.; Diaz, K.; McIntosh, E.; Rickwood, A.; Vos, S.; et al. Schizophrenia and the motivation for smoking. Perspect. Psychiatr. Care 2002, 38, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.L.; Bertrand, D. Neuronal α7 Nicotinic Receptors as a Target for the Treatment of Schizophrenia. Int. Rev. Neurobiol. 2015, 124, 79–111. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. Targeting of α7 Nicotinic Acetylcholine Receptors in the Treatment of Schizophrenia and the Use of Auditory Sensory Gating as a Translational Biomarker. Curr. Pharm. Des. 2015, 21, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.; Olincy, A.; Ross, R.G.; Waldo, M.C.; Stevens, K.E.; Adler, L.E.; Leonard, S. The genetics of sensory gating deficits in schizophrenia. Curr. Psychiatry Rep. 2003, 5, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.L.; Bertrand, D. Alpha7 neuronal nicotinic receptors as a drug target in schizophrenia. Expert Opin. Ther. Targets 2013, 17, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Espejo, E.; Viveros, M.-P.; Núñez, L.; Ellenbroek, B.A.; Rodriguez de Fonseca, F. Role of cannabis and endocannabinoids in the genesis of schizophrenia. Psychopharmacology (Berl.) 2009, 206, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Watkins, C.C.; Andrews, S.R. Clinical studies of neuroinflammatory mechanisms in schizophrenia. Schizophr. Res. 2016, 176, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Benros, M.E.; Pedersen, M.G.; Rasmussen, H.; Eaton, W.W.; Nordentoft, M.; Mortensen, P.B. A nationwide study on the risk of autoimmune diseases in individuals with a personal or a family history of schizophrenia and related psychosis. Am. J. Psychiatry 2014, 171, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Derkits, E.J. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Kayser, M.S.; Dalmau, J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr. Res. 2016, 176, 36–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.; Eaton, W.; Cascella, N.; Fasano, A.; Warfel, D.; Feldman, S.; Richardson, C.; Vyas, G.; Linthicum, J.; Santora, D.; et al. A gluten-free diet in people with schizophrenia and anti-tissue transglutaminase or anti-gliadin antibodies. Schizophr. Res. 2012, 140, 262–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaka, J.; Karakuła-Juchnowicz, H.; Morylowska-Topolska, J.; Dzikowski, M.; Juchnowicz, D.; Flis, M.; Siek, A.; Próchnicki, M. Gluten-related disorders and schizophrenia-potential linking mechanisms, diagnostic and therapeutic challenge. Curr. Probl. Psychiatry 2017, 18, 9–24. [Google Scholar] [CrossRef]
- Girgis, R.R.; Zoghbi, A.W.; Javitt, D.C.; Lieberman, J.A. The past and future of novel, non-dopamine-2 receptor therapeutics for schizophrenia: A critical and comprehensive review. J. Psychiatr. Res. 2018, in press. [Google Scholar] [CrossRef]
- Laan, W.; Grobbee, D.E.; Selten, J.-P.; Heijnen, C.J.; Kahn, R.S.; Burger, H. Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: Results from a randomized, double-blind, placebo-controlled trial. J. Clin. Psychiatry 2010, 71, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Emiliani, F.E.; Sedlak, T.W.; Sawa, A. Oxidative stress and schizophrenia: Recent breakthroughs from an old story. Curr. Opin. Psychiatry 2014, 27, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.-H.; Steullet, P.; Kraftsik, R.; Cuenod, M.; Do, K.Q. Early-life insults impair parvalbumin interneurons via oxidative stress: Reversal by N-acetylcysteine. Biol. Psychiatry 2013, 73, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Sakurai, T.; Davis, K.L.; Buxbaum, J.D. Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog. Neurobiol. 2011, 93, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rang, H.P.; Ritter, J.M. Rang and Dale’s Pharmacology. Available online: https://books.google.com.hk/books?hl=zh-TW&lr=&id=iOLTBQAAQBAJ&oi=fnd&pg=PP1&dq=RanR+and+Dale%E2%80%99s+Pharmacology&ots=bfK9_o1-Kq&sig=cEY_pWIEHyk1WN84ygU02Hc6NNd&redir_esc=y#v=onepage&q=Rang%20and%20Dale%E2%80%99s%20Pharmacology&f=false (accessed on 18 August 2018).
- Anne, B.P. British National Formulary. Available online: https://www.cambridge.org/core/journals/psychiatric-bulletin/article/british-national-formulary/06832CA19CD79FFBFB7C90844CA47B2C (accessed on 18 August 2018).
- Leucht, S.; Corves, C.; Arbter, D.; Engel, R.R.; Li, C.; Davis, J.M. Second-generation versus first-generation antipsychotic drugs for schizophrenia: A meta-analysis. Lancet Lond. Engl. 2009, 373, 31–41. [Google Scholar] [CrossRef]
- Pisani, A.; Bernardi, G.; Ding, J.; Surmeier, D.J. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007, 30, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S. Naming the drugs we use: Neuroscience-based nomenclature, a helpful innovation. Ther. Adv. Psychopharmacol. 2018, 8, 171–172. [Google Scholar] [CrossRef] [PubMed]
- López-Muñoz, F.; Alamo, C.; Cuenca, E.; Shen, W.W.; Clervoy, P.; Rubio, G. History of the discovery and clinical introduction of chlorpromazine. Ann. Clin. Psychiatry Off. J. Am. Acad. Clin. Psychiatr. 2005, 17, 113–135. [Google Scholar] [CrossRef]
- Schatzberg, A.F.; Nemeroff, C. The American Psychiatric Publishing Textbook of Psychopharmacology. Available online: https://ajp.psychiatryonline.org/doi/full/10.1176/appi.ajp.2010.10010012 (accessed on 18 August 2018).
- Brenner, G.M.; Stevens, C. Brenner and Stevens’ Pharmacology. Available online: https://books.google.com.hk/books?hl=zh-TW&lr=&id=v3g4DwAAQBAJ&oi=fnd&pg=PP1&dq=CraiC+Stevens+Brenner+and+Stevens%E2%80%99+Pharmacology&ots=57Spmo95dW&sig=VAeT0OMmOMmoiAXOq_vouBQqwE4&redir_esc=y#v=onepage&q=Craig%20Stevens%20Brenner%20and%20SteStev%E2%80%99%20Pharmacology&f=false (accessed on 18 August 2018).
- Crilly, J. The history of clozapine and its emergence in the US market: A review and analysis. Hist. Psychiatry 2007, 18, 39–60. [Google Scholar] [CrossRef] [PubMed]
- Strange, P.G. Antipsychotic drugs: Importance of dopamine receptors for mechanisms of therapeutic actions and side effects. Pharmacol. Rev. 2001, 53, 119–133. [Google Scholar] [PubMed]
- Maher, A.R.; Theodore, G. Summary of the comparative effectiveness review on off-label use of atypical antipsychotics. J. Manag. Care Pharm. JMCP 2012, 18, S1–20. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.L.; Streicher, J.M.; Meltzer, H.Y.; Bohn, L.M. Clozapine acts as an agonist at serotonin 2A receptors to counter MK-801-induced behaviors through a βarrestin2-independent activation of Akt. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2014, 39, 1902–1913. [Google Scholar] [CrossRef] [PubMed]
- Möller, H.-J. Risperidone: A review. Expert Opin. Pharmacother. 2005, 6, 803–818. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.; Fitton, A. Risperidone. A review of its pharmacology and therapeutic potential in the treatment of schizophrenia. Drugs 1994, 48, 253–273. [Google Scholar] [CrossRef] [PubMed]
- Seeger, T.F.; Seymour, P.A.; Schmidt, A.W.; Zorn, S.H.; Schulz, D.W.; Lebel, L.A.; McLean, S.; Guanowsky, V.; Howard, H.R.; Lowe, J.A. Ziprasidone (CP-88,059): A new antipsychotic with combined dopamine and serotonin receptor antagonist activity. J. Pharmacol. Exp. Ther. 1995, 275, 101–113. [Google Scholar] [PubMed]
- Pompili, M.; Verzura, C.; Trovini, G.; Buscajoni, A.; Falcone, G.; Naim, S.; Nardella, A.; Sorice, S.; Baldessarini, R.J.; Girardi, P. Lurasidone: Efficacy and safety in the treatment of psychotic and mood disorders. Expert Opin. Drug Saf. 2018, 17, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Dargani, N.V.; Malhotra, A.K. Safety profile of iloperidone in the treatment of schizophrenia. Expert Opin. Drug Saf. 2014, 13, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Marazziti, D.; Piccinni, A.; Baroni, S.; Mungai, F.; Presta, S.; Mucci, F.; Dell’Osso, L. Current Trends on Antipsychotics: Focus on Asenapine. Curr. Med. Chem. 2016, 23, 2204–2216. [Google Scholar] [CrossRef] [PubMed]
- Kemp, D.E.; Zhao, J.; Cazorla, P.; Landbloom, R.P.; Mackle, M.; Snow-Adami, L.; Szegedi, A. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J. Clin. Psychiatry 2014, 75, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, R.A.; Bruno, A.; Muscatello, M.R.A. Efficacy and safety of sertindole in schizophrenia: A clinical review. J. Clin. Psychopharmacol. 2015, 35, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.-J.; Jeong, T.C.; Noh, K.; Baek, I.-W.; Kwon, K.; Kim, E.; Yoon, Y.-R.; Kang, W. Prandial effect on the systemic exposure of amisulpride. Arch. Pharm. Res. 2014, 37, 1325–1328. [Google Scholar] [CrossRef] [PubMed]
- Salvi, V.; Mencacci, C.; Barone-Adesi, F. H1-histamine receptor affinity predicts weight gain with antidepressants. Eur. Neuropsychopharmacol. 2016, 26, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Deng, C.; Huang, X.-F. The role of hypothalamic H1 receptor antagonism in antipsychotic-induced weight gain. CNS Drugs 2013, 27, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Solismaa, A.; Kampman, O.; Lyytikäinen, L.-P.; Seppälä, N.; Viikki, M.; Mononen, N.; Lehtimäki, T.; Leinonen, E. Histaminergic gene polymorphisms associated with sedation in clozapine-treated patients. Eur. Neuropsychopharmacol. 2017, 27, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, L.; Yang, J.; Bresee, L.; Jette, N.; Patten, S.; Pringsheim, T. Second-Generation Antipsychotics and Metabolic Side Effects: A Systematic Review of Population-Based Studies. Drug Saf. 2017, 40, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Burris, K.D.; Molski, T.F.; Xu, C.; Ryan, E.; Tottori, K.; Kikuchi, T.; Yocca, F.D.; Molinoff, P.B. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J. Pharmacol. Exp. Ther. 2002, 302, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, D.A.; Renock, S.; Arrington, E.; Chiodo, L.A.; Liu, L.-X.; Sibley, D.R.; Roth, B.L.; Mailman, R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2003, 28, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Tamminga, C.A. Partial dopamine agonists in the treatment of psychosis. J. Neural Transm. Vienna Austria 1996 2002, 109, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Mailman, R.B.; Murthy, V. Third generation antipsychotic drugs: Partial agonism or receptor functional selectivity? Curr. Pharm. Des. 2010, 16, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A. Dopamine partial agonists: A new class of antipsychotic. CNS Drugs 2004, 18, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Fell, M.J.; Perry, K.W.; Falcone, J.F.; Johnson, B.G.; Barth, V.N.; Rash, K.S.; Lucaites, V.L.; Threlkeld, P.G.; Monn, J.A.; McKinzie, D.L.; et al. In vitro and in vivo evidence for a lack of interaction with dopamine D2 receptors by the metabotropic glutamate 2/3 receptor agonists 1S,2S,5R,6S-2-aminobicyclo[3.1.0]hexane-2,6-bicaroxylate monohydrate (LY354740) and (-)-2-oxa-4-aminobicyclo[3.1.0] Hexane-4,6-dicarboxylic acid (LY379268). J. Pharmacol. Exp. Ther. 2009, 331, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Masri, B.; Salahpour, A.; Didriksen, M.; Ghisi, V.; Beaulieu, J.-M.; Gainetdinov, R.R.; Caron, M.G. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. USA. 2008, 105, 13656–13661. [Google Scholar] [CrossRef] [PubMed]
- Brust, T.F.; Hayes, M.P.; Roman, D.L.; Watts, V.J. New functional activity of aripiprazole revealed: Robust antagonism of D2 dopamine receptor-stimulated Gβγ signaling. Biochem. Pharmacol. 2015, 93, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leucht, S.; Cipriani, A.; Spineli, L.; Mavridis, D.; Orey, D.; Richter, F.; Samara, M.; Barbui, C.; Engel, R.R.; Geddes, J.R.; et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: Amultiple-treatments meta-analysis. Lancet Lond. Engl. 2013, 382, 951–962. [Google Scholar] [CrossRef]
- Poyurovsky, M.; Bergman, J.; Pashinian, A.; Weizman, A. Beneficial effect of low-dose mirtazapine in acute aripiprazole-induced akathisia. Int. Clin. Psychopharmacol. 2014, 29, 296–298. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, E.L.A.; de Mendonça Lima, T.; Vieira, M.E.B.; Storpirtis, S.; Aguiar, P.M. Efficacy and safety of aripiprazole for the treatment of schizophrenia: An overview of systematic reviews. Eur. J. Clin. Pharmacol. 2018, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hope, J.; Castle, D.; Keks, N.A. Brexpiprazole: A new leaf on the partial dopamine agonist branch. Australas. Psychiatry Bull. R. Aust. N. Z. Coll. Psychiatr. 2018, 26, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Lerdrup, L.; Sugino, H.; Akazawa, H.; Amada, N.; McQuade, R.D.; Stensbøl, T.B.; Bundgaard, C.; Arnt, J.; Kikuchi, T. Brexpiprazole II: Antipsychotic-like and procognitive effects of a novel serotonin-dopamine activity modulator. J. Pharmacol. Exp. Ther. 2014, 350, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Burda-Malarz, K.; Kus, K.; Ratajczak, P.; Czubak, A.; Nowakowska, E.; Jędrzejewski, L.; Sadowski, C. Evaluation of antidepressant and memory-improving efficacy of aripiprazole and fluoxetine in alcohol-preferring rats. Acta Neuropsychiatr. 2014, 26, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Björkholm, C.; Marcus, M.M.; Konradsson-Geuken, Å.; Jardemark, K.; Svensson, T.H. The novel antipsychotic drug brexpiprazole, alone and in combination with escitalopram, facilitates prefrontal glutamatergic transmission via a dopamine D1 receptor-dependent mechanism. Eur. Neuropsychopharmacol. 2017, 27, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Ren, Q.; Yang, C.; Zhang, J.-C.; Yao, W.; Dong, C.; Ohgi, Y.; Futamura, T.; Hashimoto, K. Antidepressant effects of combination of brexpiprazole and fluoxetine on depression-like behavior and dendritic changes in mice after inflammation. Psychopharmacology (Berl.) 2017, 234, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Ren, Q.; Yang, C.; Zhang, J.-C.; Yao, W.; Dong, C.; Ohgi, Y.; Futamura, T.; Hashimoto, K. Adjunctive treatment of brexpiprazole with fluoxetine shows a rapid antidepressant effect in social defeat stress model: Role of BDNF-TrkB signaling. Sci. Rep. 2016, 6, 39209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diefenderfer, L.A.; Iuppa, C. Brexpiprazole: A review of a new treatment option for schizophrenia and major depressive disorder. Ment. Health Clin. 2017, 7, 207–212. [Google Scholar] [CrossRef] [PubMed]
- De Deurwaerdère, P. Cariprazine:New dopamine biased agonist for neuropsychiatric disorders. Drugs Today Barc. Spain 1998 2016, 52, 97–110. [Google Scholar] [CrossRef]
- Citrome, L. Cariprazine: Chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin. Drug Metab. Toxicol. 2013, 9, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Németh, G.; Laszlovszky, I.; Czobor, P.; Szalai, E.; Szatmári, B.; Harsányi, J.; Barabássy, Á.; Debelle, M.; Durgam, S.; Bitter, I.; et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: A randomised, double-blind, controlled trial. Lancet 2017, 389, 1103–1113. [Google Scholar] [CrossRef]
- Gyertyán, I.; Kiss, B.; Sághy, K.; Laszy, J.; Szabó, G.; Szabados, T.; Gémesi, L.I.; Pásztor, G.; Zájer-Balázs, M.; Kapás, M.; et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem. Int. 2011, 59, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Garnock-Jones, K.P. Cariprazine: A Review in Schizophrenia. CNS Drugs 2017, 31, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Scarff, J.R. The prospects of cariprazine in the treatment of schizophrenia. Ther. Adv. Psychopharmacol. 2017, 7, 237–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-M.; Han, C.; Lee, S.-J.; Jun, T.-Y.; Patkar, A.A.; Masand, P.S.; Pae, C.-U. Investigational dopamine antagonists for the treatment of schizophrenia. Expert Opin. Investig. Drugs 2017, 26, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Tuplin, E.W.; Holahan, M.R. Aripiprazole, A Drug that Displays Partial Agonism and Functional Selectivity. Curr. Neuropharmacol. 2017, 15, 1192–1207. [Google Scholar] [CrossRef] [PubMed]
- Martí-Solano, M.; Guixà-González, R.; Sanz, F.; Pastor, M.; Selent, J. Novel insights into biased agonism at G protein-coupled receptors and their potential for drug design. Curr. Pharm. Des. 2013, 19, 5156–5166. [Google Scholar] [CrossRef] [PubMed]
- Madariaga-Mazón, A.; Marmolejo-Valencia, A.F.; Li, Y.; Toll, L.; Houghten, R.A.; Martinez-Mayorga, K. Mu-Opioid receptor biased ligands: A safer and painless discovery of analgesics? Drug Discov. Today 2017, 22, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A. New paradigms in GPCR drug discovery. Biochem. Pharmacol. 2015, 98, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Filizola, M. Increasingly accurate dynamic molecular models of G-protein coupled receptor oligomers: Panacea or Pandora’s box for novel drug discovery? Life Sci. 2010, 86, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Selent, J.; Bauer-Mehren, A.; López, L.; Loza, M.I.; Sanz, F.; Pastor, M. A novel multilevel statistical method for the study of the relationships between multireceptorial binding affinity profiles and in vivo endpoints. Mol. Pharmacol. 2010, 77, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Targowska-Duda, K.M.; Budzyńska, B.; Biała, G.; Silva, A.G.; Castro, M. In vitro, molecular modeling and behavioral studies of 3-{[4-(5-methoxy-1H-indol-3-yl)-1,2,3,6-tetrahydropyridin-1-yl]methyl}-1,2-dihydroquinolin-2-one (D2AAK1) as a potential antipsychotic. Neurochem. Int. 2016, 96, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.-M.; Gainetdinov, R.R.; Caron, M.G. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol. Sci. 2007, 28, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-M.; Caron, M.G. Complementary roles of the DRY motif and C-terminus tail of GPCRS for G protein coupling and beta-arrestin interaction. Biochem. Biophys. Res. Commun. 2008, 366, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Brust, T.F.; Hayes, M.P.; Roman, D.L.; Burris, K.D.; Watts, V.J. Bias analyses of preclinical and clinical D2 dopamine ligands: Studies with immediate and complex signaling pathways. J. Pharmacol. Exp. Ther. 2015, 352, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Shonberg, J.; Herenbrink, C.K.; López, L.; Christopoulos, A.; Scammells, P.J.; Capuano, B.; Lane, J.R. A structure-activity analysis of biased agonism at the dopamine D2 receptor. J. Med. Chem. 2013, 56, 9199–9221. [Google Scholar] [CrossRef] [PubMed]
- Szabo, M.; Klein Herenbrink, C.; Christopoulos, A.; Lane, J.R.; Capuano, B. Structure-activity relationships of privileged structures lead to the discovery of novel biased ligands at the dopamine D2 receptor. J. Med. Chem. 2014, 57, 4924–4939. [Google Scholar] [CrossRef] [PubMed]
- Aringhieri, S.; Kolachalam, S.; Gerace, C.; Carli, M.; Verdesca, V.; Brunacci, M.G.; Rossi, C.; Ippolito, C.; Solini, A.; Corsini, G.U.; et al. Clozapine as the most efficacious antipsychotic for activating ERK 1/2 kinases: Role of 5-HT2A receptor agonism. Eur. Neuropsychopharmacol. 2017, 27, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; Donthamsetti, P.; Shonberg, J.; Draper-Joyce, C.J.; Dentry, S.; Michino, M.; Shi, L.; López, L.; Scammells, P.J.; Capuano, B.; et al. A new mechanism of allostery in a G protein-coupled receptor dimer. Nat. Chem. Biol. 2014, 10, 745–752. [Google Scholar] [CrossRef] [PubMed]
- McRobb, F.M.; Crosby, I.T.; Yuriev, E.; Lane, J.R.; Capuano, B. Homobivalent ligands of the atypical antipsychotic clozapine: Design, synthesis, and pharmacological evaluation. J. Med. Chem. 2012, 55, 1622–1634. [Google Scholar] [CrossRef] [PubMed]
- Salama, I.; Löber, S.; Hübner, H.; Gmeiner, P. Synthesis and binding profile of haloperidol-based bivalent ligands targeting dopamine D(2)-like receptors. Bioorg. Med. Chem. Lett. 2014, 24, 3753–3756. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.J.; Conn, P.J. Allosteric modulation of GPCRs: New insights and potential utility for treatment of schizophrenia and other CNS disorders. Neuron 2017, 94, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Borroto-Escuela, D.O.; Tarakanov, A.O.; Romero-Fernandez, W.; Ferraro, L.; Tanganelli, S.; Perez-Alea, M.; Di Palma, M.; Agnati, L.F. Dopamine D2 heteroreceptor complexes and their receptor-receptor interactions in ventral striatum: Novel targets for antipsychotic drugs. Prog. Brain Res. 2014, 211, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Ferré, S.; Canals, M.; Torvinen, M.; Terasmaa, A.; Marcellino, D.; Goldberg, S.R.; Staines, W.; Jacobsen, K.X.; Lluis, C.; et al. Adenosine A2A and dopamine D2 heteromeric receptor complexes and their function. J. Mol. Neurosci. MN 2005, 26, 209–220. [Google Scholar] [CrossRef]
- Guidolin, D.; Agnati, L.F.; Marcoli, M.; Borroto-Escuela, D.O.; Fuxe, K. G-protein-coupled receptor type A heteromers as an emerging therapeutic target. Expert Opin. Ther. Targets 2015, 19, 265–283. [Google Scholar] [CrossRef] [PubMed]
- González-Maeso, J.; Ang, R.L.; Yuen, T.; Chan, P.; Weisstaub, N.V.; López-Giménez, J.F.; Zhou, M.; Okawa, Y.; Callado, L.F.; Milligan, G.; et al. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 2008, 452, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunlop, J.; Brandon, N.J. Schizophrenia drug discovery and development in an evolving era: Are new drug targets fulfilling expectations? J. Psychopharmacol. Oxf. Engl. 2015, 29, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Jarskog, L.F.; Fleischhacker, W.W. Alternative pharmacologic targets for the treatment of schizophrenia: Results from phase I and II trials. Curr. Opin. Psychiatry 2013, 26, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Matosin, N.; Fernandez-Enright, F.; Frank, E.; Deng, C.; Wong, J.; Huang, X.-F.; Newell, K.A. Metabotropic glutamate receptor mGluR2/3 and mGluR5 binding in the anterior cingulate cortex in psychotic and nonpsychotic depression, bipolar disorder and schizophrenia: Implications for novel mGluR-based therapeutics. J. Psychiatry Neurosci. JPN 2014, 39, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Duinen, M.V.; Reneerkens, O.A.H.; Lambrecht, L.; Sambeth, A.; Rutten, B.P.F.; Os, J.V.; Blokland, A.; Prickaerts, J. Treatment of Cognitive Impairment in Schizophrenia: Potential Value of Phosphodiesterase Inhibitors in Prefrontal Dysfunction. Curr. Pharm. Des. 2015, 21, 3813–3828. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Antipsychotic Drugs | Drug Examples | ||
|---|---|---|---|
Phenothiazines | Side chain | Aliphatic | Chlorpromazine X = Cl; R =  |
Levomepromazine X = OCH3; R =  | |||
Promazine X = H; R =  | |||
Triflupromazine X = CF3; R =  | |||
| Piperidine | Mesoridazine X = SOCH3; R =  | ||
Pericyazine X = CN; R =  | |||
Pipotiazine X = SO2N(CH3)2; R =  | |||
Thioridazine X = SCH3; R =  | |||
| Piperazine | Fluphenazine X = CF3; R =  | ||
Perphenazine X = Cl; R =  | |||
Prochlorperazine X = Cl; R =  | |||
Trifluoperazine X = CF3; R =  | |||
Butyrophenones | Benperidol  | ||
Droperidol  | |||
Haloperidol  | |||
Thioxanthenes | Clopenthixol X = Cl; R =  mixture of cis and trans isomers | ||
Flupenthixol X = CF3; R =  mixture of cis and trans isomers | |||
Thiothixene X = SO2N(CH3)2; R =  cis isomer | |||
Zuclopenthixol X = Cl; R =  cis isomer | |||
| Dihydroindolones | Molindone  | ||
Dibenzepines | Clotiapine X = S | ||
| Loxapine X = O | |||
Diphenylbutylpiperidines | Fluspirilene  | ||
Pimozide  | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. https://doi.org/10.3390/molecules23082087
Stępnicki P, Kondej M, Kaczor AA. Current Concepts and Treatments of Schizophrenia. Molecules. 2018; 23(8):2087. https://doi.org/10.3390/molecules23082087
Chicago/Turabian StyleStępnicki, Piotr, Magda Kondej, and Agnieszka A. Kaczor. 2018. "Current Concepts and Treatments of Schizophrenia" Molecules 23, no. 8: 2087. https://doi.org/10.3390/molecules23082087