Single Laboratory Validation of a Quantitative Core Shell-Based LC Separation for the Evaluation of Silymarin Variability and Associated Antioxidant Activity of Pakistani Ecotypes of Milk Thistle (Silybum Marianum L.)


 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Chemicals
2.3. Extraction
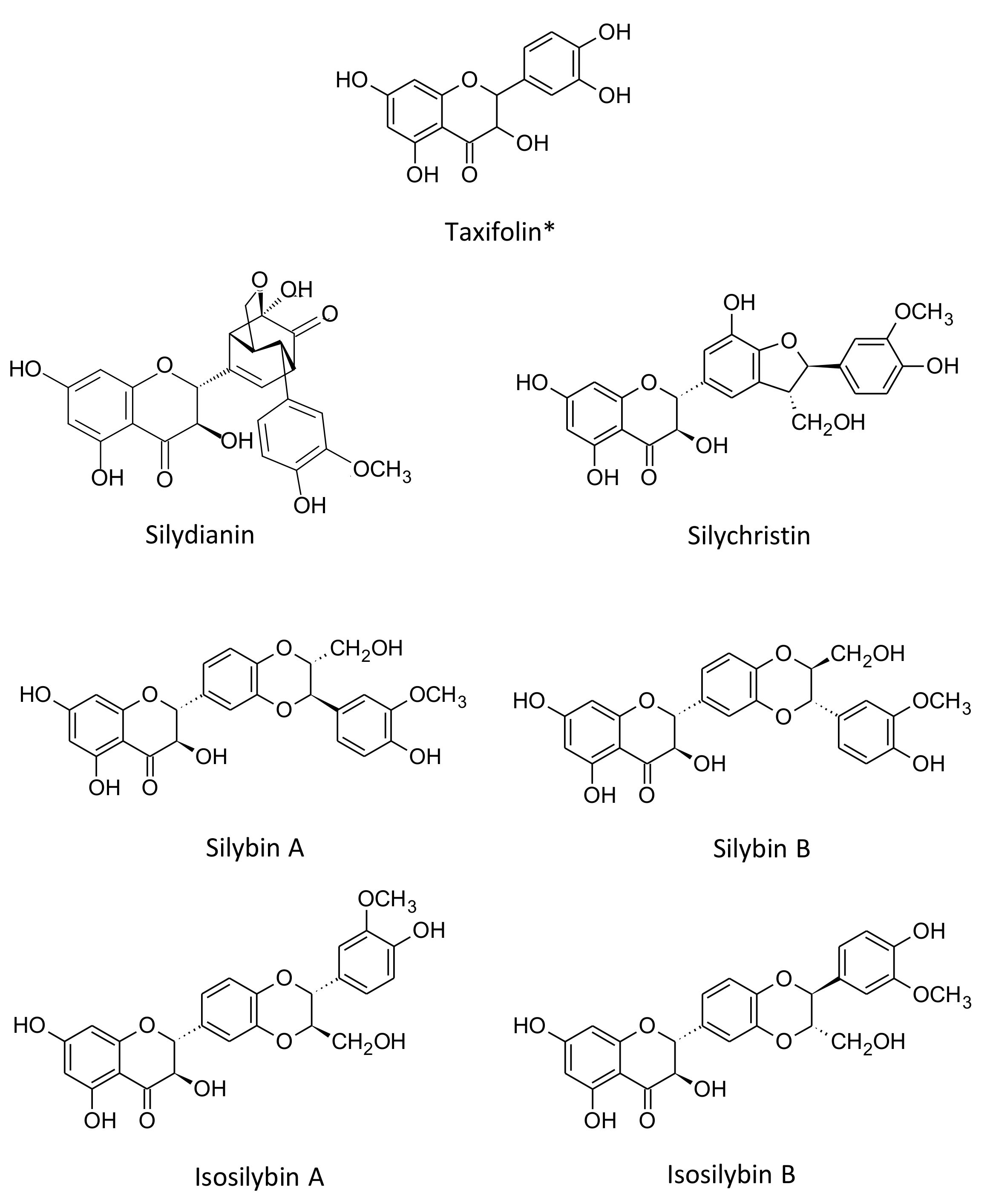
2.4. HPLC-ESI-MS Analysis
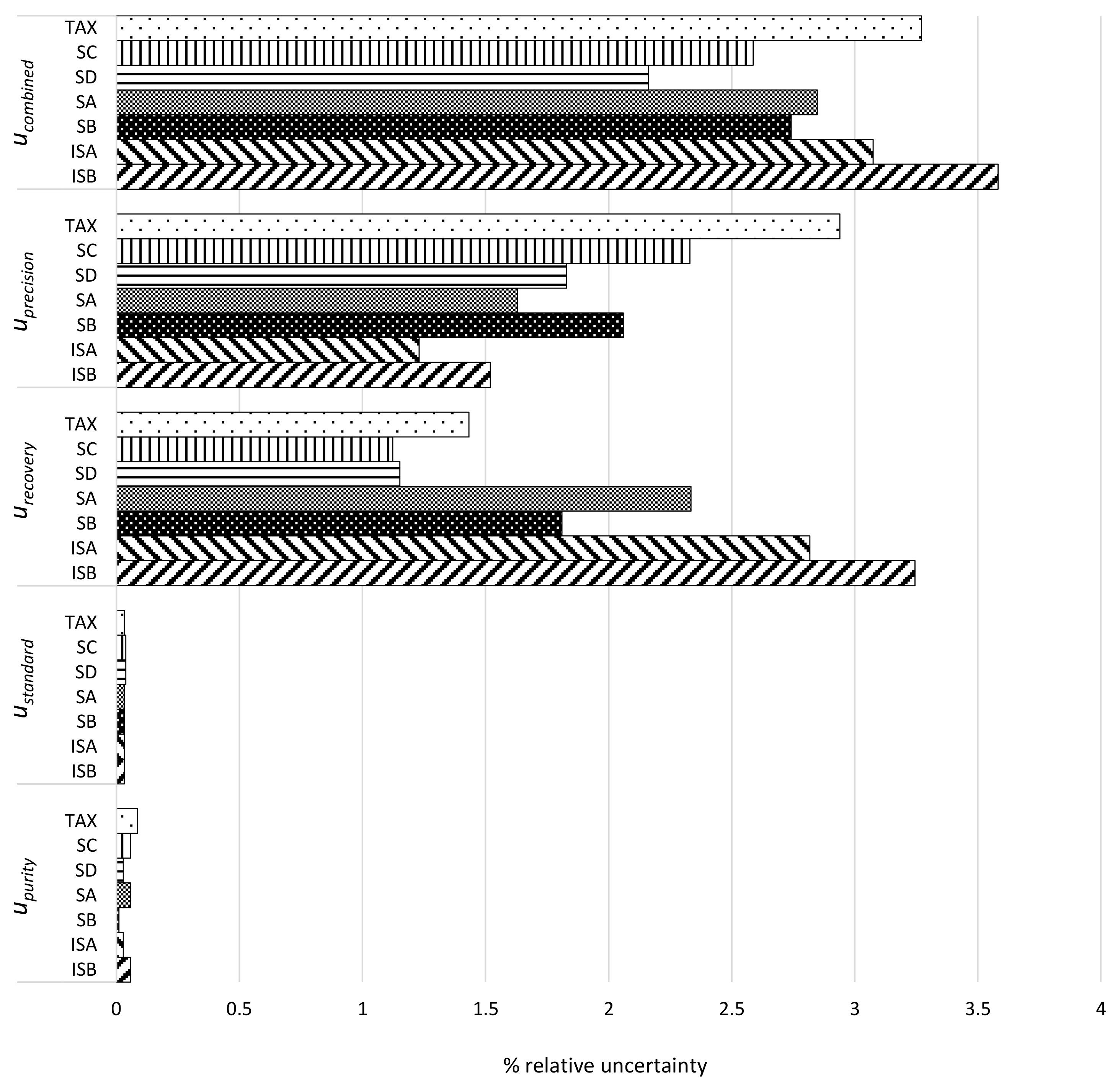
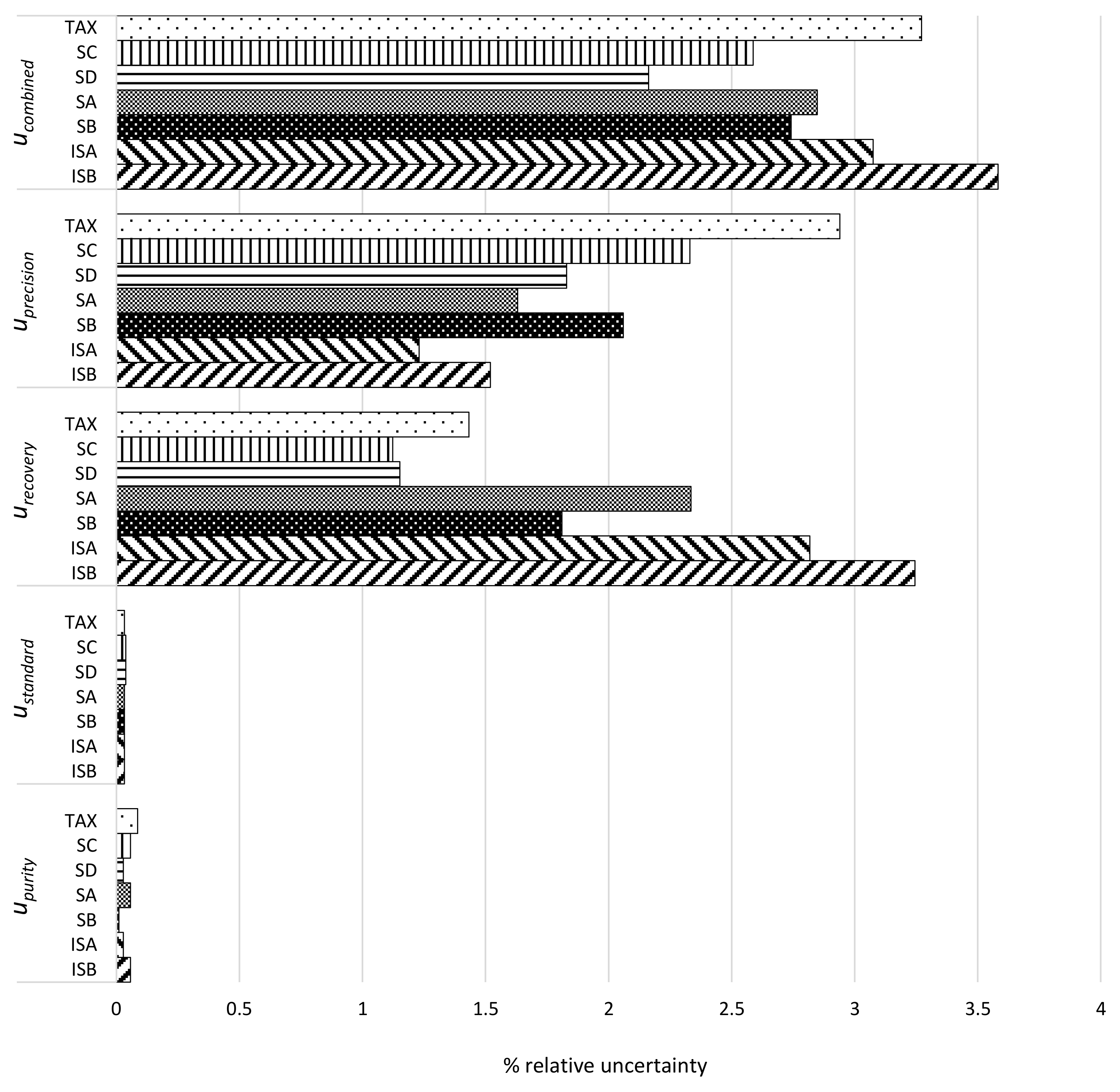
2.5. Method Validation
2.6. Determination of Total Phenolic Production
2.7. Determination of Total Flavonoid Production
2.8. Determination of Antioxidant Activity
2.9. Statistical Treatment of Data
3. Results and Discussion
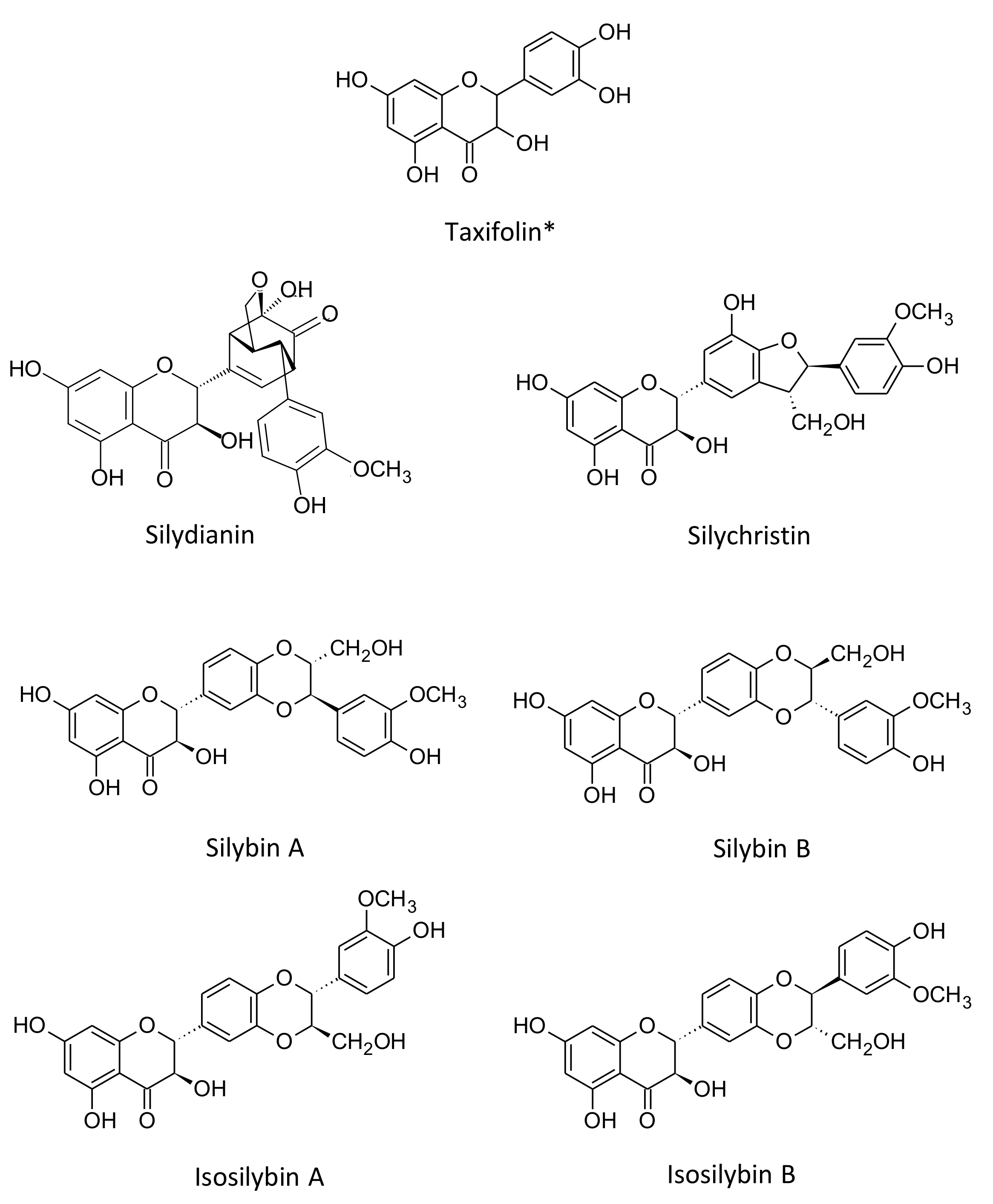
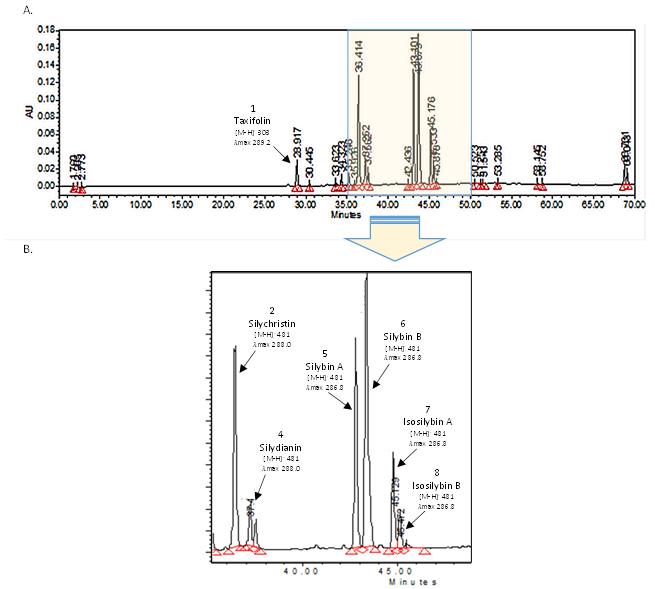
3.1. Single Laboratory Validation of a LC-Based Separation of the Silymarin Constituents
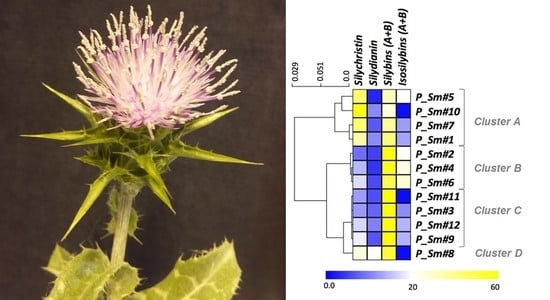
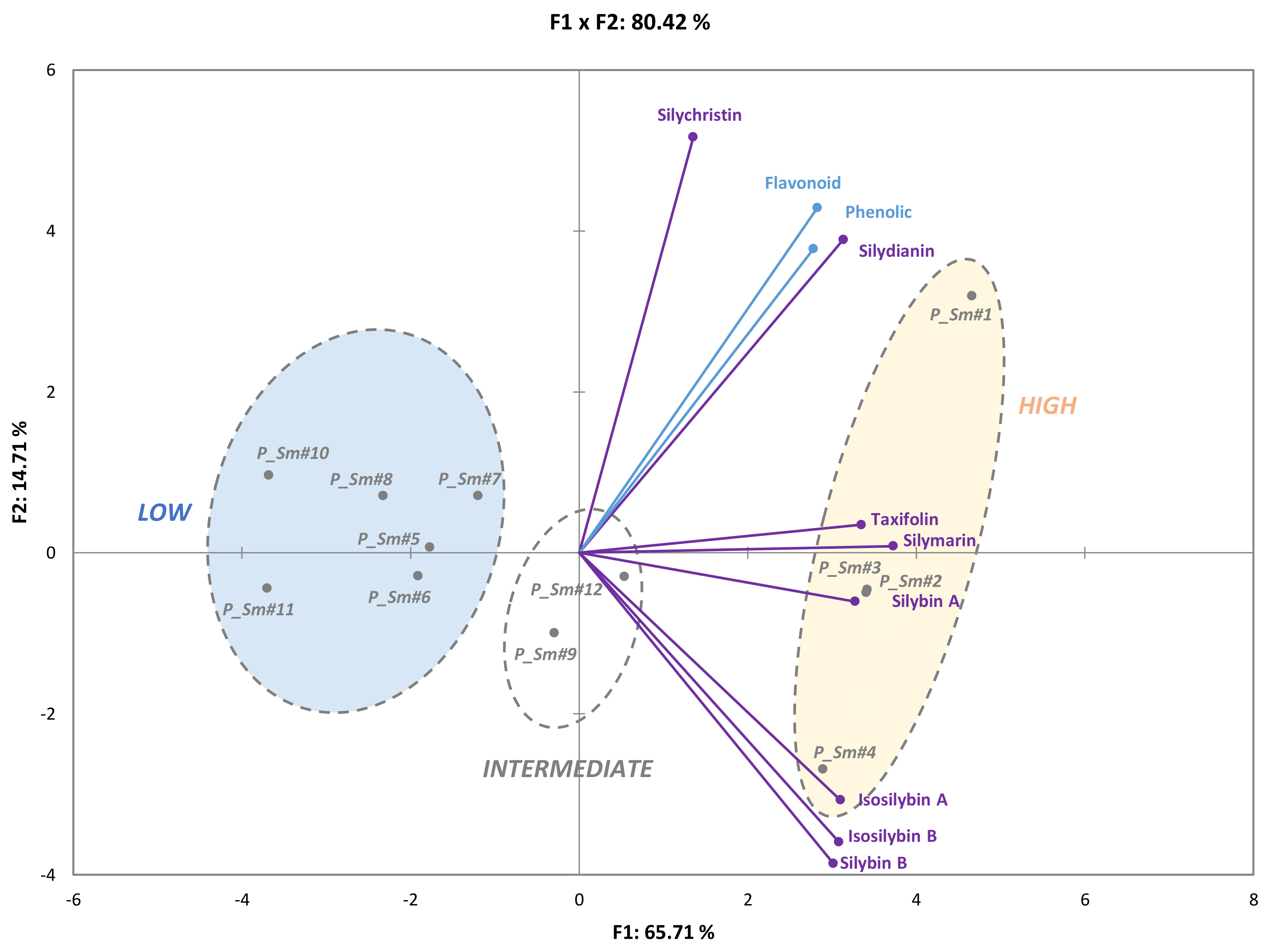
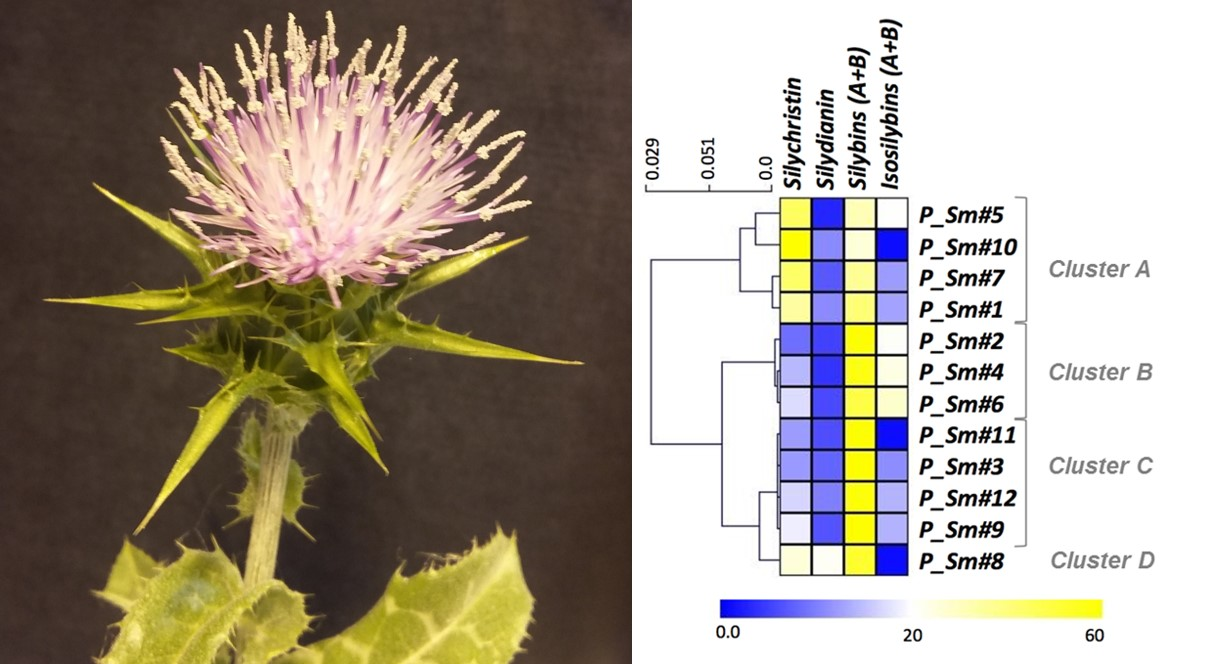
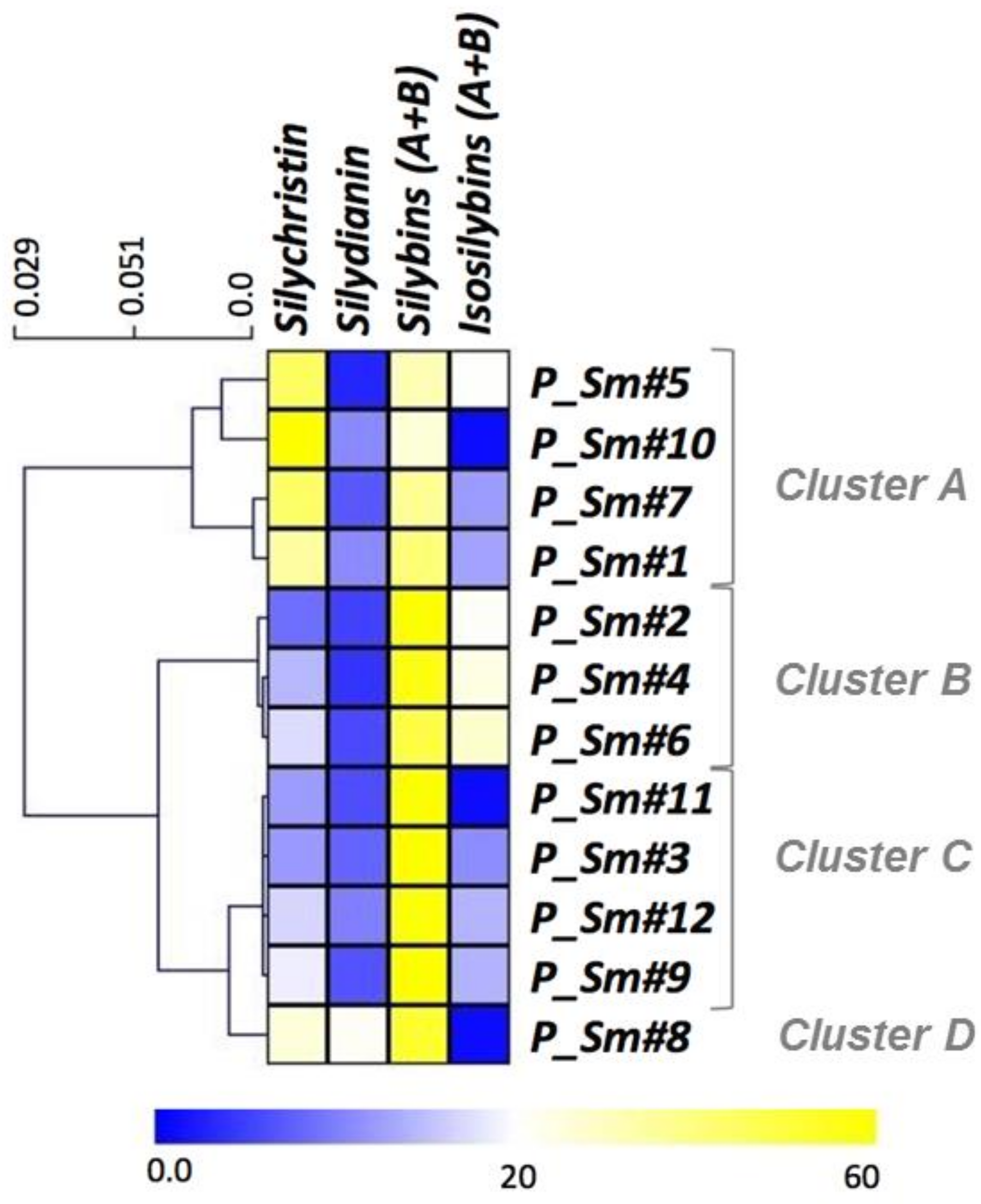
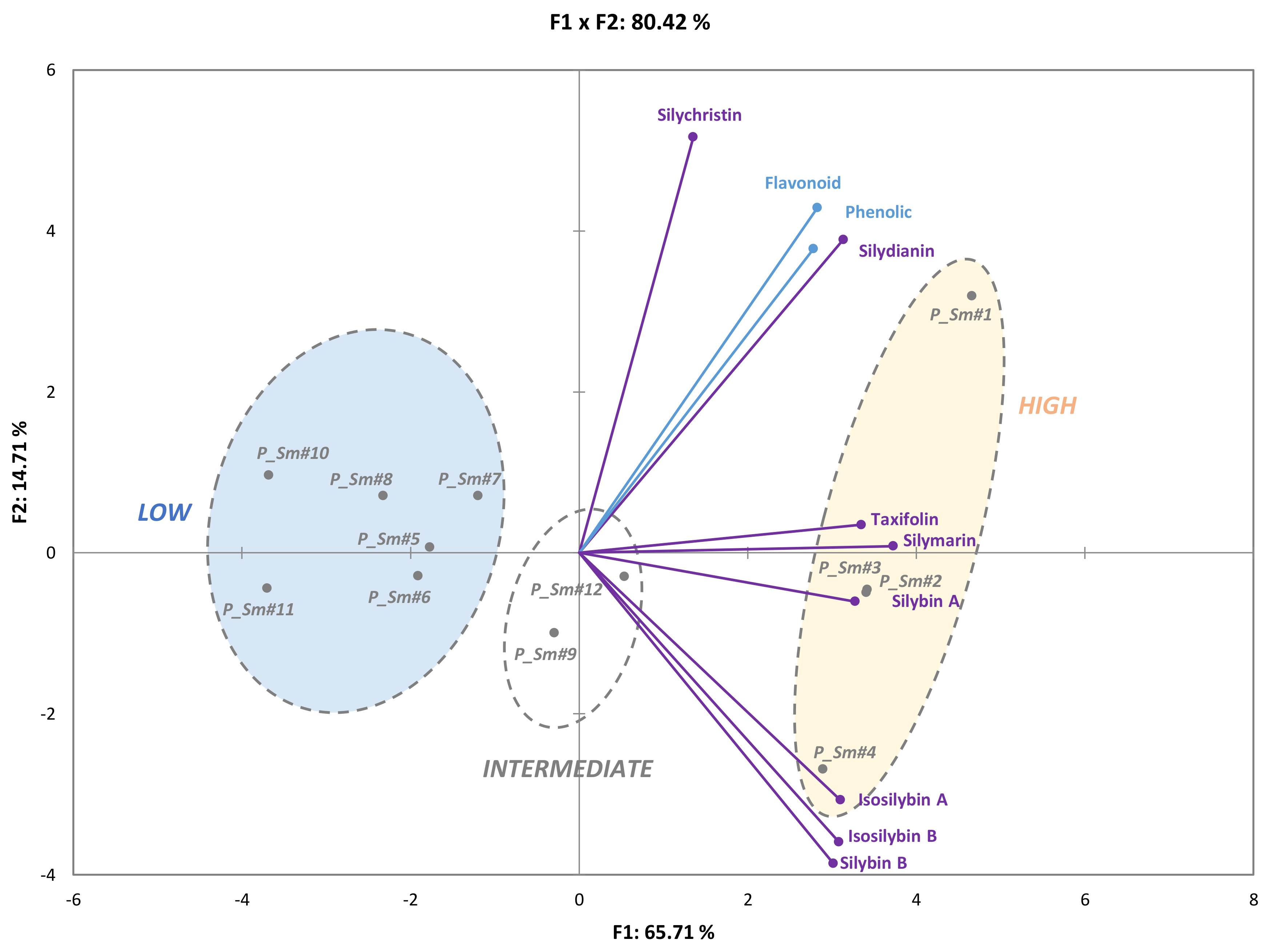
3.2. Evaluation of the Natural Variations in Silymarin Composition in Achene Extracts of Pakistani S. Marianum Accessions
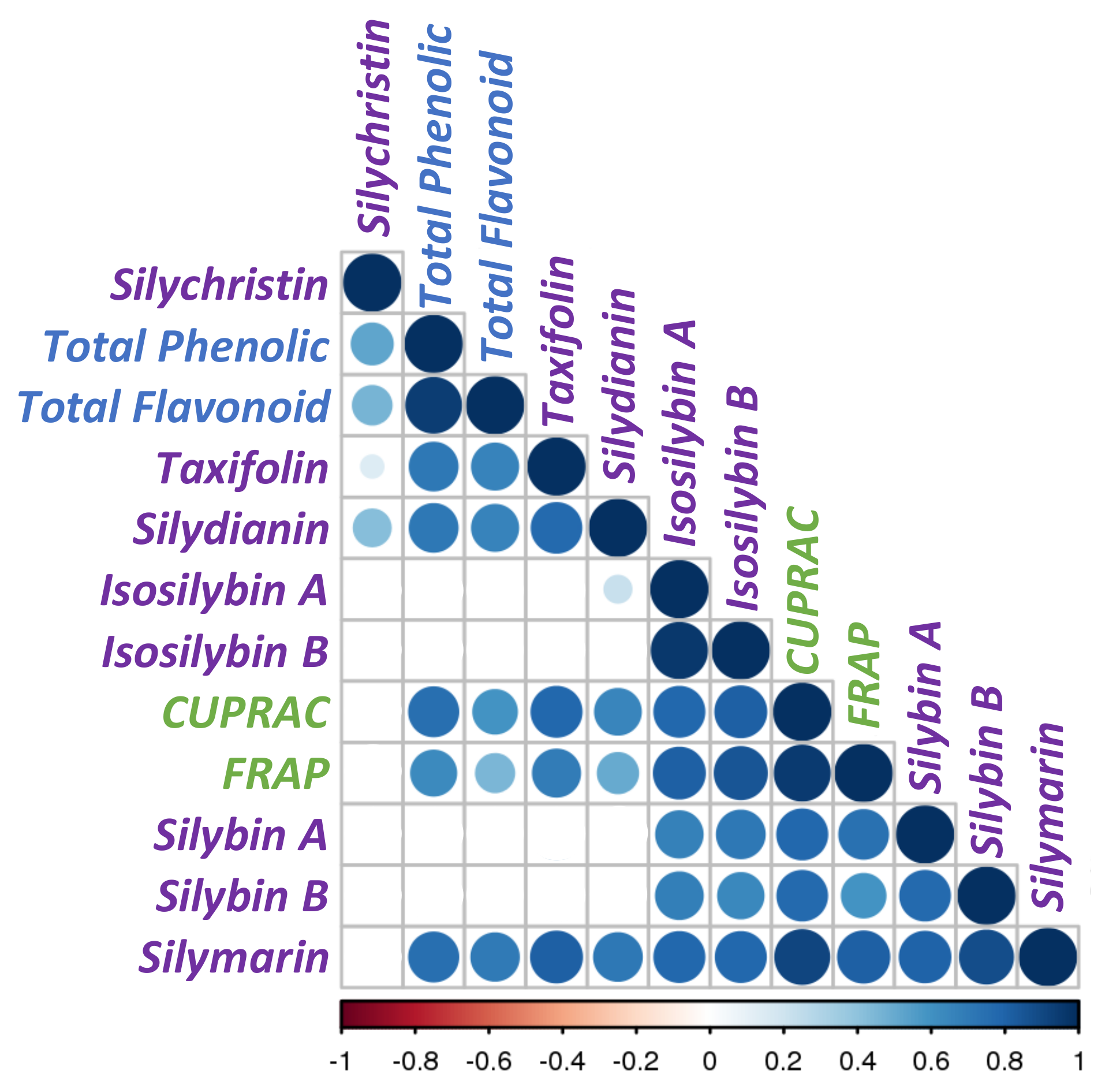
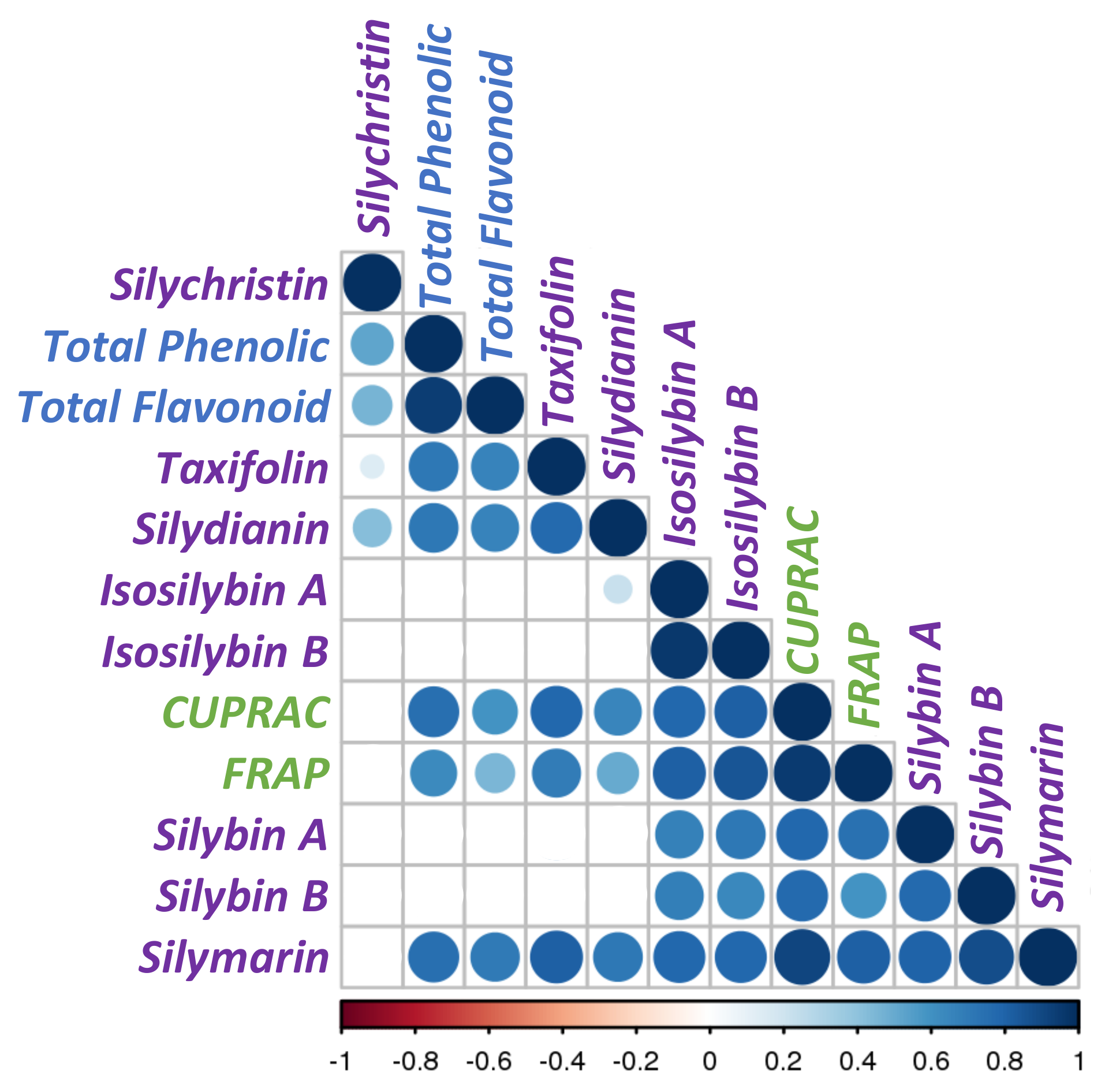
3.3. Correlations between Phytochemical Profiles and Antioxidant Activities
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, D.Y.; Liu, Y. Molecular structure and stereochemistry of Silybin A, Silybin B, Isosilybin, and Isosilybin B, isolated from Silybum marianum (Milk Thistle). J. Nat. Prod. 2003, 66, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Morazzoni, P.; Bombardelli, E. Silybum marianum (Carduus Marianus). Fitoterapia 1995, 66, 3–42. [Google Scholar]
- Madrid, E.; Corchete, P. Silymarin secretion and its elicitation by methyl jasmonate in cell cultures of Silybum marianum is mediated by phospholipase d-phosphatidic acid. J. Exp. Bot. 2010, 61, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Abouzid, S.F.; Chen, S.; Mcalpine, J.B.; Friesen, J.B.; Pauli, G.F. Silybum marianum pericarp yields enhanced silymarin products. Fitoterapia 2016, 112, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Hlangothia, D.; Abdel-rahman, F.; Nguyen, T.; Anthony, K.; Saleh, M.A. Distribution of Silymarin in the Fruit of Silybum marianum L. Pharm. Anal. Acta 2016, 7, 2. [Google Scholar] [CrossRef]
- Bijak, M. Silybin, a Major Bioactive Component of Milk Thistle (Silybum marianum L. Gaernt.)—Chemistry. Bioavailability, and Metabolism. Molecules 2017, 22, 1942. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Dallio, M.; Loguercio, C. Silymarin/Silybin and chronic liver disease: A marriage of many years. Molecules 2017, 22, 191. [Google Scholar] [CrossRef] [PubMed]
- Bijak, M.; Szelenberger, R.; Saluk, J.; Nowak, P. Flavonolignans inhibit ADP induced blood platelets activation and aggregation in whole blood. Int. J. Biol. Macromol. 2017, 95, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Bijak, M.; Saluk-bijak, J. Flavonolignans inhibit the arachidonic acid pathway in blood platelets. BMC Complement. Altern. Med. 2017, 17, 396. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.T.; Kawa, K.; Eckl, V.; Morton, C.; Stredney, R. Herbal Supplement Sales in US Increase 7.7% in 2016. HerbalGram 2017, 115, 56–65. [Google Scholar]
- Tuchweber, B.; Sieck, R.; Trost, W. Prevention by Silybin of Phalloidin-Induced Acute Hepatoxicity. Toxicolo. Appl. Pharmaco. 1979, 51, 265–275. [Google Scholar] [CrossRef]
- Tamayo, C.; Diamond, S. Review of Clinical Trials Evaluating (Silybum marianum [L.] Gaertn.). Integr. Cancer Ther. 2007, 6, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Shaker, E.; Mahmoud, H.; Mnaa, S. Silymarin, the antioxidant component and Silybum marianum extracts prevent liver damage. Food Chem. Toxicol. 2010, 48, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Polyak, S.J.; Morishima, C.; Lohmann, V.; Pal, S.; Lee, D.Y.W.; Liu, Y.; Graf, T.N.; Oberlies, N.H. Identification of hepatoprotective flavonolignans from silymarin. Proc. Natl. Acad. Sci. USA 2010, 107, 5995–5999. [Google Scholar] [CrossRef] [PubMed]
- Fraschini, F.; Demartini, G.; Esposti, D. Pharmacology of Silymarin. Clin. Drug Investig. 2002, 22, 51–65. [Google Scholar] [CrossRef]
- Valenzuela, A.; Garrido, A. Biochemical bases of the pharmacological action of the flavonoid silymarin and of its structural isomer silibinin. Biol. Res. 1994, 27, 105–112. [Google Scholar] [PubMed]
- Wang, L.; Rotter, S.; Ladurner, A.; Heiss, E.H.; Oberlies, N.H.; Dirsch, V.M.; Atanasov, A.G. Silymarin Constituents Enhance ABCA1 Expression in THP-1 Macrophages. Molecules 2016, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Anestopoulos, I.; Sfakianos, A.P.; Franco, R.; Chlichlia, K.; Panayiotidis, M.I.; Kroll, D.J.; Pappa, A. A Novel Role of Silibinin as a Putative Epigenetic Modulator in Human Prostate Carcinoma. Molecules 2017, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.K. Silymarin and skin cancer prevention : Anti-inflammatory, antioxidant and immunomodulatory effects (Review). Int. J. Oncol. 2005, 26, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Deep, G.; Agarwal, R. An overview of ultraviolet B radiation-induced skin cancer chemoprevention by Silibinin. Curr. Pharmacol. Rep. 2015, 1, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Toklu, H.Z.; Tunali-Akbay, T.; Erkanli, G.; Yüksel, M.; Ercan, F.; Şener, G. Silymarin, the antioxidant component of Silybum marianum, protects against burn-induced oxidative skin injury. Burns 2007, 33, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Fehér, P.; Ujhelyi, Z.; Vecsernyés, M.; Fenyvesi, F.; Damache, G.; Ardelean, A.; Costache, M.; Dinischiotu, A.; Hermenean, A.; Bácskay, I. Hepatoprotective effects of a self-micro emulsifying drug delivery system containing Silybum marianum native seed oil against experimentally induced liver injury. Pharmazie 2015, 70, 231–238. [Google Scholar] [PubMed]
- Shin, S.; Lee, J.A.; Kim, M.; Kum, H.; Jung, E.; Park, D. Anti-Glycation activities of phenolic constituants from Silybum marianum (Milk Thistle) flower in vitro and on human explants. Molecules 2015, 20, 3549–3564. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Agarwal, R. Mechanisms and preclinical efficacy of silibinin in preventing skin cancer. Eur. J. Cancer 2005, 41, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.S.; Hole, V.; Petrásková, L.; Biedermann, D.; Buchta, M.; Vladimír, K. The silymarin composition … and why does it matter??? Food Res. Int. 2017, 100, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Leng-Peschlow, E. Properties and medical use of flavonolignans (Silymarin) from Silybum marianum. Phyther. Res. 1996, 10, S25–S26. [Google Scholar]
- Odum, E.P. Ecology: A Bridge between Science and Society; Sinauer Associates: Georgia, UK, 1997. [Google Scholar]
- Radjabian, T.; Huseini, H.F. Anti-Hyperlipidemic and Anti-Atherosclerotic Activities of Silymarins from Cultivated and Wild Plants of Silybum marianum L. with different content of flavonolignans. Iran J. Pharmacol. Ther. 2010, 9, 63–67. [Google Scholar]
- Elwekeel, A.; Elfishawy, A.; Abouzid, S. Silymarin content in Silybum marianum fruits at different maturity stages. J. Med. Plants Res. 2013, 7, 1665–1669. [Google Scholar]
- Poppe, L.; Petersen, M. Variation in the flavonolignan composition of fruits from different Silybum marianum chemotypes and suspension cultures derived therefrom. Phytochemistry 2016, 131, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Carrier, D.J.; Crowe, T.; Sokhansanj, S.; Wahab, J.; Branka, B. Milk Thistle, Silybum marianum (L.) Gaertn., flower head development and associated marker compound profile. J. Herbs Spices Med. Plants 2003, 10, 65–74. [Google Scholar] [CrossRef]
- Khan, M.A.; Blackshaw, R.E.; Marwat, K.B. Biology of milk thistle (Silybum marianum) and the management options for growers in north-western Pakistan. Weed Biol. Manag. 2009, 9, 99–105. [Google Scholar] [CrossRef]
- Corbin, C.; Fidel, T.; Leclerc, E.A.; Barakzoy, E.; Sagot, N.; Falguiéres, A.; Renouard, S.; Blondeau, J.P.; Ferroud, C.; Doussot, J.; et al. Development and validation of an efficient ultrasound assisted extraction of phenolic compounds from flax (Linum usitatissimum L.) seeds. Ultrason. Sonochem. 2015, 26, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Sin, D.W.M.; Wong, Y.C.; Mak, C.Y.; Sze, S.T.; Yao, W.Y. Determination of five phenolic antioxidants in edible oils: Method validation and estimation of measurement uncertainty. J. Food Compost. Anal. 2006, 19, 784–791. [Google Scholar] [CrossRef]
- Dias, M.G.; Camões, M.F.G.F.C.; Oliveira, L. Uncertainty estimation and in-house method validation of HPLC analysis of carotenoids for food composition data production. Food Chem. 2008, 109, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Konieczka, P.; Namieśnik, J. Estimating uncertainty in analytical procedures based on chromatographic techniques. J. Chromatogr. A 2010, 1217, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.P.; Zapata, K.; Rojano, B.; Arias, M. Effect of light wavelength on cell growth, content of phenolic compounds and antioxidant activity in cell suspension cultures of Thevetia peruviana. J. Photochem. Photobiol. B Biol. 2016, 163, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Zhan, J.-C.; Huang, W.-D. Effects of ultraviolet C, methyl jasmonate and salicylic acid, alone or in combination, on stilbene biosynthesis in cell suspension cultures of Vitis vinifera L. cv. Cabernet Sauvignon. Plant Cell Tissue Organ Cult. 2015, 122, 197–211. [Google Scholar] [CrossRef]
- Benzie, I.; Strain, J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Apak, R.; Güçlü, K.; Ozyürek, M.; Karademir, S.E. Novel total antioxidant capacity index for dietary polyphenols and vitamins C and E, using their cupric ion reducing capability in the presence of neocuproine: CUPRAC method. J. Agric. Food Chem. 2004, 52, 7970–7981. [Google Scholar] [CrossRef] [PubMed]
- Fibigr, J.; Šatínský, D.; Solich, P. A new approach to the rapid separation of isomeric compounds in a Silybum marianum extract using UHPLC core-shell column with F5 stationary phase. J. Pharm. Biomed. Anal. 2017, 134, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Marhol, P.; Gažák, R.; Bednár, P.; Kren, V. Narrow-bore core–shell particles and monolithic columns in the analysis of silybin diastereoisomers. J. Sep. Sci. 2011, 34, 2206–2213. [Google Scholar] [CrossRef] [PubMed]
- Mudge, E.; Paley, L.; Schieber, A.; Brown, P.N. Optimization and single-laboratory validation of a method for the determination of flavonolignans in milk thistle seeds by high-performance liquid chromatography with ultraviolet detection. Anal. Bioanal. Chem. 2015, 407, 7657–7666. [Google Scholar] [CrossRef] [PubMed]
- Graf, T.N.; Cech, N.B.; Polyak, S.J.; Oberlies, N.H. A validated UHPLC-tandem mass spectrometry method for quantitative analysis of flavonolignans in milk thistle (Silybum marianum) extracts. J. Pharm. Biomed. Anal. 2016, 126, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, W. AOAC Guidelines for Single Laboratory Validation of Chemical Methods for Dietary Supplements and Botanicals; AOAC Int: Gaithersburg, MD, USA, 2002; p. 1219. [Google Scholar]
- Ellison, S.L.R.; Barwick, V.J. Estimating measurement uncertainty: reconciliation using a cause and effect approach. Accredit. Qual. Assur. 1998, 3, 101–105. [Google Scholar] [CrossRef]
- Saleh, I.A.; Vinatoru, M.; Mason, T.J.; Abdel-azim, N.S.; Aboutabl, E.A.; Hammouda, F.M. Ultrasonic-Assisted Extraction and Conventional Extraction of Silymarin from Silybum marianum seeds; A Comparison. Res. J. Pharm. Biol. Chem. Sci. 2015, 6, 709–717. [Google Scholar]
- Wianowska, D.; Wis, M. Simplified Procedure of Silymarin Extraction from Silybum marianum L. Gaertner. J. Chromatogr. Sci. 2015, 53, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Lucini, L.; Kane, D.; Pellizzoni, M.; Ferrari, A.; Trevisi, E.; Ruzickova, G.; Arslan, D. Phenolic profile and in vitro antioxidant power of different milk thistle [Silybum marianum (L.) Gaertn.] cultivars. Ind. Crop Prod. 2016, 83, 11–16. [Google Scholar] [CrossRef]
- Radjabian, T.; Rezazadeh, S.H.; Huseini, H.F. Analysis of silymarin components in the seed extracts of some milk thistle ecotypes from Iran by HPLc. Iran. J. Sci. Technol. 2008, 32, 6–8. [Google Scholar]
- Martin, R.J.; Lauren, D.R.; Smith, W.A.; Jensen, D.J.; Deo, B.; Douglas, J.A. Factors influencing silymarin content and composition in variegated thistle (Silybum marianum). New Zeal. J. Crop Hortic. Sci. 2006, 34, 239–245. [Google Scholar] [CrossRef]
- Hammoda, F.M.; Aboutabl, E.H.; Imbaby, S.I.; Nazief, H.N.; Abdallah, W.A.; Matter, M.A.; Ghareeb, H.A.; Saker, M.M.; Rady, M.R. Molecular and Phytochemical Characterization of Some Silybum Landraces Grown in Egypt. J. Genet. Eng. Biotechnol. 2009, 7, 1–6. [Google Scholar]
- Martinelli, T.; Whittaker, A.; Benedettelli, S.; Carboni, A. The study of flavonolignan association patterns in fruits of diverging Silybum marianum (L.) Gaertn. chemotypes provides new insights into the silymarin biosynthetic pathway. Phytochemistry 2017, 144, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Adzet, T.; Coll, M.R.; Iglesias, J.; Puigmacia, M. Selection and improvement of Silybum marianum. 1: Characterization of populations from different origins [silymarin, flavonolignans]. Plant Physiol. Biochem. 1987, 25, 129–135. [Google Scholar]
- Mahmood, T.; Nazar, N.; Abbasi, B.H.; Khan, M.A.; Ahmad, M.; Zafar, M. Detection of somaclonal variations using RAPD fingerprinting in Silybum marianum (L.). J. Med. Plants Res. 2010, 4, 1822–1824. [Google Scholar]
- Shokrpour, M.; Moghaddam, M.; Mohammadi, S.A.; Ziai, S.A.; Javanshir, A. Genetic properties of Milk Thistle ecotypes from Iran for morphological and flavonolignans characters. Pakistan J. Biol. Sci. 2007, 10, 3266–3271. [Google Scholar]
- Ratz-lyko, A.; Arct, J.; Pytkowska, K. Methods for evaluation of cosmetic antioxidant capacity. Ski. Res. Technol. 2012, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, T.; Potenza, E.; Moschella, A.; Zaccheria, F.; Benedettelli, S.; Andrzejewska, J. Phenotypic Evaluation of a Milk Thistle Phenotypic Evaluation of a Milk Thistle Germplasm Collection: Fruit Morphology and Chemical Composition. Crop Sci. 2016, 56, 3160–3172. [Google Scholar] [CrossRef]
- Abouzid, S.F.; Ahmed, H.S.; Moawad, A.S.; Owis, A.I.; Chen, S.; Nachtergael, A.; McAlpine, J.B.; Friesen, J.B.; Pauli, G.F. Chemotaxonomic and biosynthetic relationships between flavonolignans produced by Silybum marianum populations. Fitoterapia 2017, 119, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Davin, L.B.; Wang, H.B.; Crowell, A.L.; Bedgar, D.L.; Martin, D.M.; Sarkanen, S.; Lewis, N.G. Stereoselective bimolecular phenoxy radical coupling by an auxiliary (dirigent) protein without an active center. Science 1997, 275, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Jamali, A.; Grand, E.; Morreel, K.; Marcelo, P.; Gontier, E.; Dauwe, R. Phenylpropanoid profiling reveals a class of hydroxycinnamoyl glucaric acid conjugates in Isatis tinctoria leaves. Phytochemistry 2017, 144, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Dalisay, D.S.; Kim, K.W.; Lee, C.; Yang, H.; Rübel, O.; Bowen, B.P.; Davin, L.B.; Lewis, N.G. Dirigent Protein-Mediated Lignan and Cyanogenic Glucoside Formation in Flax Seed: Integrated Omics and MALDI Mass Spectrometry Imaging. J. Nat. Prod. 2015, 78, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples and standards presented in this study are available from the authors upon request. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Retention Time tR | Calibration Curve | LOD | LOQ | ||||
|---|---|---|---|---|---|---|---|---|
| min | RSD | RS | slope | intercept | R2 | (µg·mL−1) | (µg·mL−1) | |
| Taxifolin | 28.92 | 0.02 | 6.65 | 1292.9 | 0.7 | 0.9989 | 0.09 | 0.26 |
| Silychristin | 36.42 | 0.01 | 5.58 | 2266.4 | −12.7 | 0.9994 | 0.05 | 0.17 |
| Silydianin | 37.65 | 0.03 | 1.71 | 1649.8 | 34.8 | 0.9992 | 0.10 | 0.32 |
| Silybin A | 43.15 | 0.01 | 1.69 | 2575.0 | 5.7 | 0.9997 | 0.05 | 0.15 |
| Silybin B | 43.29 | 0.02 | 1.69 | 2515.9 | 27.3 | 0.9999 | 0.05 | 0.15 |
| Isosilybin A | 45.18 | 0.02 | 1.78 | 2726.3 | 17.8 | 0.9998 | 0.05 | 0.16 |
| Isosilybin B | 45.89 | 0.01 | 1.78 | 2861.1 | 1.9 | 0.9999 | 0.05 | 0.16 |
| Analyte | Mean (mg·g−1) | RSD (%) | HorRat |
|---|---|---|---|
| Taxifolin | 2.46 ± 0.10 | 4.17 | 1.69 |
| Silychristin | 13.31 ± 0.42 | 3.16 | 1.64 |
| Silydianin | 1.85 ± 0.05 | 2.60 | 1.01 |
| Silybin A | 10.59 ± 0.24 | 2.31 | 1.17 |
| Silybin B | 8.42 ± 0.24 | 2.92 | 1.43 |
| Isosilybin A | 5.01 ± 0.09 | 1.75 | 0.78 |
| Isosilybin B | 2.31 ± 0.05 | 2.15 | 1.69 |
| Analyte | Spiked Concentration (ppm) | Recovery (%) | Average Recovery (%) |
|---|---|---|---|
| Taxifolin | 25 | 101.85 ± 1.98 | |
| 50 | 102.77 ± 3.80 | ||
| 100 | 100.27 ± 0.35 | 101.63 ± 2.04 | |
| Silychristin | 25 | 102.25 ± 3.33 | |
| 50 | 99.70 ± 0.26 | ||
| 100 | 99.96 ± 0.45 | 100.64 ± 1.35 | |
| Silydianin | 25 | 100.52 ± 3.10 | |
| 50 | 100.78 ± 0.46 | ||
| 100 | 98.41 ± 1.46 | 99.90 ± 1.67 | |
| Silybin A | 25 | 101.75 ± 4.67 | |
| 50 | 102.45 ± 5.10 | ||
| 100 | 99.00 ± 1.12 | 101.07 ± 3.63 | |
| Silybin B | 25 | 100.66 ± 3.84 | |
| 50 | 98.09 ± 2.55 | ||
| 100 | 98.89 ± 2.87 | 99.22 ± 3.09 | |
| Isosilybin A | 25 | 97.88 ± 2.16 | |
| 50 | 100.06 ± 7.71 | ||
| 100 | 101.54 ± 2.72 | 99.83 ± 4.20 | |
| Isosilybin B | 25 | 102.17 ± 6.79 | |
| 50 | 102.93 ± 5.25 | ||
| 100 | 101.89 ± 4.60 | 102.33 ± 5.55 |
| Matrix | Taxifolin | Silychristin | Silydianin | Silybin A | Silybin B | Isosilybin A | Isosilybin B |
|---|---|---|---|---|---|---|---|
| Silymarin extract a | 98.91 ± 2.04 | 99.54 ± 2.06 | 94.42 ± 4.65 | 100.77 ± 1.53 | 100.26 ± 1.18 | 100.33 ± 1.95 | 101.96 ± 3.21 |
| Milk thistle seed extract b | 99.60 ± 2.24 | 99.15 ± 0.04 | 92.00 ± 1.68 | 99.48 ± 0.81 | 98.99 ± 1.48 | 98.22 ± 1.22 | 100.53 ± 4.77 |
| Sample | TAXa | SCa | SDa | SAa | SBa | ISAa | ISBa | SMb | Phec | Flad | CUPRACe | FRAPe |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P_Sm#1 | 2.0 ±0.3AB | 7.3 ±1.9ABC | 2.3 ±0.2A | 4.9 ±0.2AB | 3.8 ±0.4ABC | 1.4 ±0.0B | 1.3 ±0.0A | 22.9 ±2.2AB | 363.6 ±35.9A | 51.4 ±5.3A | 1257.1 ±162.2A | 1657.9 ±91.2AB |
| P_Sm#2 | 2.1 ±0.3AB | 1.7 ±0.1ABC | 1.0 ±0.2ABCD | 6.6 ±0.1A | 6.0 ±0.2ABC | 2.3 ±0.2A | 1.8 ±0.0A | 21.5 ±0.7AB | 247.0 ±5.5AB | 45.5 ±2.4A | 995.2 ±50.6AB | 1375.8 ±218.8AB |
| P_Sm#3 | 2.1 ±0.2A | 2.6 ±0.2ABC | 1.7 ±0.1AB | 6.4 ±0.4AB | 8.5 ±0.3A | 1.1 ±0.0B | 1.3 ±0.0AB | 23.6 ±0.1A | 197.6 ±11.7AB | 24. ±4.6AB | 1165.8 ±40.9A | 1509.3 ±53.6AB |
| P_Sm#4 | 1.4 ±0.2ABC | 2.8 ±0.2ABC | 0.8 ±0.2ABCD | 3.5 ±0.1BCDE | 7.5 ±0.8AB | 2.4 ±0.1A | 2.3 ±0.1A | 20.7 ±0.9AB | 121.2 ±13.1ABC | 14.0 ±1.1B | 1237.7 ±79.5A | 1843.3 ±74.9A |
| P_Sm#5 | nd D | 4.6 ±0.2AB | 0.3 ±0.0D | 1.5 ±0.1DEFG | 1.7 ±0.0DE | 1.0 ±0.0B | 1.1 ±0.0AB | 10.2 ±0.3DEF | 128.8 ±11.5ABC | 16.0 ±1.2AB | 731.0 ±44.7AB | 1235.8 ±138.8AB |
| P_Sm#6 | 1.2 ±0.1ABC | 1.8 ±0.3ABC | 0.6 ±0.3ABC | 1.8 ±0.1CDEF | 3.3 ±0.3BCDE | 1.3 ±0.0B | 1.6 ±0.1A | 11.6 ±0.9CDE | 137.1 ±9.9ABC | 16.6 ±3.0AB | 866.4 ±36.3AB | 1357.8 ±77.2AB |
| P_Sm#7 | 0.8 ±0.1BCD | 5.7 ±0.2A | 0.9 ±0.2ABCD | 1.4 ±0.0EFG | 3.4 ±0.2ABCDE | 0.8 ±0.0B | 0.8 ±0.0AB | 13.8 ±0.3CD | 81.3 ±28.7C | 19.0 ±0.6AB | 636.7 ±150.4B | 1120.0 ±259.1AB |
| P_Sm#8 | 1.4 ±0.1ABC | 1.6 ±0.1BC | 1.3 ±0.3ABC | 0.6 ±0.0G | 2.6 ±0.1CDE | nd C | nd B | 7.6 ±0.4CDEF | 86.9 ±9.4C | 15.1 ±2.7AB | 623.2 ±92.7B | 1012.3 ±150.4B |
| P_Sm#9 | 0.1 ±0.0CD | 2.0 ±0.4ABC | 0.7 ±0.0ABCD | 3.8 ±0.2ABC | 2.7 ±0.3CDE | 0.8 ±0.0B | 0.7 ±0.0AB | 11.0 ±0.2CDEF | 82.3 ±15.6C | 16.1 ±1.0AB | 702.5 ±42.2AB | 975.6 ±220.1AB |
| P_Sm#10 | nd D | 2.9 ±0.1ABC | 0.5 ±0.0BCD | 0.6 ±0.0G | 0.6 ±0.1E | nd C | nd B | 4.5 ±0.3F | 105.1 ±11.3BC | 16.5 ±1.1AB | 587.0 ±16.3B | 994.7 ±61.1B |
| P_Sm#11 | nd D | 0.6 ±0.0C | 0.3 ±0.1CD | 1.2 ±0.1FG | 2.8 ±0.2CDE | nd C | nd B | 4.9 ±0.1EF | 64.3 ±15.2C | 12.6 ±2.1B | 580.4 ±57.9B | 1041.0 ±53.5B |
| P_Sm#12 | 0.9 ±0.2ABCD | 2.5 ±0.0ABC | 1.5 ±0.2AB | 3.7 ±0.3ABCD | 5.1 ±0.5ABC | 0.9 ±0.0B | 1.2 ±0.1AB | 15.8 ±0.7ABC | 122.2 ±23.3ABC | 20.3 ±3.8AB | 878.0 ±112.8AB | 1327.8 ±102.9AB |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drouet, S.; Abbasi, B.H.; Falguières, A.; Ahmad, W.; Sumaira; Ferroud, C.; Doussot, J.; Vanier, J.R.; Lainé, E.; Hano, C. Single Laboratory Validation of a Quantitative Core Shell-Based LC Separation for the Evaluation of Silymarin Variability and Associated Antioxidant Activity of Pakistani Ecotypes of Milk Thistle (Silybum Marianum L.). Molecules 2018, 23, 904. https://doi.org/10.3390/molecules23040904
Drouet S, Abbasi BH, Falguières A, Ahmad W, Sumaira, Ferroud C, Doussot J, Vanier JR, Lainé E, Hano C. Single Laboratory Validation of a Quantitative Core Shell-Based LC Separation for the Evaluation of Silymarin Variability and Associated Antioxidant Activity of Pakistani Ecotypes of Milk Thistle (Silybum Marianum L.). Molecules. 2018; 23(4):904. https://doi.org/10.3390/molecules23040904
Chicago/Turabian StyleDrouet, Samantha, Bilal Haider Abbasi, Annie Falguières, Waqar Ahmad, Sumaira, Clothilde Ferroud, Joël Doussot, Jean Raymond Vanier, Eric Lainé, and Christophe Hano. 2018. "Single Laboratory Validation of a Quantitative Core Shell-Based LC Separation for the Evaluation of Silymarin Variability and Associated Antioxidant Activity of Pakistani Ecotypes of Milk Thistle (Silybum Marianum L.)" Molecules 23, no. 4: 904. https://doi.org/10.3390/molecules23040904
APA StyleDrouet, S., Abbasi, B. H., Falguières, A., Ahmad, W., Sumaira, Ferroud, C., Doussot, J., Vanier, J. R., Lainé, E., & Hano, C. (2018). Single Laboratory Validation of a Quantitative Core Shell-Based LC Separation for the Evaluation of Silymarin Variability and Associated Antioxidant Activity of Pakistani Ecotypes of Milk Thistle (Silybum Marianum L.). Molecules, 23(4), 904. https://doi.org/10.3390/molecules23040904