
Iron Is the Active Site in Nickel/Iron Water Oxidation Electrocatalysts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
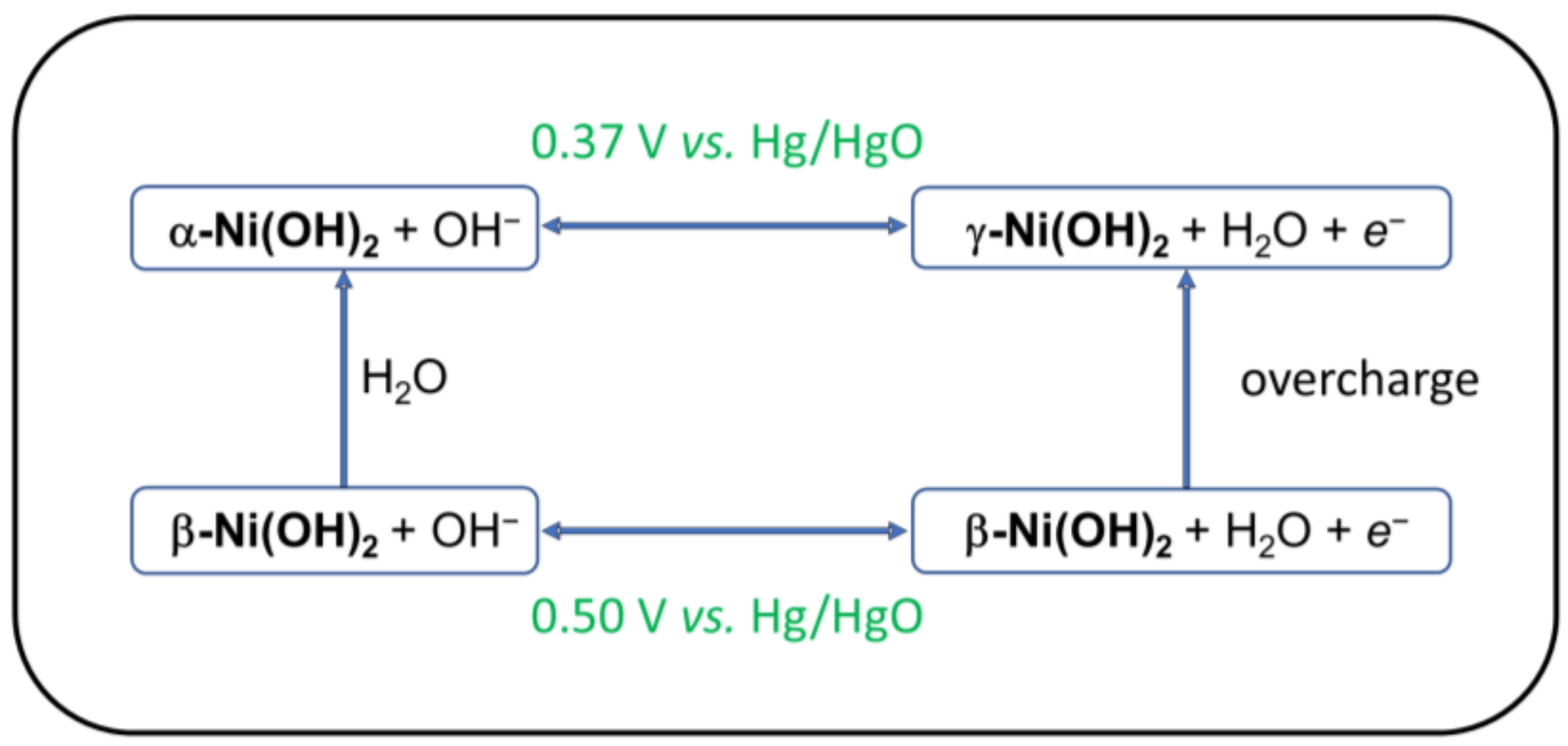
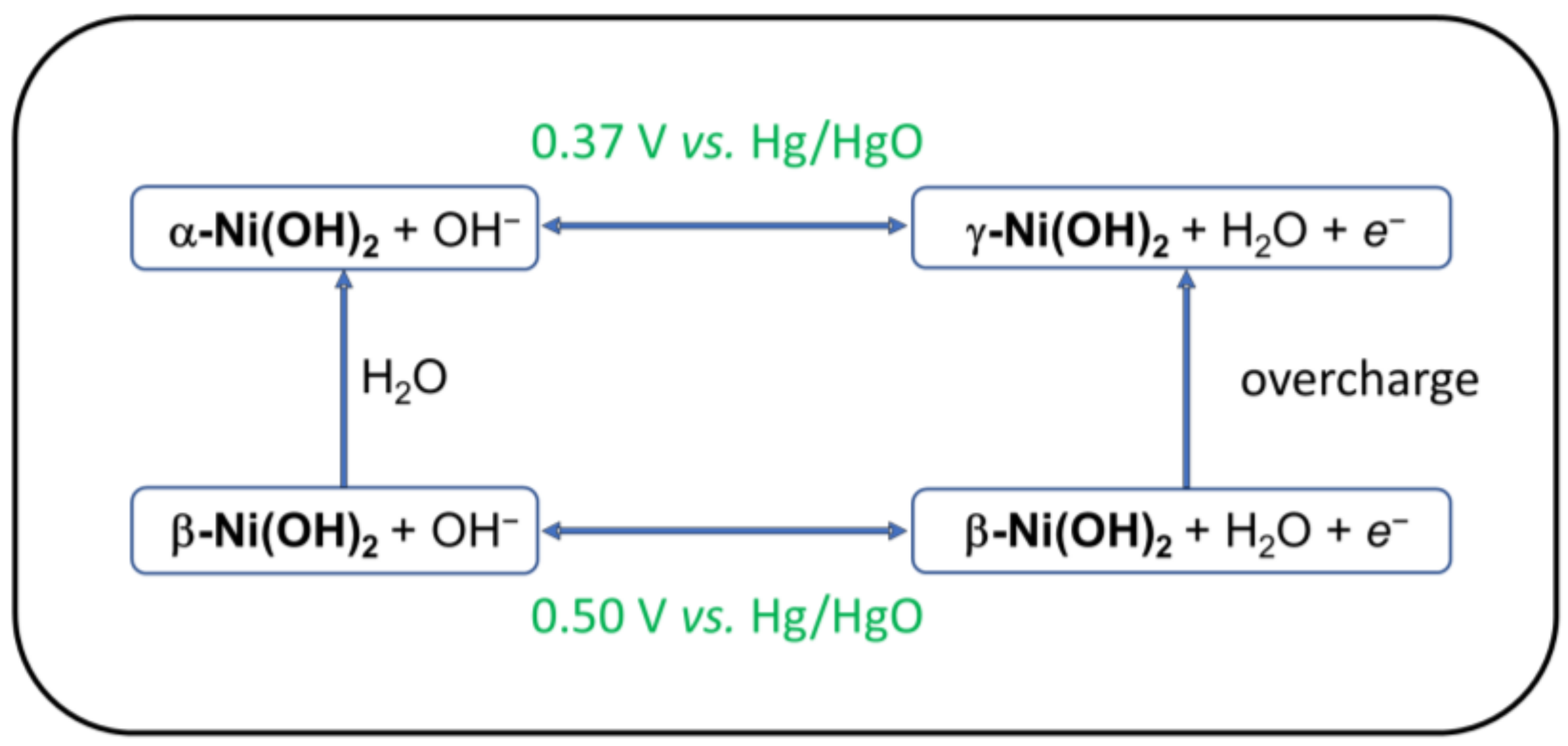
2. Nickel Hydroxide Electrodes
3. Iron Incorporation into Ni(OH)2
4. Nickel Is Not Oxidized to Ni(IV)
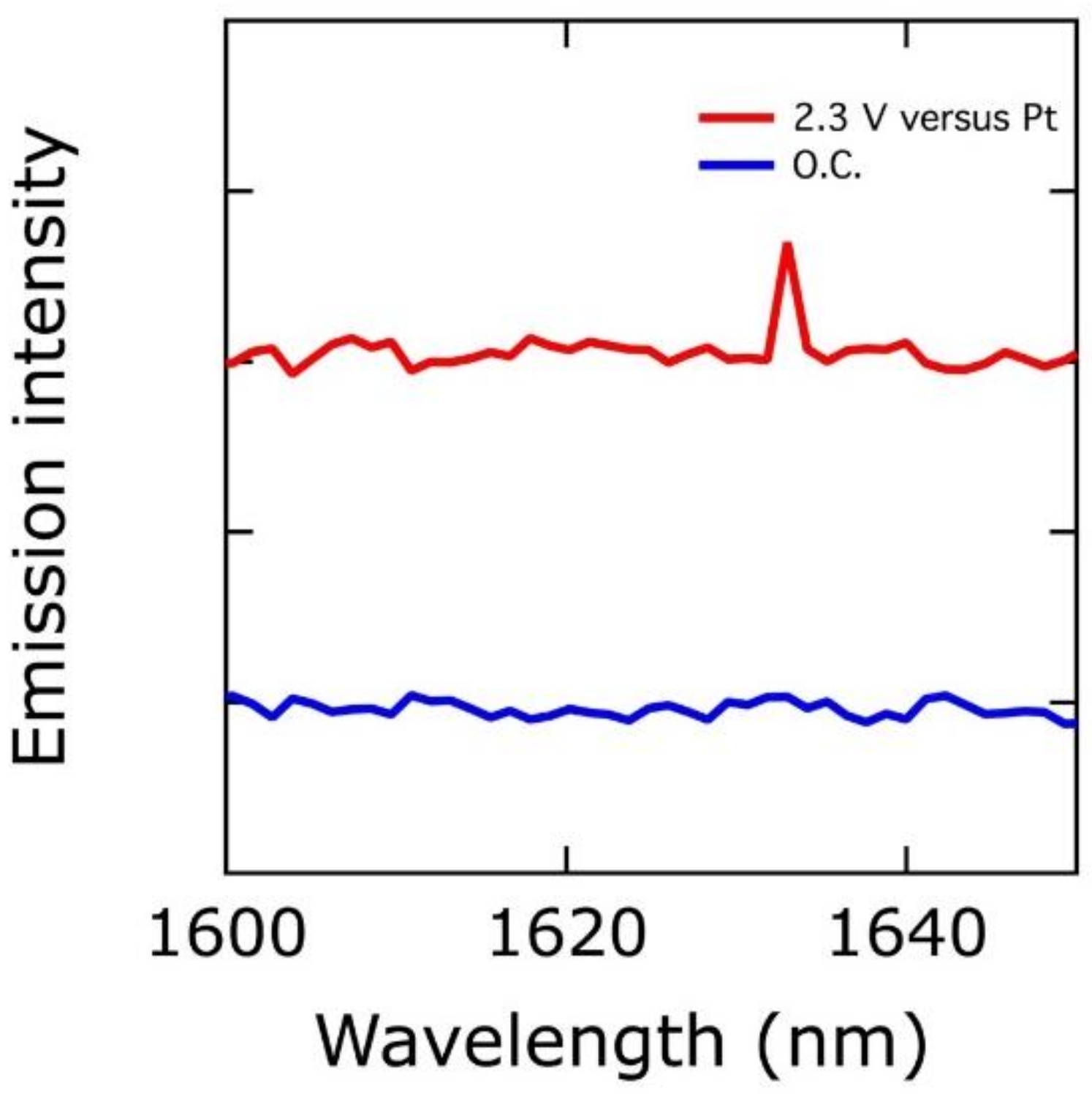
5. The Role of High-Oxidation-State Iron in OER
Acknowledgments
Conflicts of Interest
References
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Gray, H.B.; Maverick, A.W. Solar chemistry of metal complexes. Science 1981, 214, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.J. Chemical conversion of solar energy. Science 1956, 123, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Gray, H.B. Powering the planet with solar fuel. Nat. Chem. 2009, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- McKone, J.R.; Lewis, N.S.; Gray, H.B. Will solar-driven water-splitting devices see the light of day? Chem. Mater. 2014, 26, 407–414. [Google Scholar] [CrossRef]
- Hunter, B.M.; Gray, H.B.; Müller, A.M. Earth-abundant heterogeneous water oxidation catalysts. Chem. Rev. 2016, 116, 14120–14136. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.S.; Zou, S.; Enman, L.J.; Kellon, J.E.; Gabor, C.A.; Pledger, E.; Boettcher, S.W. Revised oxygen evolution reaction activity trends for first-row transition-metal (oxy)hydroxides in alkaline media. J. Phys. Chem. Lett. 2015, 6, 3737–3742. [Google Scholar] [CrossRef] [PubMed]
- Klaus, S.; Trotochaud, L.; Cheng, M.J.; Head-Gordon, M.; Bell, A.T. Experimental and computational evidence of highly active Fe impurity sites on the surface of oxidized Au for the electrocatalytic oxidation of water in basic media. ChemElectroChem 2016, 3, 66–73. [Google Scholar] [CrossRef]
- Hall, D.S.; Lockwood, D.J.; Bock, C.; MacDougall, B.R. Nickel hydroxides and related materials: A review of their structures, synthesis and properties. Proc. Royal Soc. A 2015, 471. [Google Scholar] [CrossRef] [PubMed]
- Oliva, P.; Leonardi, J.; Laurent, J.F.; Delmas, C.; Braconnier, J.J.; Figlarz, M.; Fievet, F.; de Guibert, A. Review of the structure and the electrochemistry of nickel hydroxides and oxy-hydroxides. J. Power Source 1982, 8, 229–255. [Google Scholar] [CrossRef]
- Bode, H.; Dehmelt, K.; Witte, J. Zur kenntnis der nickelhydroxidelektrode—I.Über das nickel (II)-hydroxidhydrat. Electrochim. Acta 1966, 11, 1079–1087. [Google Scholar] [CrossRef]
- Lyons, M.E.G.; Cakara, A.; O’Brien, P.; Godwin, I.; Doyle, R.L. Redox, pH sensing and electrolytic water splitting properties of electrochemically generated nickel hydroxide thin films in aqueous alkaline solution. Int. J. Electrochem. Sci. 2012, 7, 11768–11795. [Google Scholar]
- Corrigan, D.A. The catalysis of the oxygen evolution reaction by iron impurities in thin film nickel oxide electrodes. J. Electrochem. Soc. 1987, 134, 377–384. [Google Scholar] [CrossRef]
- Corrigan, D.A.; Bendert, R.M. Effect of coprecipitated metal ions on the electrochemistry of nickel hydroxide thin films: Cyclic voltammetry in 1 M KOH. J. Electrochem. Soc. 1989, 136, 723–728. [Google Scholar] [CrossRef]
- Corrigan, D.A.; Knight, S.L. Electrochemical and spectroscopic evidence on the participation of quadrivalent nickel in the nickel hydroxide redox reaction. J. Electrochem. Soc. 1989, 136, 613–619. [Google Scholar] [CrossRef]
- Stevens, M.B.; Trang, C.D.M.; Enman, L.J.; Deng, J.; Boettcher, S.W. Reactive Fe-sites in Ni/Fe (oxy)hydroxide are responsible for exceptional oxygen electrocatalysis activity. J. Am. Chem. Soc. 2017, 139, 11361–11364. [Google Scholar] [CrossRef] [PubMed]
- Trotochaud, L.; Young, S.L.; Ranney, J.K.; Boettcher, S.W. Nickel—Iron oxyhydroxide oxygen-evolution electrocatalysts: The role of intentional and incidental iron incorporation. J. Am. Chem. Soc. 2014, 136, 6744–6753. [Google Scholar] [CrossRef] [PubMed]
- Desilvestro, J.; Corrigan, D.A.; Weaver, M.J. Characterization of redox states of nickel hydroxide film electrodes by in situ surface Raman spectroscopy. J. Electrochem. Soc. 1988, 135, 885–892. [Google Scholar] [CrossRef]
- O’Grady, W.E.; Pandya, K.I.; Swider, K.E.; Corrigan, D.A. In situ X-ray absorption near-edge structure evidence for quadrivalent nickel in nickel battery electrodes. J. Electrochem. Soc. 1996, 143, 1613–1617. [Google Scholar] [CrossRef]
- Mansour, A.N.; Melendres, C.A.; Wong, J. In situ X-ray absorption spectroscopic study of electrodeposited nickel oxide films during redox reactions. J. Electrochem. Soc. 1998, 145, 1121–1125. [Google Scholar] [CrossRef]
- Hunter, B.M.; Thompson, N.B.; Müller, A.M.; Rossman, G.R.; Hill, M.G.; Winkler, J.R.; Gray, H.B. Trapping an iron(VI) water-splitting intermediate in nonaqueous media. Joule 2018. [Google Scholar] [CrossRef]
- Li, N.; Bediako, D.K.; Hadt, R.G.; Hayes, D.; Kempa, T.J.; von Cube, F.; Bell, D.C.; Chen, L.X.; Nocera, D.G. Influence of iron doping on tetravalent nickel content in catalytic oxygen evolving films. Proc. Natl. Acad. Sci. USA 2017, 114, 1486–1491. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Shao, M.; An, H.; Wang, Z.; Xu, S.; Wei, M.; Evans, D.G.; Duan, X. Fast electrosynthesis of Fe-containing layered double hydroxide arrays toward highly efficient electrocatalytic oxidation reactions. Chem. Sci. 2015, 6, 6624–6631. [Google Scholar] [CrossRef] [PubMed]
- Hunter, B.M.; Hieringer, W.; Winkler, J.R.; Gray, H.B.; Müller, A.M. Effect of interlayer anions on [NiFe]-LDH nanosheet water oxidation activity. Energy Environ. Sci. 2016, 9, 1734–1743. [Google Scholar] [CrossRef]
- Trotochaud, L.; Ranney, J.K.; Williams, K.N.; Boettcher, S.W. Solution-cast metal oxide thin film electrocatalysts for oxygen evolution. J. Am. Chem. Soc. 2012, 134, 17253–17261. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Hu, X. Exfoliation of layered double hydroxides for enhanced oxygen evolution catalysis. Nat. Commun. 2014, 5, 4477. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.S.; Kast, M.G.; Trotochaud, L.; Smith, A.M.; Boettcher, S.W. Cobalt–iron (oxy)hydroxide oxygen evolution electrocatalysts: The role of structure and composition on activity, stability, and mechanism. J. Am. Chem. Soc. 2015, 137, 3638–3648. [Google Scholar] [CrossRef] [PubMed]
- Enman, L.J.; Burke, M.S.; Batchellor, A.S.; Boettcher, S.W. Effects of intentionally incorporated metal cations on the oxygen evolution electrocatalytic activity of nickel (oxy)hydroxide in alkaline media. ACS Catal. 2016, 6, 2416–2423. [Google Scholar] [CrossRef]
- Louie, M.W.; Bell, A.T. An investigation of thin-film Ni–Fe oxide catalysts for the electrochemical evolution of oxygen. J. Am. Chem. Soc. 2013, 135, 12329–12337. [Google Scholar] [CrossRef] [PubMed]
- Vialat, P.; Leroux, F.; Mousty, C. Electrochemical properties of layered double hydroxides containing 3d metal cations. J. Solid State Electrochem. 2015, 19, 1975–1983. [Google Scholar] [CrossRef]
- Qiu, J.; Villemure, G. Anionic clay modified electrodes: Electrochemical activity of nickel(II) sites in layered double hydroxide films. J. Electroanal. Chem. 1995, 395, 159–166. [Google Scholar] [CrossRef]
- Hu, M.; Gao, X.; Lei, L.; Sun, Y. Behavior of a layered double hydroxide under high current density charge and discharge cycles. J. Phys. Chem. C 2009, 113, 7448–7455. [Google Scholar] [CrossRef]
- Winkler, J.R.; Gray, H.B. Electronic structures of oxo-metal ions. In Molecular Electronic Structures of Transition Metal Complexes I; Mingos, P.D.M., Day, P., Dahl, P.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 17–28. [Google Scholar]
- Beverskog, B.; Puigdomenech, I. Revised Pourbaix diagrams for iron at 25–300 °C. Corros. Sci. 1996, 38, 2121–2135. [Google Scholar] [CrossRef]
- Beverskog, B.; Puigdomenech, I. Revised Pourbaix diagrams for nickel at 25–300 °C. Corros. Sci. 1997, 39, 969–980. [Google Scholar] [CrossRef]
- Licht, S.; Wang, B.; Ghosh, S. Energetic iron(VI) chemistry: The super-iron battery. Science 1999, 285, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Lyons, M.D.; Brandon, M.P. The oxygen evolution reaction on passive oxide covered transition metal electrodes in alkaline solution. Part III-Iron. Int. J. Electrochem. Sci. 2008, 3, 1463–1503. [Google Scholar]
- Friebel, D.; Louie, M.W.; Bajdich, M.; Sanwald, K.E.; Cai, Y.; Wise, A.M.; Cheng, M.-J.; Sokaras, D.; Weng, T.-C.; Alonso-Mori, R.; et al. Identification of highly active Fe sites in (Ni,Fe)OOH for electrocatalytic water splitting. J. Am. Chem. Soc. 2015, 137, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Sanson, A.; Kantor, I.; Cerantola, V.; Irifune, T.; Carnera, A.; Pascarelli, S. Local structure and spin transition in Fe2O3 hematite at high pressure. Phys. Rev. B 2016, 94, 014112. [Google Scholar] [CrossRef]
- Görlin, M.; Chernev, P.; Ferreira de Araújo, J.; Reier, T.; Dresp, S.; Paul, B.; Krähnert, R.; Dau, H.; Strasser, P. Oxygen evolution reaction dynamics, faradaic charge efficiency, and the active metal redox states of Ni–Fe oxide water splitting electrocatalysts. J. Am. Chem. Soc. 2016, 138, 5603–5614. [Google Scholar] [CrossRef] [PubMed]
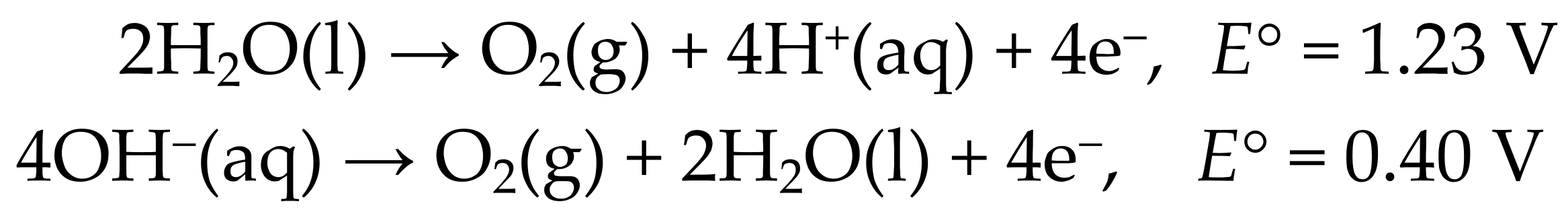

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hunter, B.M.; Winkler, J.R.; Gray, H.B. Iron Is the Active Site in Nickel/Iron Water Oxidation Electrocatalysts. Molecules 2018, 23, 903. https://doi.org/10.3390/molecules23040903
Hunter BM, Winkler JR, Gray HB. Iron Is the Active Site in Nickel/Iron Water Oxidation Electrocatalysts. Molecules. 2018; 23(4):903. https://doi.org/10.3390/molecules23040903
Chicago/Turabian StyleHunter, Bryan M., Jay R. Winkler, and Harry B. Gray. 2018. "Iron Is the Active Site in Nickel/Iron Water Oxidation Electrocatalysts" Molecules 23, no. 4: 903. https://doi.org/10.3390/molecules23040903
APA StyleHunter, B. M., Winkler, J. R., & Gray, H. B. (2018). Iron Is the Active Site in Nickel/Iron Water Oxidation Electrocatalysts. Molecules, 23(4), 903. https://doi.org/10.3390/molecules23040903