
Selective Double Addition Reaction of an E‒H Bond (E = Si, B) to a C≡N Triple Bond of Organonitriles


Department of Chemistry, Graduate School of Science, Osaka City University, Sumiyoshi-ku, Osaka 558-8585, Japan
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(11), 2769; https://doi.org/10.3390/molecules23112769
Submission received: 21 August 2018
/
Revised: 22 October 2018
/
Accepted: 23 October 2018
/
Published: 25 October 2018
(This article belongs to the Special Issue Indium in Organic Synthesis)
Abstract
:The catalytic double hydrometalation such as hydrosilylation and hydroborylation of organonitriles has attracted considerable attention because the obtained products are widely used in organic synthesis and it is thought to be one of the effective methods for reduction of organonitriles. However, the examples of these reactions are quite limited to date. This paper summarizes the development of selective double hydrosilylation, double hydroborylation, and dihydroborylsilylation of organonitriles, including their reaction mechanisms and the role of the metal species in the catalytic cycle.
1. Introduction
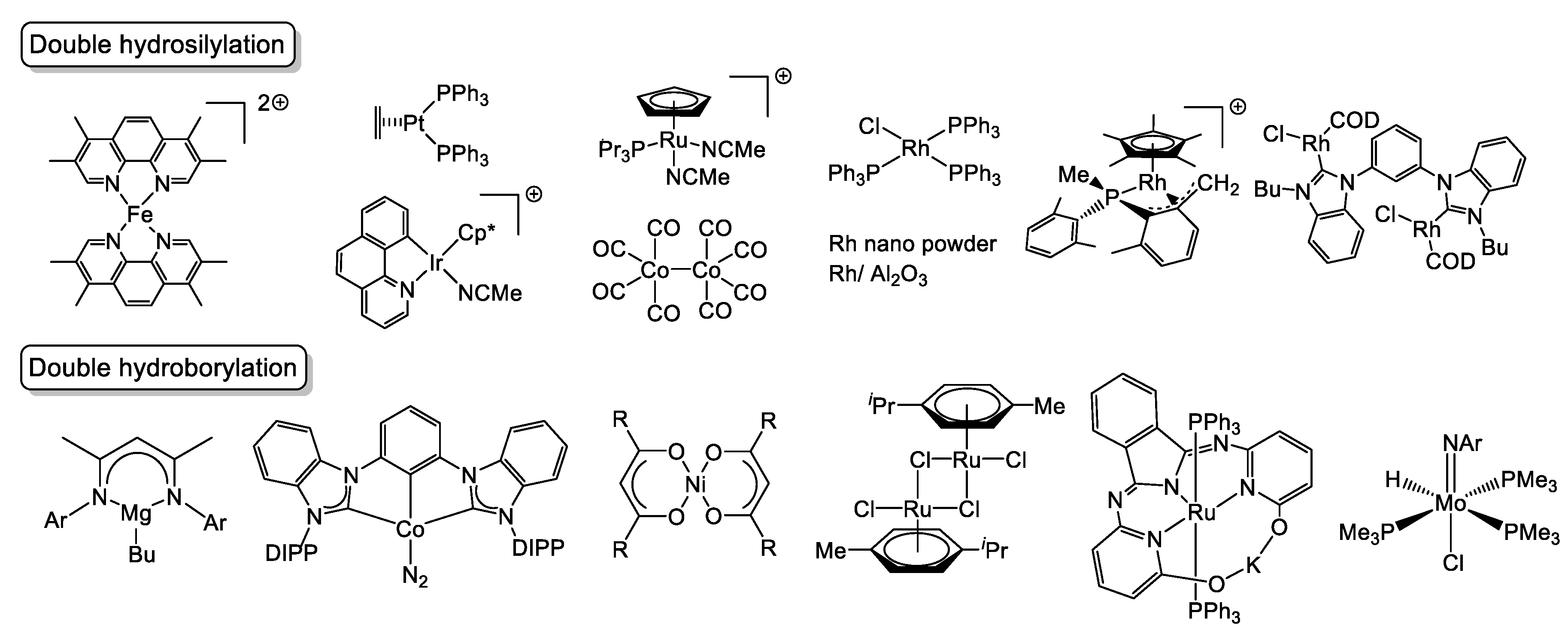
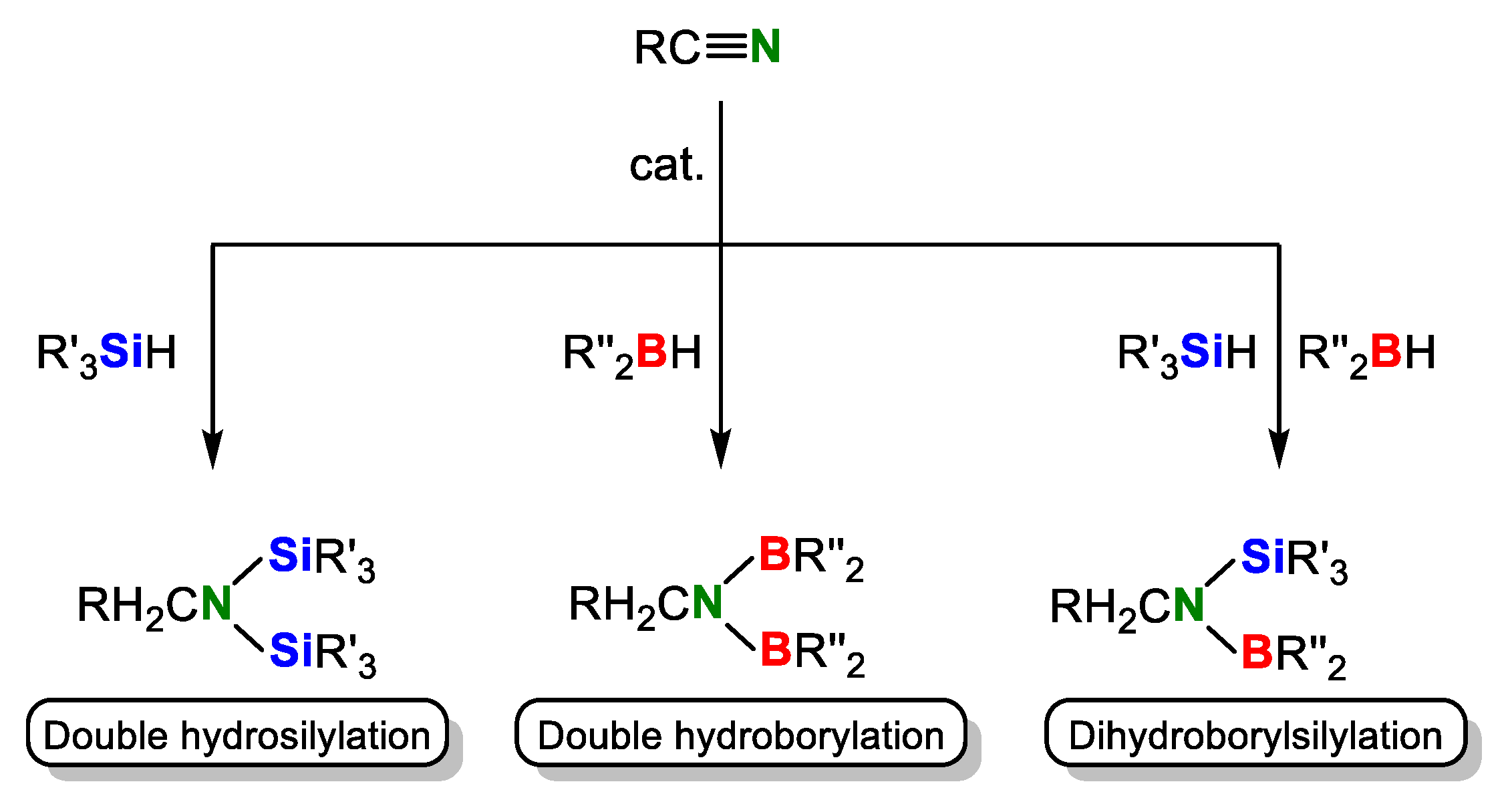
The catalytic hydrosilylation and hydroborylation of the carbon-nitrogen triple bond (C≡N bond) in organonitriles is becoming important in the synthetic chemistry. Although the term “hydroboration” is also widely used, we use “hydroborylation” in this paper from comparison with hydrosilylation. There is an advantage that these reactions do not generate by-products theoretically and the compounds with an N−Si or N−B bond thus obtained are useful products for the synthetic intermediates in organic chemistry. For examples, disilylamines (double hydrosilylation product) act as precursors for the production of Si,N-containing polymers [1,2,3,4], amine ligands for organometallic compounds [5,6], and silylating [7] and coupling [8] agents. Borylamines (hydroborylation products) have been reported to show a unique reactivity as iminium ion generators [9]. In addition, it is known that hydrosilylation and hydroborylation of a C≡N bond are one of effective methods to reduce organonitriles [10,11,12,13]. However, these hydrometalations do not occur under typical reaction conditions for hydrosilylation [14] and hydroborylation [15] because of the strong C≡N bond dissociation energy (179.3 kcal/mol, 750.0 kJ/mol) [16]. Actually, examples of catalytic double hydrometalation of the carbon-nitrogen triple bond (C≡N bond) in organonitriles are limited: one example of Fe [12], Pt [17], Ir [18], and Ru [19], two examples of Co [20,21], four examples of Rh [22,23,24,25], and main group elements and fluoride [13,26,27,28] for double hydrosilylation and one example of Mg [29], Co [30], and Ni [31], and two examples of Ru [32,33] and Mo [34,35] for double hydroborylation have been reported to date. Those metal catalysts are depicted in Figure 1. Although it is known that borylsilylamines are advantageous precursors for obtaining B/Si/N/C ceramics having a highly heat-resistant property [36,37], catalytic dihydroborylsilylation of organonitriles has not been achieved yet. (Scheme 1). In addition, the dual catalyst having both hydrosilylation and hydroborylation activities has not been found.
2. Double Hydrosilylation of Organonitriles
In 1982, Corriu and co-workers reported that the reaction of 1,4-dicyanobutane with 1,2-bis(dimethylsilyl)benzene in the presence of a catalytic amount of RhCl(PPh3)3 afforded a mixture of trans-N,N-disilylenamines (major product) and N,N-disilylamines (minor product) (Equation (1)) [23]. In the case of benzonitrile, only double hydrosilylation product was obtained, although the yield of the product was low (Equation (2)):
![Molecules 23 02769 i001]()


The proposed reaction mechanism of Rh-catalyzed double hydrosilylation of organonitriles with 1,2-bis(dimethylsilyl)benzene is shown in Scheme 2. Intermediates A and B are generated via the first hydrosilylation and they may be in equilibrium. The second hydrosilylation of A takes place to give the N,N-disilylamine (double hydrosilylation product) C. On the other hand, intermediate B is converted into the N,N-disilylenamine D as a result of aminolysis.
Murai’s group found the double hydrosilylation of aromatic and aliphatic nitriles catalyzed by a cobalt carbonyl Co2(CO)8 in 1985 and 1990 [20,21]. The desired products were obtained in the reaction of various aromatic nitriles with 10 equiv. of hydrosilane at 60 °C for 20 h in the presence of Co2(CO)8 (Table 1). The system possesses an excellent degree of functional group tolerance for the functionalized benzonitriles with electron-withdrawing or -donating groups such as Me, OMe, Cl, NMe2, CN, and CO2Me in the para position on the aryl ring. A Me group in meta position shows good reactivity, whereas that in the ortho position shows low reactivity.
Furthermore, aliphatic nitriles are adaptable to this reaction system and gave the corresponding products in moderate to excellent yields when PPh3 is added to the reaction system (Table 2). It is thought that a silylcobalt complex R3SiCo(CO)4, which is prepared by the reaction of Co2(CO)8 with R3SiH, is an important catalytic active spices in this system.
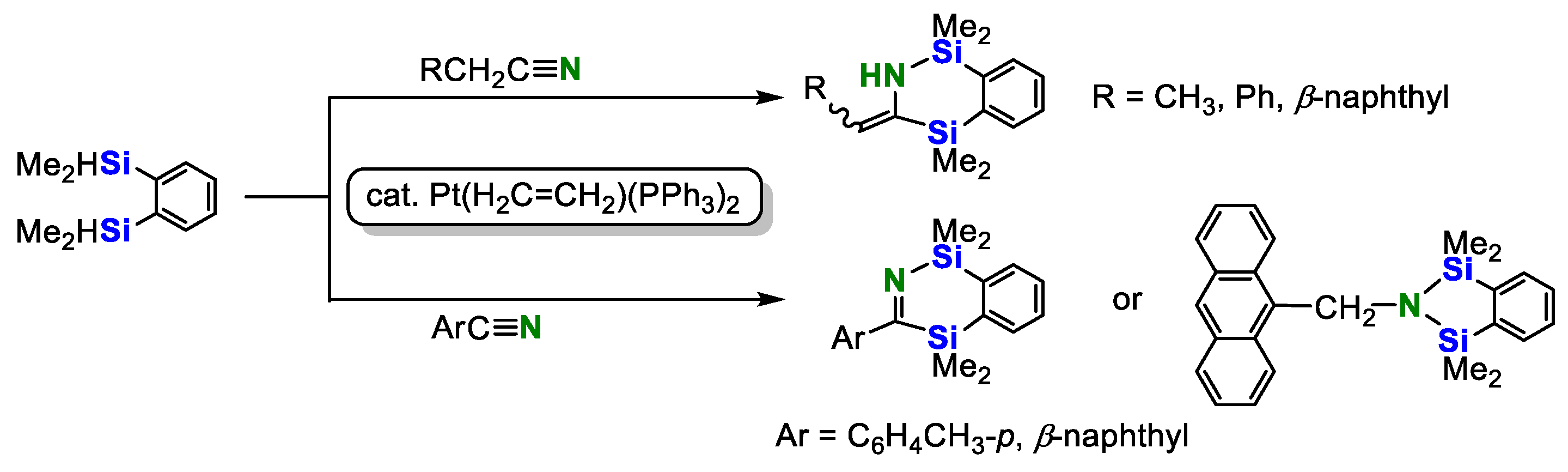
A platinum-catalyzed reaction of various nitriles with 1,2-bis(dimethylsilyl)benzene was reported by Tanaka’s group in 1992 (Scheme 3) [17]. In the presence of Pt(H2C=CH2)(PPh3)2 catalyst, reactions of aliphatic nitriles with 1,2-bis(dimethylsilyl)benzene gave the N-silyl enamies, while aryl nitriles were converted into the corresponding imines in high to excellent yields. The double hydrosilylation product was yielded in 64% when 9-anthroylnitrile was used.
In 1999, double hydrosilylation of arylnitriles catalyzed by heterogenous Rh powder and rhodium on γ-alumina was achieved by Pertici and co-workers (Table 3) [22]. The tendency of the reaction is similar to the Murai’s report [20]. The desired product was not obtained when the substrate with a Me group in the ortho position on the aryl ring was used. In addition, the yields decreased when HSi(OEt)3 as a hydrosilane or rhodium on γ-alumina instead of Rh powder was used.
Selective catalytic hydrosilylation of nitriles was found by Nikonov and Gutsulyak in 2010 [19]. The reaction of organonitriles with HSiMe2Ph in a 1:1 molar ratio afforded the corresponding imines. In addition, the N,N-disilylamines were produced by the reaction of organonitriles with 2.5 equiv. of HSiMe2Ph although a long reaction time was required (Table 4). In the case of isobutyronitrile, the mixture of N,N-disilyenamine (57%) and N,N- disilylamines (43%) were yielded.
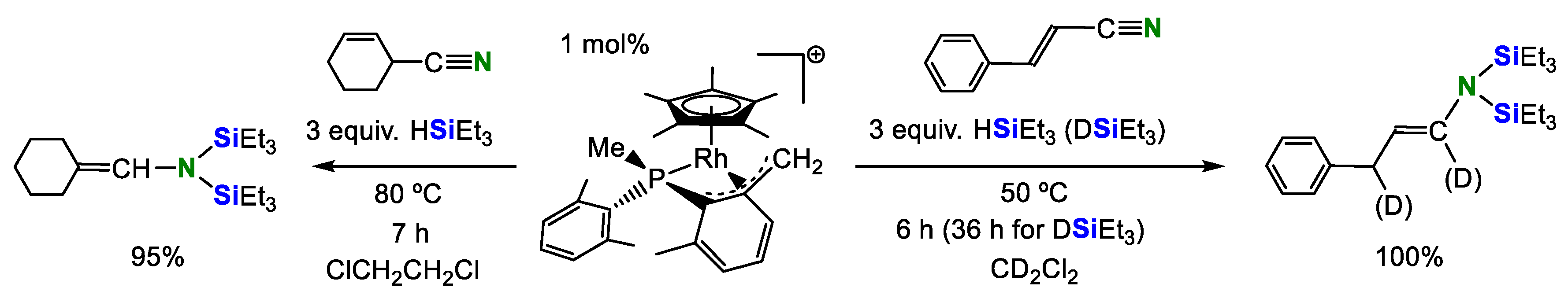
Rhodium-catalyzed hydrosilylation of α,β-unsaturated nitriles into vinylamines was achieved by Carmona’s group in 2011 (Scheme 4) [24]. Acetonitrile showed low activity (<40%) and benzonitrile did not undergo hydrosilylation in this catalytic system.
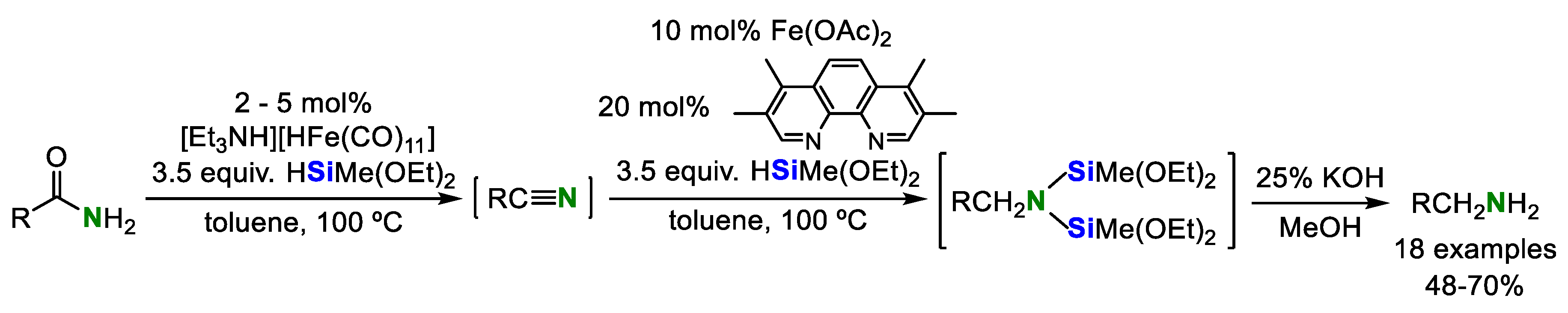
Beller’s group achieved the conversion of aromatic and aliphatic primary amides into amines catalyzed by two iron cooperative catalytic system in 2012 [12]. In this system, the combination of Fe(OAc)2 and phenanthroline ligand acts as a catalyst for the double hydrosilylation of aromatic and aliphatic nitriles, which are prepared by reduction of amides catalyzed by an iron complex [Et3NH][HFe3(CO)11] (Scheme 5).
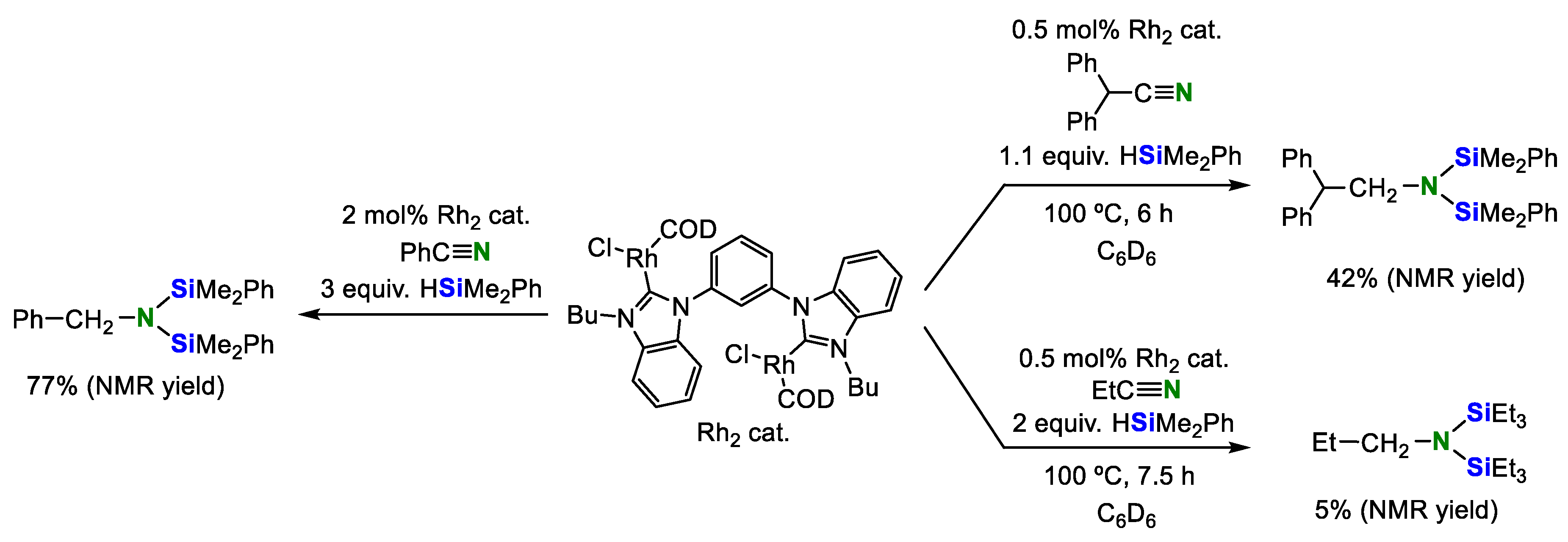
In 2013, Hollis and co-workers reported that a homobimetallic Rh complex having an NHC ligand also acted as a catalyst for the double hydrosilylation (Scheme 6) [25]. Benzonitrile was converted into the corresponding product in good yield. For aliphatic nitriles, the activity of diphenylacetonitrile was higher than that of propionitrile (the yields of the corresponding disilylamines were 42% and 5%, respectively).
In 2017, Djukic and co-workers reported an iridacycle complex as a catalyst for the conversion of organonitriles into N,N-disilylamines by the double hydrosilylation [18]. The reaction was adaptable to a wide variety of aromatic nitriles (Table 5). In this system, Cl and F groups in ortho position on the aryl ring did not disturb the double hydrosilylation, whereas a nitrile having a coordination-feasible substituent did the reaction.
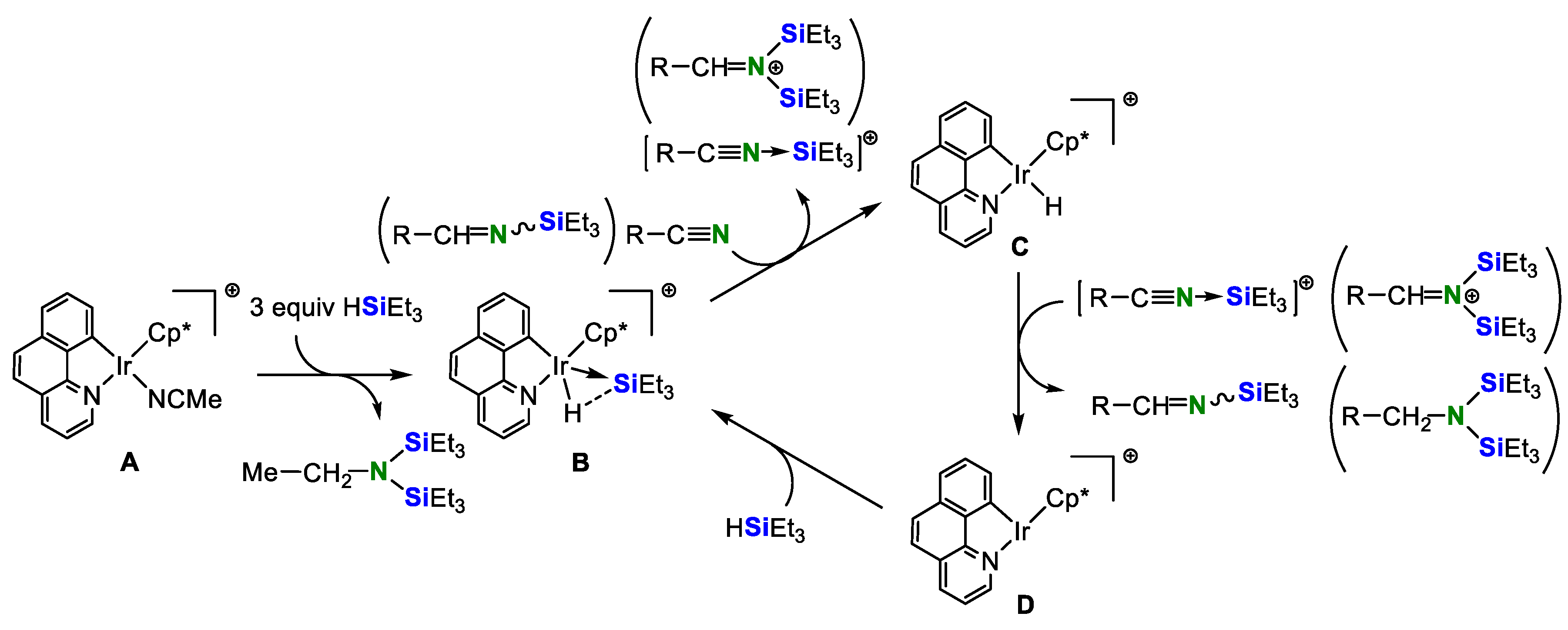
Scheme 7 depicts a plausible reaction pathway of the double hydrosilylation of nitriles catalyzed by a cationic iridium complex A. The reaction of A with 3 equiv. of HSiEt3 affords the silane−iridacycle adduct B and EtN(SiEt3)2 as a result of electrophilic and heterolytic activation of the Si−H bond. Subsequently, the abstraction of the SiEt3 group in B by nitrile gives the hydrido complex C and the N-silylnitrilium cation and then the hydrido transfer from C to the N-silylnitrilium cation produces the N-silylimine and unsaturated Ir complex D. Finally, D reacts with HSiEt3 to regenerate B. A similar reaction proceeds once again to give the desired N,N-disilylamine.
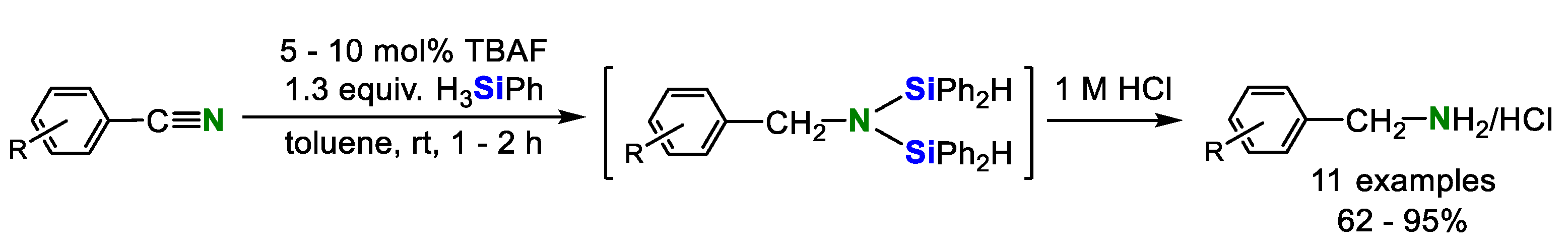
Recently, some metal-free hydrosilylation reactions of organonitriles were achieved. Beller’s group reported TBAF catalyzed hydrosilylation for the reduction of aromatic nitriles in 2013 (Scheme 8) [13]. Various aryl nitriles were converted into the corresponding benzylamines via N,N-disilylamines. Heterocyclic nitriles such as 3-thiophenecarbonitrile and picolinonitrile, as well as hexanenitrile showed no activity.
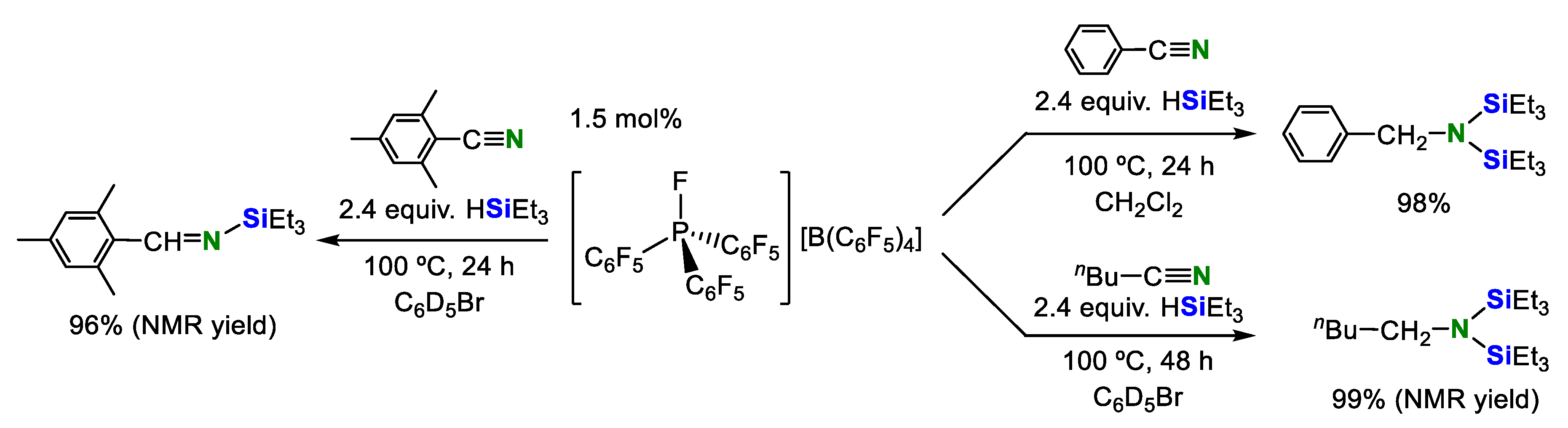
In 2015, Grimme, Stephan and co-workers found that an electrophilic phosphonium salt, [(C6F5)3PF][B(C6F5)4] acted as a catalyst for the double hydrosilylation of organonitriles. In this system, benzonitrile and propionitrile were converted into the corresponding N,N-disilylamines in quantitative yields. The N-silylimine was selectively formed in excellent yield when sterically bulky mesityl nitrile was used (Scheme 9) [26].
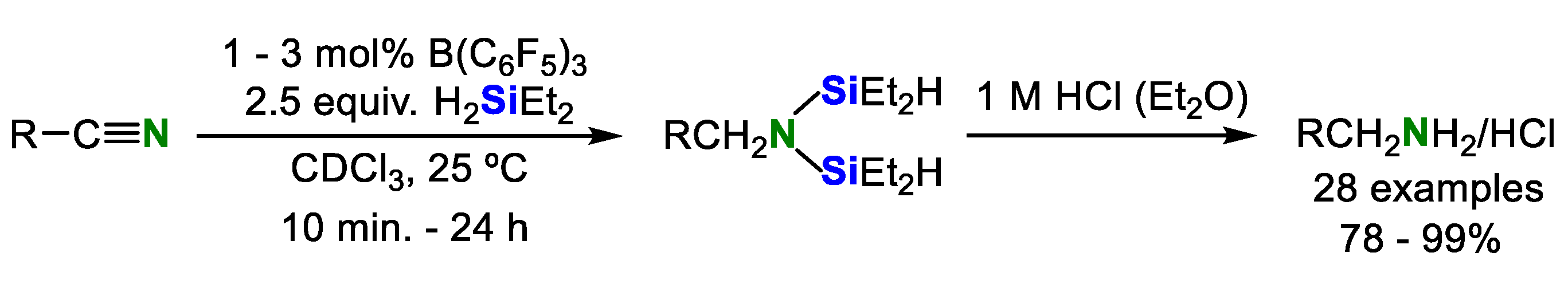
In the same year, Chang’s group reported that tris(pentafluorophenyl)borane [B(C6F5)3]-catalyzed silylative reduction of conjugated nitriles to β-silyl amines as a result of selective double hydrosilylation of the C≡N bond and hydrosilylation of the C=C bond [27]. Table 6 summarizes the scope and limitation of substrates for this reaction. The system possesses a good degree of functional group tolerance for the functionalized conjugated nitrile with an electron-withdrawing or -donating group on the aryl ring. Furthermore, this catalytic system was also applicable to aryl and alkyl nitriles (Scheme 10) [28].
In the cases of the catalytic double hydrosilylation, an excess amount of the hydrosilane over the organonitrile was required for the selective formation of the desired disilylamines. Therefore, a new synthetic strategy without using an excess amount of the hydrosilane and a new catalyst have been demanded.
In order to create a new approach to double hydrosilylation of organonitriles, we focused on transition metal complexes with Z-type ligand(s). The interaction of a Z-type ligand (acts as a two-electron acceptor, a Lewis acid) with a late transition metal (acts as a two-electron donor, a Lewis base) has attracted considerable attention as a new approach for controlling the electronic characteristics and reactivity of the metal center [38,39,40,41,42,43,44,45,46,47]. This approach is becoming increasingly important in the field of catalysts [48,49,50,51,52,53,54,55,56]. Previously, we reported that triruthenium dodecacarbonyl Ru3(CO)12 reacted with InX3 (X = Cl, Br) to yield the first ruthenium(0) indane complex, fac-[Ru(NCCH3)3(CO)2(InX3)] (X = Cl (1Cl), Br (1Br)). In addition, the reaction of 1Cl and 1Br with 1 equiv. of PPh3 afforded cis,cis,trans-[Ru(NCCH3)2(CO)2(InX3)(PPh3)] (X = Cl (2Cl), Br (2Br)) as a result of selective replacement of CH3CN(trans to InX3) by PPh3 (Equation (3)) [57]. It is considered that the InX3 in 1Cl, 1Br, 2Cl, and 2Br is a Z-type ligand.

On the other hand, the reaction of triiron dodecacarbonyl Fe3(CO)12 with InX3 afforded the iron complex containing indium ligands [Fe(NCCH3)6][cis-Fe(CO)4(InX3)2] (X = Cl (3Cl), Br (3Br), I (3I)) (Equation (4)) [58]. These complexes represent the first example of transition metal complexes containing two terminal indium fragments. For the anionic iron complex [cis-Fe(CO)4(InCl3)]2–, the 57Fe Mössbauer and IR data suggest that the Fe0(CO)4 has two [Fe–In–X3]− portions like [InX4]− called as “indate”.

As there had been no organic reactions catalyzed by a combination of an iron complex and an indium source, the catalytic ability of the iron complex was examined for double hydrosilylation of organonitriles, and it was found interesting knowledge. The results were described in a bit more detail below.
The reaction of CH3CN with HSiMe2Ph in the presence of a catalytic amount of 3Cl produced CH3CH2N(SiMe2Ph)2 in 85% yield (Equation (5)) [59]. It should be noted that this catalytic reaction provided the double hydrosilylation product selectively in spite of using an excess amount of acetonitrile over the hydrosilane.

Complex 3Cl was a better catalyst than 3B and 3I. The catalytic activity of a mixture of dodecacarbonyltriiron Fe3(CO)12 and indium trichloride (InCl3) was similar to that of 3Cl, whereas the double hydrosilylation did not proceed when either Fe3(CO)12 or InCl3 was used. It was revealed that not the cationic iron complex [Fe(NCCH3)6]2+ but the anionic iron complex [cis-Fe(CO)4(InCl3)]2- of 3Cl played a crucial role in the double hydrosilylation. Various aliphatic and aromatic nitriles (RC≡N, for which R = Me, Et, iPr, iBu, Ph, p-Tol, m-Tol or o-Tol) underwent the double hydrosilylation without the formation of the single hydrosilylation compound (Table 7). In the double hydrosilylation of propanenitrile (EtCN), the expected product EtCH2N(SiMe2Ph)2 was obtained as the main product along with a little amount of MeCH2N(SiMe2Ph)2 (2% yield as determined by NMR). The latter product is considered to be derived from the dissociated CH3CN obtained by the MeCN/EtCN ligand exchange on the iron center of the cationic part in 3Cl. The yields of the products decreased when going from p- to m- and o-tolunitrile (55, 49, and 41%, respectively), presumably due to steric effects. This catalytic system was also applicable to 4-pyridinecarbonitrile although the yield of the corresponding disilylamine was low (21%). No reaction occurred for tBuCN, CCl3CN, and C6F5CN with HSiMe2Ph. These results indicate that organonitriles having a bulky or an electro-withdrawing group are unfavorable for the double hydrosilylation. The double hydrosilylation reaction of MeCN did not proceed when a bulkier hydrosilane (HSiMePh2) was used. Instead, the reactions of HSiMe2Fc with p-TolCN and H2SiMePh with MeCN provided the double hydrosilylation compounds in 43% and 76% yields, respectively.
3. Double Hydroborylation of Organonitriles
Nikonov and co-worker found that the imido-hydrido Mo(IV) complex acted as a catalyst for the double hydroborylation of organonitriles in 2012 [34] and 2015 [35]. The reaction of organonitriles RCN (R = Me, Ph, tBu) with 2 equiv. of HBcat (catecholborane) in the presence of a catalytic amount of imido-hydrido Mo(IV) complex afforded the corresponding N,N-diborylamines in good to excellent yields (Scheme 11).
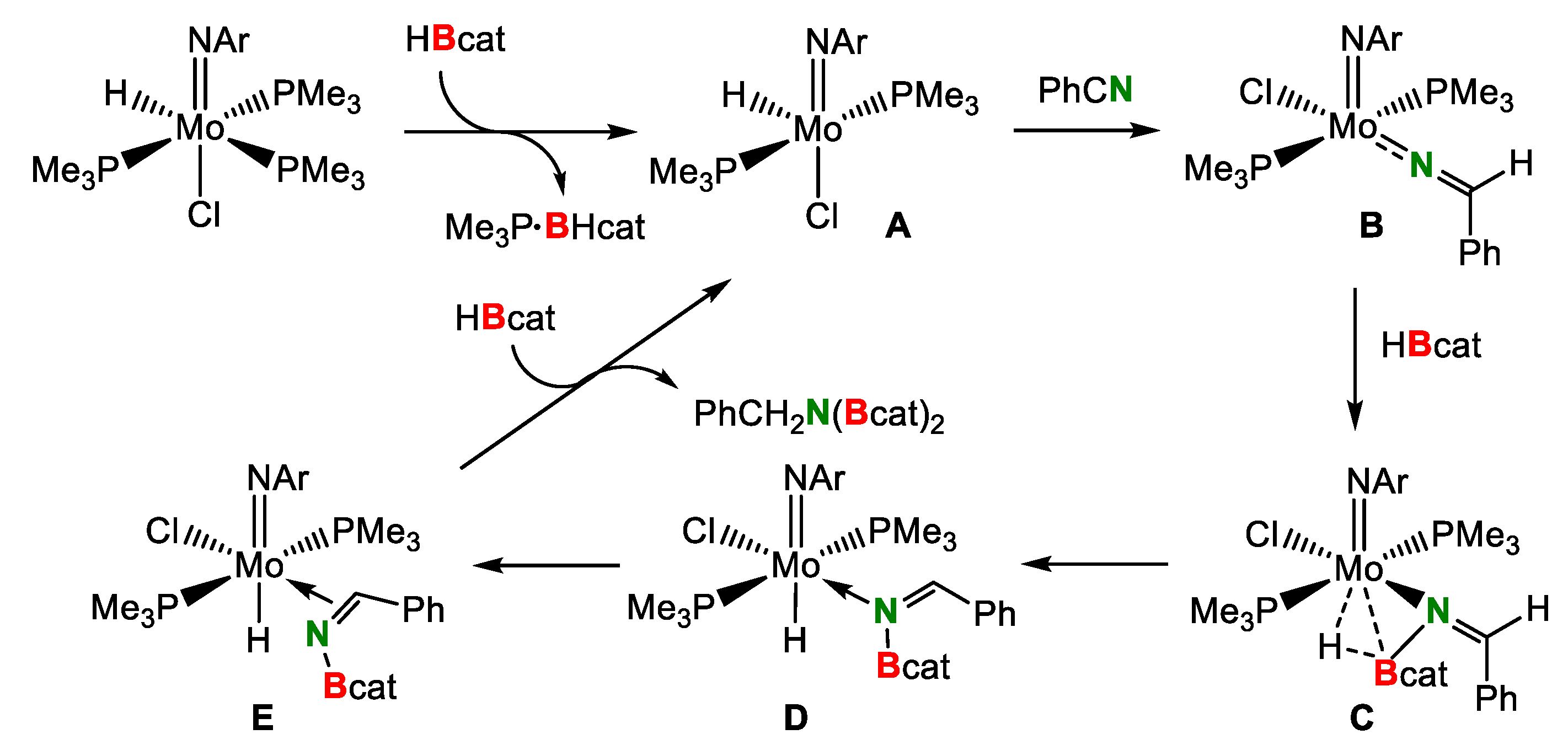
A plausible reaction pathway of the double hydroborylation of nitriles catalyzed by the imido-hydrido Mo(IV) complex was shown in Scheme 12. The abstraction of the coordinated PMe3 ligand by HBcat results in the formation of unsaturated Mo complex A. Subsequently, the reaction of A with PhCN affords benzylideneamide complex B and then B reacts with HBcat to yield agostic amido-borane adduct complex C. Complex C is converted into borylimine complex E through N-coordinated borylimine complex D. Finally, the elimination of the desired N,N-diborylamine from the Mo center in E regenerates catalytic intermediate A to complete the catalytic cycle.
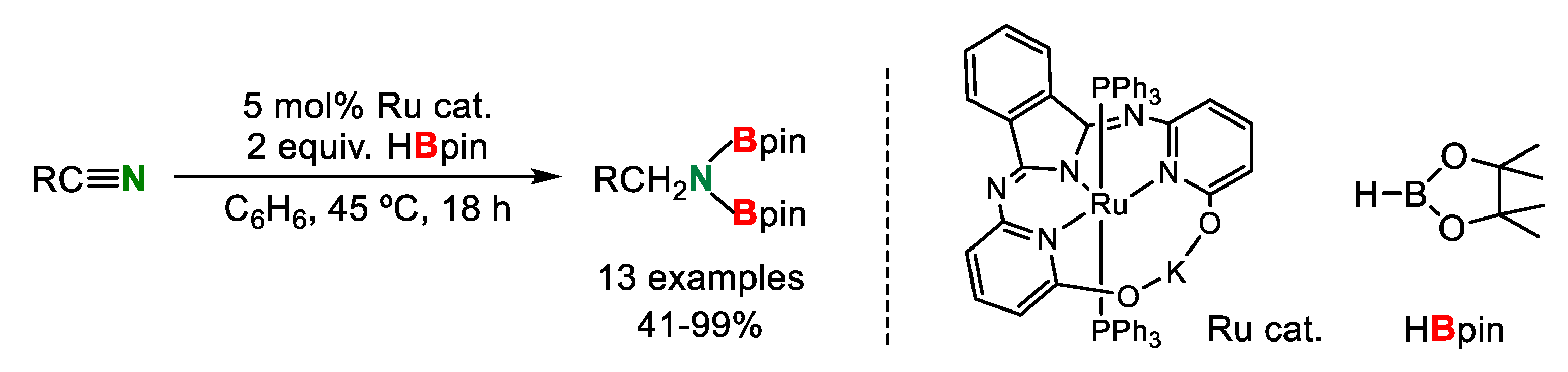
In 2015, Szymczak’s group reported that catalytic nitrile hydroborylation using a ruthenium complex having a bifunctional pincer ligand took place for several p-substituted aryl nitriles with HBpin (pinacolborane) to give the corresponding diborylamines in moderate to excellent yields (Scheme 13) [33].
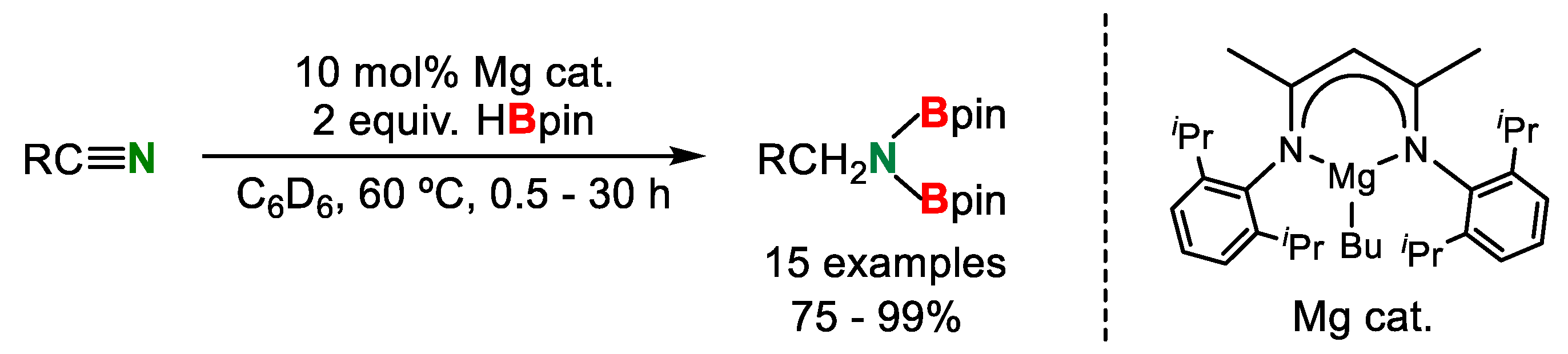
The double hydroborylation of organonitriles by HBpin was also reported by Hill’s group in 2016. In this reaction, a β-diketiminato n-butylmagnesium complex was found to be an efficient catalyst and the desired products were obtained in good to excellent yields (Scheme 14) [29]. This reaction showed good functional group tolerance. In addition, benzonitrile having a Me group in the ortho position on the aryl ring also showed good reactivity (86%).
At almost the same time, Gunanathan’s group reported the selective conversion of nitriles into amines by double hydroborylayion [32]. Various organonitriles reacted with 2 equiv. of HBpin in the presence of a catalytic amount of homobimetallic Ru complex [Ru(p-cymene)Cl2]2 to obtain the correspsonding N,N-diborylamines in good to excellent yields (Scheme 15). It was thought that a boryl hydrido complex [Ru(p-cymene)H(Bpin)], which was prepared by the reaction of [Ru(p-cymene)Cl2]2 with HBpin, was an important catalytically active species in this system.
In 2017, Fout’s group reported the double hydroborylation of organonitriles catalyzed by a Co(I) complex [30]. In this system, alkyl and (hetero)aryl nitriles were converted into the desired N,N-diborylamines in moderate to high yields (Table 8).
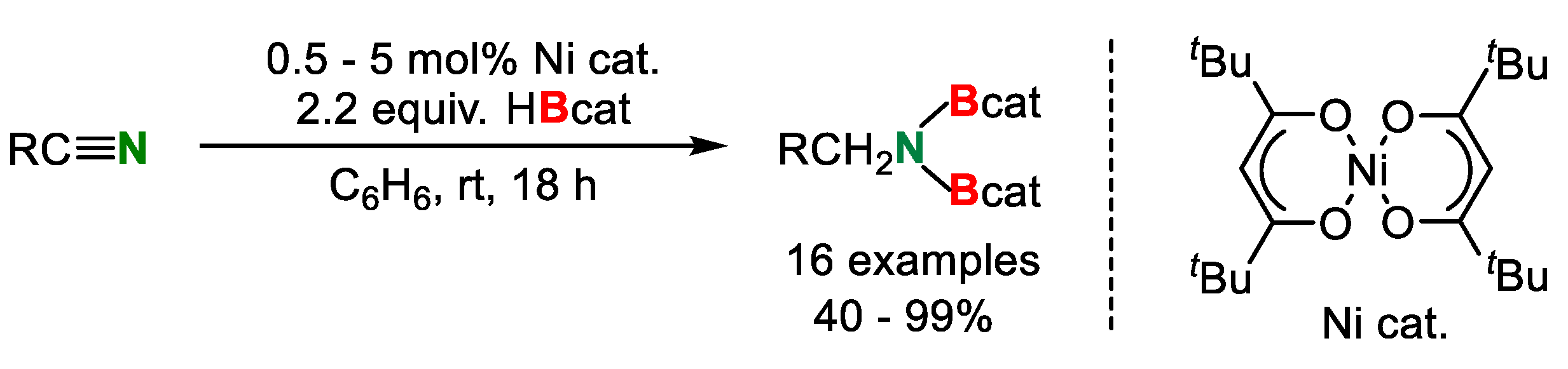
In the same year, a nickel catalyzed double hydroborylation of organonitriles was achieved by Nakajima, Shimada and co-workers [31]. The reaction of organonitriles with 2.2 equiv. of HBcat yielded N,N-diborylamines in moderate to excellent yields (Scheme 16). The reaction was applicable to a wide variety of nitriles whereas benzonitrile having a Me group in the ortho position on aryl ring (40%) and 2-thienyl nitrile (47%) showed lower reactivities.
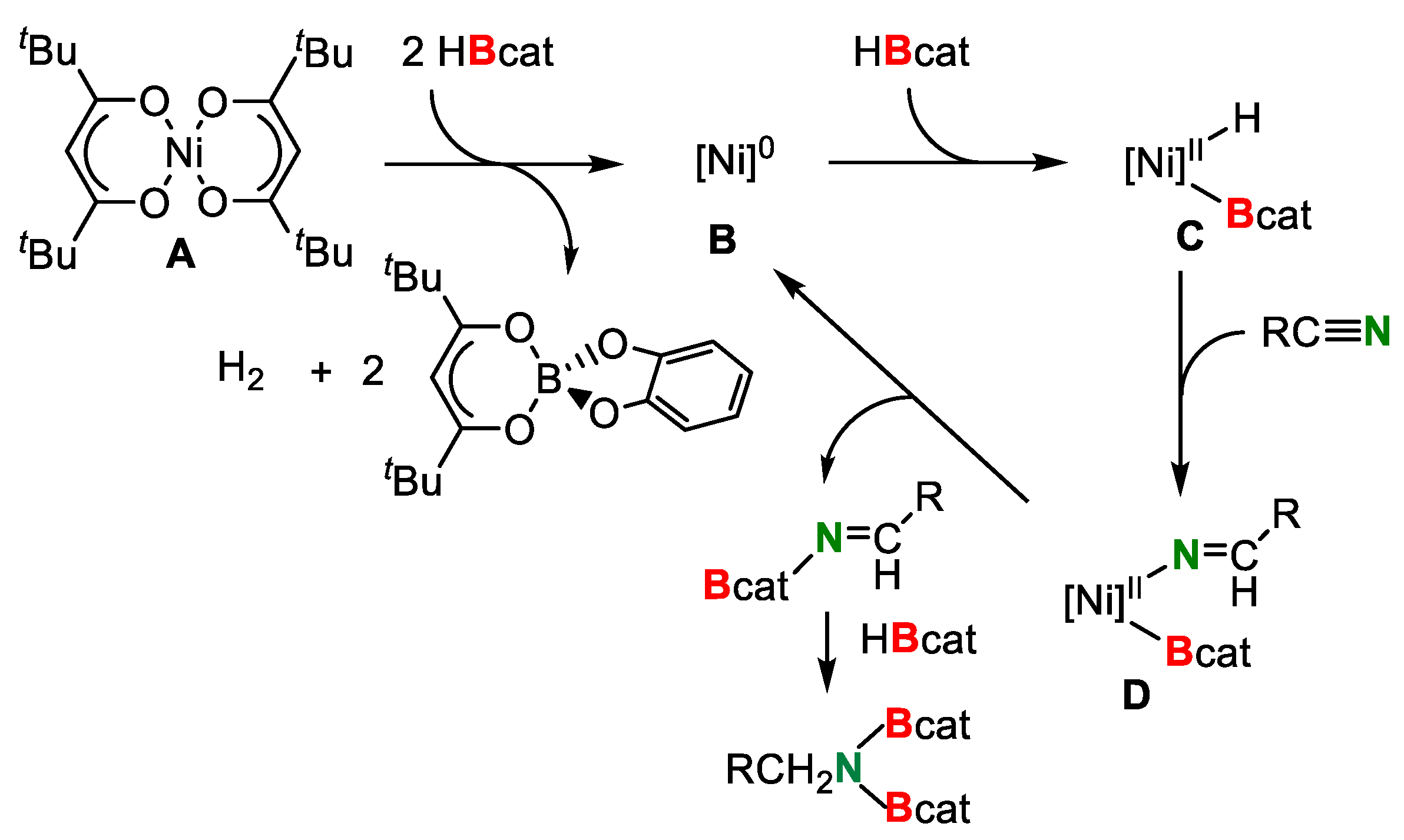
A proposed mechanism is shown in Scheme 17. The reduction of A by 2 equiv. of HBcat produces the active Ni(0) species B. Oxidative addition of H–Bcat toward the Ni(0) center gives boryl hydrido intermediate C. Insertion of a nitrile into the Ni–H bond in C affords D. The subsequent reductive elimination of the borylimine from D regenerates an intermediate B to complete the catalytic cycle. The obtained borylimine further reacts with HBcat to give the N,N-diborylamine.
We also reported the catalytic activity of 3Cl for the double hydroborylation of organonitriles (Table 9) [60]. The tendency of the double hydroborylation by 3Cl was similar to that of the double hydrosilylation. In the double hydroborylation, tBuCN was also converted into the corresponding product in good yield. No reaction occurred for CCl3CN, C6F5CN, and 4-PyCN with HBpin, suggesting that a strong electron-withdrawing substituent, or a coordination-feasible substituent on the nitrile carbon retards or disturbs the double hydroborylation of the nitrile portion. The molecular structures of EtN(Bpin)2 and PhCH2N(Bpin)2 were confirmed by single-crystal X-ray structure diffraction analyses. These structures showed the formation of diborylamine as results of the selective double hydroborylation of organonitriles.
4. Dihydroborylsilylation of Acetonitrile
With the hope of selective formation of borylsilylamine in the Fe-In cooperative catalytic system, the reaction of acetonitrile with both hydrosilane and hydroborane was investigated [60]. The mixture of acetonitrile (4.0 mmol), HSiMe2Ph (0.4 mmol), HBpin (0.4 mmol), and 3Cl (0.04 mmol) was stirred at 80 °C for 24 h under an argon atmosphere (Equation (6)). The desired borylsilylamine EtN(SiMe2Ph)(Bpin) was obtained with high selectivity although diborylamine was also generated in 6% NMR yield. The isolation of EtN(SiMe2Ph)(Bpin) in 81% yield was achieved by the distillation using a Kugelrohr in a glove box. This reaction is the first one-pot synthesis of borylsilylamine via catalytic hydrosilylation and hydroborylation.

In order to obtain insight into the reaction pathway of our catalytic system, we checked the double hydrosilylation under the similar reaction conditions in Entry 1 in Table 1 in the presence of 5 equiv. of InCl3 (Equation (7)). The expected double hydrosilylation product was not obtained. Therefore, we thought that the dissociation of InCl3 from the iron center in 3Cl was one of the key steps in our system.

Baba and co-worker reported that the indium trihalide InX3 reacted with hydrosilane to give indium hydride HInX2 and this compound acted as a radical [61]. If the elimination of InCl3 occurs from the iron center in 3Cl, the released InCl3 seems to react with hydrosilane to yield the corresponding indium hydride HInCl2. Therefore, we examined our reaction system in the presence of TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) as a radical scavenger and found that the trace amount of the disilylamine was yielded (Equation (8)). This result showed that HInCl2 was involved in the reaction pathway:
![Molecules 23 02769 i008]()

When deuterated acetonitrile (CD3CN) was used in place of CH3CN under the same reaction conditions in Entry 1 in Table 1, CD3CH2N(SiMe2Ph)2 was obtained in 73% yield (Equation (9)):
![Molecules 23 02769 i009]()

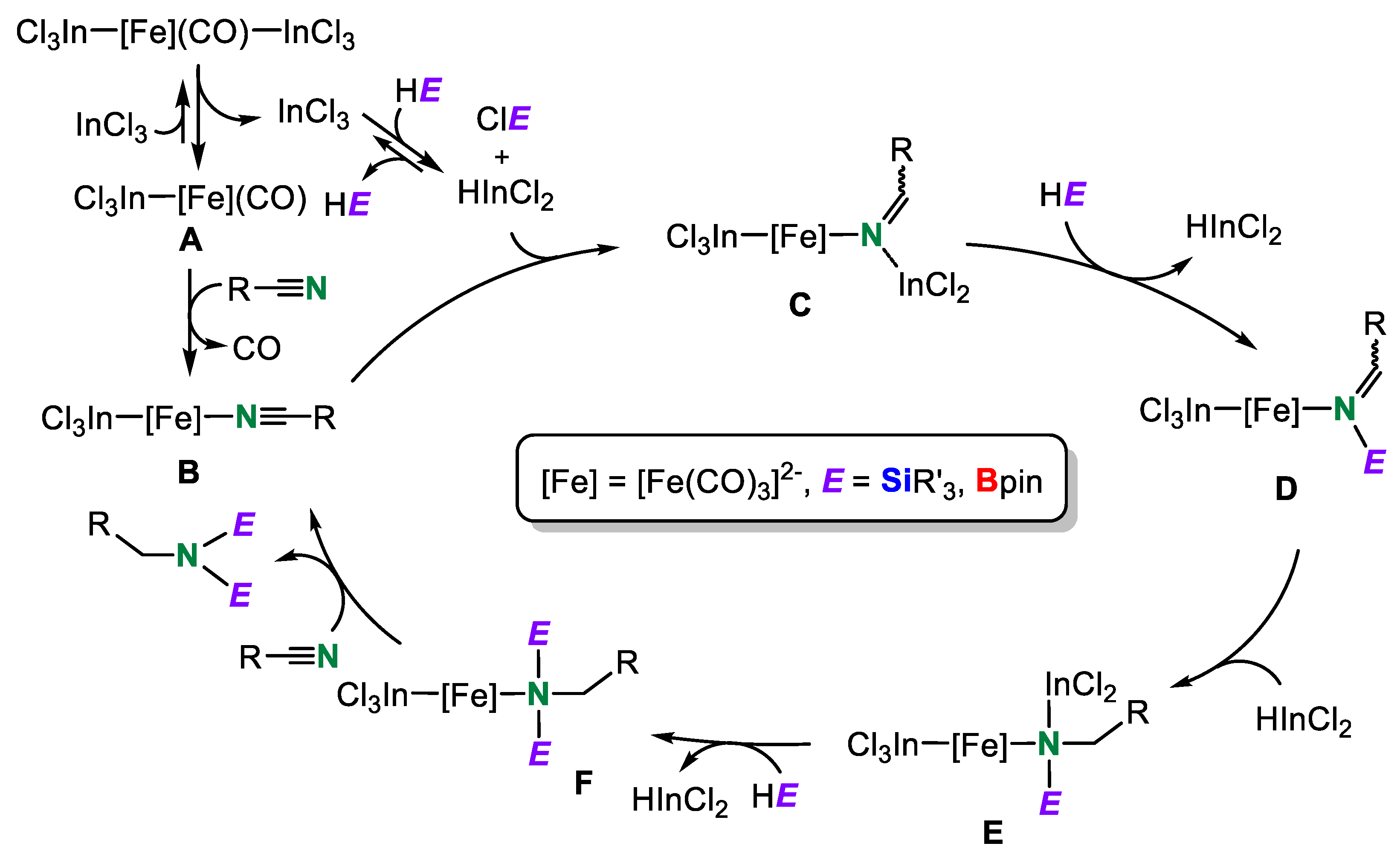
Based on the results mentioned above, we proposed a tentative catalytic cycle for the double hydrosilylation, double hydroborylation, and dihydroborylsilylation of organonitriles in the presence of 3Cl (Scheme 18). Dissociation of one of the coordinated InCl3 ligands from [Fe(InCl3)2(CO)4]2– occurs to give free InCl3 and monoindium-iron complex [Fe(InCl3)(CO)4]2– A. Then, the reaction of the eliminated InCl3 with HE (E = SiR’3, Bpin) provides HInCl2 and ClE. The formation of ClBpin was confirmed by the NMR measurement of the reaction mixture of HBpin and InCl3 in acetnitrile-d3. On the other hand, release of one carbonyl ligand from A and successive coordination of nitrile takes place to form nitrile complex [Fe(InCl3)(CO)3(NCR)]2– B. Complex B reacts with HInCl2 to generate indylimine iron intermediate C, followed by the reaction with HE to yield imine iron complex D and HInCl2. A similar reaction proceeds once again to give indane amine iron complex F through E. Finally, the elimination of the corresponding amine compound from the iron center in F and then recoordination of an organonitrile to the iron center yields catalytic intermediate B to complete the catalytic cycle. We believe that the imine moiety in D may not dissociate, causing selective formation of the corresponding amine compounds in this catalytic system.
5. Conclusions
There is growing interest in the selective double addition reaction of an E‒H bond (E = Si, B) to a C≡N triple bond of organonitriles because two N‒Si bonds or two N‒B bonds can be generated in one pot. Great efforts to establish catalytic system of such double addition by many research groups have resulted in several outstanding findings to date. Although some reaction mechanisms have been proposed, there are many unclear points from a mechanistic point of view.
We also have been engaged in creation of new catalytic systems for double hydrosilylation and double hydroborylation of organonitriles, and found a new catalytic system in which both iron and indium serve cooperatively. In addition, we found that this catalytic system could be applicable to the first single-step synthesis of borylsilylamine. The consideration of the reaction mechanism suggested that the anionic iron complex [cis-Fe(CO)4(InCl3)]2- was an important catalytic precursor.
Selective double addition of an E‒H bond to a C≡C triple bond and a C≡E triple bond (not only a C≡N triple bond but also other C≡heteroatom triple bonds) is becoming promising. More investigation concerning creation of new catalytic systems and elucidation of reaction mechanisms are expected to be continued.
Author Contributions
M.I. and H.N. wrote the paper.
Funding
This work was supported by a Grant-in-Aid for Scientific Research from JSPS (Category C, no. 16K05728 (M.I.)), and by a Grant-in Aid for Scientific Research on Innovation Area “Stimuli-responsive Chemical Species for the Creation of Functional Molecules” (No. 15H00957 (H.N.)) from JSPS, Japan.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Birot, M.; Pillot, J.-P.; Dunogues, J. Comprehensive Chemistry of Polycarbosilanes, Polysilazanes, and Polycarbosilazanes as Precursors of Ceramics. Chem. Rev. 1995, 95, 1443–1477. [Google Scholar] [CrossRef]
- Colombo, P.; Mera, G.; Riedel, R.; Sorarù, G.D. Polymer-Derived Ceramics: 40 Years of Research and Innovation in Advanced Ceramics. J. Am. Ceram. Soc. 2010, 93, 1805–1837. [Google Scholar] [CrossRef]
- Meng, L.; Zhang, X.; Tang, Y.; Su, K.; Kong, J. Hierarchically porous silicon−carbon−nitrogen hybrid materials towards highly efficient and selective adsorption of organic dyes. Sci. Rep. 2015, 5, 7910. [Google Scholar] [CrossRef] [PubMed]
- Viard, A.; Miele, P.; Bernard, S. Polymer-derived ceramics route toward SiCN and SiBCN fibers: From chemistry of polycarbosilazanes to the design and characterization of ceramic fibers. J. Ceram. Soc. Jpn. 2016, 124, 967–980. [Google Scholar] [CrossRef]
- Jansen, M.; Jäschke, T. Crystal Structure and Spectroscopic Characterisation of Hexamethyldisilazane-trichloroaluminum [(H3C)3Si]2NH·AlCl3. Z. Naturforsch. B J. Chem. Sci. 2000, 55, 763–767. [Google Scholar] [CrossRef]
- Ayed, T.; Barthelat, J.-C.; Tangour, B.; Pradère, C.; Donnadieu, B.; Grellier, M.; Sabo-Etienne, S. Structure and Bonding in a Disilazane Ruthenium Complex. Catalytic Selective Deuteration of Disilazane. Organometallics 2005, 24, 3824–3826. [Google Scholar] [CrossRef]
- Tanabe, Y.; Misaki, T.; Kurihara, M.; Iida, A.; Nishii, Y. Silazanes/catalytic bases: Mild, powerful and chemoselective agents for the preparation of enol silyl ethers from ketones and aldehydes. Chem. Commun. 2002, 0, 1628–1629. [Google Scholar] [CrossRef]
- Shimizu, K.; Minami, Y.; Goto, O.; Ikehira, H.; Hiyama, T. Silicon-based C–N Cross-coupling Reaction. Chem. Lett. 2014, 43, 438–440. [Google Scholar]
- Suginome, M.; Uehlin, L.; Murakami, M. Aminoboranes as “Compatible” Iminium Ion Generators in Aminative C−C Bond Formations. J. Am. Chem. Soc. 2004, 126, 13196–13197. [Google Scholar] [CrossRef] [PubMed]
- Nixon, T.D.; Whittlesey, M.K.; Williams, J.M.J. Ruthenium-catalysed transfer hydrogenation reactions with dimethylamine borane. Tetrahedron Lett. 2011, 52, 6652–6654. [Google Scholar] [CrossRef]
- Laval, S.; Dayoub, W.; Favre-Reguillon, A.; Berthod, M.; Demonchaux, P.; Mignani, G.; Lemaire, M. A mild and efficient method for the reduction of nitriles. Tetrahedron Lett. 2009, 50, 7005–7007. [Google Scholar] [CrossRef]
- Das, S.; Wendt, B.; Möller, K.; Junge, K.; Beller, M. Two Iron Catalysts are Better than One: A General and Convenient Reduction of Aromatic and Aliphatic Primary Amides. Angew. Chem. Int. Ed. 2012, 51, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Bornschein, C.; Werkmeister, S.; Junge, K.; Beller, M. TBAF-catalyzed hydrosilylation for the reduction of aromatic nitriles. New J. Chem. 2013, 37, 2061–2065. [Google Scholar] [CrossRef]
- Marciniec, B.; Guliński, J.; Urbaniak, W.; Kornetka, Z.W. Comprehensive Handbook on Hydrosilylation; Pergamon: Oxford, UK, 1992. [Google Scholar]
- Dhillon, R.S. Hydroboration and Organic Synthesis. 9-Borabicyclo[3.3.1]Nonane (9-BBN); Springer: Berlin, Germany, 2007. [Google Scholar]
- Luo, Y.-R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Reddy, N.P.; Uchimaru, Y.; Lautenschlager, H.-J.; Tanaka, M. Platinum-Catalyzed Novel Reactions of Nitriles and an Azirine with o-Bis(dimethylsilyl)benzene. Chem. Lett. 1992, 21, 45–48. [Google Scholar] [CrossRef]
- Hamdaoui, M.; Desrousseaux, C.; Habbita, H.; Djukic, J.-P. Iridacycles as Catalysts for the Autotandem Conversion of Nitriles into Amines by Hydrosilylation: Experimental Investigation and Scope. Organometallics 2017, 36, 4864–4882. [Google Scholar] [CrossRef]
- Gutsulyak, D.V.; Nikonov, G.I. Chemoselective Catalytic Hydrosilylation of Nitriles. Angew. Chem. Int. Ed. 2010, 49, 7553–7556. [Google Scholar] [CrossRef] [PubMed]
- Murai, T.; Sakane, T.; Kato, S. Cobalt carbonyl catalyzed reduction of aromatic nitriles with a hydrosilane leading to N,N-disilylamines. Tetrahedron Lett. 1985, 26, 5145–5148. [Google Scholar] [CrossRef]
- Murai, T.; Sakane, T.; Kato, S. Cobalt carbonyl catalyzed hydrosilylation of nitriles: A new preparation of N,N-disilylamines. J. Org. Chem. 1990, 55, 449–453. [Google Scholar] [CrossRef]
- Caporusso, A.M.; Panziera, N.; Pertici, P.; Pitzalis, E.; Salvadori, P.; Vitulli, G.; Martra, G. Hydrosilylation of aromatic nitriles promoted by solvated rhodium atom-derived catalysts. J. Mol. Catal. A 1999, 150, 275–285. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Moreau, J.J.E.; Pataud-Sat, M. Reactions de l’ortho-bis(dimethylsilyl)benzene avec les nitriles catalysees par des complexes du rhodium. J. Organomet. Chem. 1982, 228, 301–308. [Google Scholar] [CrossRef]
- Campos, J.; Rubio, M.; Esqueda, A.C.; Carmona, E. Large-scale preparation and labelling reactions of deuterated silanes. J. Label. Compd. Radiopharm. 2012, 55, 29–38. [Google Scholar] [CrossRef]
- Huckaba, A.J.; Hollis, T.K.; Reilly, S.W. Homobimetallic Rhodium NHC Complexes as Versatile Catalysts for Hydrosilylation of a Multitude of Substrates in the Presence of Ambient Air. Organometallics 2013, 32, 6248–6256. [Google Scholar] [CrossRef]
- Pérez, M.; Qu, Z.-W.; Caputo, C.B.; Podgorny, V.; Hounjet, L.J.; Hansen, A.; Dobrovetsky, R.; Grimme, S.; Stephan, D.W. Hydrosilylation of Ketones, Imines and Nitriles Catalysed by Electrophilic Phosphonium Cations: Functional Group Selectivity and Mechanistic Considerations. Chem. Eur. J. 2015, 21, 6491–6500. [Google Scholar] [CrossRef] [PubMed]
- Gandhamsetty, N.; Jeong, J.; Park, J.; Park, S.-W.; Chang, S. Boron-Catalyzed Silylative Reduction of Nitriles in Accessing Primary Amines and Imines. J. Org. Chem. 2015, 80, 7281–7287. [Google Scholar] [CrossRef] [PubMed]
- Gandhamsetty, N.; Park, J.; Jeong, J.; Park, S.-W.; Park, S.; Chang, S. Chemoselective Silylative Reduction of Conjugated Nitriles under Metal-Free Catalytic Conditions: β-Silyl Amines and Enamines. Angew. Chem. Int. Ed. 2015, 54, 6832–6836. [Google Scholar] [CrossRef] [PubMed]
- Weetman, C.; Anker, M.D.; Arrowsmith, M.; Hill, M.S.; Kociok-Kohn, G.; Liptrot, D.J.; Mahon, M.F. Magnesium-catalysed nitrile hydroboration. Chem. Sci. 2016, 7, 628–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, A.D.; Entsminger, S.W.; Fout, A.R. Insights into a Chemoselective Cobalt Catalyst for the Hydroboration of Alkenes and Nitriles. ACS Catal. 2017, 7, 3730–3734. [Google Scholar] [CrossRef]
- Nakamura, G.; Nakajima, Y.; Matsumoto, K.; Srinivas, V.; Shimada, S. Nitrile hydroboration reactions catalysed by simple nickel salts, bis(acetylacetonato)nickel(II) and its derivatives. Catal. Sci. Technol. 2017, 7, 3196–3199. [Google Scholar] [CrossRef]
- Kaithal, A.; Chatterjee, B.; Gunanathan, C. Ruthenium-Catalyzed Selective Hydroboration of Nitriles and Imines. J. Org. Chem. 2016, 81, 11153–11161. [Google Scholar] [CrossRef] [PubMed]
- Geri, J.B.; Szymczak, N.K. A Proton-Switchable Bifunctional Ruthenium Complex That Catalyzes Nitrile Hydroboration. J. Am. Chem. Soc. 2015, 137, 12808–12814. [Google Scholar] [CrossRef] [PubMed]
- Khalimon, A.Y.; Farha, P.; Kuzmina, L.G.; Nikonov, G.I. Catalytic hydroboration by an imido-hydrido complex of Mo(IV). Chem. Commun. 2012, 48, 455–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalimon, A.Y.; Farha, P.M.; Nikonov, G.I. Imido−hydrido complexes of Mo(IV): Catalysis and mechanistic aspects of hydroboration reactions. Dalton Trans. 2015, 44, 18945–18956. [Google Scholar] [CrossRef] [PubMed]
- Baldus, P.; Jansen, M.; Sporn, D. Ceramic Fibers for Matrix Composites in High-Temperature Engine Applications. Science 1999, 285, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, M.; Kroschel, M.; Jäschke, T.; Nuss, J.; Jansen, M.; Kolios, G.; Morillo, A.; Tellaeche, C.; Nieken, U. Towards continuous processes for the synthesis of precursors of amorphous Si/B/N/C ceramics. J. Mater. Chem. 2008, 18, 1810–1818. [Google Scholar] [CrossRef]
- Shriver, D.F. Transition metal basicity. Acc. Chem. Res. 1970, 3, 231–238. [Google Scholar] [CrossRef]
- Parkin, G.A. Simple Description of the Bonding in Transition-Metal Borane Complexes. Organometallics 2006, 25, 4744–4747. [Google Scholar] [CrossRef]
- Hill, A.F. An Unambiguous Electron-Counting Notation for Metallaboratranes. Organometallics 2006, 25, 4741–4743. [Google Scholar] [CrossRef]
- Bouhadir, G.; Amgoune, A.; Bourissou, D. Chapter 1 phosphine-boranes and related ambiphilic compounds: Synthesis, structure, and coordination to transition metals. In Advances in Organometallic Chemstry; Hill, A.F., Fink, M.J., Eds.; Elsevier: London, UK, 2010; Volume 58, pp. 1–107. ISBN 978-0-12-374784-6. [Google Scholar]
- Amgoune, A.; Bourissou, D. σ-Acceptor, Z-type ligands for transition metals. Chem. Commun. 2011, 47, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, H.; Dewhurst, R.D. Transition metals as Lewis bases: “Z-type” boron ligands and metal-to-boron dative bonding. Dalton Trans. 2011, 40, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kameo, H.; Nakazawa, H. Recent Developments in the Coordination Chemistry of Multidentate Ligands Featuring a Boron Moiety. Chem. Asian J. 2013, 8, 1720–1734. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.S.; Gabbaï, F.P. Coordination- and Redox-Noninnocent Behavior of Ambiphilic Ligands Containing Antimony. Acc. Chem. Res. 2016, 49, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.S.; Gabbaï, F.P. Coordination and Redox Non-innocent Behavior of Hybrid Ligands Containing Tellurium. Chem. Lett. 2016, 45, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Bouhadir, G.; Bourissou, D. Complexes of ambiphilic ligands: Reactivity and catalytic applications. Chem. Soc. Rev. 2016, 45, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, M.V.; Xie, J.; Lu, C.C. Stable Dihydrogen Complexes of Cobalt(−I) Suggest an Inverse trans-Influence of Lewis Acidic Group 13 Metalloligands. J. Am. Chem. Soc. 2017, 139, 6570–6573. [Google Scholar] [CrossRef] [PubMed]
- Kameo, H.; Kawamoto, T.; Sakaki, S.; Bourissou, D.; Nakazawa, H. Transition-Metal-Mediated Cleavage of Fluoro-Silanes under Mild Conditions. Chem. Eur. J. 2016, 22, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- Kameo, H.; Ikeda, K.; Bourissou, D.; Sakaki, S.; Takemoto, S.; Nakazawa, H.; Matsuzaka, H. Transition-Metal-Mediated Germanium–Fluorine Activation: Inverse Electron Flow in σ-Bond Metathesis. Organometallics 2016, 35, 713–719. [Google Scholar] [CrossRef]
- Cammarota, R.C.; Lu, C.C. Tuning Nickel with Lewis Acidic Group 13 Metalloligands for Catalytic Olefin Hydrogenation. J. Am. Chem. Soc. 2015, 137, 12486–12489. [Google Scholar] [CrossRef] [PubMed]
- Schindler, T.; Lux, M.; Peters, M.; Scharf, L.T.; Osseili, H.; Maron, L.; Tauchert, M.E. Synthesis and Reactivity of Palladium Complexes Featuring a Diphosphinoborane Ligand. Organometallics 2015, 34, 1978–1984. [Google Scholar] [CrossRef]
- Fong, H.; Moret, M.-E.; Lee, Y.; Peters, J.C. Heterolytic H2 Cleavage and Catalytic Hydrogenation by an Iron Metallaboratrane. Organometallics 2013, 32, 3053–3062. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.S.; Rittle, J.; Peters, J.C. Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 2013, 501, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, W.-C.; Gu, W.; MacInnis, M.C.; Timpa, S.D.; Bhuvanesh, N.; Zhou, J.; Ozerov, O.V. Facile Insertion of Rh and Ir into a Boron–Phenyl Bond, Leading to Boryl/Bis(phosphine) PBP Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 2086–2089. [Google Scholar] [CrossRef] [PubMed]
- You, D.; Yang, H.; Sen, S.; Gabbaï, F.P. Modulating the σ-Accepting Properties of an Antimony Z-type Ligand via Anion Abstraction: Remote-Controlled Reactivity of the Coordinated Platinum Atom. J. Am. Chem. Soc. 2018, 140, 9644–9651. [Google Scholar] [CrossRef] [PubMed]
- Itazaki, M.; Ito, M.; Nakazawa, H. Synthesis, Structure and Reactivity of Ruthenium(0) Indane Complex fac-[Ru(NCMe)3(CO)2(InX3)] (X = Cl, Br). Eur. J. Inorg. Chem. 2015, 2015, 2033–2036. [Google Scholar] [CrossRef]
- Itazaki, M.; Ito, M.; Nakashima, S.; Nakazawa, H. Synthesis and Characterization of [Fe(NCCH3)6][cis-Fe(InX3)2(CO)4] (X = Cl, Br, I) Containing Two Terminal Indium Fragments. Dalton Trans. 2016, 45, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Itazaki, M.; Nakazawa, H. Selective Double Hydrosilylation of Nitriles Catalyzed by an Iron Complex Containing Indium Trihalide. ChemCatChem 2016, 8, 3323–3325. [Google Scholar] [CrossRef]
- Ito, M.; Itazaki, M.; Nakazawa, H. Selective Double Hydroboration and Dihydrobrylsilylation of Organonitriles by an Iron-indium Cooperative Catalytic System. Inorg. Chem. 2017, 56, 13709–13714. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.; Shibata, I. Dihaloindium hydride as a novel reducing agent. Chem. Rec. 2005, 5, 323–335. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Double hydrosilylation, double hydroborylation, and dihydroborylsilylation of organonitriles.
Scheme 1.
Double hydrosilylation, double hydroborylation, and dihydroborylsilylation of organonitriles.

Figure 1.
Metal catalysts for double hydrosilylation and double hydroborylation of organonitriles.

Scheme 2.
Proposed mechanism for double hydrosilylation of organonitriles with 1,2-bis(dimethylsilyl)benzene.
Scheme 2.
Proposed mechanism for double hydrosilylation of organonitriles with 1,2-bis(dimethylsilyl)benzene.

Scheme 3.
A platinum-catalyzed reaction of various nitriles with 1,2-bis(dimethylsilyl)benzene.

Scheme 4.
Rh-catalyzed double hydrosilylation of α,β-unsaturated nitriles to vinyl amines.

Scheme 5.
Reductions of aromatic and aliphatic primary amides to amines catalyzed by two iron complexes.
Scheme 5.
Reductions of aromatic and aliphatic primary amides to amines catalyzed by two iron complexes.

Scheme 6.
Homobimetallic Rh complex-catalyzed hydrosilylation of nitriles.

Scheme 7.
Plausible reaction pathway of the double hydrosilylation of nitriles catalyzed by a cationic iridium complex.
Scheme 7.
Plausible reaction pathway of the double hydrosilylation of nitriles catalyzed by a cationic iridium complex.

Scheme 8.
TBAF catalyzed hydrosilylation for the reduction of aromatic nitriles.

Scheme 9.
[(C6F5)3PF][B(C6F5)4]-catalyzed hydrosilylation of nitriles.

Scheme 10.
[B(C6F5)3]-catalyzed double hydrosilylation of aryl and alkyl nitriles.

Scheme 11.
Double hydroborylation of nitriles catalyzed by an imido-hydrido Mo(IV) complex.

Scheme 12.
Plausible reaction pathway of the double hydroborylation of nitriles catalyzed by an imido-hydrido Mo(IV) complex.
Scheme 12.
Plausible reaction pathway of the double hydroborylation of nitriles catalyzed by an imido-hydrido Mo(IV) complex.

Scheme 13.
Ru-catalyzed double hydroborylation of nitriles.

Scheme 14.
Double hydroborylation of nitriles catalyzed by a Mg complex.

Scheme 15.
Homobimetallic Ru-catalyzed hydroborylation of nitriles.

Scheme 16.
Ni complex-catalyzed hydroborylation of nitriles.

Scheme 17.
Proposed mechanism for Ni-catalyzed double hydroborylation of nitriles.

Scheme 18.
Proposed catalytic cycle for double hydrosilylation, double hydroborylation, and dihydroborylsilylation of organonitriles in the presence of 3Cl.
Scheme 18.
Proposed catalytic cycle for double hydrosilylation, double hydroborylation, and dihydroborylsilylation of organonitriles in the presence of 3Cl.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
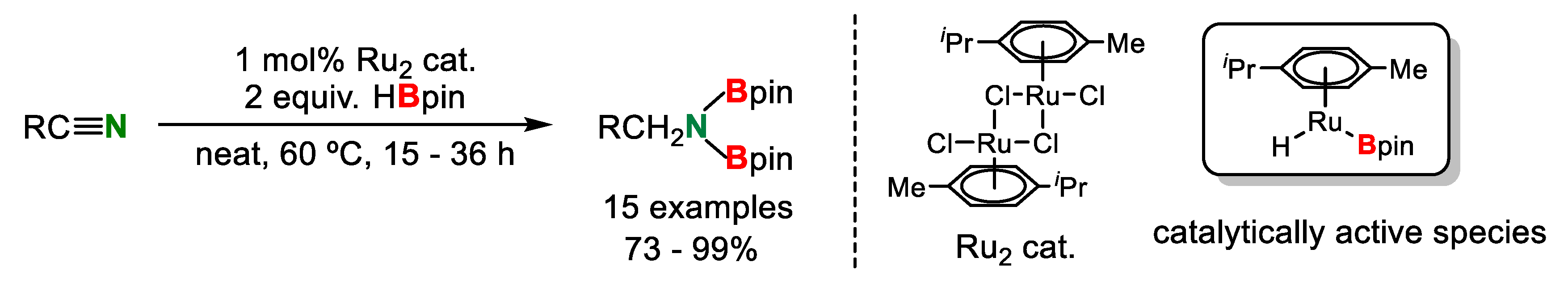{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Cobalt carbonyl catalyzed double hydrosilylation of aromatic nitriles a,b.

a Reaction conditions: nitrile (2.5 mmol), HSiMe3 (25 mmol), Co2(CO)8 (0.2 mmol), toluene (10 mL). b GLC yields in parentheses. c 40 h. d 48 h. e Co2(CO)8 (0.625 mmol) was used.
Table 2.
Cobalt carbonyl catalyzed double hydrosilylation of aliphatic nitriles a,b.

a Reaction conditions: nitrile (2.5 mmol), HSiMe3 (25 mmol), Co2(CO)8 (0.2 mmol), PPh3 (0.4 mmol), toluene (10 mL). b The yields of enamine in parentheses. c HSi(OEt)3 (10 mmol) and toluene (5 mL) were used. d P(OEt)3 (0.4 mmol) was used.
Table 3.
Double hydrosilylation of aromatic nitriles promoted by rhodium metal particles a,b.

a Reaction conditions: nitrile (9.8 mmol), hydrosilane (49 mmol), rhodium (0.1 mg atom). b GLC conversion of the nitriles in parentheses. c 4 h. d 7 h.
Table 4.
Ru-catalyzed double hydrosilylation of nitriles a.

a Reaction conditions: nitrile (0.13 mmol), HSiMe2Ph (0.33 mmol), Ru cat. (7.0 mg). b Nitrile (0.08 mmol), HSiMe2Ph (0.19 mmol), Ru cat. (3.0 mg), and CD3Cl instead of CD2Cl2 were used.
Table 5.
Ir-catalyzed double hydrosilylation of aryl nitriles a.

a Conditions: (a) nitrile (1.1 mmol), HSiEt3 (0.35 mL, 2.1 mmol), Ir cat. (6.8 mg, 4.8 mmol [0.5 mol %] or 13.7 mg, 9.7 mmol [1 mol %]), 70 °C; (b) aryl nitrile (0.8 mmol), HSiEt3 (0.3 mL, 1.8 mmol), Ir cat. ([0.5 mol %] or [1 mol %]), 70 °C; (c) conditions similar to those in (b), except that instead of 2.2 equiv. of HSiEt3, 4.4 equiv. (0.60 mL, 3.7 mmol) was used; (d) conditions similar to those in (b), except that instead of 2.2 equiv. of HSiEt3, 8.8 equiv. (1.20 mL, 7.5 mmol) was used. Yields were determined by 1H NMR spectroscopy using 1,3,5-tri-tert-butylbenzene as internal reference.
Table 6.
[B(C6F5)3]-catalyzed hydrosilylation of conjugated nitriles a.

a Conditions: Substrate (0.5 mmol), silane (4 equiv), and B(C6F5)3 (5 mol%). Yield of isolated product given and value within parentheses is the yield of the initially formed β-silyl-N,N-disilylamine using 1,1,2,2-tetrachloroethane as an internal standard. b B(C6F5)3 (7 mol%).
Table 7.
Double hydrosilylation of organonitriles in the presence of 3Cl a,b,c.

a Reaction conditions: organonitrile (4.0 mmol), hydrosilane (0.80 mmol), 3Cl (0.040 mmol); b Isolated yield; c Fc stands for a ferrocenyl group; d Little amounts of by-product EtN(SiMe2Ph)2 were removed by distillation.
Table 8.
Double hydroborylation of organonitriles in the presence of a Co complex.

Table 9.
Double hydroborylation of organonitriles in the presence of 3Cl a,b.

a Reaction conditions: organonitrile (4.0 mmol), HBpin (0.80 mmol), 3Cl (0.040 mmol); b Isolated yield; c 10 mol% 3Cl was used; d Catechol borane was used at room temperature for 18 h.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Itazaki, M.; Nakazawa, H. Selective Double Addition Reaction of an E‒H Bond (E = Si, B) to a C≡N Triple Bond of Organonitriles. Molecules 2018, 23, 2769. https://doi.org/10.3390/molecules23112769
AMA Style
Itazaki M, Nakazawa H. Selective Double Addition Reaction of an E‒H Bond (E = Si, B) to a C≡N Triple Bond of Organonitriles. Molecules. 2018; 23(11):2769. https://doi.org/10.3390/molecules23112769
Chicago/Turabian StyleItazaki, Masumi, and Hiroshi Nakazawa. 2018. "Selective Double Addition Reaction of an E‒H Bond (E = Si, B) to a C≡N Triple Bond of Organonitriles" Molecules 23, no. 11: 2769. https://doi.org/10.3390/molecules23112769