Metal-Catalyzed Cross-Coupling Reactions on Azaindole Synthesis and Functionalization


LAQV@REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Campus de Caparica, 2829-516 Caparica, Portugal
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(10), 2673; https://doi.org/10.3390/molecules23102673
Submission received: 28 September 2018
/
Revised: 11 October 2018
/
Accepted: 13 October 2018
/
Published: 17 October 2018
(This article belongs to the Special Issue Frontiers in Metal-Catalysed Cross-Coupling Reactions for the Synthesis and Functionalisation of Heterocycles)
Abstract
:Azaindoles are rare in nature but extremely attractive for drug discovery programs. Azaindoles can be obtained by diverse methods, including those involving metal-catalyzed reactions. This important core has been fascinating the scientific community due to their challenging synthesis and relevant bioactivity. This paper highlights the diverse synthetic methodologies developed to date involving metal-catalyzed reaction to attain azaindoles and its functionalization.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
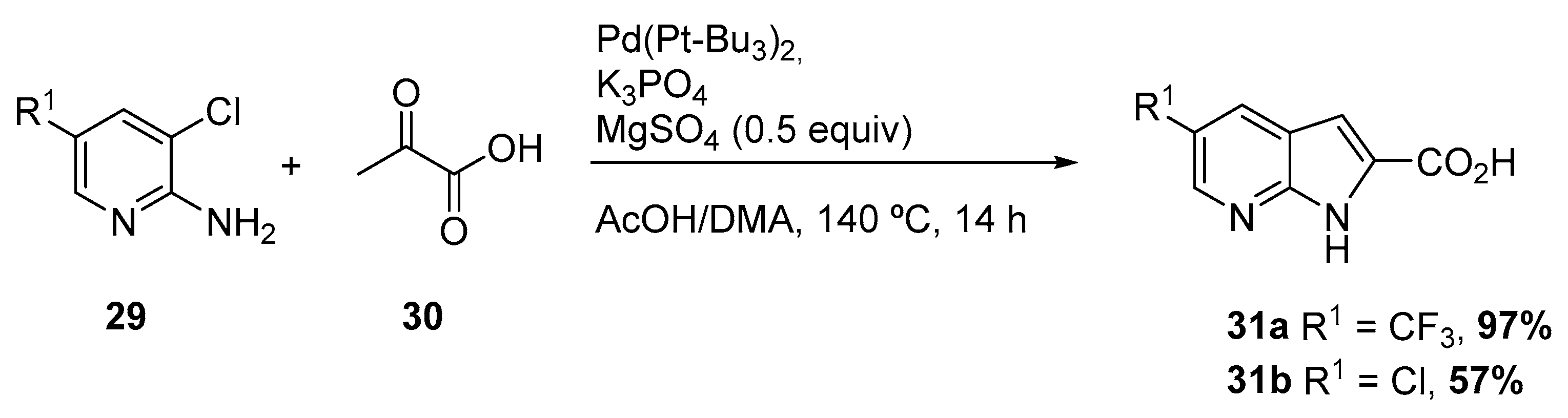{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Azaindoles are heterocyclic structures that have enticed the interest of the scientific community, since they are bio-isosteres of the indole and considered privileged structures in medicinal chemistry. This nucleus when properly functionalized can have a wide range of medicinal applications. Thus, substituted azaindoles unlike other heterocycles, can have their properties modulated by changing the substitution pattern or the position of the endocyclic nitrogen [1].
Usually azaindoles are synthesized starting from aminopyridines followed by building up of the pyrrole ring. This approach parallels the indole synthesis from anilines, however, due to the electron-deficient nature of pyridine ring that alters the electronic properties of the conjugate system, many classic indole synthetic methods are not as efficient or just do not work, though constituting a synthetic challenge [2].
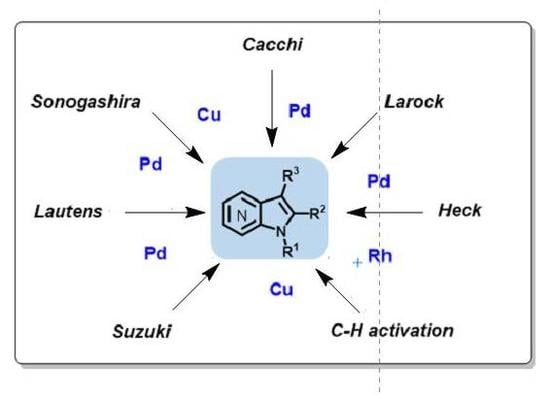
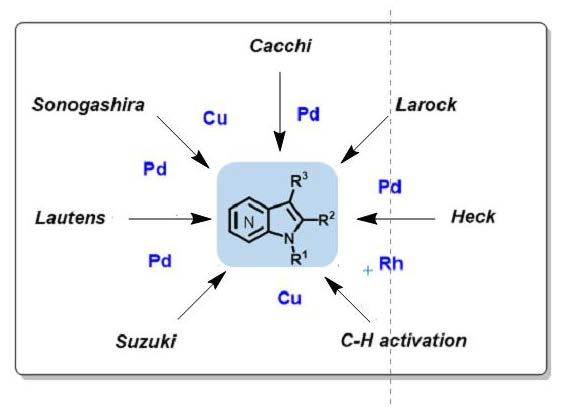
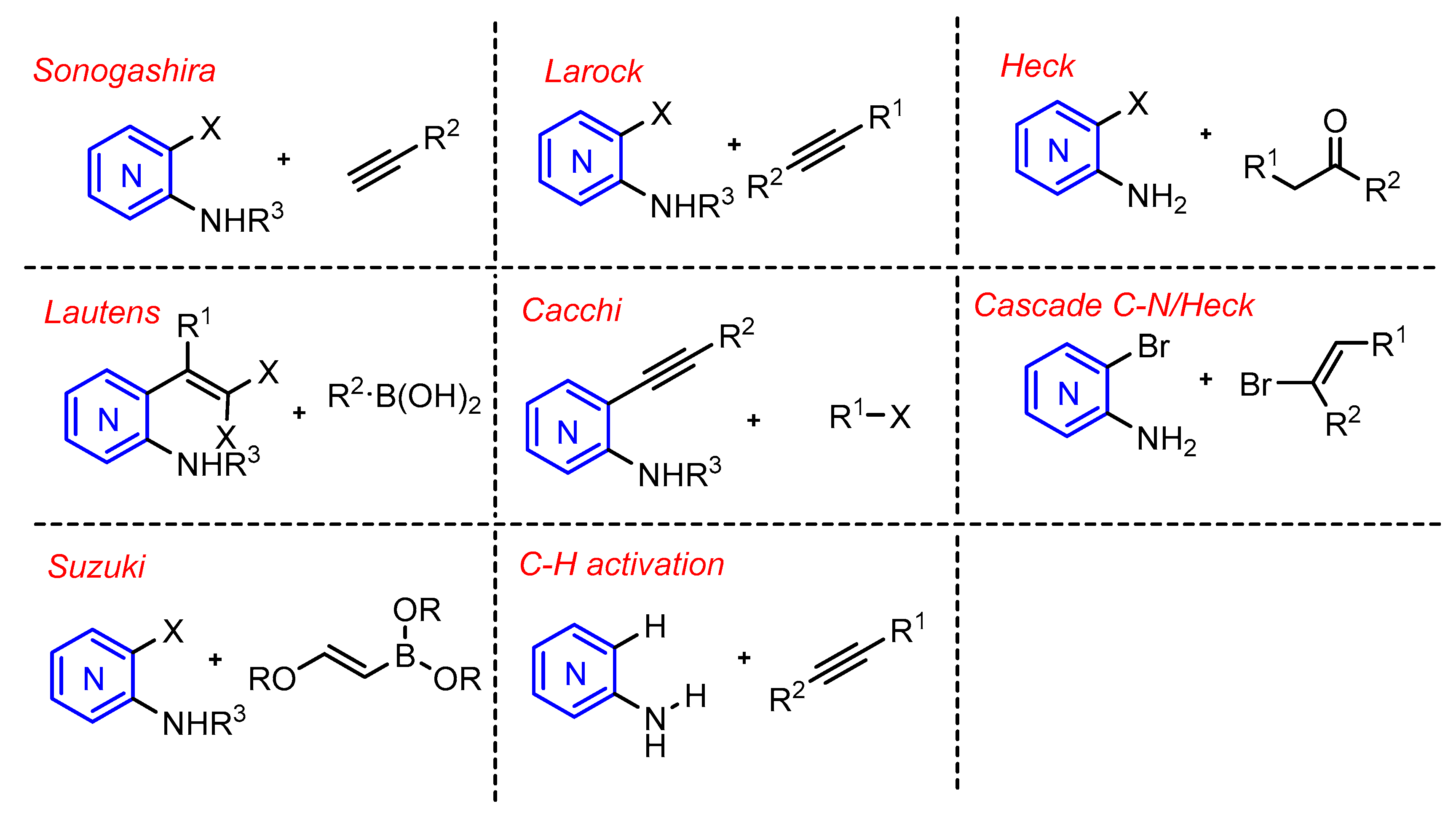
Metal-catalyzed cross-coupling reactions, constitute a very modern topic in organic synthesis, and are highly useful for the construction and derivation of these aminopyridine-containing heterocycles (Scheme 1). In particular, the well-known Sonogashira, Heck, and Suzuki cross-couplings have been used on the synthesis of azaindoles. Other methods involving metal-catalyzed reactions have also been described such as the Cacchi and Lautens methods. The metal-catalyzed C–H activation reaction has been scarcely explored in azaindole synthesis, as well as its functionalization.
2. Metal-Catalyzed Cross-Coupling Reactions
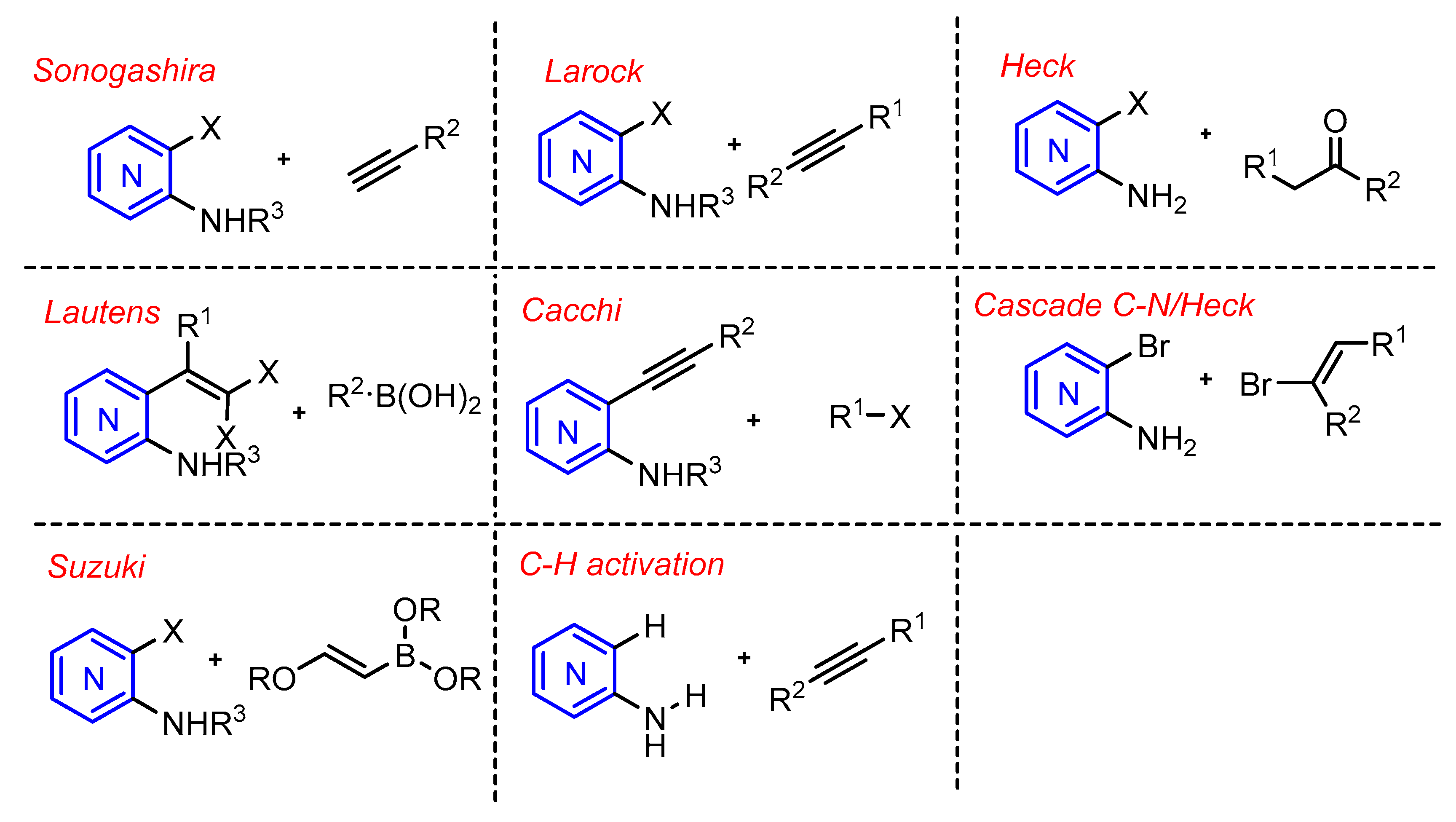
2.1. Sonogashira Reaction
The Sonogashira cross-coupling reaction was first reported by K. Sonogashira in 1975 and was established as a method for C–C bond formation via palladium-catalyzed coupling of terminal alkynes with aryl halides or allyl halides.
When applied to azaindole synthesis, the Sonogashira reaction usually involves amino-halopyridines that can be coupled with terminal alkynes by Sonogashira coupling, followed by ring formation, in the presence of strong bases like potassium hydride or copper-mediated cyclization.
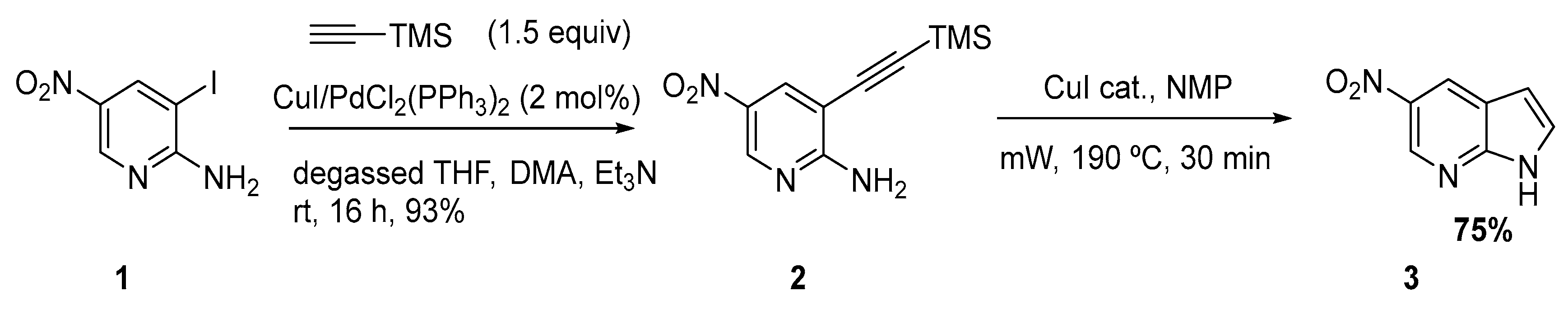
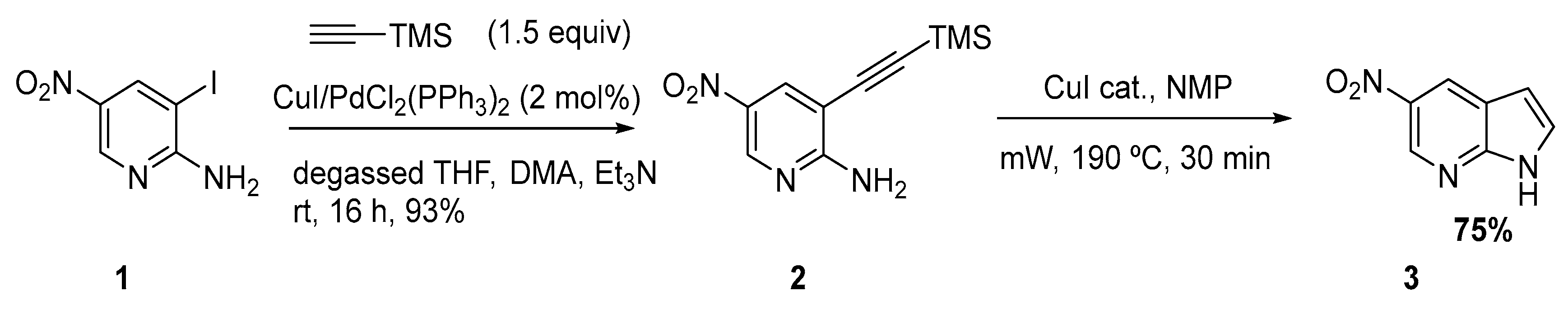
In order to explore microwave assisted copper-mediated cyclization S. Pearson reported the performance of several Sonogashira reactions with nitro-substituted aminopyridines [3]. The first step involved a Sonogashira reaction of 2-amino-3-iodo-5-nitropyridine 1 with TMSA in a THF/dimethylacetamide (DMA) mixture, followed by cyclization to the azaindole structure 3 using catalytic CuI under microwave irradiation (Scheme 2).
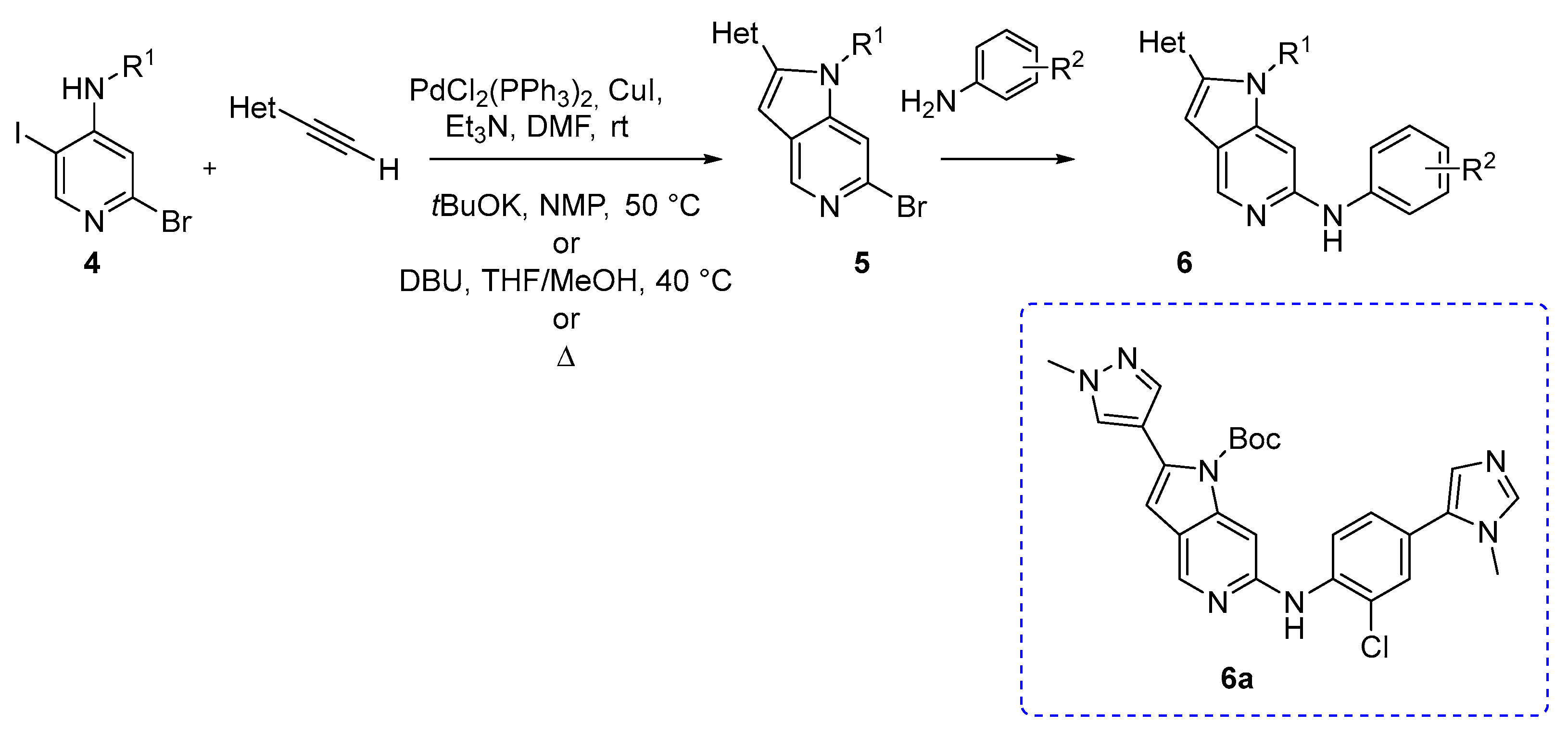
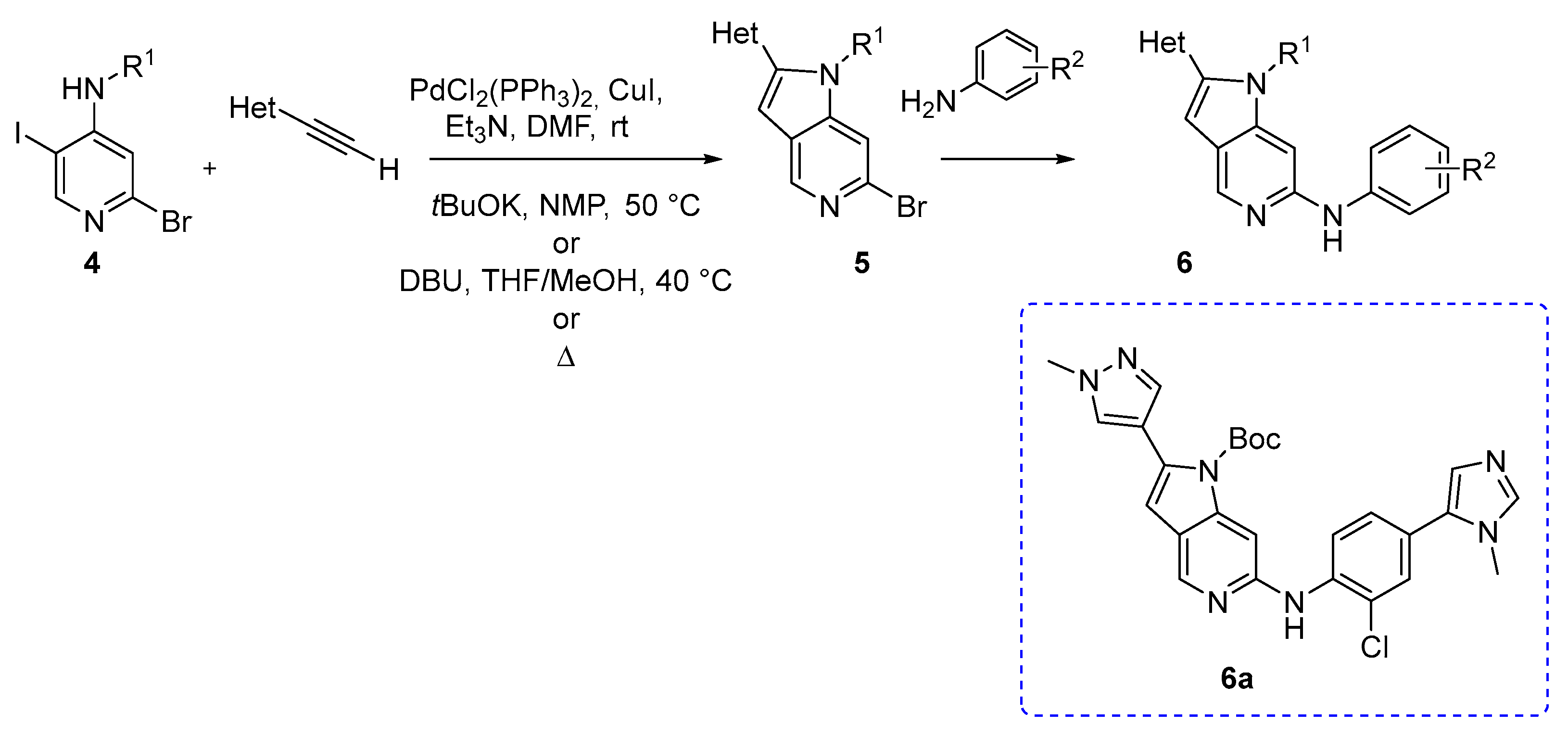
S. Naud and coworkers reported the synthesis of azaindole derivatives as possible inhibitors of mitotic kinase monopolar spindle 1 (MPS1), which is usually overexpressed in many human cancers. The authors used an appropriately substituted 4-amino-2-bromo-5-iodopyridine (4) and an alkyne, and synthesized several key intermediates using a palladium-mediated Sonogashira coupling, followed by intramolecular cyclization. These intermediates 5 were then functionalized via a C–N cross-coupling reaction (Scheme 3) [4].
Sonogashira coupling conditions were optimized according to the starting material. The Sonogashira reactions were carried using PdCl2(PPh3)2, CuI, and Et3N in DMF and the temperature was RT or 60 °C, according to the synthetic approach (Scheme 3). Compound 6a was the most promising since it demonstrated a good oral pharmacokinetic profile in mouse and rat as well as inhibition of MPS1 activity in vivo following oral administration [4].
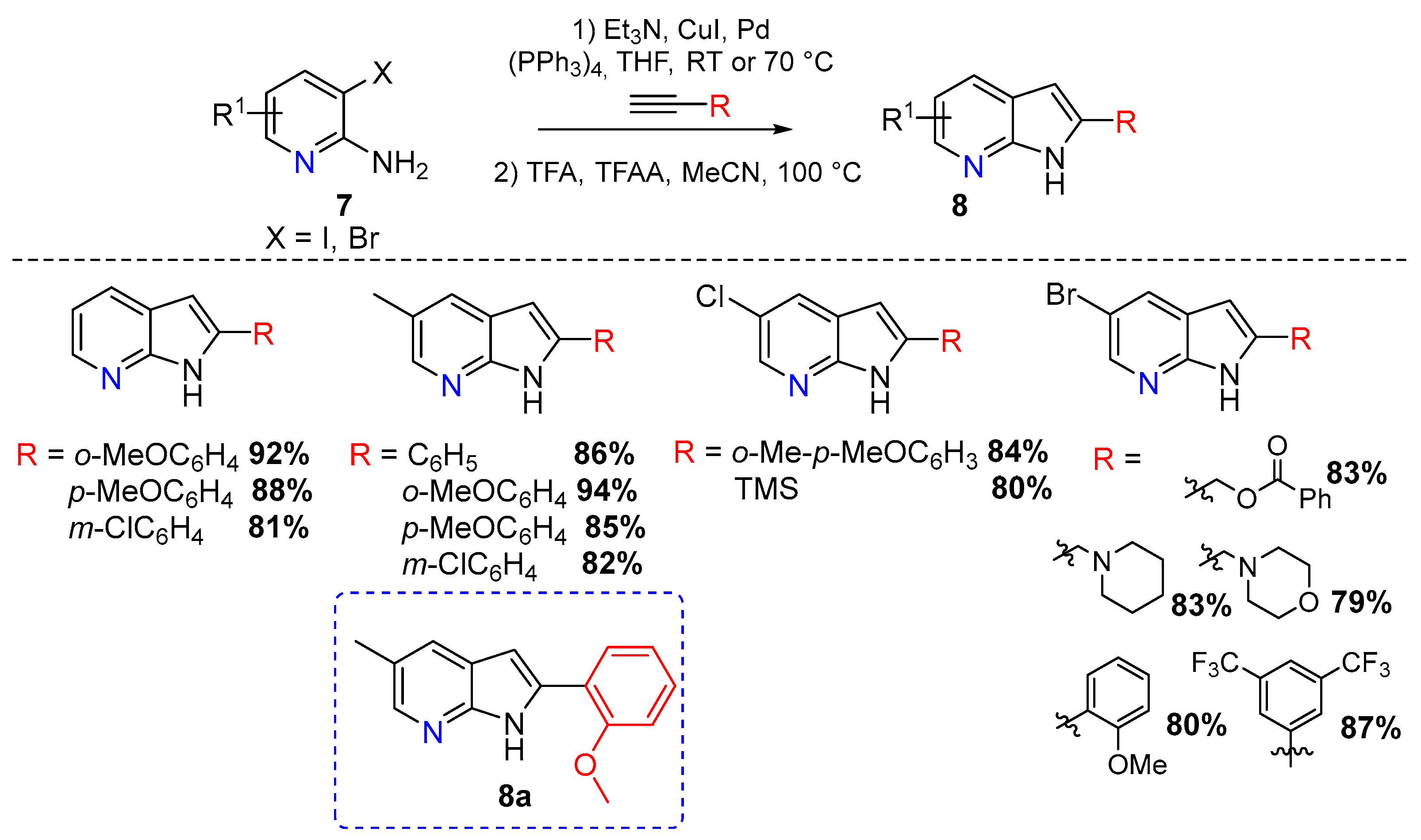
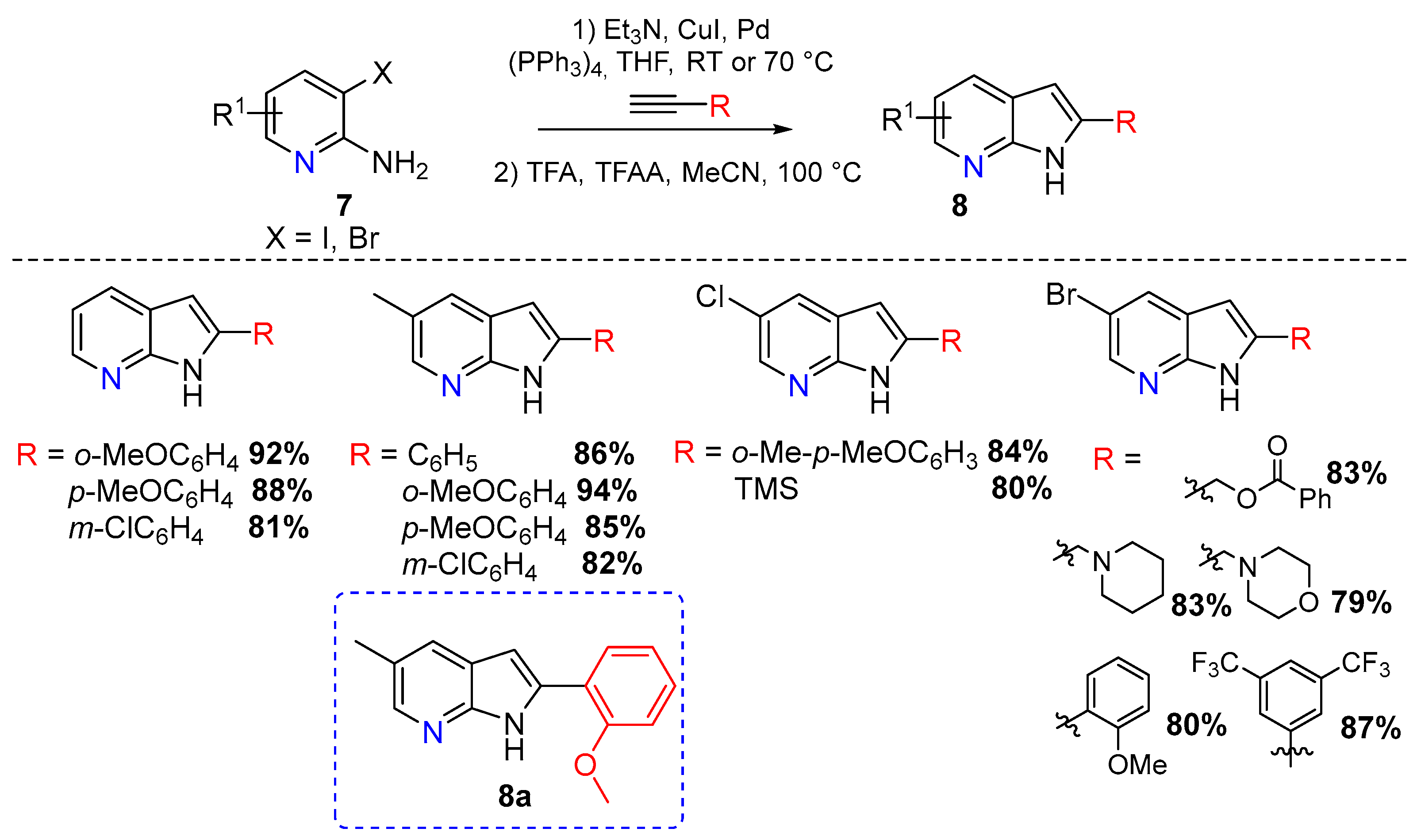
The acid-catalyzed synthesis of 7-azaindoles was reported by T. Leboho and coworkers in 2014. The authors synthesized several 7-azaindole derivatives 8 from 3-alkynyl-2-aminopyridines (Scheme 4) [5].
The Sonogashira coupling reaction was performed using a CuI/Pd(PPh3)4 catalytic system, followed by ring closure under acidic conditions. A wide range of acids was used including HCl, H2SO4, AcOH, and trifluoroacetic acid (TFA). The best yields were obtained using 1 equiv of TFA and 1.3 equiv of TFAA in MeCN and heating to reflux for 8 h (Scheme 4). These structures were then evaluated for their antimicrobial activity with compound 8a being the most active against Pseudomonas aeruginosa [5].
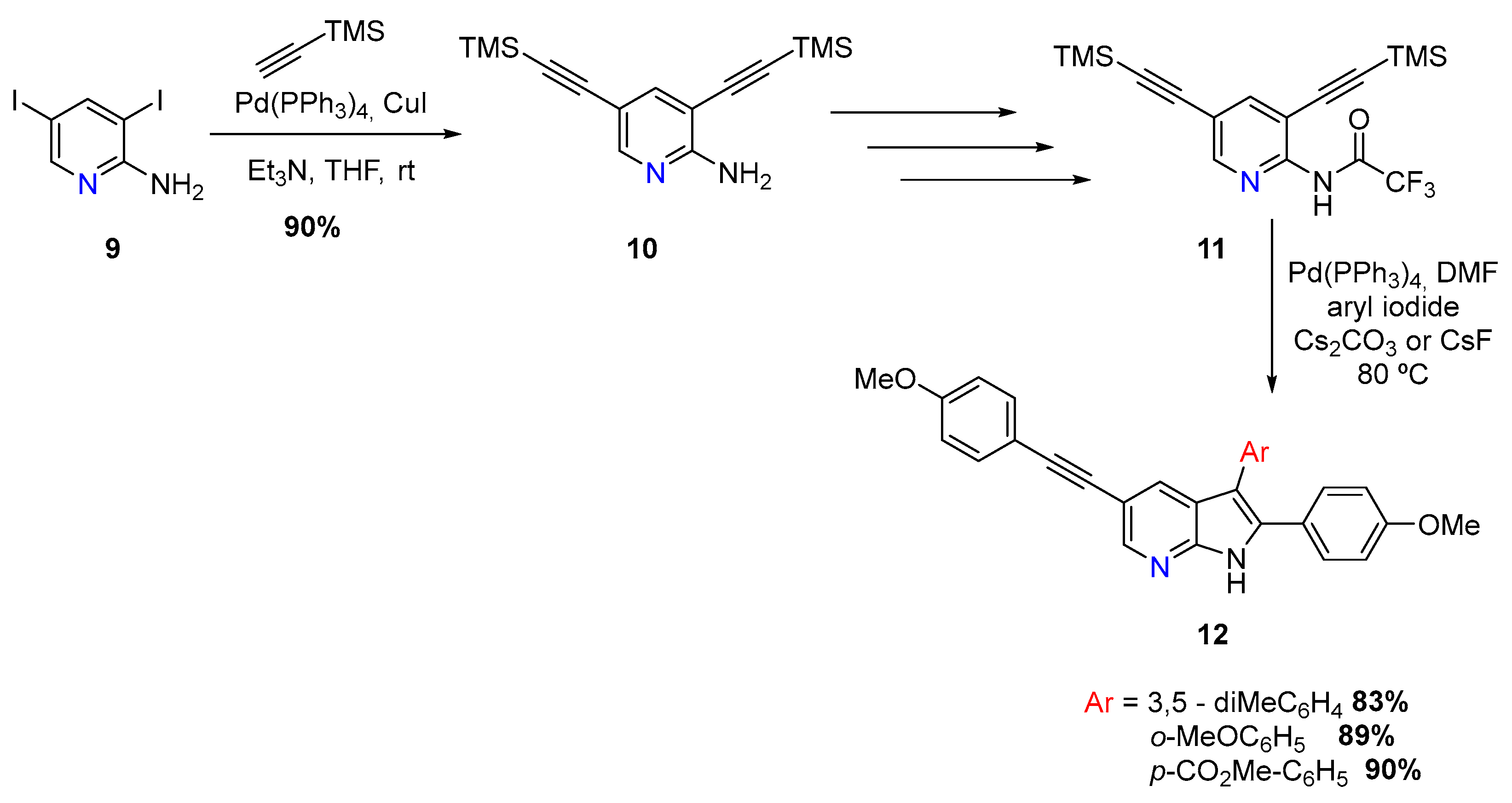
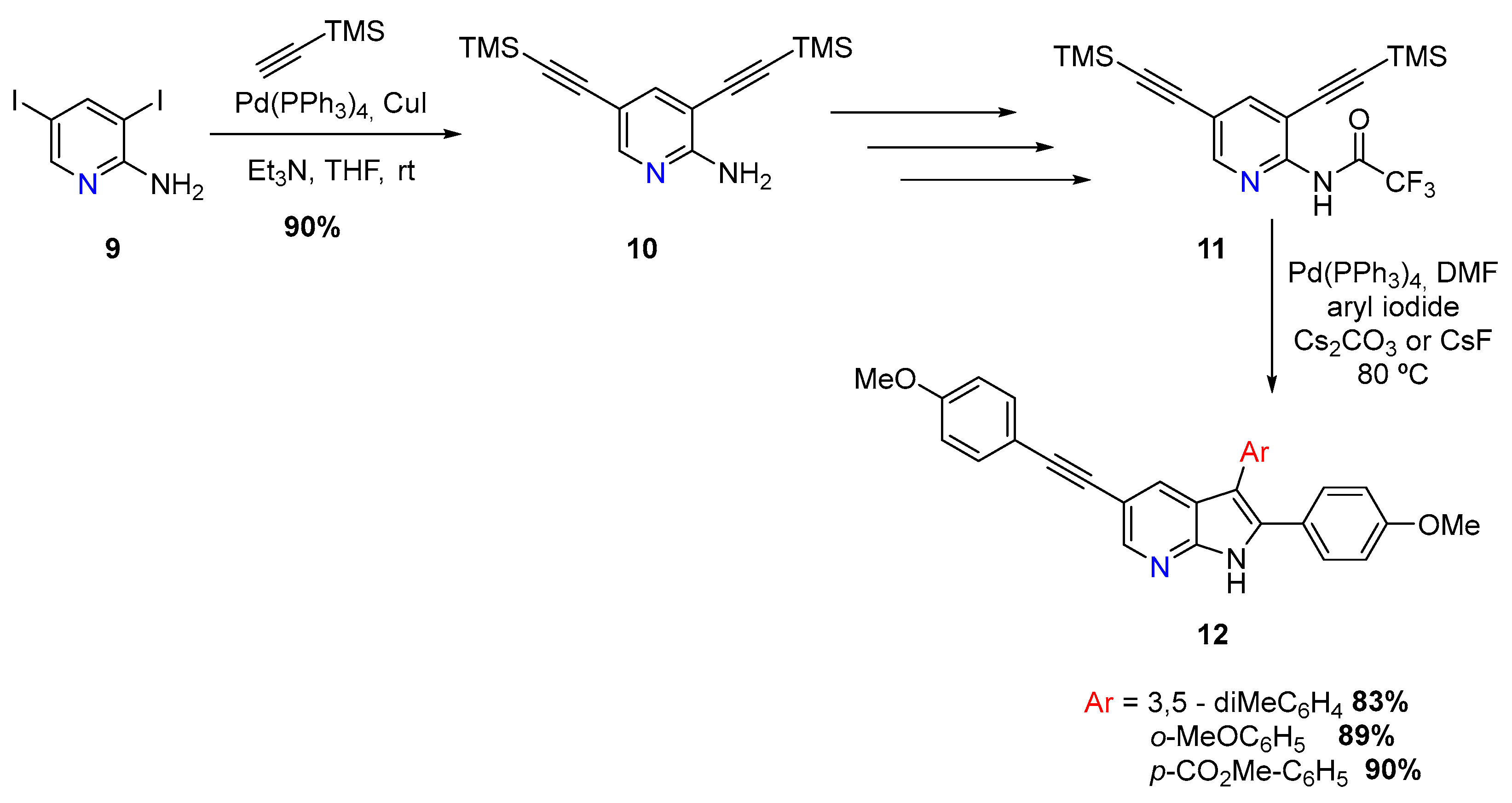
New 7-azaindole derivatives (12) were discovered as promising inhibitors for the gastrointestinal protozoal parasite Giardia duodenalis by T. Leboho and coworkers, via a double Sonogashira reaction, using dihalogenated aminopyridines (9) [6]. Two paths were explored, one starting with 3,5-diiodoaminopyridine and other with 5-bromo-3-iodoaminopyridine (Scheme 5).
The first approach involved a double Sonogashira using 3,5-diiodoaminopyridine (9) and a Pd(PPh3)4/CuI catalytic system, followed by removal of trimethylsilyl with TBAF. Furthermore, another double Sonogashira coupling was employed using 4-methoxyiodobenzene. This aminopyridine was then converted into the trifluoroacetamide derivative 11 and subjected to a Cacchi reaction. With the use of several aromatic iodides, and a Pd(PPh3)4 catalytic system, with Cs2CO3 or CsF as base, it was possible to synthesize three 2,3,5-trisubstituted azaindoles (12) (Scheme 5) [6].
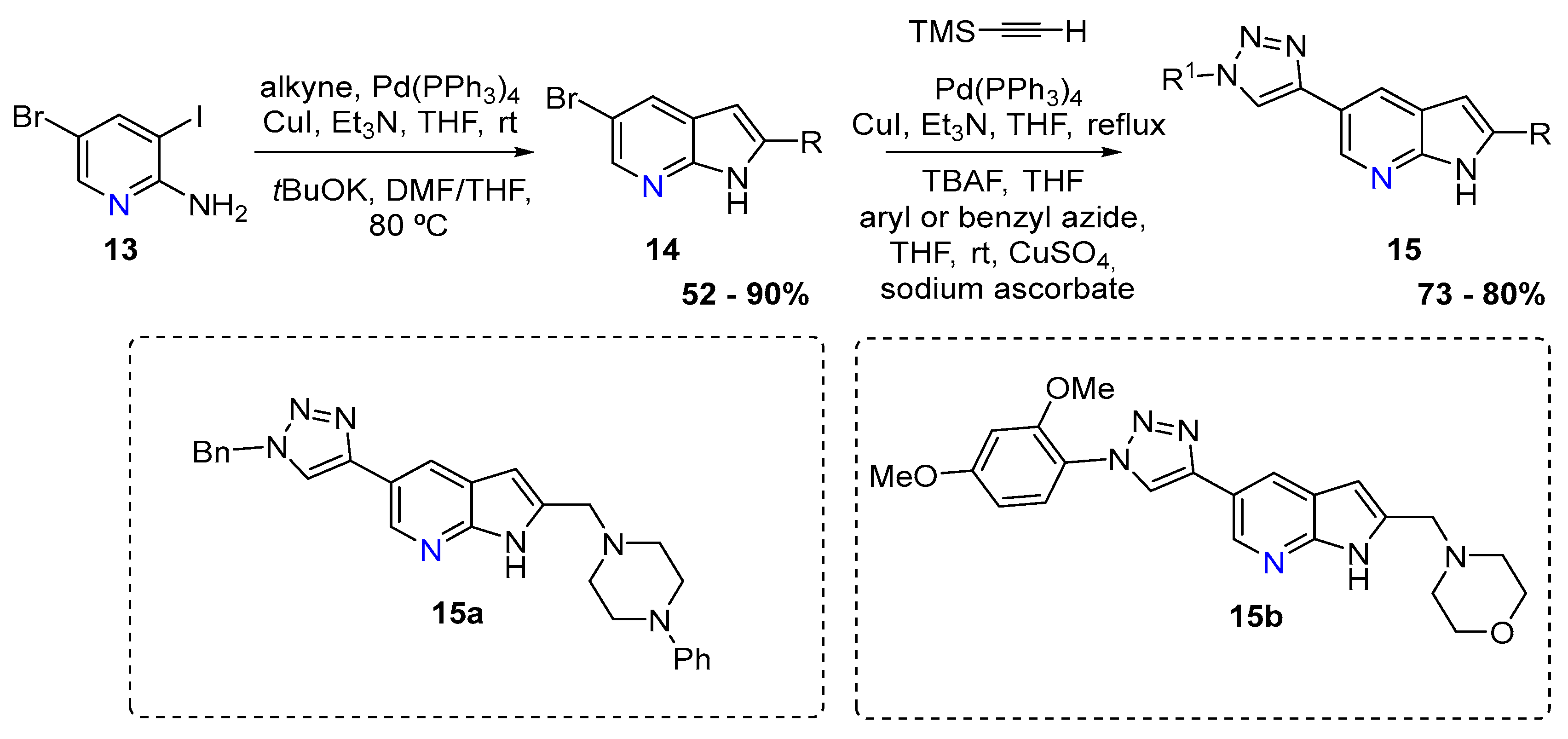
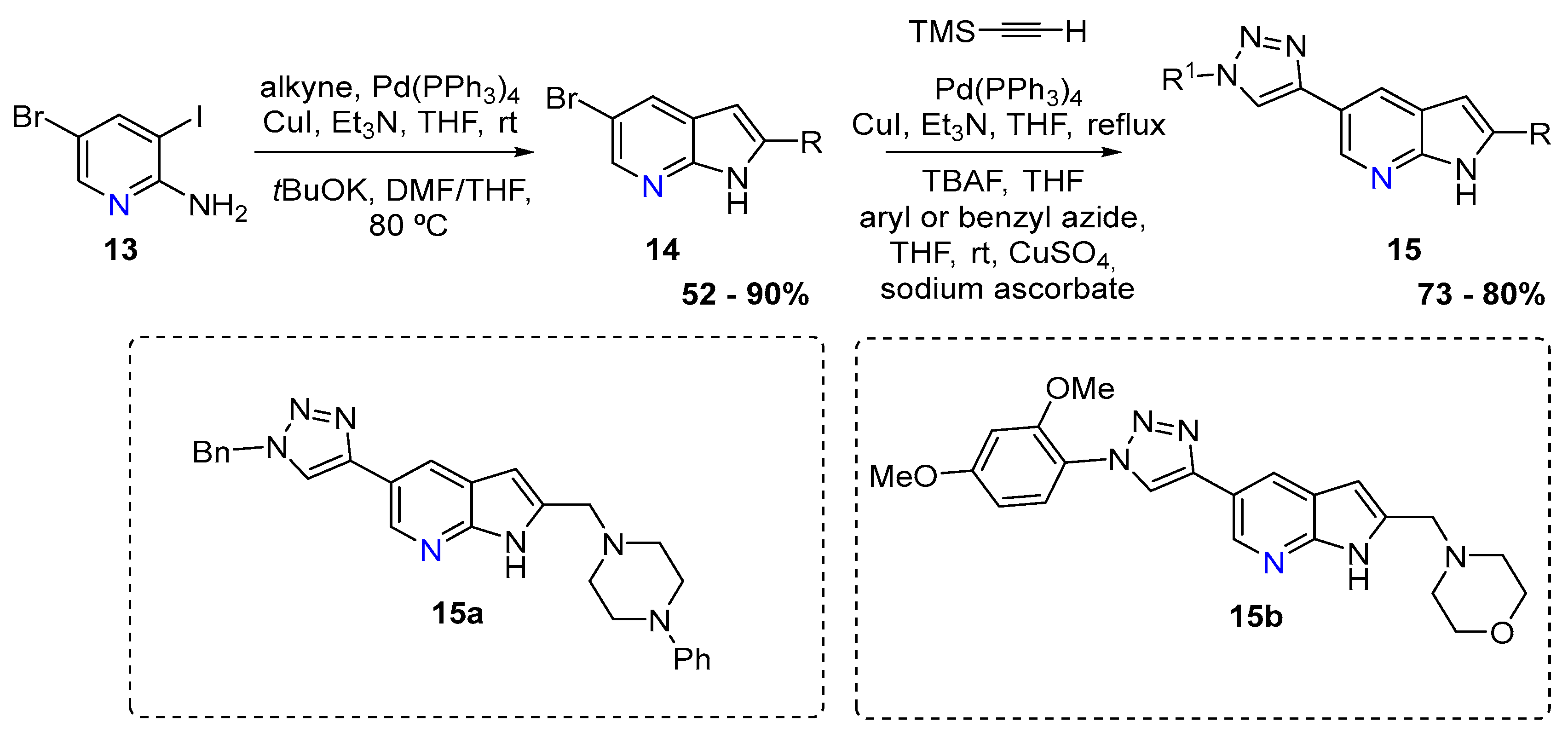
The second approach relied on a double Sonogashira using 5-bromo-3-iodoaminopyridine (13) that afforded several derivatives that were then treated with t-BuOK, DMF/THF at 80 °C. The azaindoles (14) were then functionalized to afford several 7-azaindoles (15) bearing triazole and quinoxaline moieties, with yield up to 87%. Compound 15a and 15b showed the best activity against the Giardia duodenalis trophozoites (Scheme 6) [6].
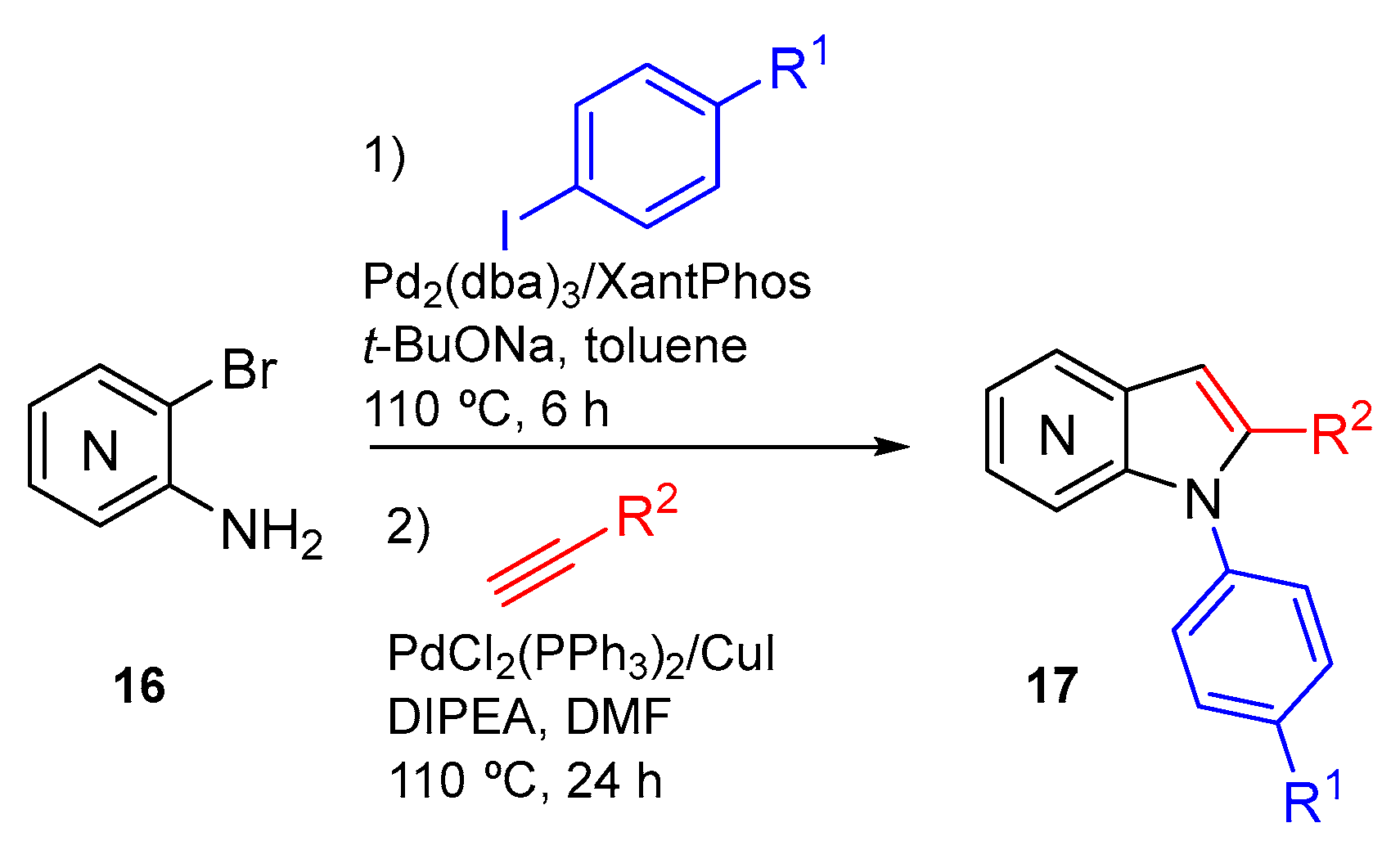
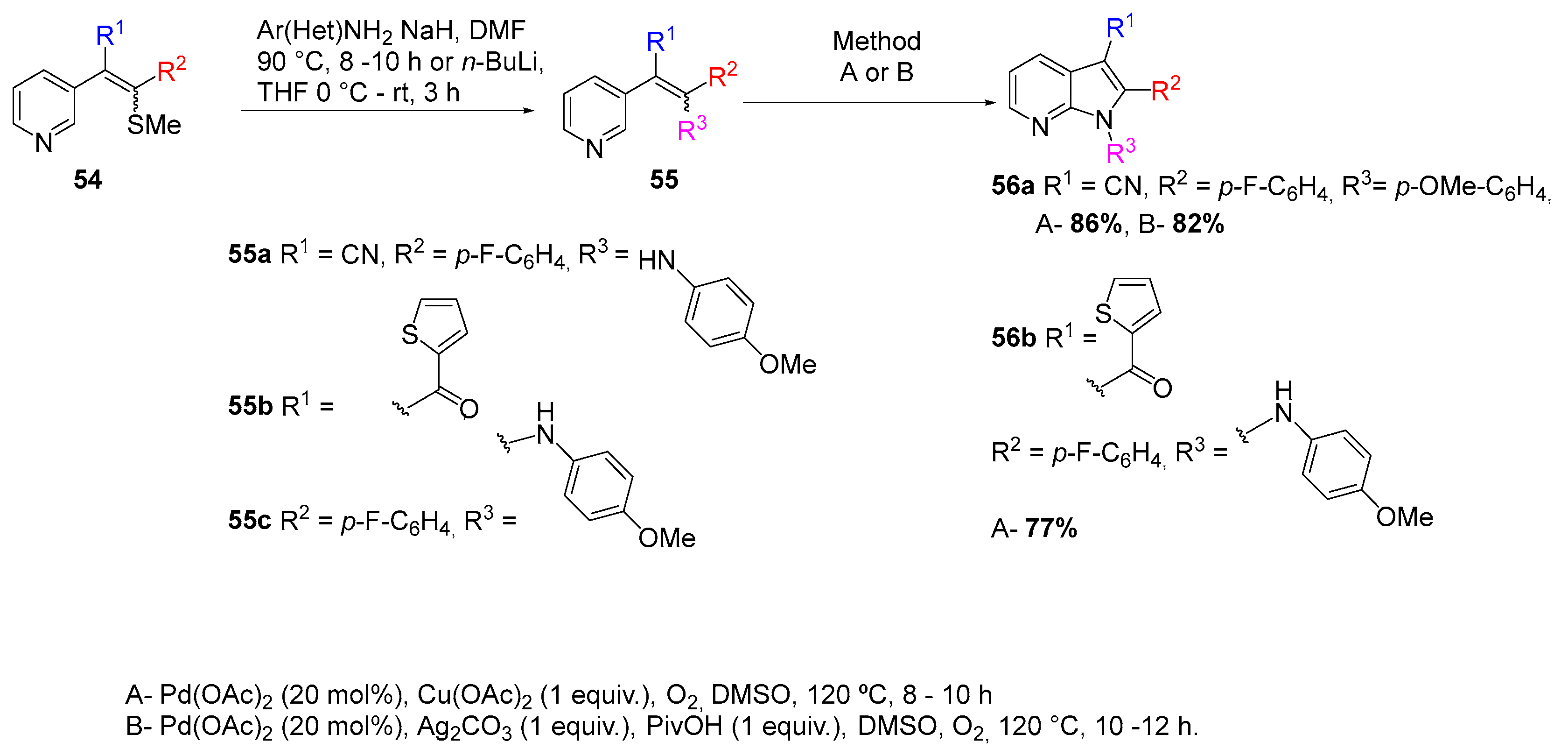
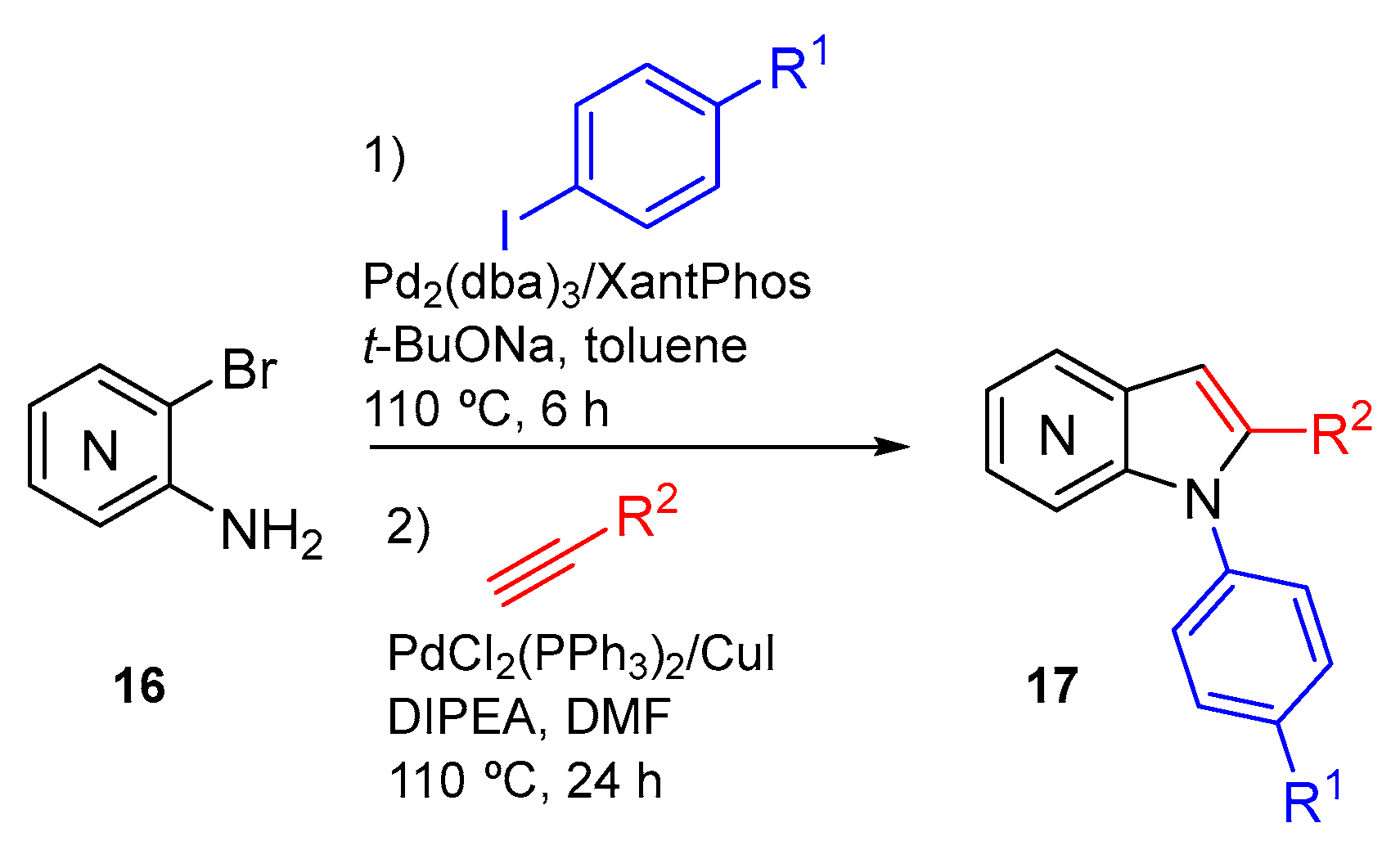
In 2017 we reported a one-pot approach for azaindole synthesis that involved N-arylation and Sonogashira coupling reaction followed by in situ cyclization (Scheme 7). This methodology uses amino-halopyridines as starting materials and allows the synthesis of 1,2-disubstituted 4-, 5-, 6- and 7-azaindoles [7].
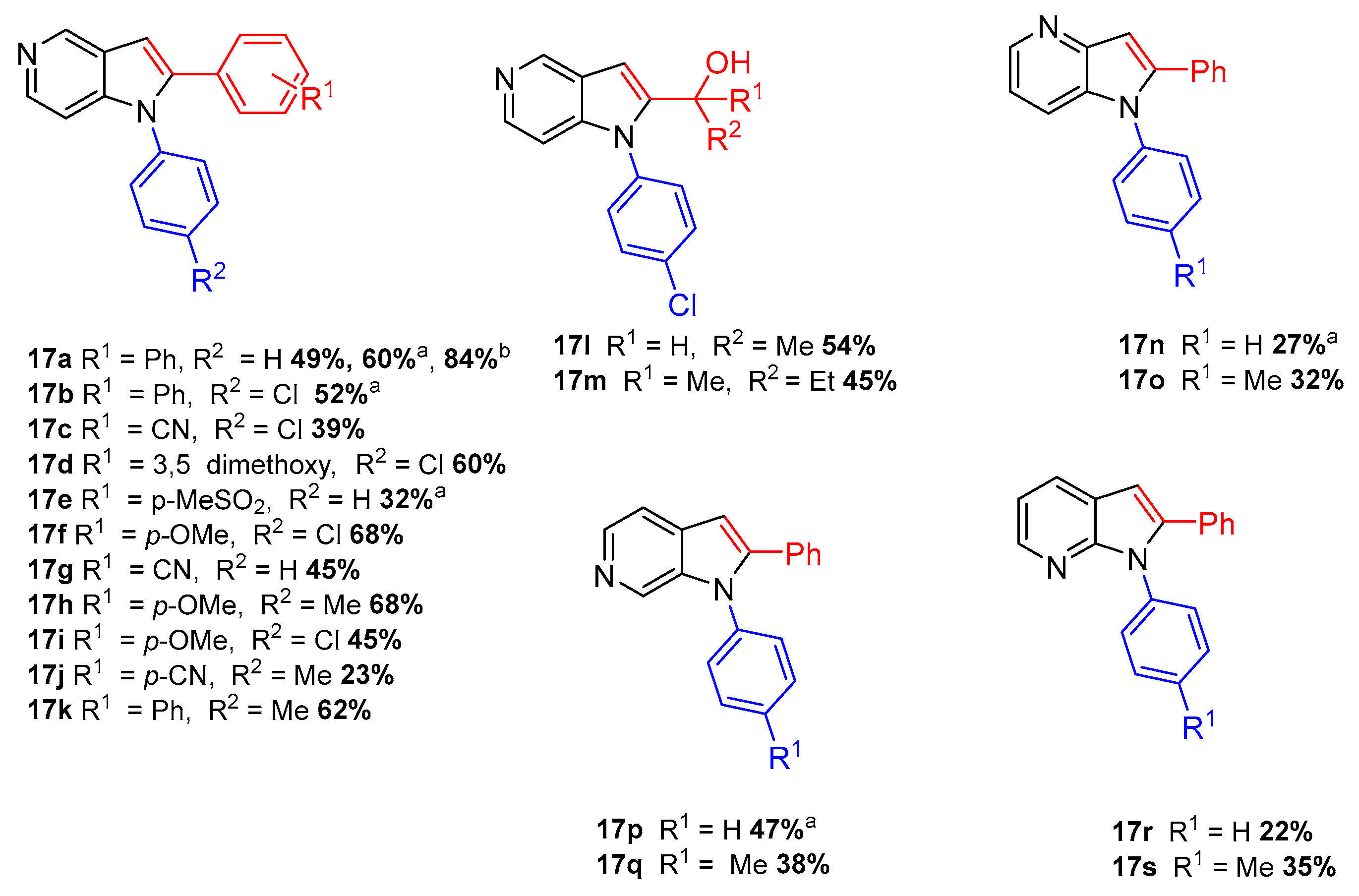
In order to study the reaction scope, several iodides were employed in the N-arylation reaction as well as several alkynes in the Sonogashira reactions (Scheme 8). The results obtained demonstrate that this methodology exhibits a wide scope and compatibility with electron-withdrawing and electron-donating groups.
2.2. Larock Reaction
Recently, transition metal-catalyzed approaches to prepare azaindoles from appropriately substituted pyridines and terminal alkynes have been reported [8,9]. These include a coupling/cyclization process involving palladium or copper catalysis, an intramolecular Heck reaction of enamine derivatives [10], and a heteroannulation of internal alkynes [11,12,13,14]; according to the procedure developed by Larock for the synthesis of indoles [15,16].
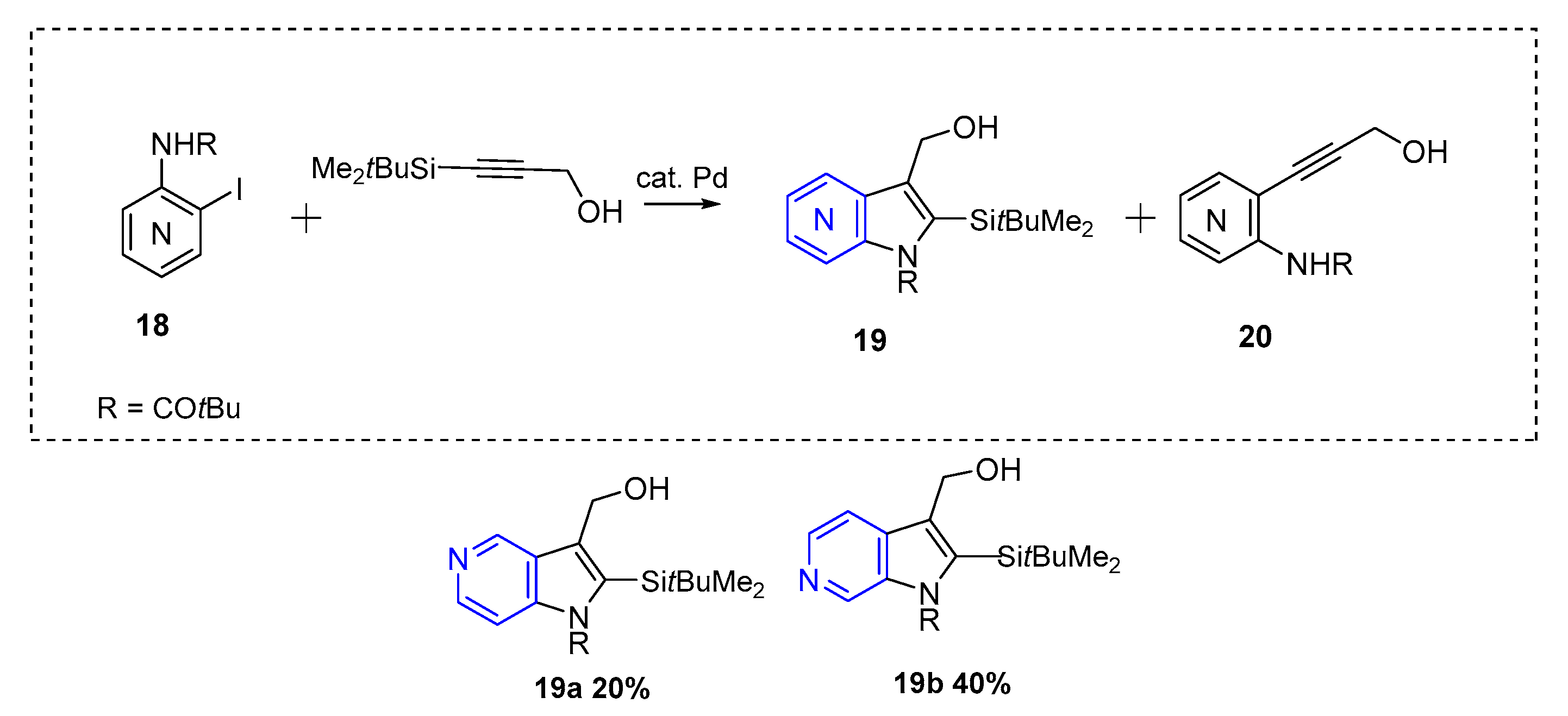
The first azaindole synthesis employing Larock methodology was reported in 1993 by Gronowitz et al. This method afforded substituted 5 and 6-azaindole (19) in moderate yields (up to 40% in the case of 19b, Scheme 9) [14]. The palladium source used was Pd(OAc)2 (5 mol %) in the presence of KOAc as base (5 equiv).
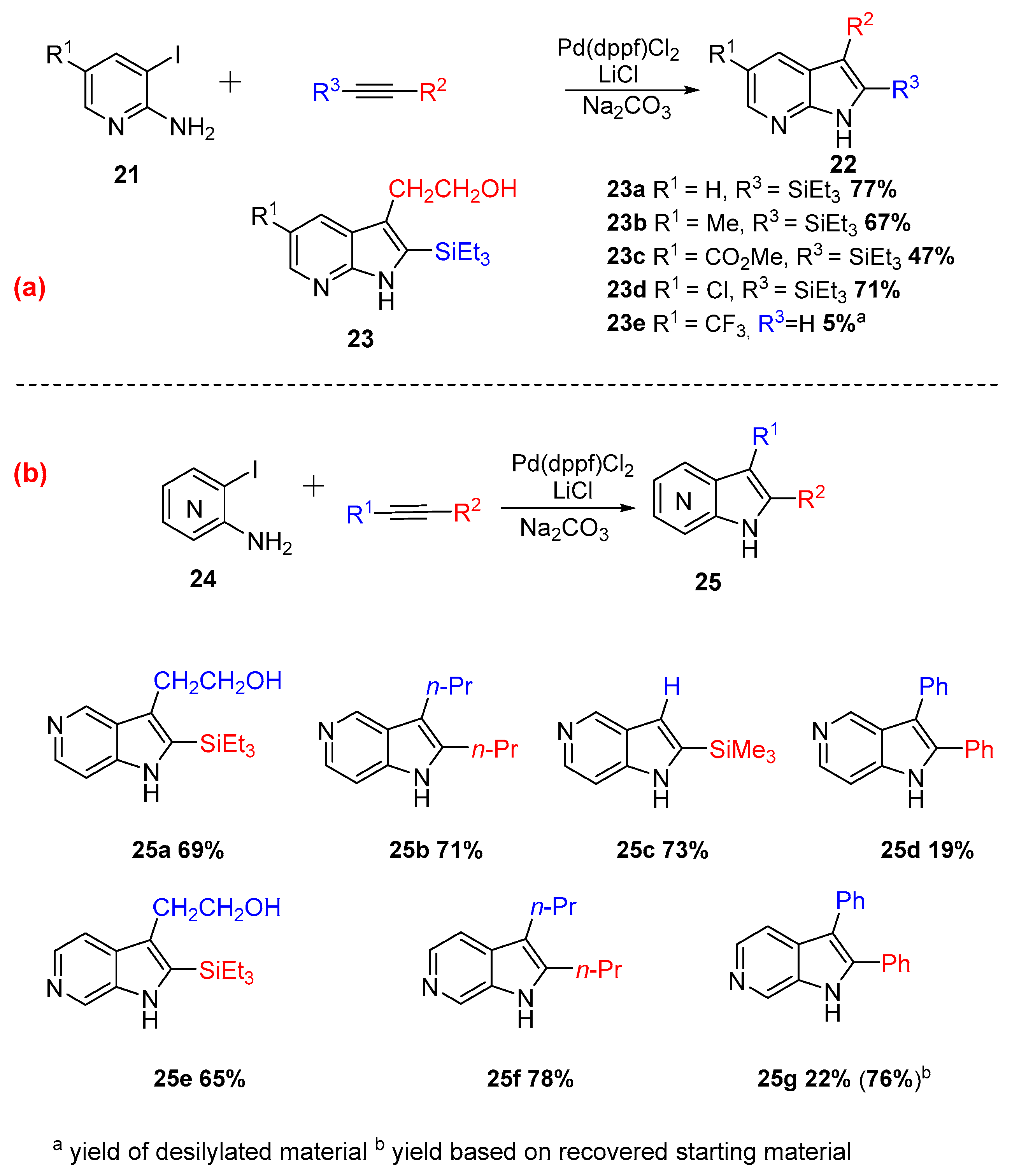
In 1998, Ujjainwalla et. al. pursued a methodology to access azaindoles substituted in the pyridine ring. This method gave access to 2,3,5-trisubstituted-7-azaindoles (Scheme 10a), 2,3-disubstituted-5-azaindoles (Scheme 10b), and 2,3-disubstituted-6-azaindoles (Scheme 10b) with very good yields (up to 77%). The catalytic system was changed to Pd(dppf)Cl2 rather than Pd(OAc)2, affording higher regioselectivity, reproducibility, and improved yield [12].
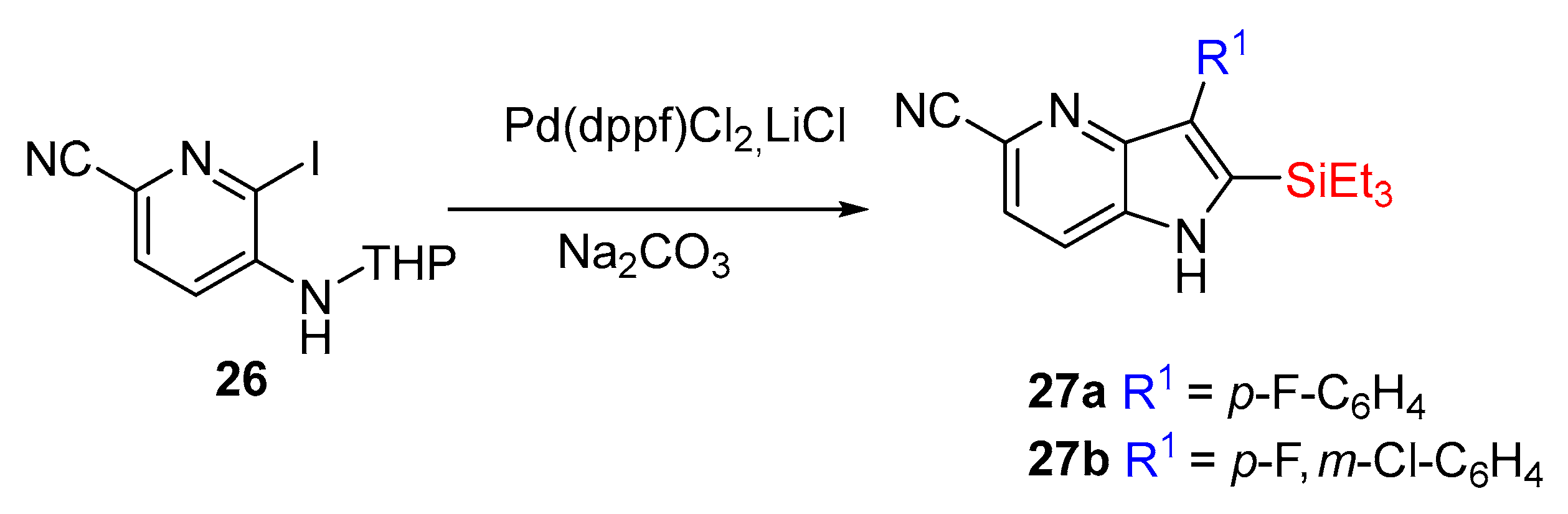
Inspired by these discoveries, H. Koolman et al. executed a complementary route employing Larock methodology to synthesize a tyrosine kinase inhibitor, a 4-azaindole core attached to a diaryl substitution in the C-2 and C-3 position of 27 (yields from 48 to 66%, over two steps). These products were not isolated since they were a part of an extensive synthesis (Scheme 11) [17].
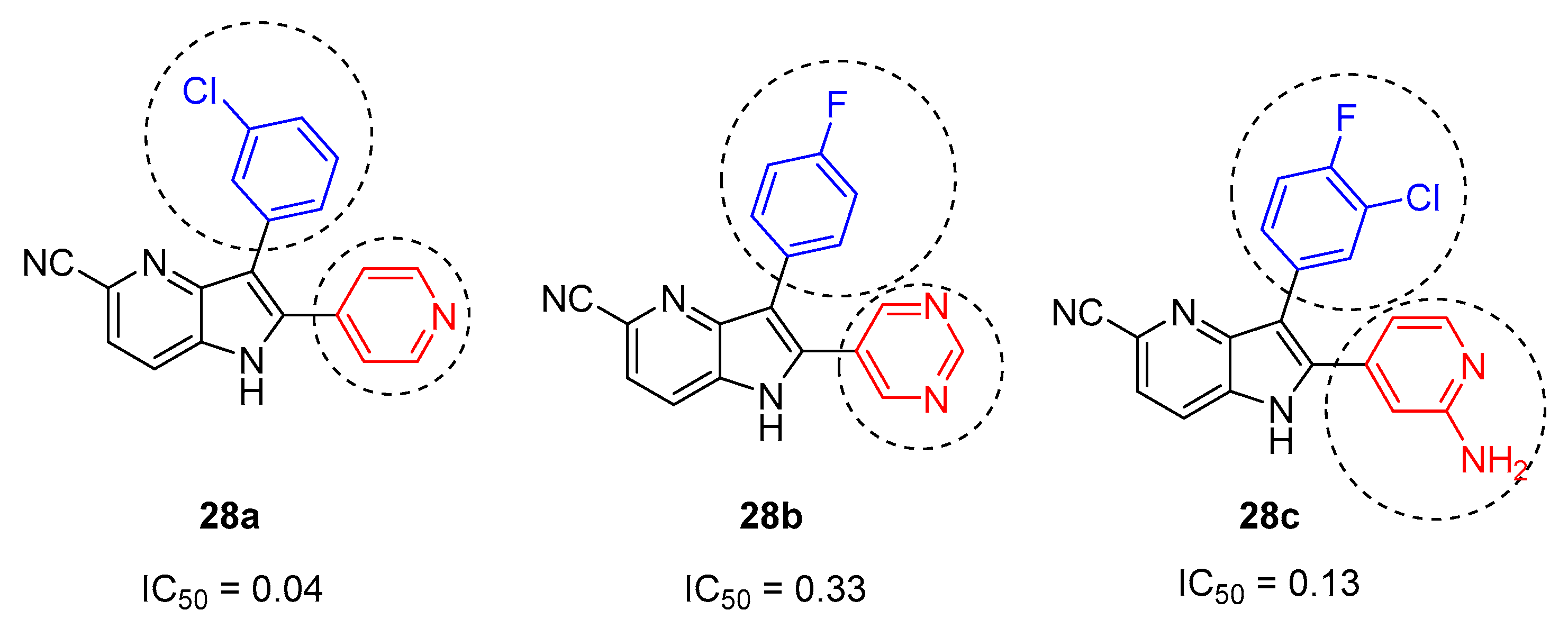
The objective of these studies was the synthesis of the compounds 28a, 28b, and 28c (Scheme 12) in order to measure their inhibitory activity of c-Met (tyrosine-protein kinase Met).
2.3. Heck Reaction
In 1999, the first synthesis of azaindoles via Heck reaction was demonstrated by Blache and coworkers [18]. This approach consisted of enamine formation in the presence of Pd(Ph3)4 and NaHCO3 in HMPA at 140 °C, consisting of a Hegedus–Mori–Heck reaction. However, the reaction only led to low yields and high amounts of recovered starting enamines from 2-aminopyridine.
Later, in 2004, Nazaré and coworkers reported a one-step palladium-catalyzed annulation procedure for the synthesis of substituted, polyfunctionalized 4- and 7-azaindoles 31, by reaction of amino ortho-chloropyridines 29 with a variety of pyruvic acid derivatives 30, under mild conditions (Scheme 13) [19]. The protocol involved an enamine formation followed by Heck reaction. The protocol consisted on the treatment of a functionalized 2-amino ortho-chloropyridine with 3 equiv of an acyclic ketone in the presence of Pd(Pt-Bu3)2, a base, and MgSO4 as a water scavenger. The method revealed to be applicable both for indoles and azaindoles, starting from the corresponding ortho-chloro anilines and amino ortho-chloropyridines, respectively.
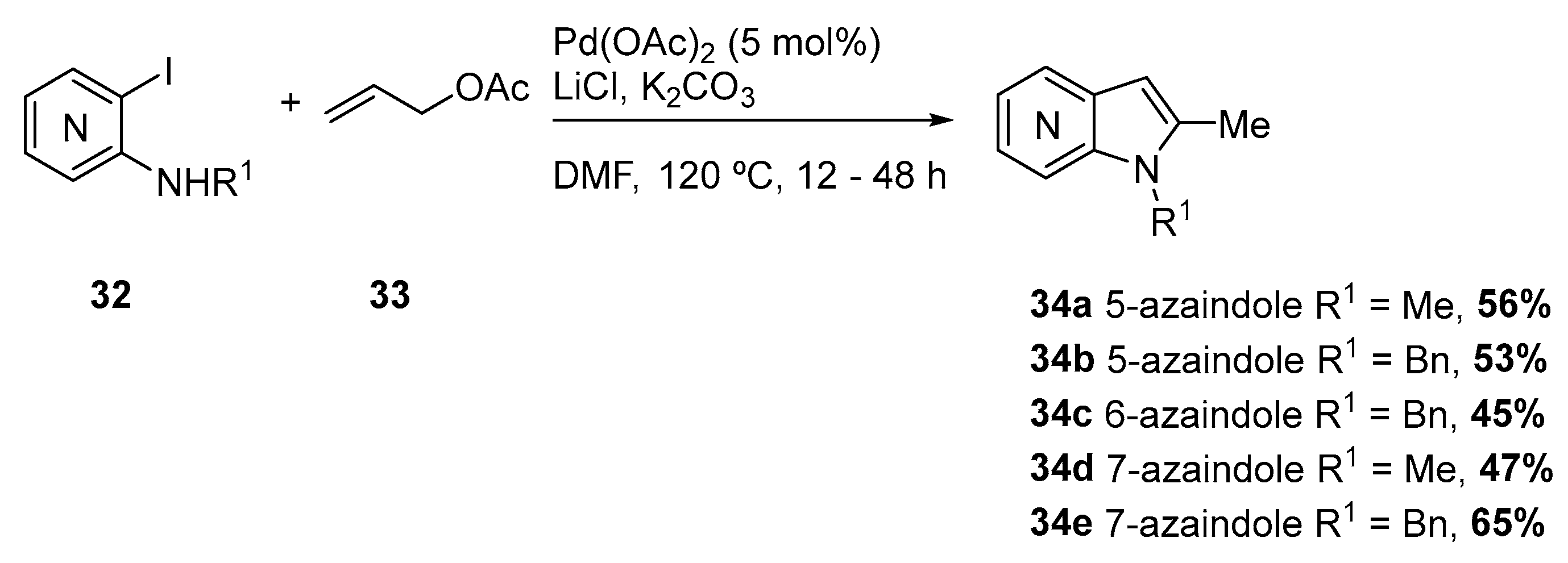
The synthesis of 2-methyl 5-, 6-, and 7-azaindoles (34) via palladium-catalyzed annulation was reported by Yum et al. that described the reaction of ortho-iodoarylamines (32) with allyl acetate under Pd(OAc)2 (5 mol %), LiCl (1 equiv), K2CO3 (3 equiv), allyl acetate (33) (and 2 equiv) in DMF at 120 °C [20]. The authors extended their protocol to other aromatic ring fused pyrrole derivatives with several ortho-iodoarylamines (32) with allyl acetate (33) under the optimized reaction conditions, such as indoles and pyrrolo-quinolines. Higher yields were obtained when N-protected substrates were used however, the azaindoles were obtained in moderate yields (Scheme 14).
The authors proposed that the mechanism of the reaction proceeds via formation of a π-allyl complex followed by intermolecular nucleophilic attack generating the pyrrole ring and regenerating Pd(0).
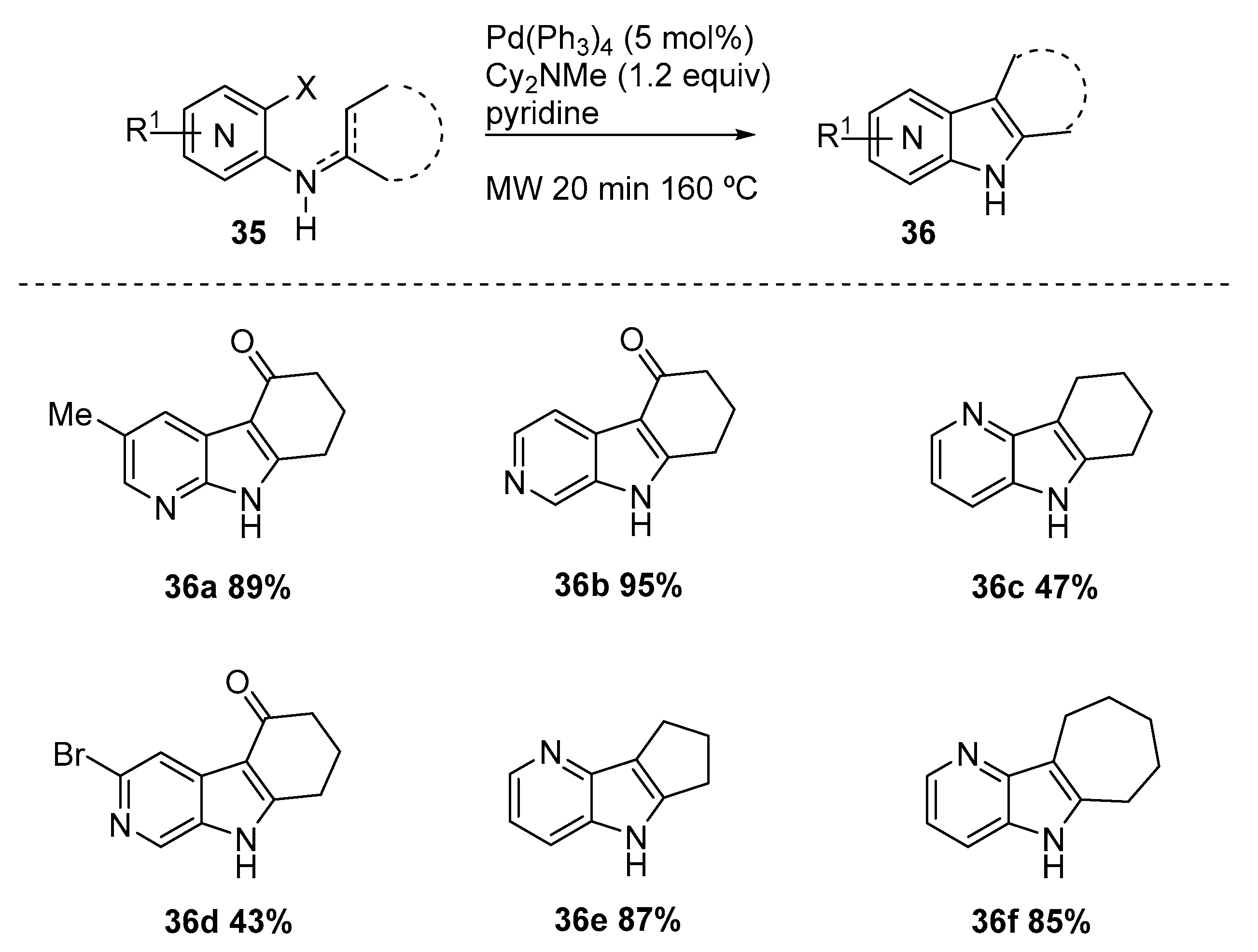
An intramolecular Heck reaction (Hegedus–Mori–Heck reaction) was reported for the preparation of several azaindoles by Lachance and coworkers [21]. The authors described the intramolecular Heck reaction of imines/enamines (35) under microwave conditions in the presence of Pd(Ph3)4 and Cy2NMe in pyridine, and obtained good yields of azaindoles (36) (Scheme 15). The protocol revealed to be compatible with the presence of bromine, ketone, and esters. All isomers of azaindoles were prepared directly from the corresponding amino-ortho-halogenated pyridines (halogen= iodo, bromo, and chloro) in the presence of a cyclic ketone or an acyclic aromatic ketone, in up to 80% yield. The use of microwave irradiation allowed to reduce the reaction time and promoted the palladium coupling.
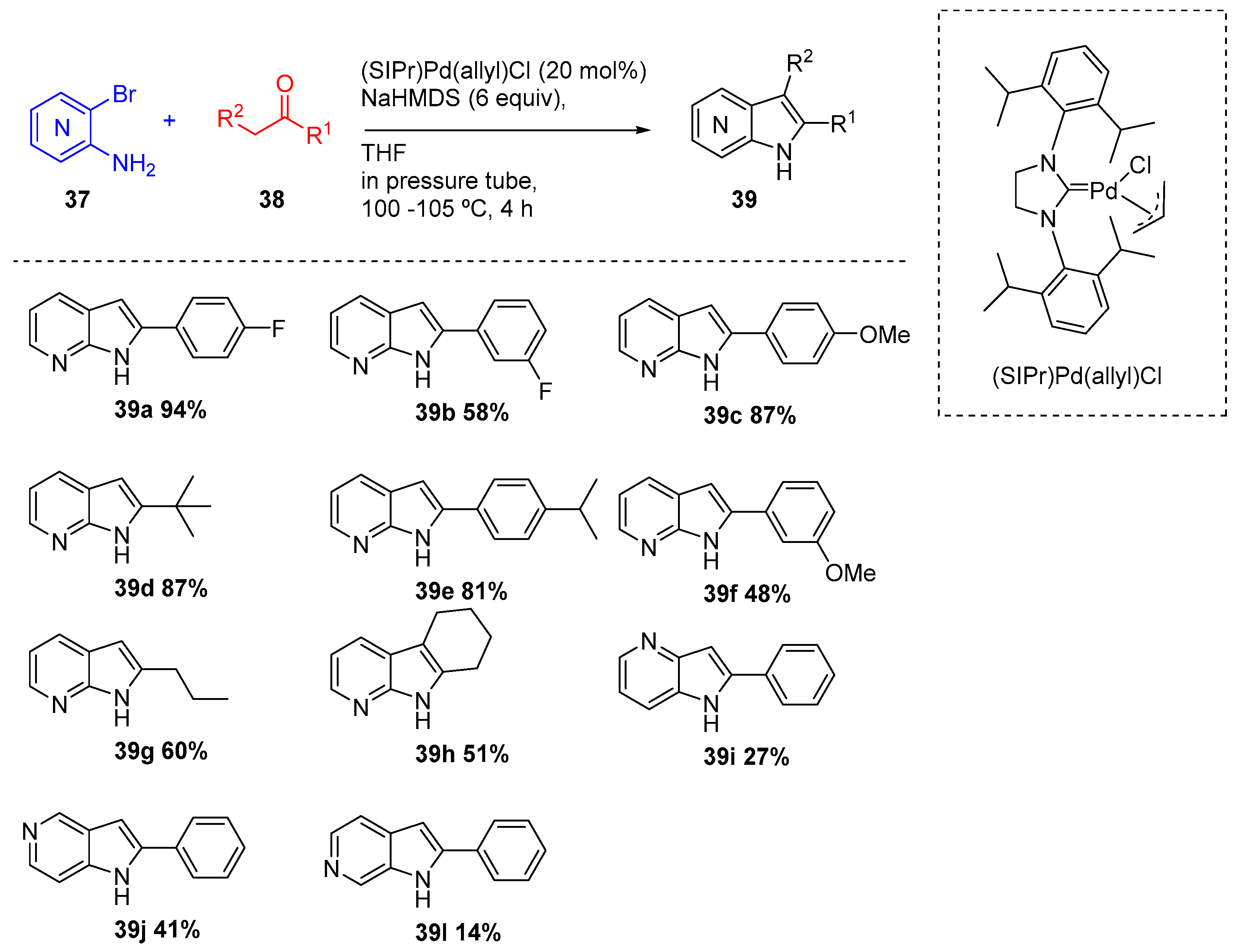
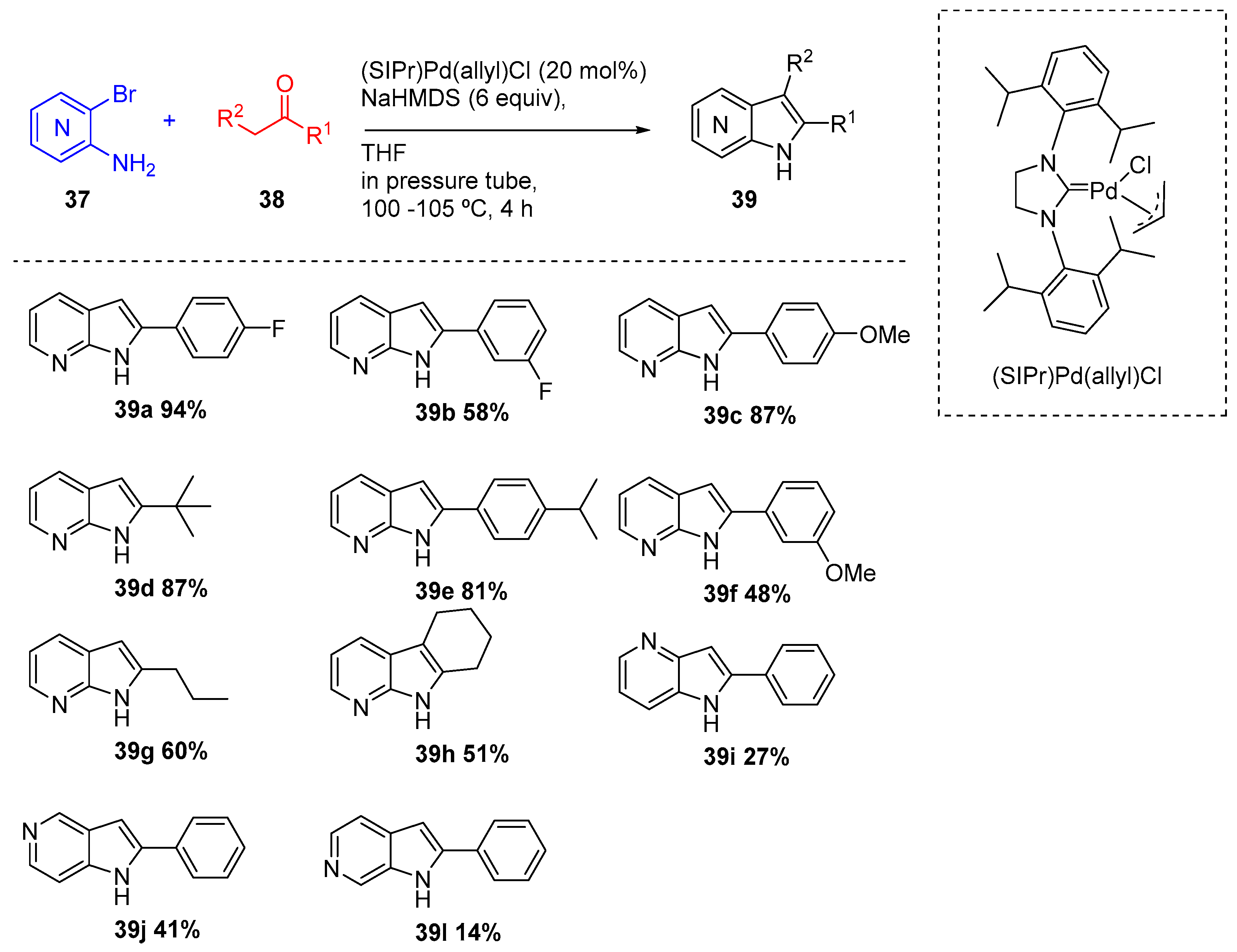
In 2010, Spergel and coworkers reported a one-pot method for the construction of a variety of azaindoles (39) using simple ketones (38) and amino-ortho-halopyridines (37) via a palladium-catalyzed α-heteroarylation of ketone enolates (Scheme 16) [22]. A N-heterocyclic carbene palladium catalyst, the (SIPr)Pd(allyl)Cl described by Nolan [23], was examined and revealed to be suitable to convert the amino-ortho-bromopyridines (37) in the corresponding azaindoles (39) in low to high yields. The method allowed preparation of a variety of azaindoles (all regioisomers) from unsymetric ketones.
2.4. Suzuki Coupling and Lautens Reaction
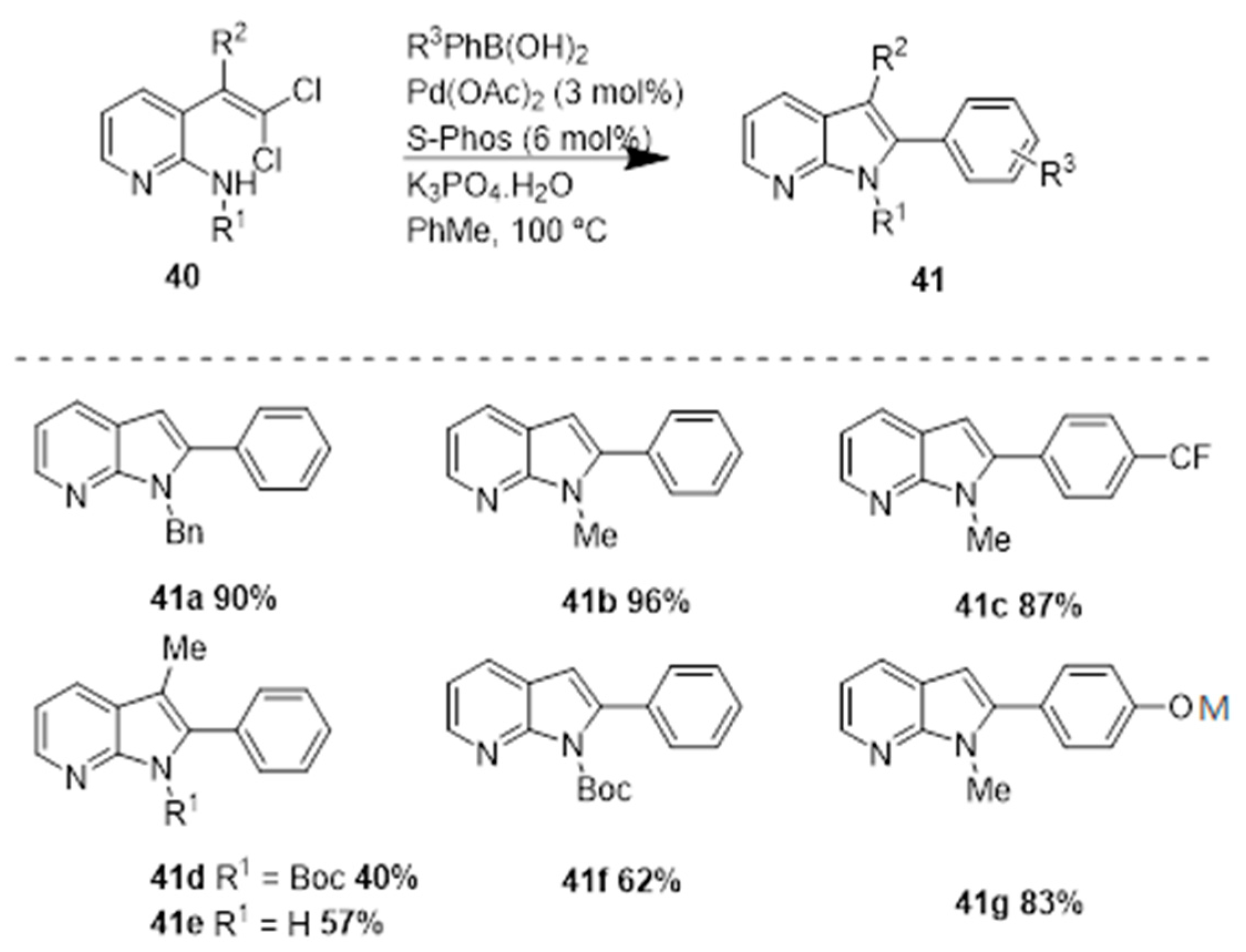
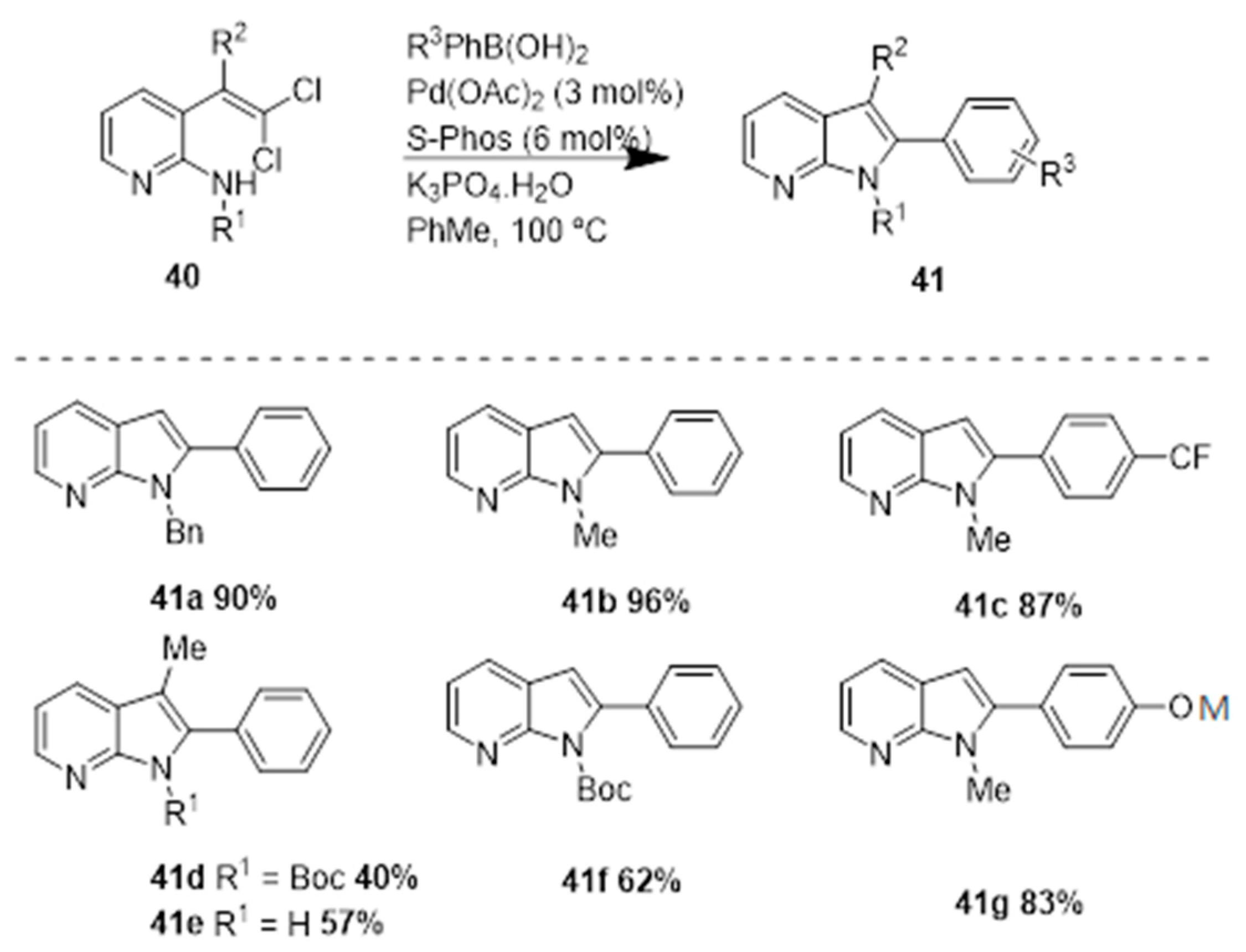
In 2007, Mark Lautens and coworkers [24] reported a palladium-catalyzed reaction of gem-dichloroolefins and a boronic acid via a tandem intramolecular C-N and intramolecular Suzuki coupling process. The group initiated their studies with indole synthesis via a tandem C-N/Suzuki coupling of gem-dibromovinylaniline. However, the authors did not observe 7-azaindole formation under these conditions when a dibromovinyl aminopyridine was used as substrate, presumably due to catalyst poisoning. To overcome this problem, the authors used N-protected substrates, that led to successful reactions; the best yields were obtained with N-alkyl substrates (N-Me), e.g., 41b, 41c, and 41g were obtained in high yields (Scheme 17).
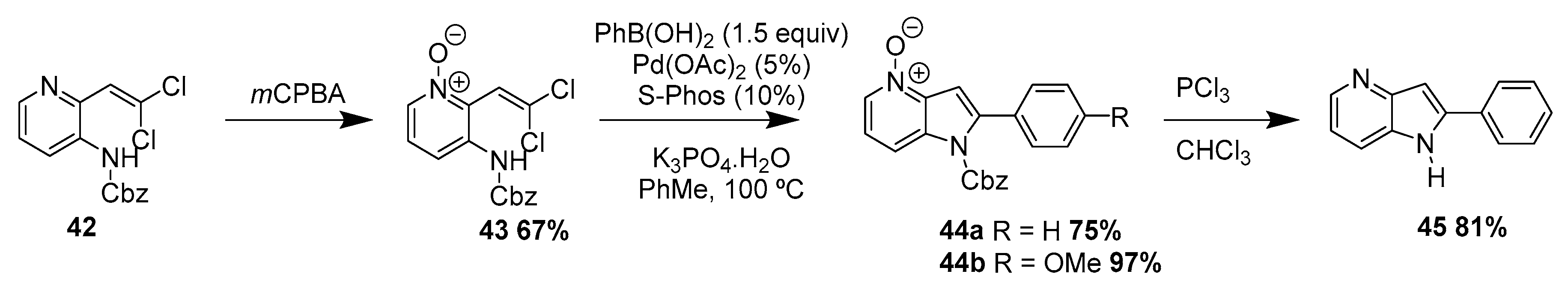
The authors also prepared 6-azaindoles using N-Boc protected substrates. Concerning the 5-azaindoles, the authors observed formation of the bis-Suzuki coupling product along with the desired product. Thus, in order to avoid the formation of this mixture the authors reverted the properties of the substrate by using the pyridyl group protected as a N-oxide. A complex mixture of products was also observed by the authors while preparing the 4-azaindoles, this might be due to the coordination of the pyridyl nitrogen that retards the C–N bond formation. The authors overcame this problem by protecting the pyridyl nitrogen as the N-oxide (43), and observed the formation of the corresponding products in good to high yields of 44a and 44b, respectively (Scheme 18). This approach added to more steps in the synthetic sequence, and the final azaindole (45c) was obtained in 81% yield. This method represents a very flexible protocol to access all four isomers of azaindole in good to excellent yield.
The Suzuki reaction was also applied to the construction of azaindoles by Swen Hoelder and coworkers [25]. The authors described an efficient two-step route, starting from chloroamino-N-heterocycles, to prepare a wide range of aza- and diazaindoles, avoiding the use of protecting groups. The method involved an optimized Suzuki–Miyaura coupling with (2-ethoxyvinyl)borolane using 3 mol % of SPhos/Pd(OAc)2 (2.5:1) as catalyst in refluxing MeCN/H2O (3:2) with K3PO4 as base, followed by acetic acid-catalyzed cyclization.
2.5. Cacchi Reaction
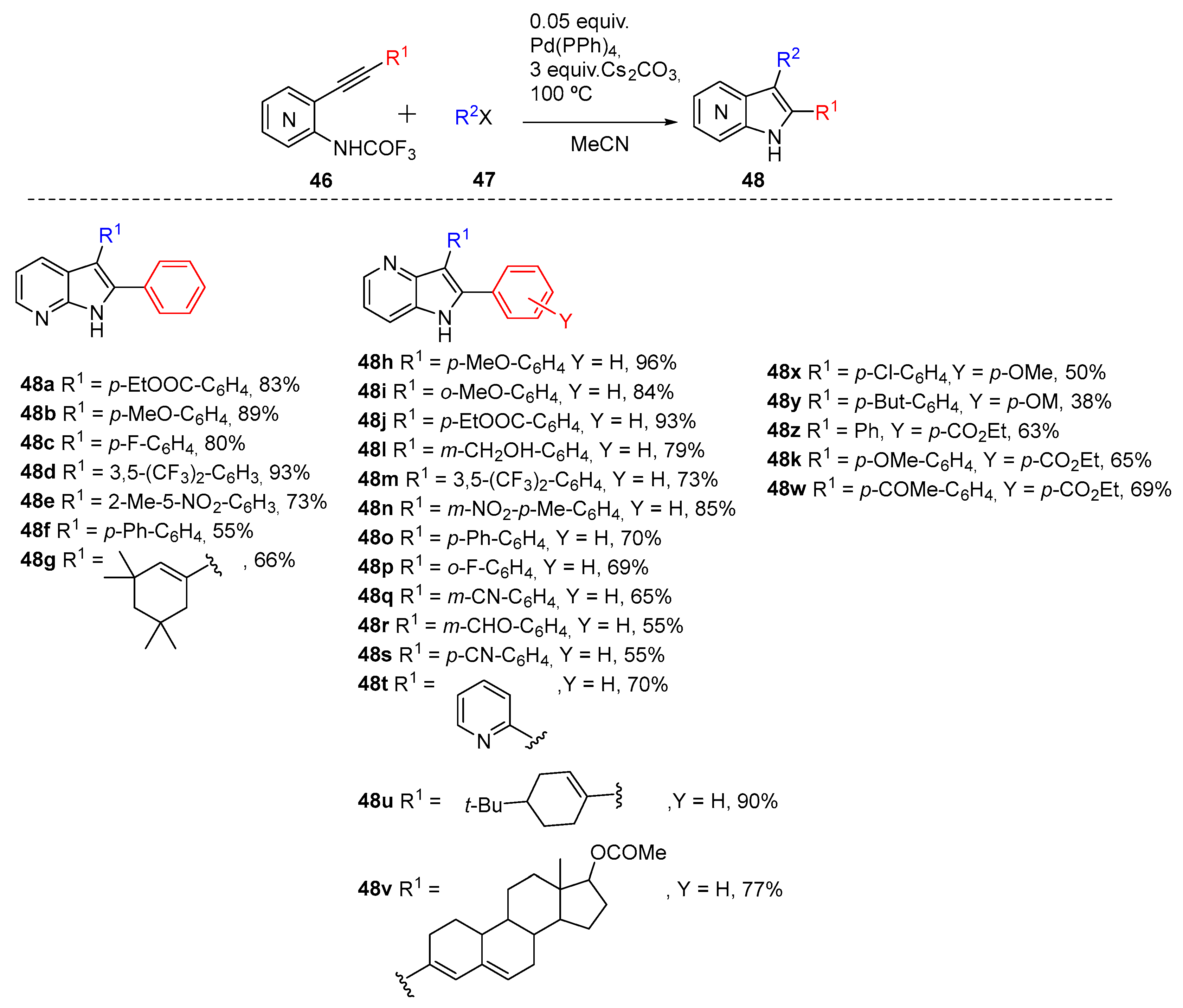
In 2005, Cacchi et al. adapted their protocol that accessed indoles to build the azaindole core [26]. The procedure describes an aminopalladation-reductive elimination protocol to develop a solution-phase synthesis of free N-H 2,3-disubstituted azaindoles. Previously the authors showed that the basicity of the nitrogen of the starting acetanilides plays a crucial role in the synthesis of indoles, via the aminopalladation-reductive elimination process [27]. Furthermore, the pyridine moiety plays a beneficial role in favoring the formation of the free N–H pyrrole ring, since when o-(phenylethynyl)acetanilide was subjected to the same cyclization conditions the starting alkyne was recovered in 98% yield. Therefore, they examined the use of the trifluoroacetamido derivative (46), which produced the corresponding azaindole (48) in 3.5 h under the same conditions, confirming the crucial role of the trifluoroacetyl group in this type of cyclization (Scheme 19).
2.6. C-H Activation Reaction
This reaction constitutes a great advance in metal-catalysis, however there are still some challenges, such as the regioselectivity [28] (C–H sp2 and sp3 bonds are ubiquous), the low reactivity due to a great energy barrier to break a C–H bond (104 kcal/mol) and at last the chemoselectivity [29,30].
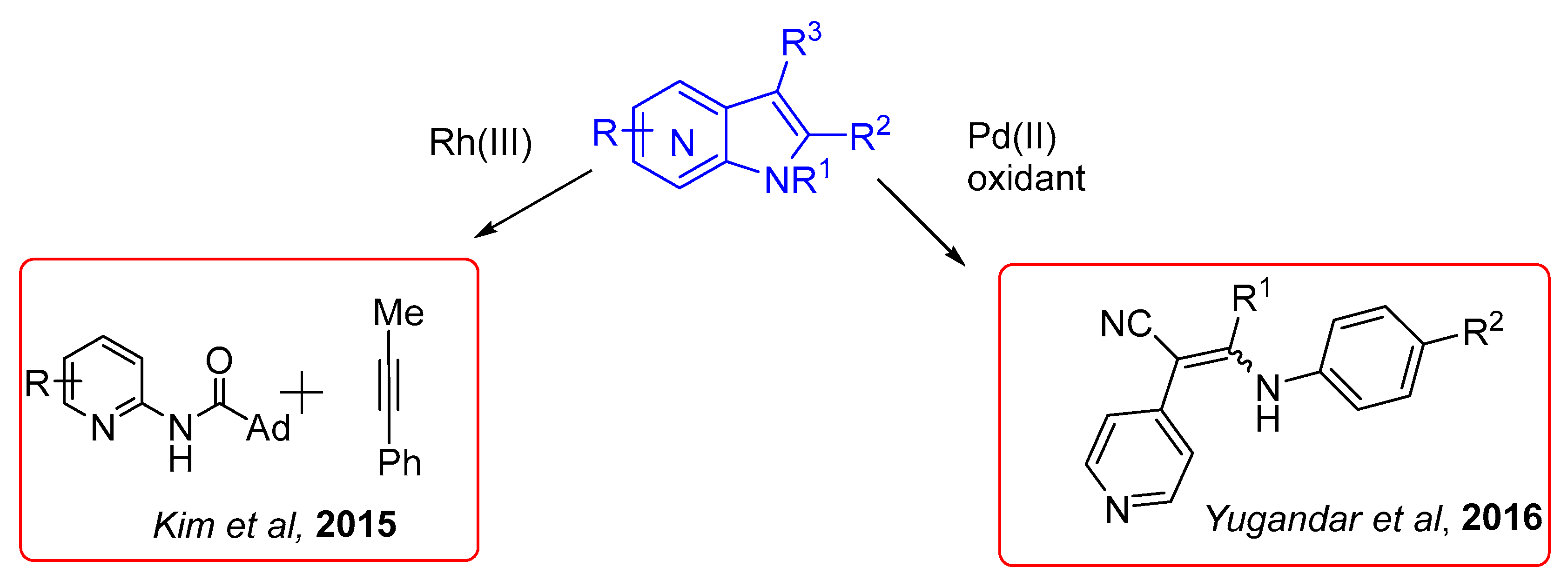
Regarding azaindole synthesis, there are still few reports on C–H activation reaction applied to afford these compounds. One of such approaches relies on the use of rhodium and palladium catalysis affording the 7-azaindole isomer (Scheme 20).
Organic synthesis has changed hugely mainly due to the introduction of metal-catalyzed reactions. However, these approaches require the presence of reactive functional groups in one of both coupling partners. Consequently, the reducing number of necessary functionalities in coupling reactions, like in the case of C–C coupling thorough C–H activation, emerged as an attractive alternative [31,32,33].
2.6.1. C–H via Rhodium Catalysis
The low reactivity of aminopyridines complicates the building up of the azaindole ring. These difficulties make substrate prefunctionalization crucial in synthetic methods such as Larock. These methods require the use of amino ortho-halogenated pyridines to access different substitution patterns.
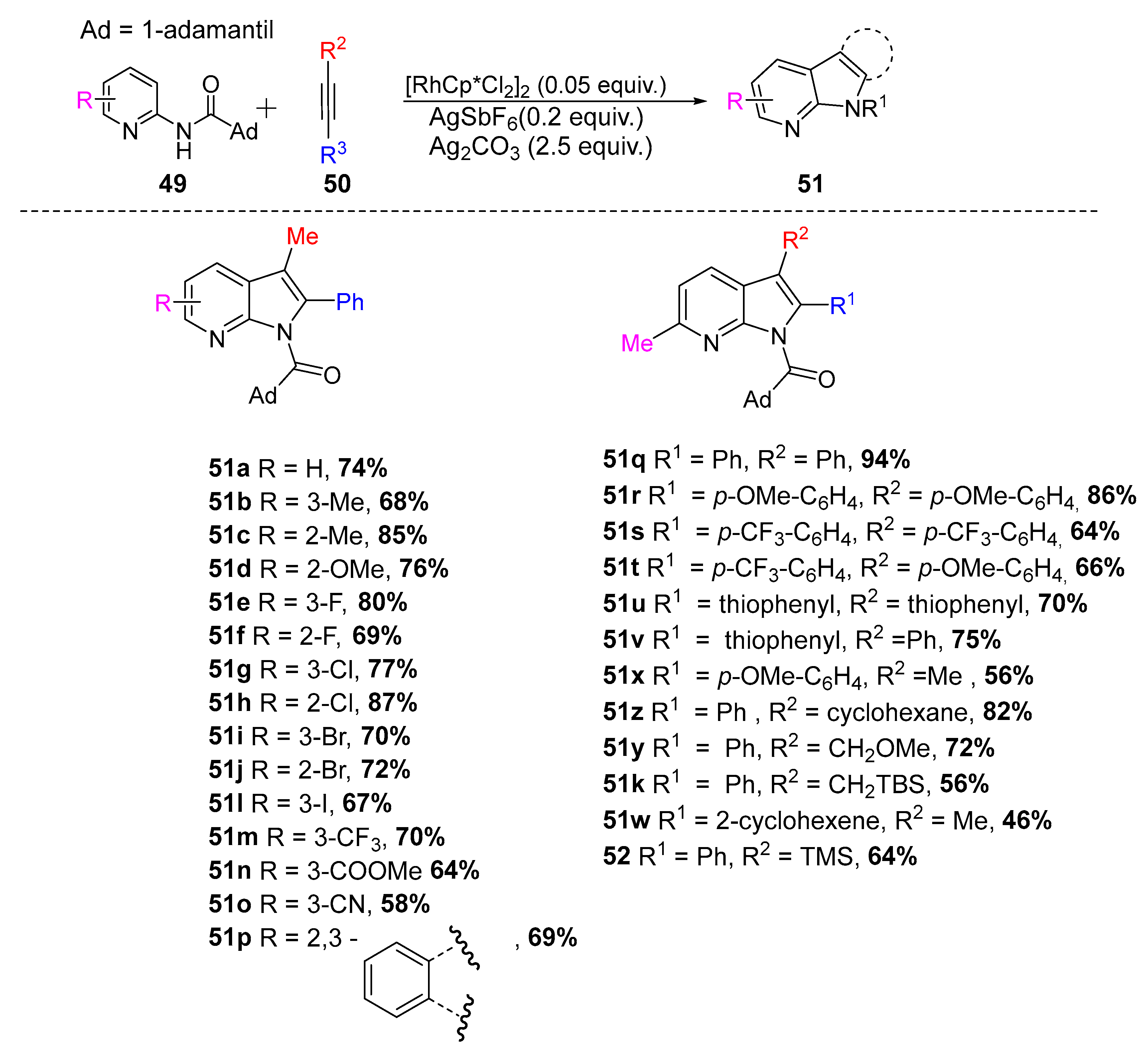
In 2015, Kim et al. created a strategy envisioning C–H activation with rhodium(III) catalysis. This approach was based in the use of aminopyridines and alkynes. Since the aminopyridine ring can be considered a Lewis base, a Lewis acid (Ag2CO3) was strategically used to coordinate with the N atom of the pyridine ring, facilitating the annulation process (Scheme 21) [34].
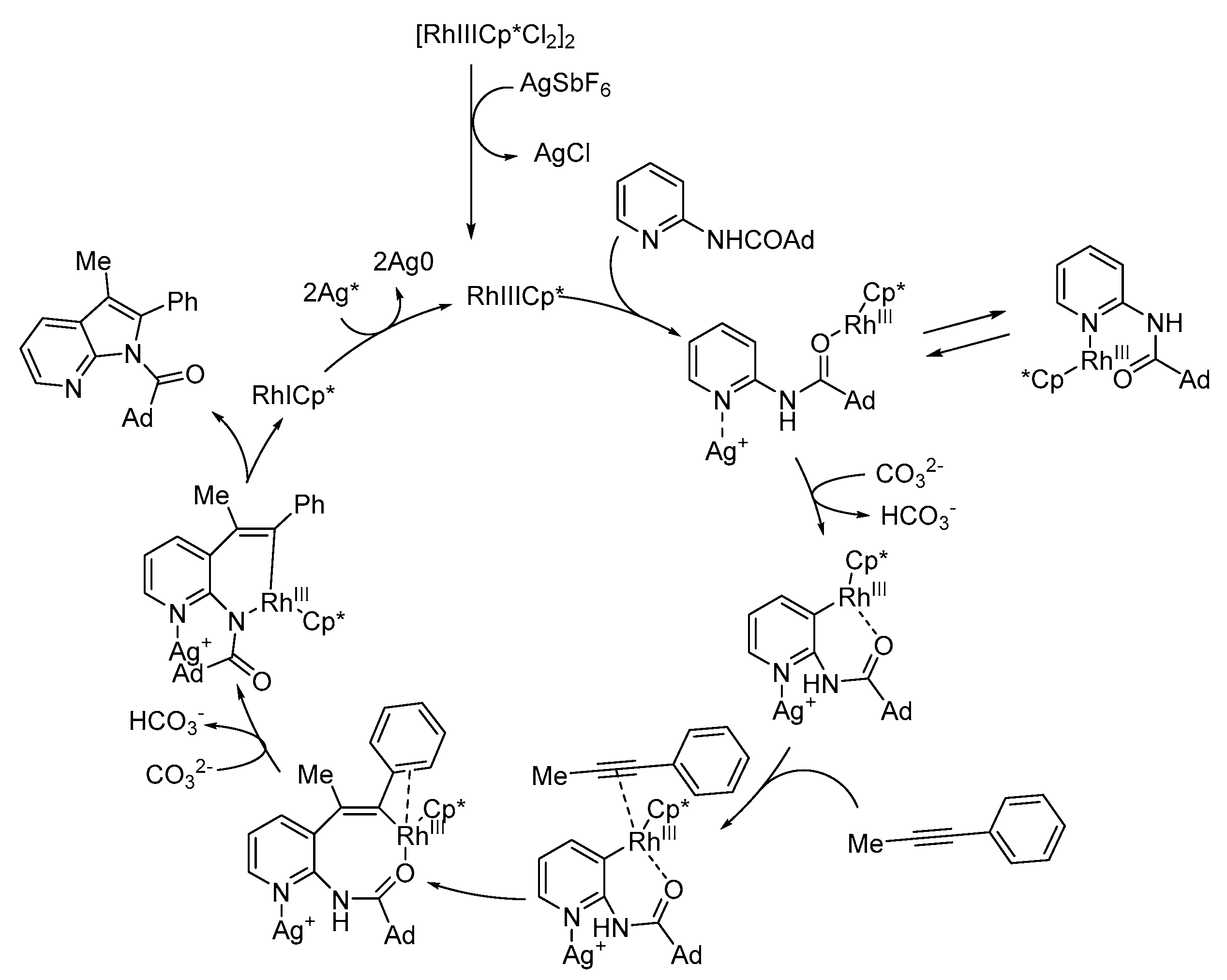
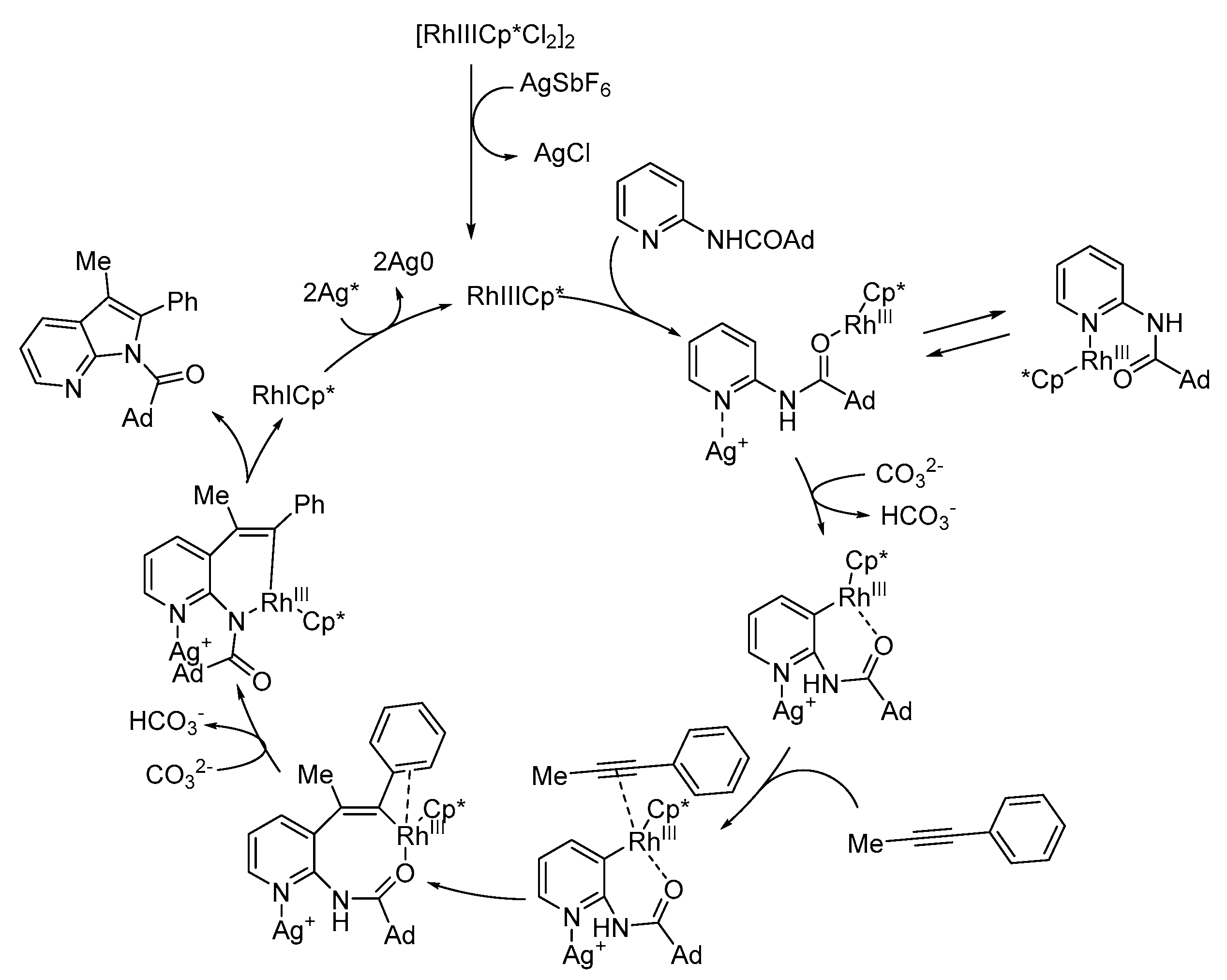
The authors proposed a plausible mechanism for the rhodium-catalyzed 7-azaindole synthesis, as depicted in Scheme 22. The authors propose that the silver ions coordinate to the pyridyl nitrogen atom and facilitate the C–H bond cleavage of the aminopyridine.
2.6.2. C-H Activation Reaction via Palladium Catalysis
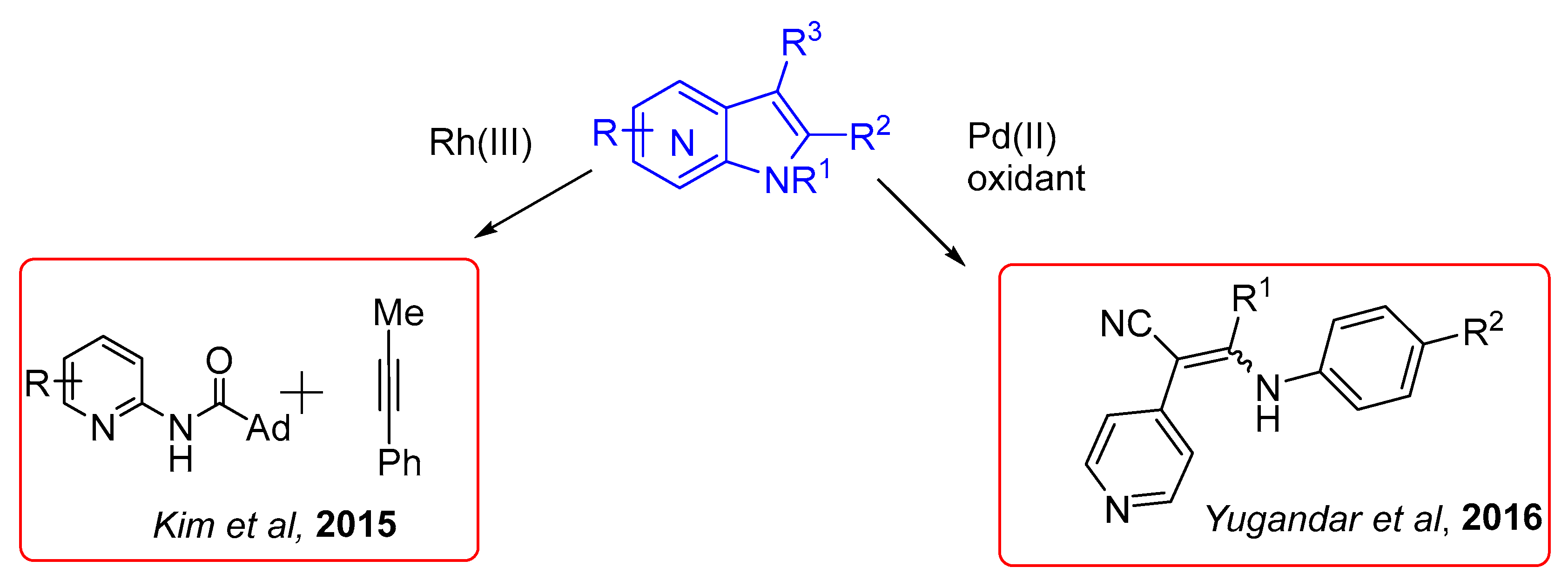
Recently, following Buchwald’s pioneering report of carbazole synthesis via Pd(II)-catalyzed intramolecular C–H activation/C–N bond formation, attention has been turned on the C–H functionalization reactions for the construction of various heterocyclic ring systems [35,36,37]. These methods have also been applied to the indole synthesis and intramolecular cross dehydrogenative coupling (CDC) has become a promising protocol for the synthesis of indoles from enamines and imines involving C3–C3a bond formation [38,39,40,41,42,43,44]. In continuation of these studies, Yugandar et al. disclosed an efficient route to 1-N-aryl/NH-2-(het)aryl/alkyl-3-cyano/aroylindoles and their heterofused analogs by palladium-catalyzed intramolecular oxidative C–H functionalization–amination of readily available 2,3-(het)aryl-3-N-aryl/acylenaminonitriles and enaminones (Scheme 23) [45].
This reaction displays high regioselectivity and good functional group tolerance at various positions of azaindole skeleton along with high yields in this cyclization reaction. The reaction represents one of the few examples, in which an aryl C–H bond is activated by an aminoaryl directing group, that subsequently acts as the reaction partner in the same process [46].
3. Conclusions
Azaindoles can be prepared from aminopyridines, similarly to the indole synthesis from anilines. The most recent synthetic approaches rely on the use of metal-catalyzed reactions. Metal-catalyzed cross-coupling reactions, which constitute a very modern and emergent topic in organic synthesis, can be highly useful for the construction and derivation of these aminopyridine-containing heterocycles. However, the challenging nature of aminopyridines difficults the application of metal-catalyzed reactions. Despite this, great advances have been achieved on the use of metal-catalyzed reactions to prepare and functionalyse azaindoles. The main difficulty is to develop a procedure that is wide in scope, allowing access to all isomers; is high yielding; uses mild conditions; and avoids the use of protecting groups, especially at the amino group.
So far several synthetic routes have been reported for the preparation of azaindoles from aminopyridines, including Sonogashira, Larock-type, Heck, Lautens, Suzuki coupling, Cacchi, and more recently C–H activation. These methods constitute a major advance in the development of new synthetic methods to attain azaindoles.
It is expected that, in a near future, new approaches consisting of one-pot protocols involving metal-catalyzed reactions will emerge to improve the synthesis of functionalized azaindoles having a high impact in industry.
Author Contributions
All the authors contributed to this overview.
Funding
We thank the FC&T for fellowship SFRH/BD/1428767/2018. This work was supported by the Associated Laboratory for Sustainable Chemistry-Clean Processes and Technologies- LAQV which is financed by national funds from FCT/MEC (UID/QUI/50006/2013) and cofinanced by the ERDF under the PT2020 Partnership Agreement (POCI-01-0145-FEDER-007265). The NMR spectrometers are part of The National NMR Facility, supported by FC&T (RECI/BBB-BQB/0230/2012).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mérour, J.Y.; Buron, F.; Plé, K.; Bonnet, P.; Routier, S. The azaindole framework in the design of kinase inhibitors. Molecules 2014, 19, 19935–19979. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Reeves, J.T.; Gallou, F.; Tan, Z.; Yee, N.K.; Senanayake, C.H. Organometallic methods for the synthesis and functionalization of azaindoles. Chem. Soc. Rev. 2007, 36, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Pearson, S.E.; Nandan, S. A practical, efficient synthesis of 5-amino-7-azaindole. Synthesis (Stuttg) 2005, 2503–2506. [Google Scholar] [CrossRef]
- Naud, S.; Westwood, I.M.; Faisal, A.; Sheldrake, P.; Bavetsias, V.; Atrash, B.; Cheung, K.M.J.; Liu, M.; Hayes, A.; Schmitt, J.; et al. Structure-based design of orally bioavailable 1 h -Pyrrolo[3,2-c]pyridine inhibitors of mitotic kinase monopolar spindle 1 (MPS1). J. Med. Chem. 2013, 56, 10045–10065. [Google Scholar] [CrossRef] [PubMed]
- Leboho, T.C.; Van Vuuren, S.F.; Michael, J.P.; De Koning, C.B. The acid-catalysed synthesis of 7-azaindoles from 3-alkynyl-2- aminopyridines and their antimicrobial activity. Org. Biomol. Chem. 2014, 12, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Leboho, T.C.; Giri, S.; Popova, I.; Cock, I.; Michael, J.P.; De Koning, C.B. Double Sonogashira reactions on dihalogenated aminopyridines for the assembly of an array of 7-azaindoles bearing triazole and quinoxaline substituents at C-5: Inhibitory bioactivity against Giardia duodenalis trophozoites. Bioorg. Med. Chem. 2015, 23, 4943–4951. [Google Scholar] [CrossRef] [PubMed]
- Purificação, S.I.; Pires, M.J.D.; Rippel, R.; Santos, A.S.; Marques, M.M.B. One-Pot Synthesis of 1,2-Disubstituted 4-, 5-, 6-, and 7-Azaindoles from Amino- o -halopyridines via N-Arylation/Sonogashira/Cyclization Reaction. Org. Lett. 2017, 19, 5118–5121. [Google Scholar] [CrossRef] [PubMed]
- Dority, J.A.; Bacon, E.R.; Lesher, G.Y.; Kumar, V.; Singh, B. Synthesis of 7-Azaindole and 7-Azaoxindole Derivatives through a Palladium-Catalyzed Cross-Coupling Reaction. J. Org. Chem. 1992, 57, 6995–6998. [Google Scholar] [CrossRef]
- Xu, L.; Lewis, I.R.; Davidsen, S.K.; Summers, J.B. Transition metal catalyzed synthesis of 5-azaindoles. Tetrahedron Lett. 1998, 39, 5159–5162. [Google Scholar] [CrossRef]
- Trejo, A.; Arzeno, H.; Browner, M.; Chanda, S.; Cheng, S.; Comer, D.D.; Dalrymple, S.A.; Dunten, P.; Lafargue, J.A.; Lovejoy, B.; et al. Design and Synthesis of 4-Azaindoles as Inhibitors of p38 MAP Kinase. J. Med. Chem. 2003, 46, 4702–4713. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.M.; Choi, J.K.; Yum, E.K.; Chi, D.Y. Palladium-catalyzed functionalization of 5- and 7-azaindoles. Tetrahedron Lett. 2000, 41, 919–922. [Google Scholar] [CrossRef]
- Ujjainwalla, F.; Warner, D. Synthesis of 5-, 6- and 7-azaindoles via palladium-catalyzed heteroannulation of internal alkynes. Tetrahedron Lett. 1998, 39, 5355–5358. [Google Scholar] [CrossRef]
- Park, S.S.; Choi, J.K.; Yum, E.K.; Ha, D.C. A facile synthesis of 2,3-disubstituted pyrrolo[2,3-b]pyridines via palladium-catalyzed heteroannulation with internal alkynes. Tetrahedron Lett. 1998, 39, 627–630. [Google Scholar] [CrossRef]
- Wensbo, D.; Eriksson, A.; Jeschke, T.; Annby, U.; Gronowitz, S.; Cohen, L.A. Palladium-catalysed synthesis of heterocondensed pyrroles. Tetrahedron Lett. 1993, 34, 2823–2826. [Google Scholar] [CrossRef]
- Larock, R.C.; Yum, E.K.; Refvik, M.D. Synthesis of 2,3-disubstituted indoles via palladium-catalyzed annulation of internal alkynes. J. Org. Chem. 1998, 63, 7652–7662. [Google Scholar] [CrossRef]
- Larock, R.C.; Yum, E.K. Synthesis of Indoles via Palladium-Catalyzed Heteroannulation of Internal Alkynes. J. Am. Chem. Soc. 1991, 113, 6689–6690. [Google Scholar] [CrossRef]
- Koolman, H.; Heinrich, T.; Böttcher, H.; Rautenberg, W.; Reggelin, M. Syntheses of novel 2,3-diaryl-substituted 5-cyano-4-azaindoles exhibiting c-Met inhibition activity. Bioorg. Med. Chem. Lett. 2009, 19, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Blache, Y.; Sinibaldi-Troin, M.E.; Hichour, M.; Benezech, V.; Chavignon, O.; Gramain, J.C.; Teulade, J.C.; Chapat, J.P. Heterocyclic enaminones: Photochemical synthesis of 6,7,8,9-tetrahydro- 5H-pyrido[2,3-b]indol-9-ones. Tetrahedron 1999, 55, 1959–1970. [Google Scholar] [CrossRef]
- Nazaré, M.; Schneider, C.; Lindenschmidt, A.; Will, D.W. A flexible, palladium-catalyzed indole and azaindole synthesis by direct annulation of chloroanilines and chloroaminopyridines with ketones. Angew. Chem. Int. Ed. 2004, 43, 4526–4528. [Google Scholar] [CrossRef] [PubMed]
- Kgun Yum, E.; Sung Hong, C.; Yong Seo, J.; Sung, N.-D. Synthesis of Aromatic Ring Fused Pyrrole Derivatives by Palladium-catalyzed Annulation of o-Iodoarylamines with Allyl Acetate. Heterocycles 2004, 63, 631. [Google Scholar] [CrossRef]
- Lachance, N.; April, M.; Joly, M.-A. Rapid and Efficient Microwave-Assisted Synthesis of 4-, 5-, 6- and 7-Azaindoles. Synthesis (Stuttg) 2005, 2005, 2571–2577. [Google Scholar] [CrossRef]
- Spergel, S.H.; Okoro, D.R.; Pitts, W. One-pot synthesis of azaindoles via palladium-catalyzed α-heteroarylation of ketone enolates. J. Org. Chem. 2010, 75, 5316–5319. [Google Scholar] [CrossRef] [PubMed]
- Viciu, M.S.; Germaneau, R.F.; Nolan, S.P. Well-Defined, Air-Stable (NHC)Pd(Allyl)Cl (NHC = N-Heterocyclic Carbene) Catalysts for the Arylation of Ketones. Org. Lett. 2002, 4, 4053–4056. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.Q.; Yuen, J.; Lautens, M. A general modular method of azaindole and thienopyrrole synthesis via Pd-catalyzed tandem couplings of gem-dichloroolefins. J. Org. Chem. 2007, 72, 5152–5160. [Google Scholar] [CrossRef] [PubMed]
- Whelligan, D.K.; Thomson, D.W.; Taylor, D.; Hoelder, S. Two-step synthesis of aza- and diazaindoles from chloroamino-N-heterocycles using ethoxyvinylborolane. J. Org. Chem. 2010, 75, 11–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacchi, S.; Fabrizi, G.; Parisi, L.M. The aminopalladation-reductive elimination process as a tool for the solution-phase synthesis of 2,3-disubstituted azaindole libraries. J. Comb. Chem. 2005, 7, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Antonio, A.; Sandro, C.; Fabio, M. A versatile approach to 2,3-disubstituted indoles through the palladium-catalysed cyclization of o-alkynyltrifluoroacetanilides with vinyl triflates and aryl halides. Tetrahedron Lett. 1992, 33, 3915–3918. [Google Scholar] [CrossRef]
- Shul’Pin, G.B. Selectivity enhancement in functionalization of C-H bonds: A review. Org. Biomol. Chem. 2010, 8, 4217–4228. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.S.; Goldberg, K.I. Organometallic C-H Bond Activation: An Introduction. In Activation and Functionalization of C-H Bonds; American Chemical Society: Washington, DC, USA, 2004; pp. 1–43. ISBN 0097-6156. [Google Scholar]
- Dantignana, V.; Milan, M.; Cussó, O.; Company, A.; Bietti, M.; Costas, M. Chemoselective Aliphatic C-H Bond Oxidation Enabled by Polarity Reversal. ACS Cent. Sci. 2017, 3, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Kitamura, T.; Fujiwara, Y. Catalytic functionalization of arenes and alkanes via C-H bond activation. Acc. Chem. Res. 2001, 34, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Dyker, G. Palladium-Catalyzed C-H Activation of tert-Butyl Groups: A Simple Synthesis of 1,2-Dihydrocyclobutabenzene Derivatives. Angew. Chem. Int. Ed. Engl. 1994, 33, 103–105. [Google Scholar] [CrossRef]
- Ritleng, V.; Sirlin, C.; Pfeffer, M. Ru-, Rh-, and Pd-catalyzed C-C bond formation involving C-H activation and addition on unsaturated substrates: Reactions and mechanistic aspects. Chem. Rev. 2002, 102, 1731–1769. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Hong, S. Rh(III)-catalyzed 7-azaindole synthesis via C-H activation/annulative coupling of aminopyridines with alkynes. Chem. Commun. 2015, 51, 11202–11205. [Google Scholar] [CrossRef] [PubMed]
- Stokes, B.J.; Driver, T.G. Transition metal-catalyzed formation of N-heterocycles via aryl-or vinyl C-H bond amination. Eur. J. Org. Chem. 2011, 2011, 4071–4088. [Google Scholar] [CrossRef]
- Kim, H.; Chang, S. Transition-Metal-Mediated Direct C-H Amination of Hydrocarbons with Amine Reactants: The Most Desirable but Challenging C-N Bond-Formation Approach. ACS Catal. 2016, 6, 2341–2351. [Google Scholar] [CrossRef]
- Zhang, M. Construction of heterocycle scaffolds via transition metal-catalyzed sp2 C-H functionalization. Adv. Synth. Catal. 2009, 351, 2243–2270. [Google Scholar] [CrossRef]
- Li, C.J. Cross-dehydrogenative coupling (CDC): Exploring C-C bond formations beyond functional group transformations. Acc. Chem. Res. 2009, 42, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhao, L.; Li, C.J. Palladium-catalyzed direct oxidative Heck-Cassar-Sonogashira type alkynylation of indoles with alkynes under oxygen. Chem. Commun. 2010, 46, 4184–4186. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, X.; Glorius, F. Palladium-catalyzed selective dehydrogenative cross-couplings of heteroarenes. Angew. Chem. Int. Ed. 2011, 50, 7479–7481. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Hua, R. Rhodium(III)-catalyzed c-h activation and indole synthesis with hydrazone as an auto-formed and auto-cleavable directing group. Chem. A Eur. J. 2014, 20, 2352–2356. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Han, J.; Chen, J.; Deng, H.; Shao, M.; Zhang, H.; Cao, W. Mild and Efficient One-Pot Synthesis of 2-(Perfluoroalkyl)indoles by Means of Sequential Michael-Type Addition and Pd(II)-Catalyzed Cross-Dehydrogenative Coupling (CDC) Reaction. Org. Lett. 2015, 17, 3283–3285. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.B.; Zhang, J.L.; Liu, Y.Y.; Liu, B.; Yang, X.H.; Li, J.H. Metal-free nitrative cyclization of N-aryl imines with tert-butyl nitrite: Dehydrogenative access to 3-nitroindoles. Chem. Commun. 2015, 51, 1886–1888. [Google Scholar] [CrossRef] [PubMed]
- Vijay Kumar, S.; Saraiah, B.; Parameshwarappa, G.; Ila, H.; Verma, G.K. Synthesis of N-functionalized/NH-multisubstituted indoles, thienopyrroles, pyrroloindoles, and pyrazolopyrroles via sequential one-pot base-mediated and copper-catalyzed inter- and intramolecular amination of 2-[2-bromo(het)aryl]-3-(het)aryl-3-(methylthio. J. Org. Chem. 2014, 79, 7961–7978. [Google Scholar] [CrossRef] [PubMed]
- Haffemayer, B.; Gulias, M.; Gaunt, M.J. Amine directed Pd(II)-catalyzed C-H bond functionalization under ambient conditions. Chem. Sci. 2011, 2, 312–315. [Google Scholar] [CrossRef]
Scheme 1.
Metal-catalyzed methods reported for the construction of the azaindole core.
Scheme 2.
Sonogashira mediated synthesis of 5-nitro-7-azaindole [3].
Scheme 2.
Sonogashira mediated synthesis of 5-nitro-7-azaindole [3].
Scheme 3.
Synthesis of 5-azaindole derivatives as promising mitotic kinase monopolar spindle 1 inhibitors [4].
Scheme 3.
Synthesis of 5-azaindole derivatives as promising mitotic kinase monopolar spindle 1 inhibitors [4].
Scheme 4.
Acid-catalyzed synthesis of 7-azaindoles 8 using amino-halopyridines and amino-dihalopyridines [5].
Scheme 4.
Acid-catalyzed synthesis of 7-azaindoles 8 using amino-halopyridines and amino-dihalopyridines [5].
Scheme 5.
Synthesis of azaindoles 12 using double Sonogashira coupling starting from 3,5-diiodoaminopyridine 9 [6].
Scheme 5.
Synthesis of azaindoles 12 using double Sonogashira coupling starting from 3,5-diiodoaminopyridine 9 [6].
Scheme 6.
Double Sonogashira using 5-iodo-3-iodoaminopyridine (13) [6].
Scheme 6.
Double Sonogashira using 5-iodo-3-iodoaminopyridine (13) [6].
Scheme 7.
General scheme for the one-pot synthesis of 1,2-disubstituted 4-,5-,6- and 7-azaindoles 17 [7].
Scheme 7.
General scheme for the one-pot synthesis of 1,2-disubstituted 4-,5-,6- and 7-azaindoles 17 [7].
Scheme 8.
Scope of the one-pot N-arylation/Sonogashira/cyclization [7]. a Reaction carried stepwise. b Reaction carried out at 1 mmol scale.
Scheme 8.
Scope of the one-pot N-arylation/Sonogashira/cyclization [7]. a Reaction carried stepwise. b Reaction carried out at 1 mmol scale.

Scheme 9.
Pd-Catalyzed reactions of aryl iodides with propargyl alcohols [14].
Scheme 9.
Pd-Catalyzed reactions of aryl iodides with propargyl alcohols [14].
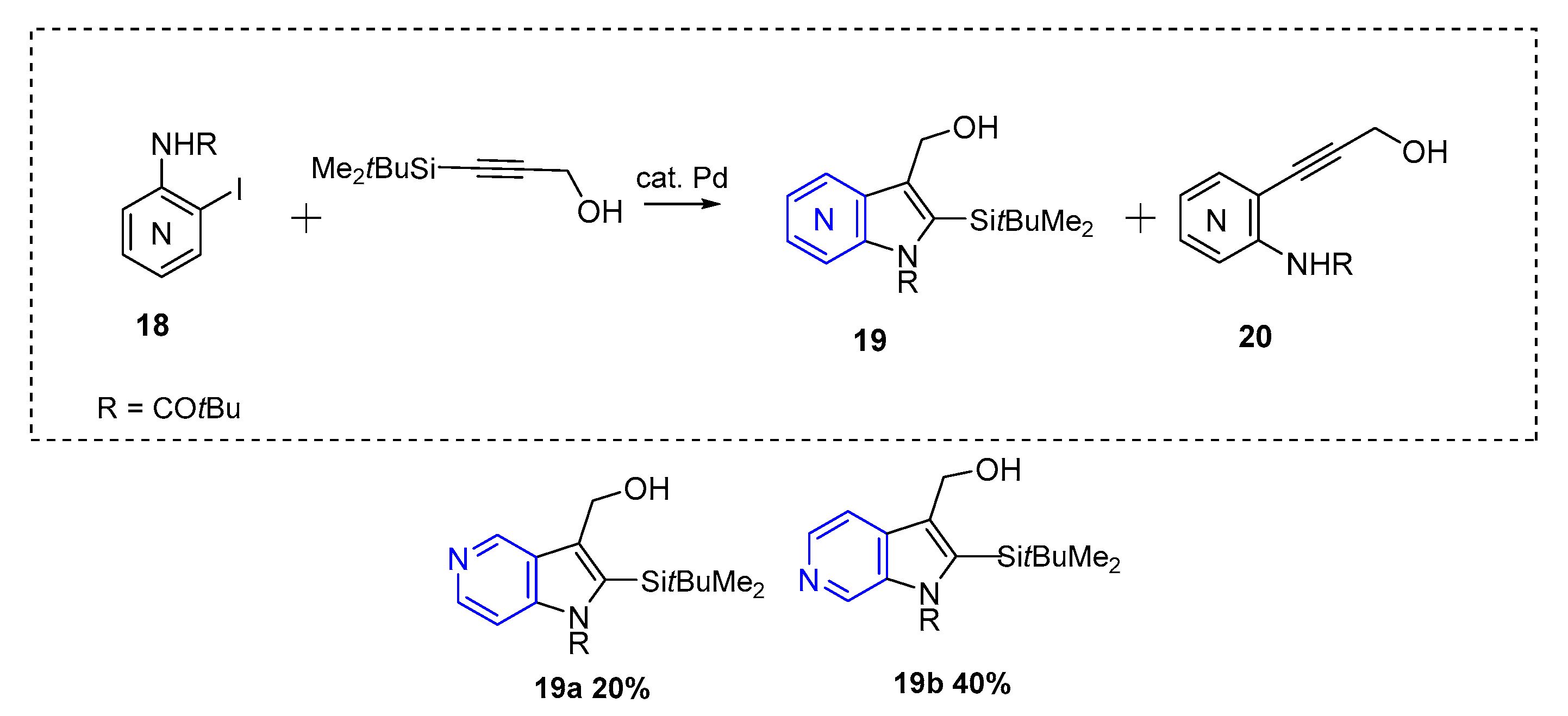
Scheme 10.
Synthesis of 2,3-disubstituted-5-azaindoles, 2,3,5-trisubstituted-7-azaindoles, and 2,3-disubstituted-6-azaindoles.
Scheme 10.
Synthesis of 2,3-disubstituted-5-azaindoles, 2,3,5-trisubstituted-7-azaindoles, and 2,3-disubstituted-6-azaindoles.
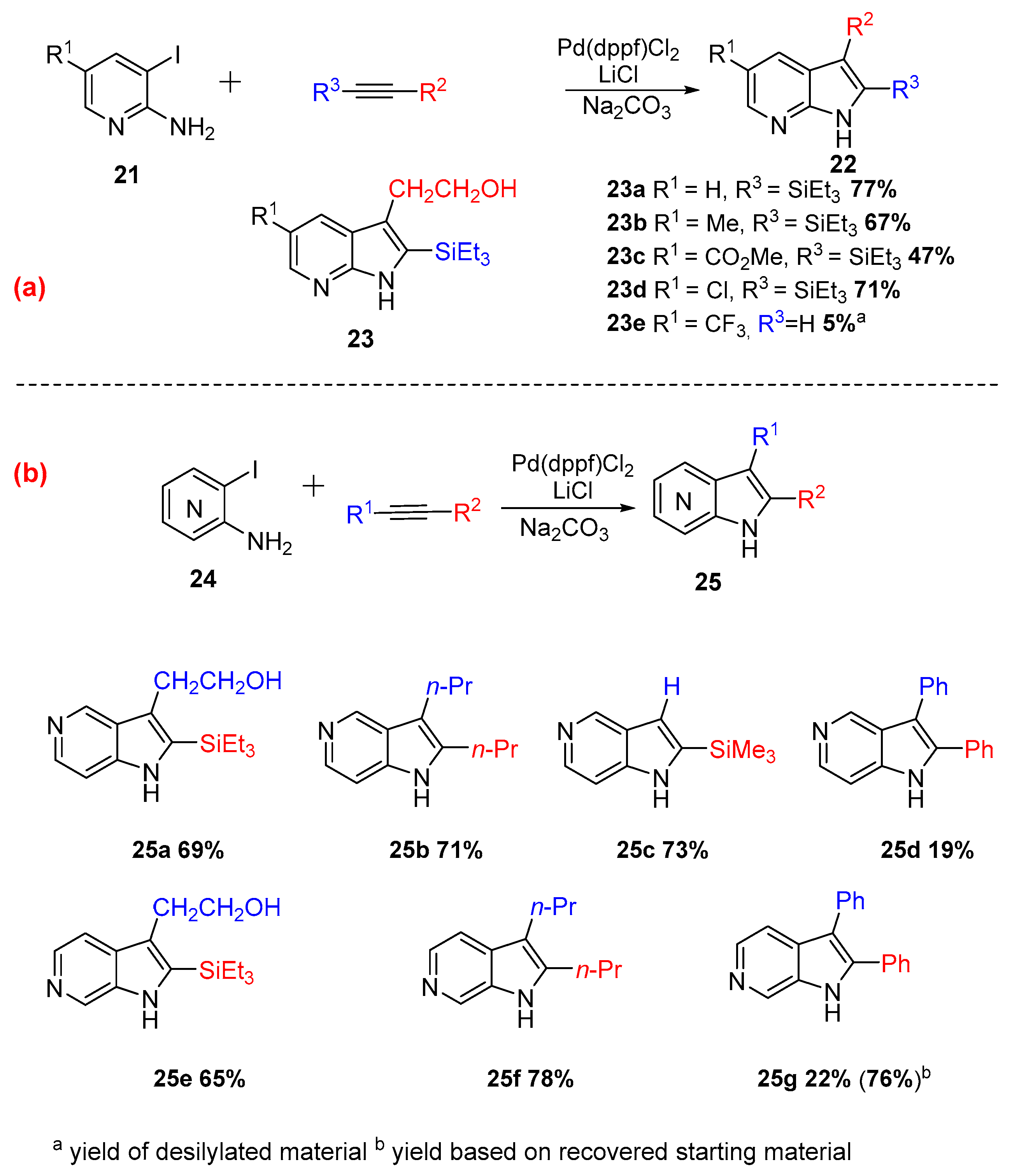
Scheme 11.
Synthesis of 2,3-disubstituted-4-azaindoles 27.
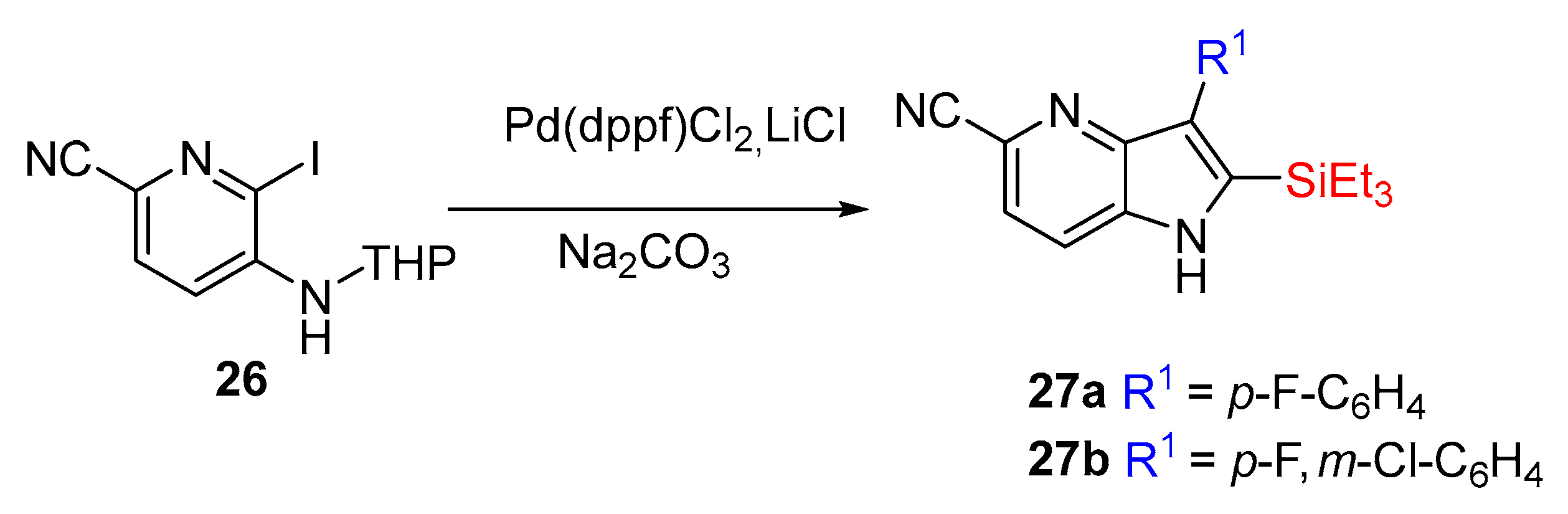
Scheme 12.
Important bioactive compounds 28a, 28b, and 28c and their respective IC50.
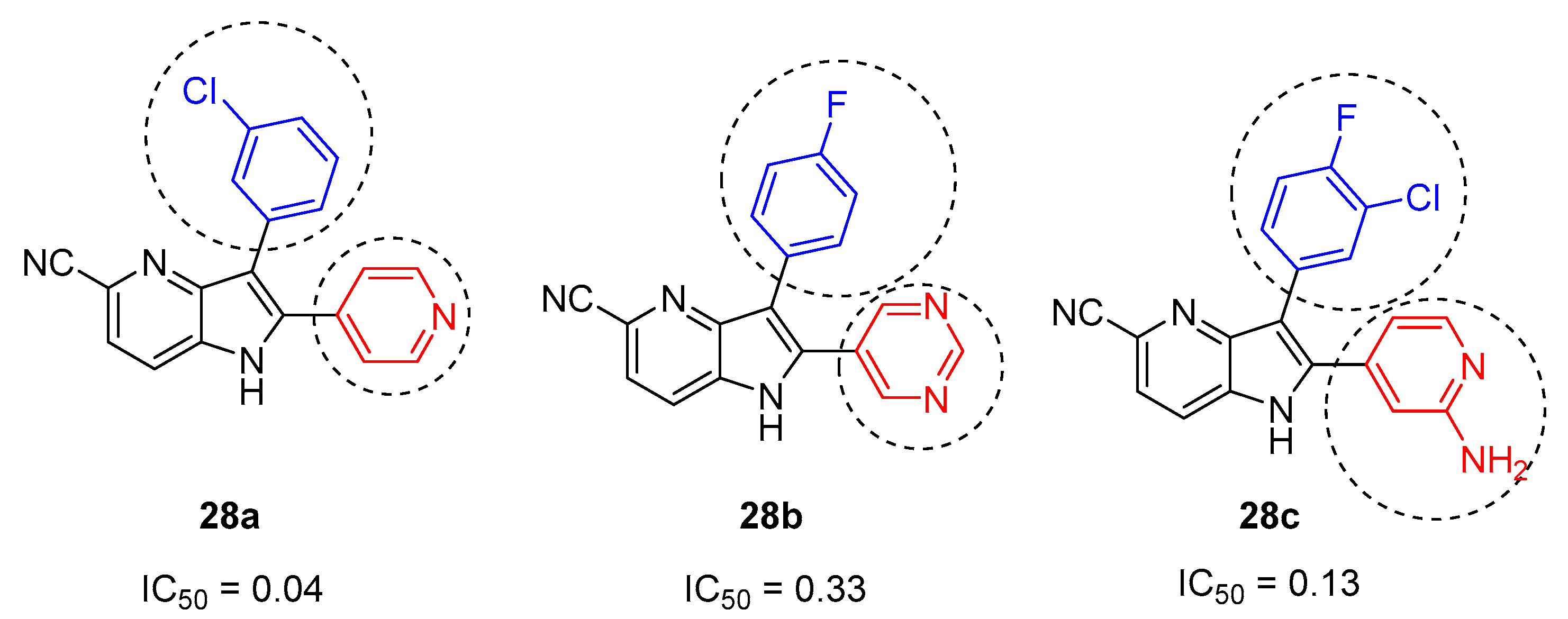
Scheme 13.
Synthesis of 7-azaindole via enamine formation and subsequent Heck reaction [19].
Scheme 13.
Synthesis of 7-azaindole via enamine formation and subsequent Heck reaction [19].
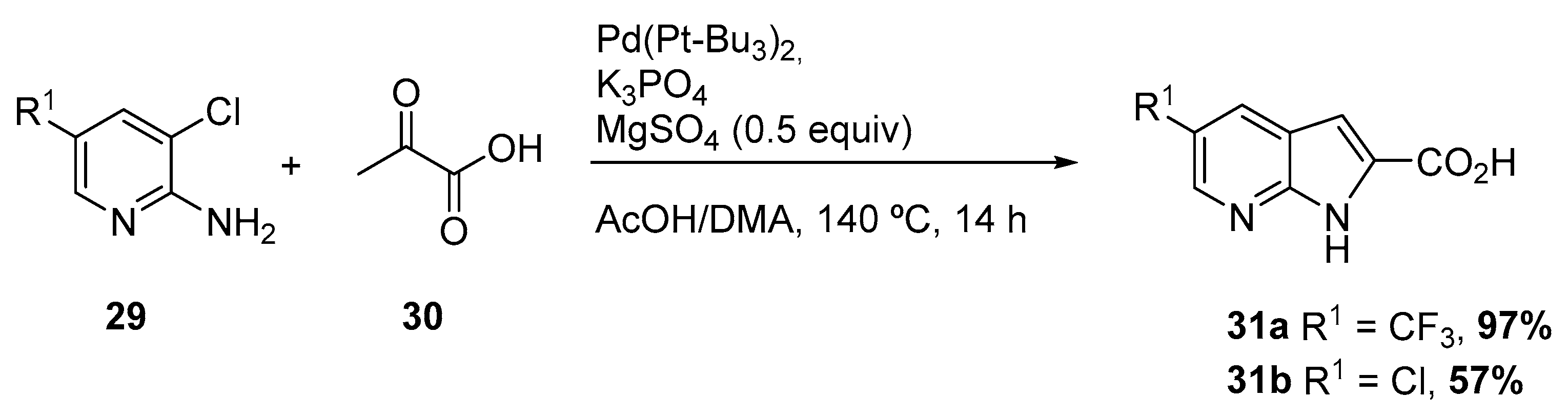
Scheme 14.
Synthesis of 2-methyl 5-, 6-, and 7-azaindoles 34 via palladium-catalyzed annulation of amino-ortho-iodopyridines 32 with allyl acetate 33 [20].
Scheme 14.
Synthesis of 2-methyl 5-, 6-, and 7-azaindoles 34 via palladium-catalyzed annulation of amino-ortho-iodopyridines 32 with allyl acetate 33 [20].

Scheme 15.
Synthesis of azaindoles (36) via an intramolecular Heck reaction under microwave conditions [21].
Scheme 15.
Synthesis of azaindoles (36) via an intramolecular Heck reaction under microwave conditions [21].

Scheme 16.
Synthesis azaindoles (39) from ketones (38) and amino-ortho-halopyridines (37).
Scheme 17.
Synthesis of azaindoles (41) by a Pd-catalyzed reaction of gem-dichloroolefins and a boronic acid, via a tandem intramolecular C–N and intramolecular Suzuki coupling process [24].
Scheme 17.
Synthesis of azaindoles (41) by a Pd-catalyzed reaction of gem-dichloroolefins and a boronic acid, via a tandem intramolecular C–N and intramolecular Suzuki coupling process [24].
Scheme 18.
Synthesis azaindole 45 by a Pd-catalyzed reaction of gem-dichloroolefins and a boronic acid using the pyridyl N-oxide 43 as substrate [24].
Scheme 18.
Synthesis azaindole 45 by a Pd-catalyzed reaction of gem-dichloroolefins and a boronic acid using the pyridyl N-oxide 43 as substrate [24].

Scheme 19.
Aminopalladation-reductive elimination reaction for 4- and 7-azaindole synthesis (48) from 2-Alkynyl-3-trifluoroacetamidopyridines 46.
Scheme 19.
Aminopalladation-reductive elimination reaction for 4- and 7-azaindole synthesis (48) from 2-Alkynyl-3-trifluoroacetamidopyridines 46.

Scheme 20.
C–H activation to access the azaindole core.
Scheme 21.
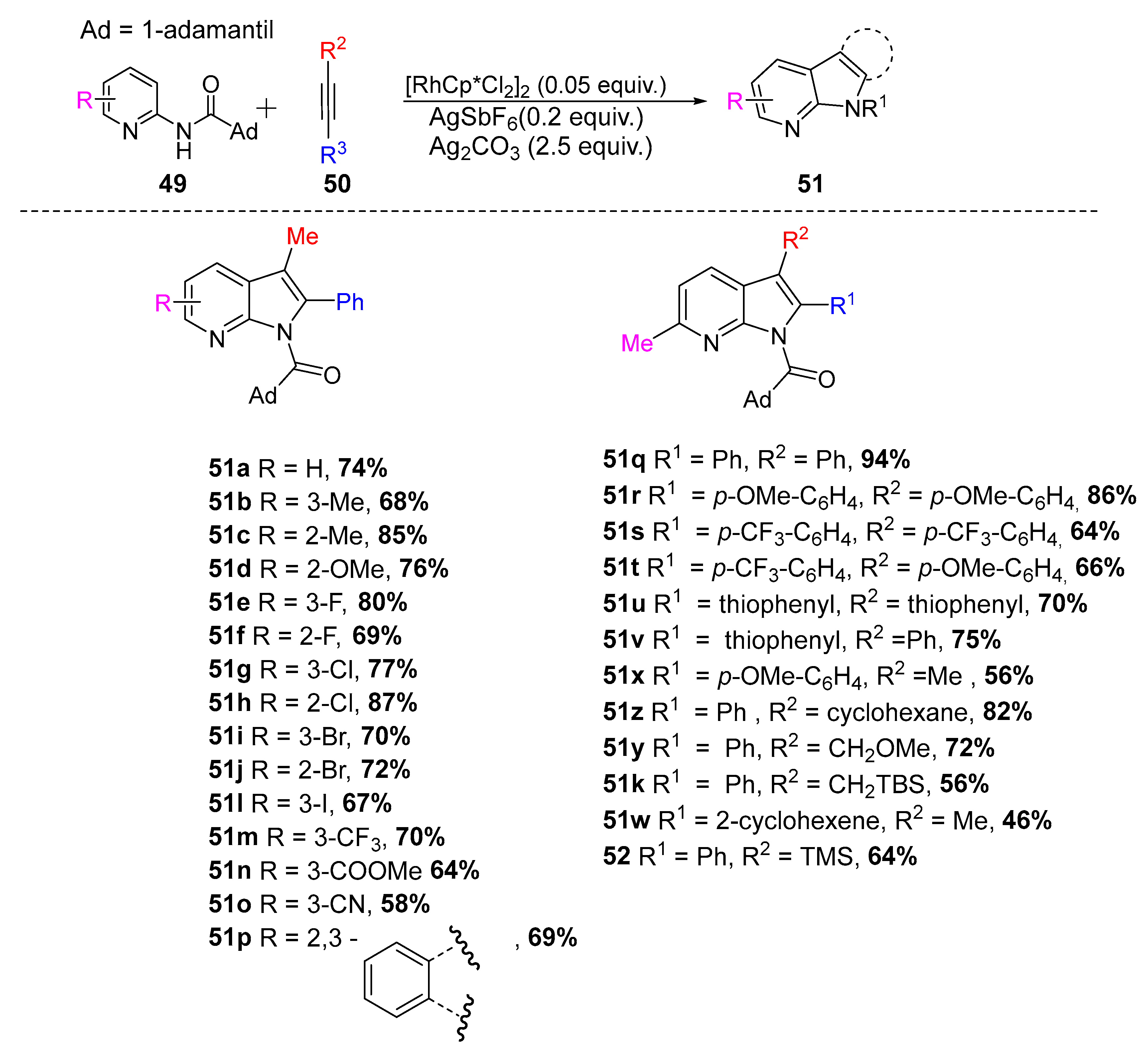
Synthesis of azaindoles (51) from alkynes and N-substituted aminopyridines (49) using Rh(III) catalysis [34].
Scheme 21.
Synthesis of azaindoles (51) from alkynes and N-substituted aminopyridines (49) using Rh(III) catalysis [34].
Scheme 22.
Plausible reaction pathway of Rh(III) catalysis for azaindole synthesis [34].
Scheme 22.
Plausible reaction pathway of Rh(III) catalysis for azaindole synthesis [34].
Scheme 23.
Synthesis of 7-azaindole core from 2,3-(het)aryl-3-N-aryl/acylenaminonitriles and enaminones.
Scheme 23.
Synthesis of 7-azaindole core from 2,3-(het)aryl-3-N-aryl/acylenaminonitriles and enaminones.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Santos, A.S.; Mortinho, A.C.; Marques, M.M.B. Metal-Catalyzed Cross-Coupling Reactions on Azaindole Synthesis and Functionalization. Molecules 2018, 23, 2673. https://doi.org/10.3390/molecules23102673
AMA Style
Santos AS, Mortinho AC, Marques MMB. Metal-Catalyzed Cross-Coupling Reactions on Azaindole Synthesis and Functionalization. Molecules. 2018; 23(10):2673. https://doi.org/10.3390/molecules23102673
Chicago/Turabian StyleSantos, A. Sofia, Ana C. Mortinho, and M. Manuel B. Marques. 2018. "Metal-Catalyzed Cross-Coupling Reactions on Azaindole Synthesis and Functionalization" Molecules 23, no. 10: 2673. https://doi.org/10.3390/molecules23102673