
Application of HPCCC Combined with Polymeric Resins and HPLC for the Separation of Cyclic Lipopeptides Muscotoxins A–C and Their Antimicrobial Activity

 , , , ,
, , , ,
Abstract
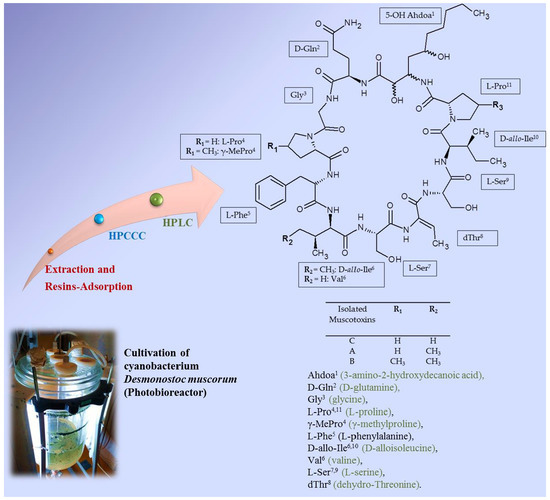
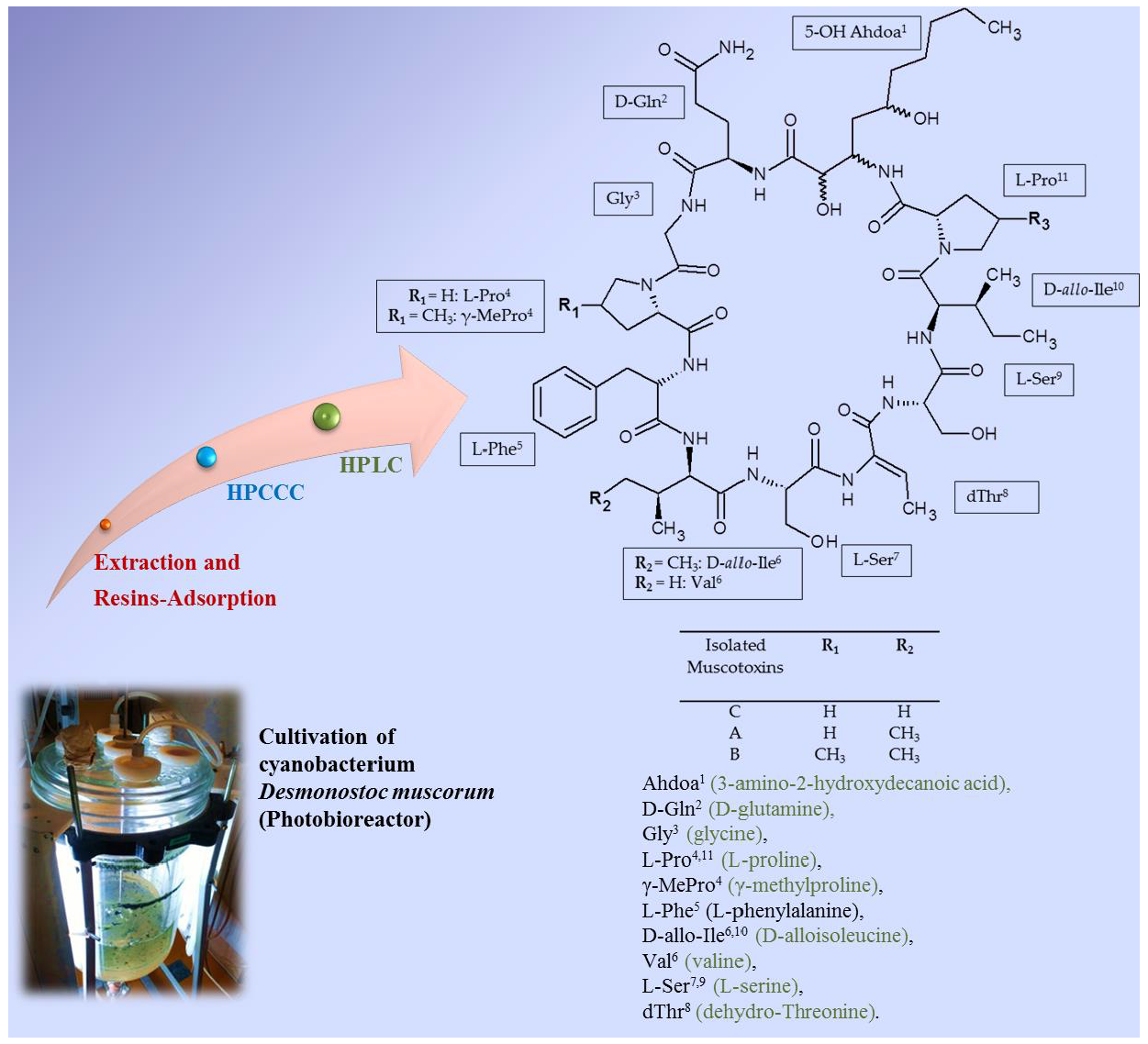
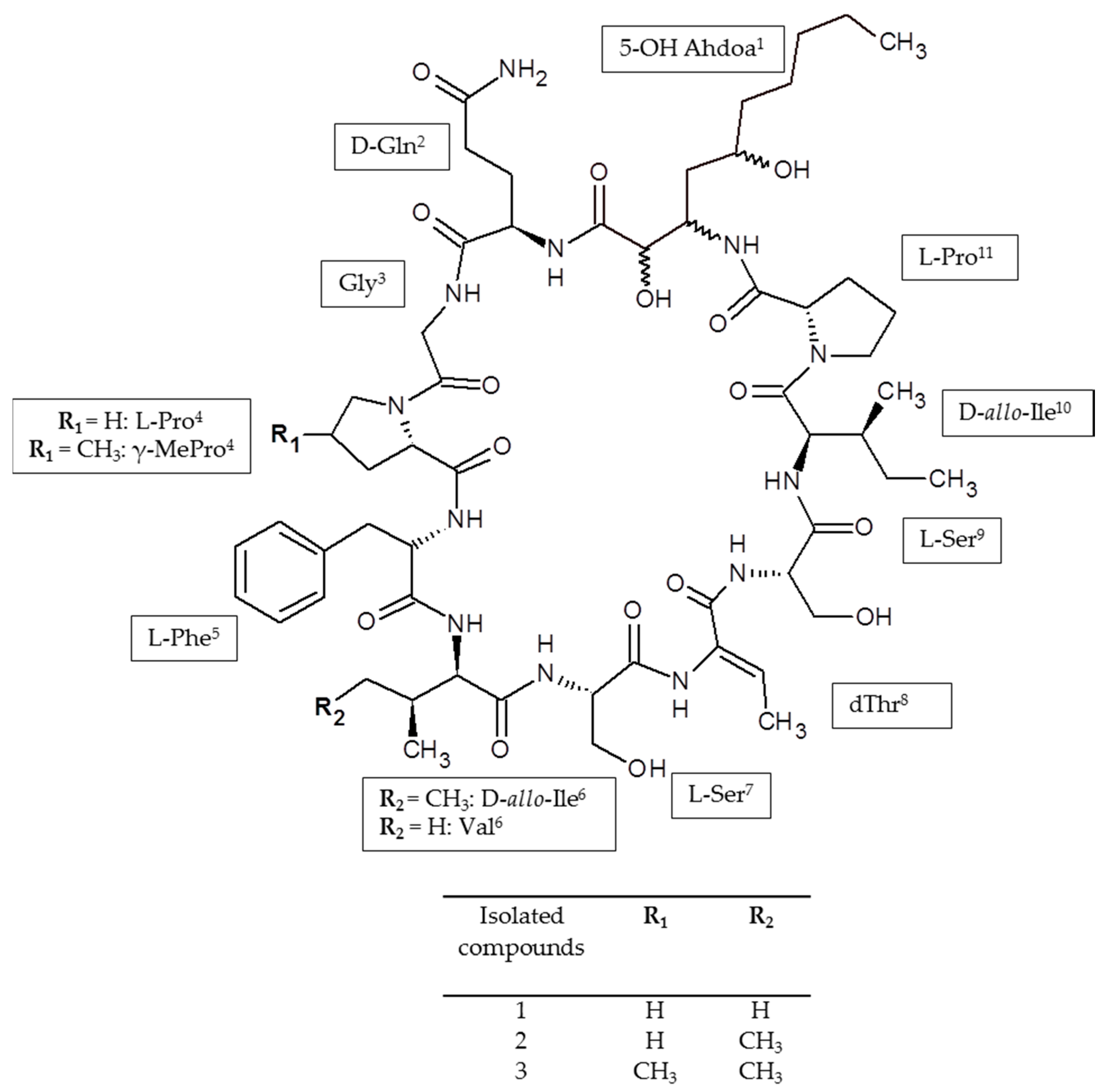
:
1. Introduction
2. Results and Discussion
2.1. HPLC-ESI-HRMS Analysis of the Crude Extract
2.2. Enrichment of the Crude Extract by Adsorption Resins
2.3. Optimization of the HPCCC Conditions
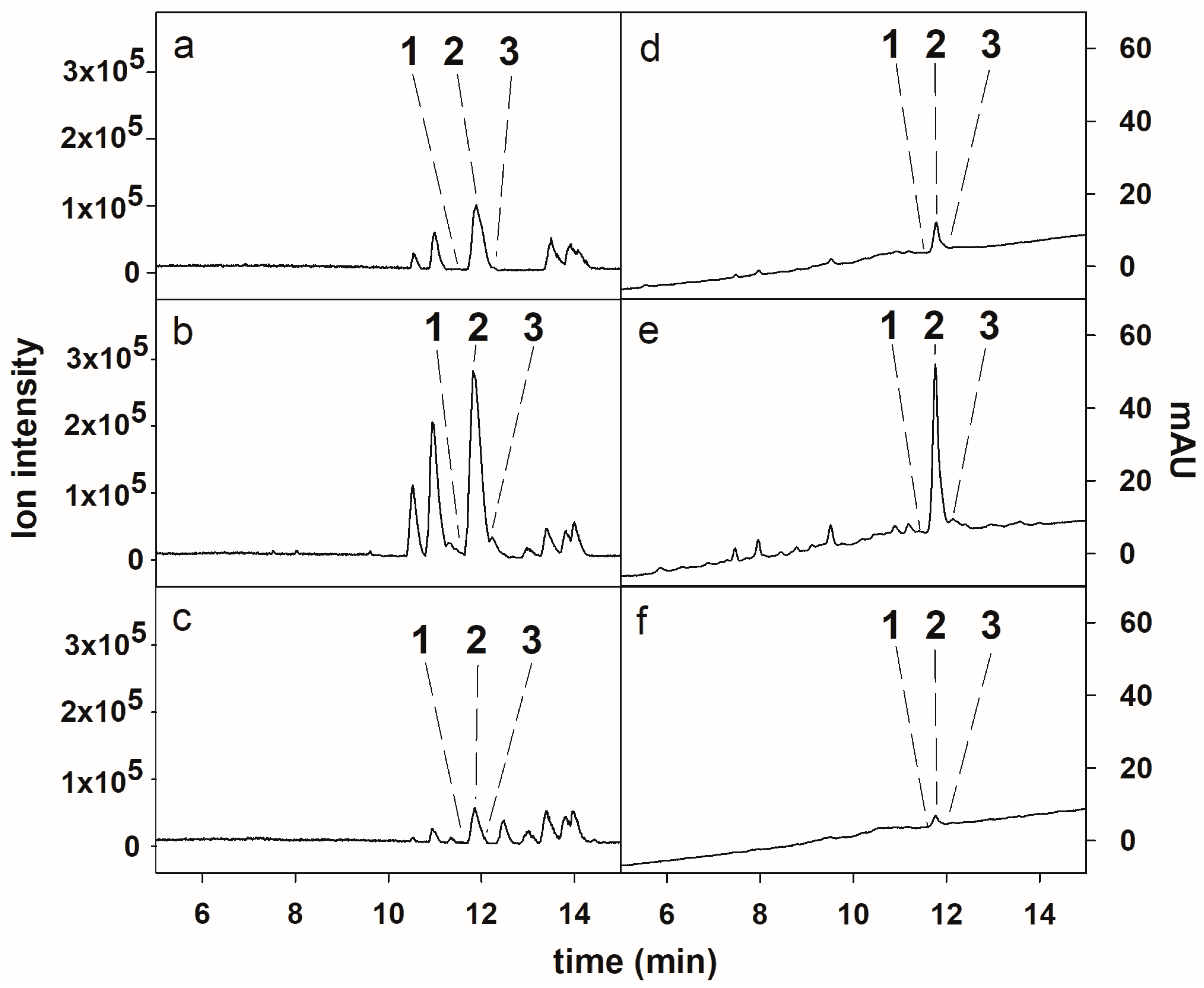
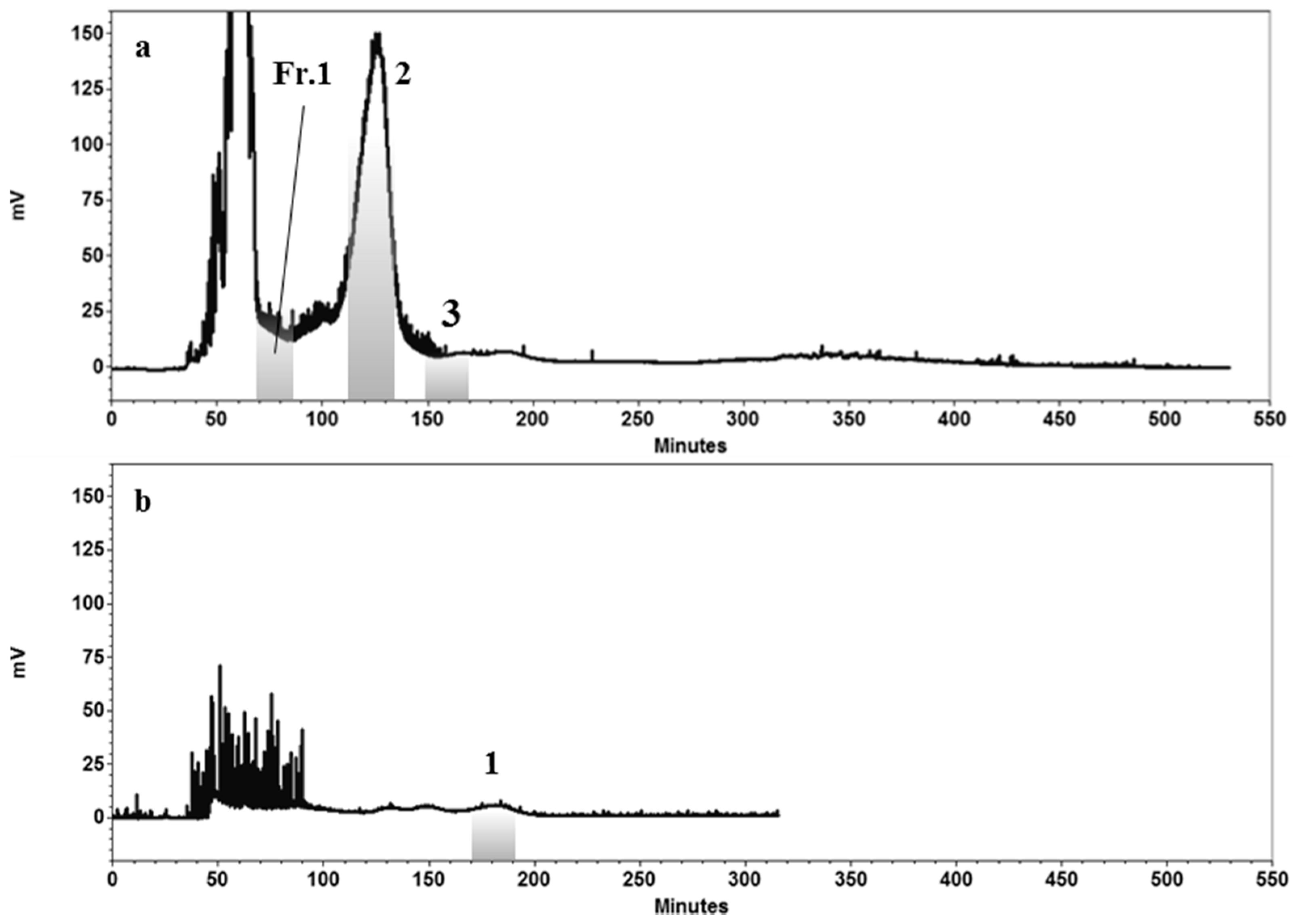
2.4. HPCCC Separation of Compounds 1–3
2.5. HPLC Purification of the Compounds Obtained from HPCCC
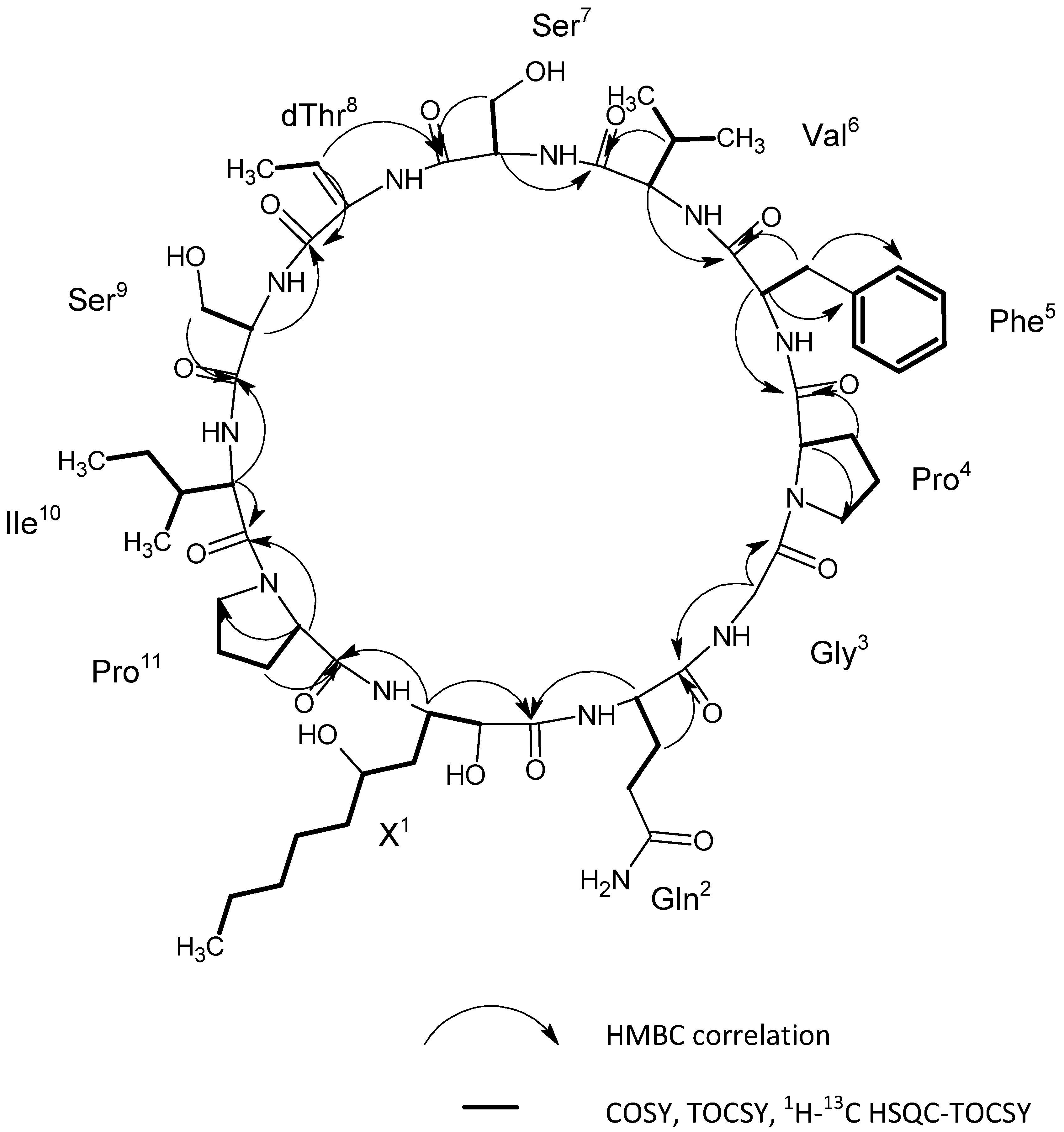
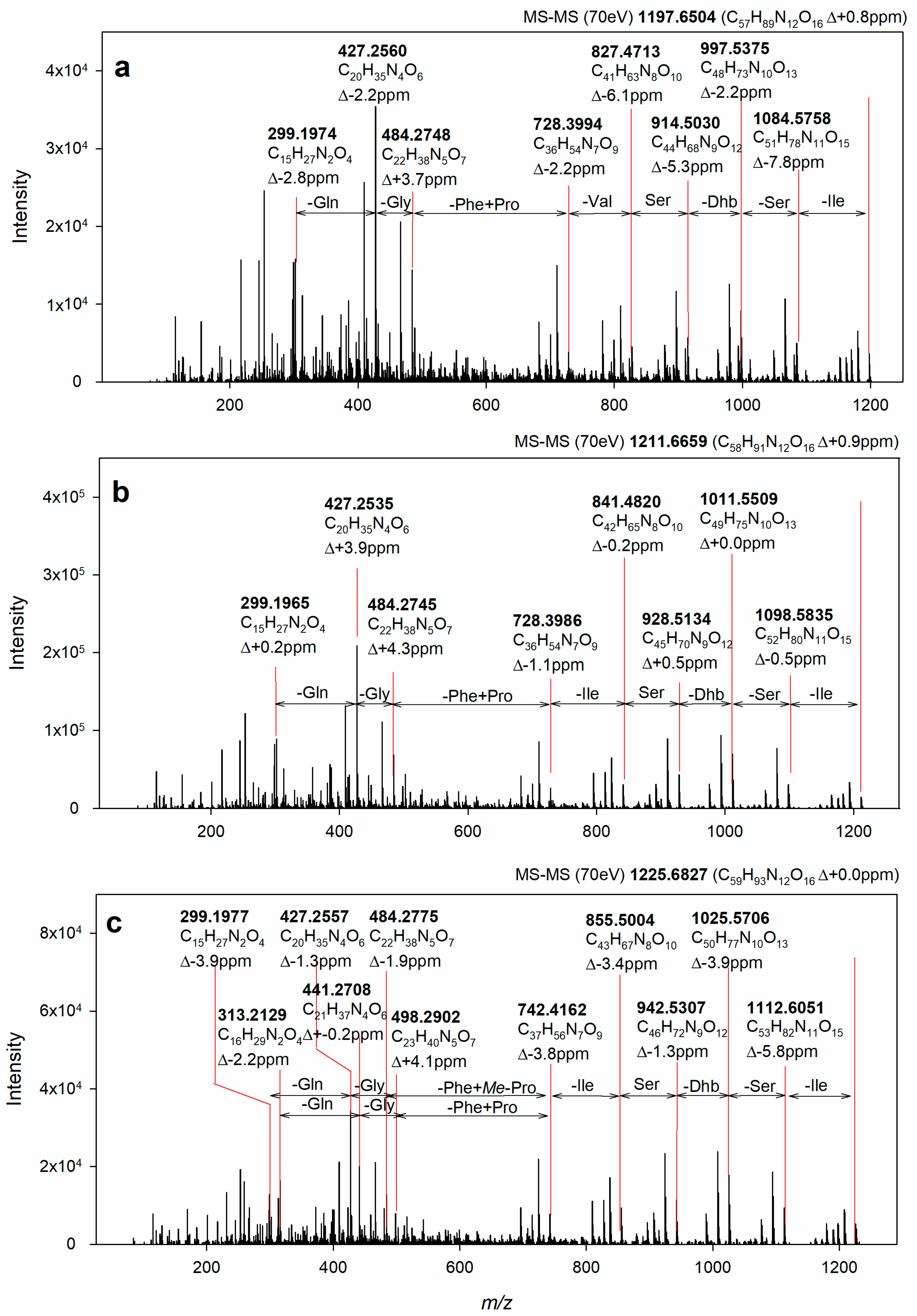
2.6. Identification of the Target Compounds
2.7. In Vitro Antimicrobial Activity
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Culture Growth Conditions and Biomass Extraction
3.3. Enrichment of Crude Extract by Non-Ionic Polymeric Resins
3.4. HPCCC Separation
3.4.1. HPCCC Apparatus
3.4.2. Selection of the Two-Phase Solvent System
3.4.3. Preparation of the Two-Phase Solvent System and Sample Solution
3.4.4. HPCCC Separation Procedure
3.5. Subsequent Purification of HPCCC Peak Fractions by Using Semipreparative HPLC
3.6. HPLC-ESI-HRMS Analysis of Extracts and HPCCC Fractions
3.7. Structural Identification of the Isolated Target Compounds
3.8. In Vitro Antimicrobial Bioassay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chlipala, G.E.; Mo, S.; Orjala, J. Chemodiversity in freshwater and terrestrial cyanobacteria—A source for drug discovery. Curr. Drug Targets 2011, 12, 1654–1673. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Sanchez, C.; Shen, B. Hybrid peptide-polyketide natural products: Biosynthesis and prospects toward engineering novel molecules. Met. Eng. 2001, 3, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Vitullo, D.; Senatore, M.; Lima, G.; Lanzotti, V. Antifungal cyclic lipopeptides from Bacillus amyloliquefaciens strain BO5A. J. Nat. Prod. 2013, 76, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.H.; Sørensen, D.; Tobiasen, C.; Andersen, J.B.; Christophersen, C.; Givskov, M.; Sørensen, J. Antibiotic and biosurfactant properties of cyclic lipopeptides produced by fluorescent Pseudomonas spp. from the sugar beet rhizosphere. Appl. Environ. Microbiol. 2002, 68, 3416–3423. [Google Scholar] [CrossRef] [PubMed]
- Hrouzek, P.; Kuzma, M.; Černý, J.; Novák, P.; Fišer, R.; Simek, P.; Lukešová, A.; Kopecký, J. The cyanobacterial cyclic lipopeptides puwainaphycins F/G are inducing necrosis via cell membrane permeabilization and subsequent unusual actin relocalization. Chem. Res. Toxicol. 2012, 25, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Krunic, A.; Shen, Q.; Swanson, S.M.; Orjala, J. Minutissamides A–D, antiproliferative cyclic decapeptides from the cultured cyanobacterium Anabaena minutissima. J. Nat. Prod. 2011, 74, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Sturdy, M.; Krunic, A.; Kim, H.; Shen, Q.; Swanson, S.M.; Orjala, J. Minutissamides E–L, antiproliferative cyclic lipodecapeptides from the cultured freshwater cyanobacterium cf. Anabaena sp. Bioorg. Med. Chem. 2012, 20, 6134–6143. [Google Scholar] [CrossRef] [PubMed]
- Maschmeyer, G.; Glasmacher, A. Pharmacological properties and clinical efficacy of a recently licensed systemic antifungal, caspofungin. Mycoses 2005, 48, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Felnagle, E.A.; Jackson, E.E.; Chan, Y.A.; Podevels, A.M.; Berti, A.D.; McMahon, M.D.; Thomas, M.G. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol. Pharm. 2008, 5, 191–211. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Müller, A.; Miess, H.; Gross, H. Cyclic lipopeptides as antibacterial agents—Potent antibiotic activity mediated by intriguing mode of actions. Int. J. Med. Microbiol. 2014, 304, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Joo, S.H. Cyclic peptides as therapeutic agents and biochemical tools. Biomol. Ther. 2012, 20, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Liskamp, R.M.J.; Rijkers, D.T.S.; Bakker, S.E. Bioactive macrocyclic peptides and peptide mimics. In Modern Supramolecular Chemistry: Strategies for Macrocycle Synthesis; Diederich, F., Stang, P.J., Tykwinski, R.R., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 1–27. ISBN 9783527318261. [Google Scholar]
- Tomek, P.; Hrouzek, P.; Kuzma, M.; Sýkora, J.; Fiser, R.; Cerný, J.; Novák, P.; Bártová, S.; Simek, P.; Hof, M.; et al. Cytotoxic lipopeptide muscotoxin A, isolated from soil cyanobacterium Desmonostoc muscorum, permeabilizes phospholipid membranes by reducing their fluidity. Chem. Res. Toxicol. 2015, 28, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Destandau, E.; Elfakir, C. New advances in countercurrent chromatography and centrifugal partition chromatography: Focus on coupling strategy. Anal. Bioanal. Chem. 2014, 406, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Tapia, A.; Cheel, J.; Theoduloz, C.; Rodríguez, J.; Schmeda-Hirschmann, G.; Gerth, A.; Wilken, D.; Jordan, M.; Jiménez-González, E.; Gomez-Kosky, R.; et al. Free radical scavengers from Cymbopogon citratus (DC.) Stapf. plants cultivated in bioreactors by the temporary immersion (TIS)-principle. Z. Naturforsch. C 2007, 62, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Spórna-Kucab, A.; Ignatova, S.; Garrard, I.; Wybraniec, S. Versatile solvent systems for the separation of betalains from processed Beta vulgaris L. juice using counter-current chromatography. J. Chromatogr. B 2013, 941, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.; Garrard, I.; da Silva, A.J.; Leitão, G.G. Changes in the mobile phase composition on a stepwise counter-current chromatography elution for the isolation of flavonoids from Siparuna Glycycarpa. J. Sep. Sci. 2013, 36, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Li, H.B.; Wong, R.N.; Ji, B.; Jiang, Y. Isolation and purification of the bioactive carotenoid zeaxanthin from the microalga Microcystis aeruginosa by high-speed counter-current chromatography. J. Chromatogr. A 2005, 1064, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Li, H.B.; Fan, K.W.; Chen, F. Isolation and purification of canthaxanthin from the microalga Chlorella zofingiensis by high-speed counter-current chromatography. J. Sep. Sci. 2006, 29, 699–703. [Google Scholar] [CrossRef]
- Cheel, J.; Urajová, P.; Hájek, J.; Hrouzek, P.; Kuzma, M.; Bouju, E.; Faure, K.; Kopecký, J. Separation of cyclic lipopeptide puwainaphycins from cyanobacteria by countercurrent chromatography combined with polymeric resins and HPLC. Anal. Bioanal. Chem. 2017, 409, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.E.; Bornemann, V.; Niemczura, W.P.; Gregson, J.M.; Chen, J.L.; Norton, T.R.; Patterson, G.M.L.; Helms, G.L. Puwainaphycin C, a cardioactive cyclic peptide from the blue-green alga Anabaena BQ-16-1. Use of two-dimensional 13C-13C and 13C-15N correlation spectroscopy in sequencing the amino acid units. J. Am. Chem. Soc. 1989, 111, 6128–6132. [Google Scholar] [CrossRef]
- Martin, N.I.; Hu, H.; Moake, M.M.; Churey, J.J.; Whittal, R.; Worobo, R.W.; Vederas, J.C. Isolation, structural characterization, and properties of mattacin (polymyxin M), a cyclic peptide antibiotic produced by Paenibacillus kobensis M. J. Biol. Chem. 2003, 278, 13124–13132. [Google Scholar] [CrossRef] [PubMed]
- Sasse, F.; Steinmetz, H.; Schupp, T.; Petersen, F.; Memmert, K.; Hofmann, H.; Heusser, C.; Brinkmann, V.; von Matt, P.; Höfle, G.; et al. Argyrins, immunosuppressive cyclic peptides from myxobacteria. I. Production, isolation, physico-chemical and biological properties. J. Antibiot. 2002, 55, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Sasse, F.; Steinmetz, H.; Höfle, G.; Reichenbach, H. Archazolids, new cytotoxic macrolactones from Archangium gephyra (Myxobacteria). Production, isolation, physico-chemical and biological properties. J. Antibiot. 2003, 56, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Conway, W.D. Experimental observations of the hydrodynamic behavior of solvent systems in high-speed counter-current chromatography. III. Effects of physical properties of the solvent systems and operating temperature on the distribution of two-phase solvent systems. J. Chromatogr. A 1984, 301, 405–414. [Google Scholar] [CrossRef]
- Berthod, A.; Maryutina, T.; Spivakov, B.; Shpigun, O.; Sutherland, I.A. Countercurrent chromatography in analytical chemistry. Pure Appl. Chem. 2009, 81, 355–387. [Google Scholar] [CrossRef]
- Meena, K.R.; Kanwar, S.S. Lipopeptides as the antifungal and antibacterial agents: Applications in food safety and therapeutics. Biomed. Res. Int. 2015, 2015, 473050. [Google Scholar] [CrossRef] [PubMed]
- Smykalova, I.; Soukup, A.; Ondráčková, E.; Hrouzek, P. Soubor Laboratorních in Vitro Biotestů Pro Testování Bioaktivních Látek Z Mikrořas; Agritec, výzkum, šlechtění a služby, s.r.o.: Šumperk, Czech Republic, 2016. [Google Scholar]
- Espinel-Ingroff, A.; Fothergill, A.; Ghannoum, M.; Manavathu, E.; Ostrosky-Zeichner, L.; Pfaller, M.A.; Rinaldi, M.G.; Schell, W.; Walsh, T.J. Quality control and reference guidelines for CLSI broth microdilution method (M38-A document) for susceptibility testing of anidulafungin against molds. J. Clin. Microbiol. 2007, 45, 2180–2182. [Google Scholar] [CrossRef] [PubMed]
- Borman, A.M.; Fraser, M.; Palmer, M.D.; Szekely, A.; Houldsworth, M.; Patterson, Z.; Johnson, E.M. MIC distributions and evaluation of fungicidal activity for amphotericin B, itraconazole, voriconazole, posaconazole and caspofungin and 20 species of pathogenic filamentous fungi determined using the CLSI broth microdilution method. J. Fungi 2017, 3, 27. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent Systems | Composition | Relative Proportions of Solvents (v/v/v/v) | Phase Volume Ratio (UP/LP) | Settling Time (s) | Density Difference (LP-UP, g/mL) | Partition Coefficient (K) of Muscotoxins | ||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||||
| 1 | n-Hex–EtOH–H2O | 10:5:5 | 1.22 | 7 | 0.272 | 0.00 | 0.03 | 0.01 |
| 2 | n-Hex–EtOAc–EtOH–H2O | 9:1:5:5 | 1.00 | 10 | 0.226 | 0.00 | 0.03 | 0.01 |
| 3 | n-Hex–EtOAc–EtOH–H2O | 8:2:5:5 | 1.00 | 11 | 0.197 | 0.00 | 0.01 | 0.00 |
| 4 | n-Hex–EtOAc–EtOH–H2O | 7:3:5:5 | 0.85 | 14 | 0.229 | 0.01 | 0.06 | 0.02 |
| 5 | n-Hex–EtOAc–EtOH–H2O | 6:4:5:5 | 0.82 | 18 | 0.197 | 0.01 | 0.09 | 0.04 |
| 6 | n-Hex–EtOAc–EtOH–H2O | 5:5:5:5 | 0.82 | 33 | 0.183 | 0.00 | 0.01 | 0.01 |
| 7 | n-Hex–EtOAc–EtOH–H2O | 4:5:4:5 | 1.00 | 33 | 0.133 | 0.02 | 0.14 | 0.05 |
| 8 | n-Hex–EtOAc–EtOH–H2O | 3:5:3:5 | 1.00 | 21 | 0.152 | 0.08 | 0.32 | 0.27 |
| 9 | n-Hex–EtOAc–EtOH–H2O | 2:5:2:5 | 1.12 | 18 | 0.139 | 0.40 | 0.97 | 1.22 |
| 10 | n-Hex–EtOAc–EtOH–H2O | 1:5:1:5 | 1.00 | 13 | 0.112 | 0.36 | 0.94 | 1.41 |
| 11 | n-Hex–EtOAc–EtOH–H2O–AcOH | 1:5:1:5:1 | 0.97 | 24 | 0.119 | 1.09 | 1.97 | 2.45 |
| 10 | Atom | δC [ppm] | m | δH [ppm] | nH | m | JHH [Hz] |
|---|---|---|---|---|---|---|---|
| X1 | 1 | 174.68 | s | - | 0 | - | - |
| 2 | 73.12 | d | 4.166 | 1 | d | 2.2 | |
| 3 | 51.09 | d | 4.330 | 1 | m | - | |
| 4 | 40.57 | t | 1.82H | 1 | m | - | |
| 1.66H | 1 | m | - | ||||
| 5 | 69.67 | d | 3.61H | 1 | m | - | |
| 6 | 38.16 | t | 1.53H | 1 | m | - | |
| 1.42H | 1 | m | - | ||||
| 7 | 26.41 | t | 1.50H | 1 | m | - | |
| 1.35H | 1 | m | - | ||||
| 8 | 33.10 | t | 1.31H | 2 | m | - | |
| 9 | 23.76 | t | 1.34H | 2 | m | - | |
| 10 | 14.44 | q | 0.904 | 3 | m | - | |
| Gln2 | C=O | 173.76 | s | - | 0 | - | - |
| α | 54.24 | d | 4.479 | 1 | m | - | |
| β | 29.02 | t | 2.24H | 1 | m | - | |
| 1.96H | 1 | m | - | ||||
| γ | 32.96 | t | 2.298 | 1 | m | - | |
| 2.24H | 1 | m | - | ||||
| δ | 178.48 | s | - | 0 | - | - | |
| Gly3 | C=O | 170.70 | s | - | 0 | - | - |
| α | 43.46 | t | 4.118 | 1 | d | 17.3 | |
| 3.951 | 1 | d | 17.3 | ||||
| Pro4 | C=O | 174.59 | s | - | 0 | - | - |
| α | 62.33 | d | 4.289 | 1 | m | - | |
| β | 30.55 | t | 2.12H | 1 | m | - | |
| 1.65H | 1 | m | - | ||||
| γ | 25.56 | t | 1.87H | 1 | m | - | |
| 1.66H | 1 | m | - | ||||
| δ | 48.01 | t | 3.62H | 1 | m | - | |
| 3.53H | 1 | m | - | ||||
| Phe5 | C=O | 173.79 | s | - | 0 | - | - |
| α | 56.33 | d | 4.62H | 1 | m | - | |
| β | 37.42 | t | 3.29H | 1 | m | - | |
| 2.952 | 1 | dd | 10.3, 14.0 | ||||
| ipso | 138.87 | s | - | 0 | - | - | |
| ortho | 130.15 | d | 7.231 | 2 | m | - | |
| meta | 129.69 | d | 7.288 | 2 | m | - | |
| para | 127.94 | d | 7.209 | 1 | m | - | |
| Val6 | C=O | 174.83 | s | - | 0 | - | - |
| α | 61.02 | d | 4.139 | 1 | d | 7.8 | |
| β | 30.93 | d | 2.21H | 1 | m | - | |
| γ | 19.10 | q | 0.89H | 3 | m | - | |
| β-Me | 19.80 | q | 0.842 | 3 | d | 6.7 | |
| Ser7 | C=O | 171.89 | s | - | 0 | - | - |
| α | 57.97 | d | 4.43H | 1 | m | - | |
| β | 62.80 | t | 3.922 | 1 | dd | 5.2, 11.3 | |
| 3.882 | 1 | dd | 4.9, 11.3 | ||||
| dThr8 | C=O | 167.07 | s | - | 0 | - | - |
| α | 130.78 | s | - | 0 | - | - | |
| β | 127.80 | d | 5.912 | 1 | q | 7.4 | |
| γ | 13.68 | q | 1.921 | 3 | d | 7.4 | |
| Ser9 | C=O | 172.40 | s | - | 0 | - | - |
| α | 57.14 | d | 4.610 | 1 | m | - | |
| β | 63.45 | t | 4.000 | 1 | dd | 5.3, 11.5 | |
| 3.797 | 1 | dd | 4.1, 11.5 | ||||
| Ile10 | C=O | 172.25 | s | - | 0 | - | - |
| α | 56.53 | d | 4.640 | 1 | d | 7.2 | |
| β | 38.44 | d | 1.93H | 1 | m | - | |
| γ | 27.42 | t | 1.42H | 1 | m | - | |
| 1.170 | 1 | m | - | ||||
| δ | 12.13 | q | 0.948 | 3 | t | 7.3 | |
| β-Me | 15.12 | q | 0.928 | 3 | d | 6.8 | |
| Pro11 | C=O | 173.53 | s | - | 0 | - | - |
| α | 61.69 | d | 4.408 | 1 | m | - | |
| β | 30.48 | t | 2.04H | 2 | m | - | |
| γ | 25.56 | t | 1.99H | 2 | m | - | |
| δ | 48.9H | t | 3.83H | 1 | m | - | |
| 3.62H | 1 | m | - |
| Tested Microorganisms | (µg/mL) | |
|---|---|---|
| Bacteria | MIC | MIC95 |
| Staphylococcus aureus | NA | ND |
| Bacillus subtilis | 37.5 (4) | ≤300 |
| Streptococcus sanguinis | NA | ND |
| Pseudomonas aeroginosa | NA | ND |
| Escherichia coli | NA | ND |
| Fungi | MFC | |
| Candida friedrichii | 75 (8) | <300 |
| Aspergillus fumigatus | 2.34 (32) | 37.5 |
| Fusarium oxysporum | 75 (8) | <300 |
| Trichoderma harzianum | 37.5 (4) | 300 |
| Bipolaris sorokiniana | NA | ND |
| Monographella cucumerina | 2.34 (2) | 75 |
| Chaetomium globosum | 18.75 (2) | <300 |
| Alternaria alternata | 0.58 (2) | 75 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheel, J.; Hájek, J.; Kuzma, M.; Saurav, K.; Smýkalová, I.; Ondráčková, E.; Urajová, P.; Vu, D.L.; Faure, K.; Kopecký, J.; et al. Application of HPCCC Combined with Polymeric Resins and HPLC for the Separation of Cyclic Lipopeptides Muscotoxins A–C and Their Antimicrobial Activity. Molecules 2018, 23, 2653. https://doi.org/10.3390/molecules23102653
Cheel J, Hájek J, Kuzma M, Saurav K, Smýkalová I, Ondráčková E, Urajová P, Vu DL, Faure K, Kopecký J, et al. Application of HPCCC Combined with Polymeric Resins and HPLC for the Separation of Cyclic Lipopeptides Muscotoxins A–C and Their Antimicrobial Activity. Molecules. 2018; 23(10):2653. https://doi.org/10.3390/molecules23102653
Chicago/Turabian StyleCheel, José, Jan Hájek, Marek Kuzma, Kumar Saurav, Iva Smýkalová, Eliška Ondráčková, Petra Urajová, Dai Long Vu, Karine Faure, Jiří Kopecký, and et al. 2018. "Application of HPCCC Combined with Polymeric Resins and HPLC for the Separation of Cyclic Lipopeptides Muscotoxins A–C and Their Antimicrobial Activity" Molecules 23, no. 10: 2653. https://doi.org/10.3390/molecules23102653