1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications
by
 , and
, and
Jakub Adamek
1,2,* ,
,
Anna Węgrzyk
1,2,
Justyna Kończewicz
1,2,
Krzysztof Walczak
1 and
Karol Erfurt
3 1
Department of Organic Chemistry, Bioorganic Chemistry and Biotechnology, Silesian University of Technology, B. Krzywoustego 4, 44-100 Gliwice, Poland
2
Biotechnology Center of Silesian University of Technology, B. Krzywoustego 8, 44-100 Gliwice, Poland
3
Department of Chemical Organic Technology and Petrochemistry, Silesian University of Technology, B. Krzywoustego 4, 44-100 Gliwice, Poland
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(10), 2453; https://doi.org/10.3390/molecules23102453
Submission received: 31 August 2018
/
Revised: 19 September 2018
/
Accepted: 23 September 2018
/
Published: 25 September 2018
(This article belongs to the Special Issue Organophosphorus Chemistry 2018)
Abstract
:The α-amidoalkylating properties of 1-(N-acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength are discussed and examined. It is demonstrated that such type of phosphonium salts reacts smoothly with a diverse array of carbon- and heteroatom-based nucleophiles, including 1-morpholinocyclohexene, 1,3-dicarbonyl compounds, benzotriazole sodium salt, p-toluenesulfinate sodium salt, benzylamine, triarylphosphines, and other P-nucleophiles. Reactions are conducted at room temperature, in a short time (5–15 min) and mostly without catalysts. Simple work-up procedures result in good or very good yields of products. The structures of known compounds were established by spectroscopic methods and all new compounds have been fully characterized using 1H-, 13C-, 31P-NMR, IR spectroscopy, and high-resolution mass spectrometry. Mechanistic aspects of described transformations are also performed and discussed. It was demonstrated that unique properties make 1-(N-acylamino)alkyl-triarylphosphonium salts with weakened Cα-P+ bond strength interesting building blocks with great potential, especially in α-amidoalkylation reactions.
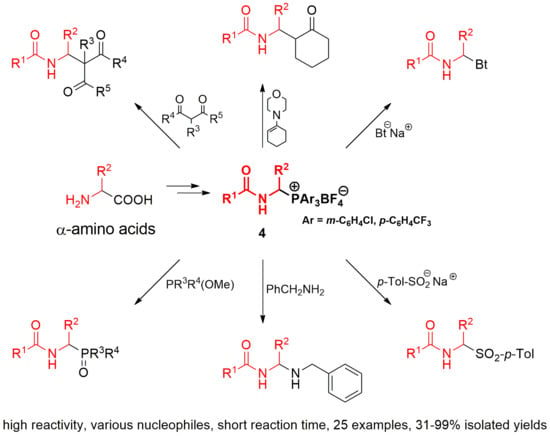
1. Introduction
Specific structural features of 1-(N-acylamino)alkylphosphonium salts make them very interesting reagents. The presence of a positively charged nucleofugal phosphonium moiety in the close surroundings of the N-acyl group determines its unique chemical properties such as high reactivity in α-amidoalkylations [1,2,3,4,5,6,7,8,9,10]. This type of reactions has enjoyed unflagging interest for years as a synthetic method with great potential, especially valuable for C-C and C-heteroatom bond formation [1,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37].
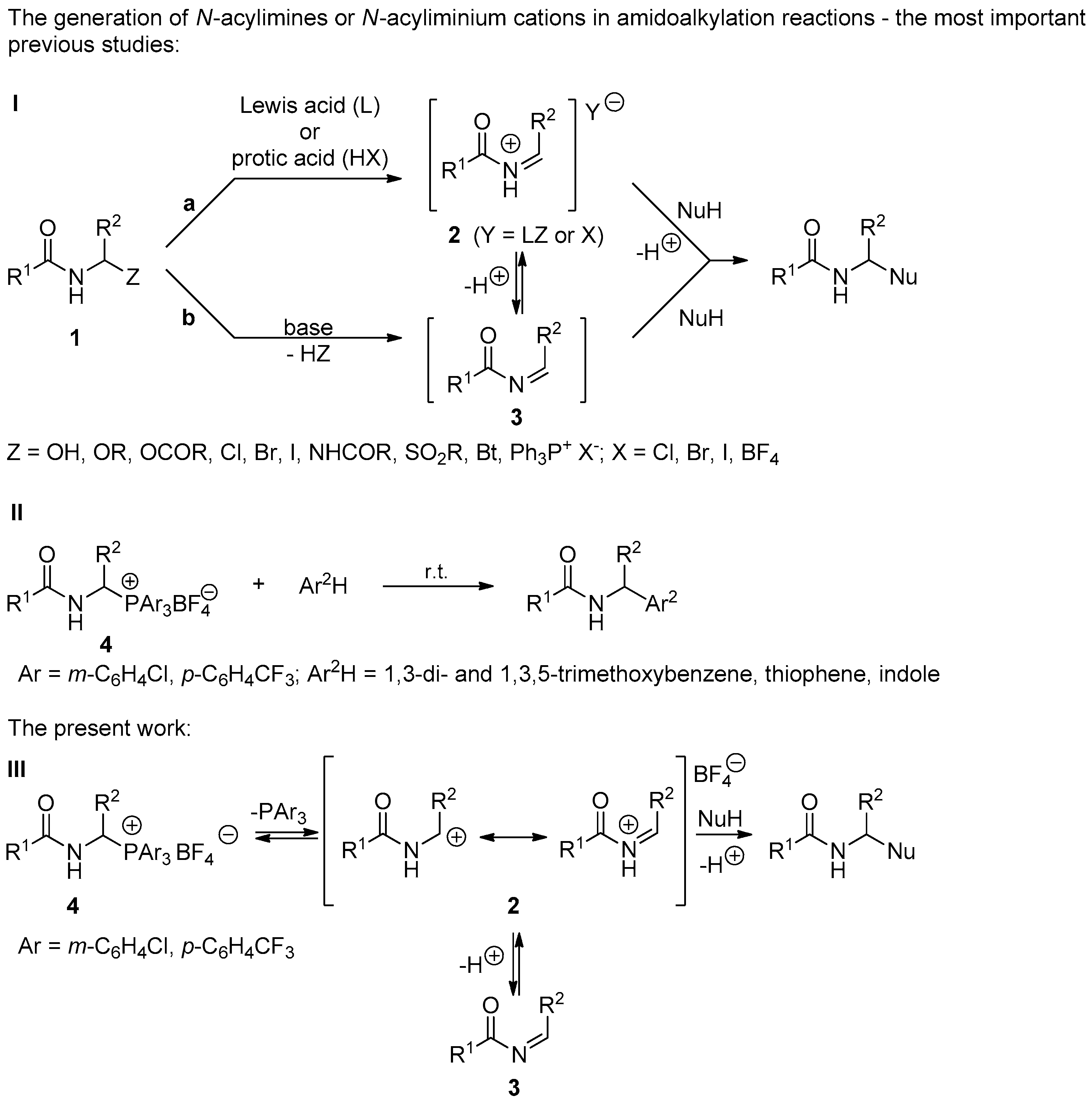
In general, the reactivity of α-amidoalkylating agents 1 depends on the efficiency of the generation of N-acyliminium cation 2 or N-acylimine 3 from its precursor and the equilibrium constant of this reaction. Of course, the reactivity of N-acyliminium cation 2 or N-acylimine 3 toward a nucleophile is also significant [1,2,3]. To produce the proper α-amidoalkylating agents, for instance N-acyliminium cation 2 or N-acylimine 3 from the most popular precursors such as α-amido sulfones (Z = SO2Ar), N-(1-benzotriazolil)alkylamides (Z = Bt), and N-(1-alkoxyalkyl)amides (Z = OR), it is necessary to use catalysts, mainly Lewis acids (route a, Scheme 1 (I)) [18,19,20,21,22,23,24,25,29,30,31,32,33,34,35,36,37]. In contrast, 1-(N-acylamino)alkyltriphenylphosphonium salts 1 (Z = Ph3P+ X−) do not require the use of acidic catalysts because of the permanent positive charge on the phosphonium group. However, the relatively high stability of Cα-P+ bond forces the use of a base catalyst (e.g., Hünig’s base, DBU, TBD; route b, Scheme 1 (I)) and sometimes microwave radiation [1,4,5,6,7,8,9,10].
Recently, we have proven that the use of 1-(N-acylamino)alkyltriarylphosphonium salts 4 derived from the EWG-substituted triarylphosphines facilitates the cleavage of Cα-P+ bond and thereby the generation of N-acyliminium cation (Scheme 1 (II)) [2,3]. This phenomenon significantly increases the reactivity of 1-(N-acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength and allows us to conduct α-amidoalkylation without the need for any catalyst. In this work, we demonstrate that the abovementioned phosphonium salts 4 react smoothly with various nucleophiles in a short time under mild conditions and create new carbon-carbon or carbon-heteroatom bonds with good or very good yields (Scheme 1 (III)).
2. Results and Discussion
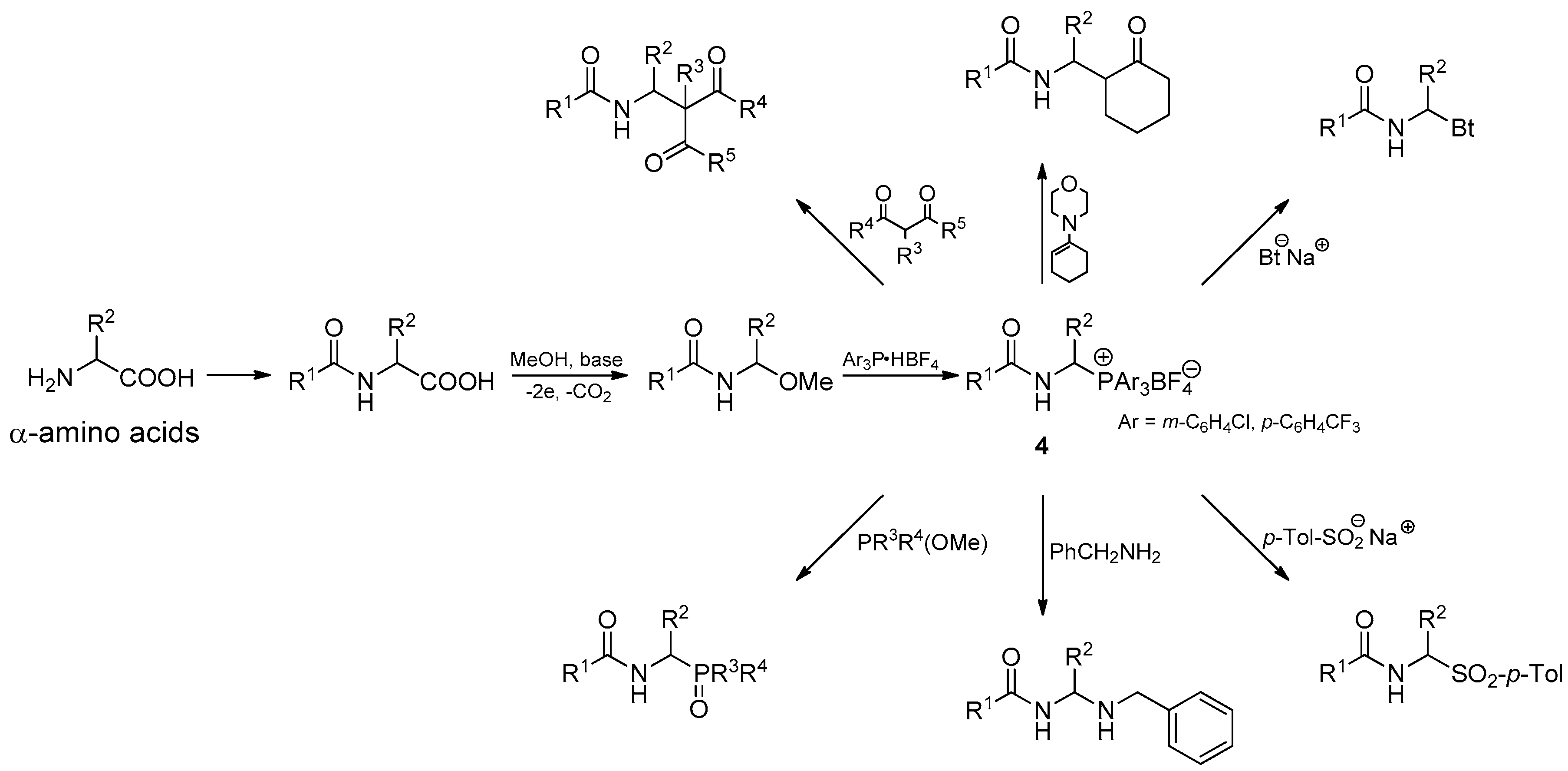
In this section, we focus our studies on the reactivity of phosphonium salts 4 with weakened Cα-P+ bond strength in α-amidoalkylation of various types of carbon- and heteronucleophiles, as shown in Scheme 2. 1-Morpholinocyclohexene and 1,3-dicarbonyl compounds, such as dimethyl malonate, diethyl malonate, and ethyl acetoacetate, are used as carbon nucleophiles. In the case of heteronucleophiles, the reaction toward benzotriazole sodium salt, p-toluenesulfinate sodium salt, benzylamine, triarylphosphines, trimethyl phosphite, dimethyl phenylphosphonite, and methyl diphenylphosphinite is examined.
The synthesis of 1-(N-acylamino)alkyltriarylphosphonium salts 4 from α-amino acids was performed according to our previously described procedure, for which the electrochemical decarboxylative α-methoxylation of N-acyl-α-amino acids and substitution of the methoxy group by triarylphosphonium moiety are crucial steps (see Scheme 2) [2,9].
2.1. 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength in the Selected Reaction of C-C Bond Formation
One of the crucial issues of organic synthesis is the formation of new C-C bonds. The possibility of an effective extension of the carbon skeleton is important in many fields, especially in medicinal chemistry, agrochemical synthesis, or in the synthesis of natural products.
Several years ago, we had proved that 1-(N-acylamino)alkyltriphenylphosphonium salts 4 (Ar = Ph) reacted quite easily with 1,3-dicarbonyl compounds in the presence of a base such as DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), DBN (1,5-diazabicyclo[4.3.0]non-5-ene), or TBD (1,5,7-triaza- bicyclo[4.4.0]dec-5-ene) under microwave irradiation at 60 °C. However, the use of organic bases (DBU, DBN, TBD) complicates the course of α-amidoalkylation reaction due to the formation of amidinium and guanidinium salts [10]. To avoid this, in the current work, we used lithium diisopropylamide (LDA) as a base to produce enolate anions from the corresponding 1,3-dicarbonyl compounds. In our protocol, α-amidoalkylation was conducted under argon, in THF, at room temperature. A THF solution of enolate anions generated using LDA was introduced into the THF solution of the phosphonium salt 4. The reaction was performed for 15 min, and the use of microwave irradiation was not necessary. In addition, it was found that the most favorable molar ratio of phosphonium salt 4: 1,3-dicarbonyl compound: LDA was 1:8:1 (compare experiments 1 and 2, Table 1).
Under the abovementioned conditions, phosphonium salts with weakened Cα-P+ bond strength 4 react with 1,3-dicarbonyl compounds, including diethyl and dimethyl malonate and ethyl acetoacetate, to give the corresponding products 6 with good yields, regardless of whether tris(4-trifluoromethylphenyl)- or tris(3-chlorophenyl)phosphonium salts were used as substrates. Only in the case of phosphonium salts with the benzyloxy carbamate protective group, the yields were significantly lower and did not exceed 30% (entry 8, Table 1).
α-Amidoalkylation of carbon nucleophiles was successfully extended to enamines. Based on the recently described protocol [8], we have demonstrated that 1-(N-acylamino)alkyltriarylphosphonium salts 4 with weakened Cα-P+ bond strength react with 1-morpholinocyclohexene in Stork-type enamination to give the expected products 8 with good yields. We have proved that this reaction can be conducted in acetonitrile, at room temperature and without using any base catalysts. The optimized reaction time was 60 min, and the molar ratio of phosphonium salt to 1-morpholinocyclohexene was 1:2. We were able to separate the major diastereoisomer from the mixture using column chromatography and crystallization technique. Furthermore, the use of phosphonium salts with the benzyloxy carbamate protective group resulted in the decrease in reaction efficiency, as already observed for reactions with 1,3-dicarbonyl compounds (see Table 2).
2.2. 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength in α-Amidoalkylation of Selected Heteronucleophiles
Applications of 1-(N-acylamino)alkyltriphenylphosphonium salts 4 (Ar = Ph) in the α-amidoalkylation of heteronucleophiles have been reported many times in the literature [5,6,7,8,10]. Usually in these types of reactions, it was necessary to use base catalysts, elevated temperature, and microwave irradiation. The effect of the use of 1-(N-acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength 4 (R = m-C6H4Cl, p-C6H4CF3) on α-amidoalkylation has not been investigated so far. Therefore, we selected several heteronucleophiles such as benzotriazole sodium salt, p-toluenesulfinate sodium salt, benzylamine, and triphenylphosphine, and examined their α-amidoalkylation by phosphonium salts with weakened Cα-P+ bond strength. As it was expected, reactions of salts with a modified structure of the phosphonium group occurred much faster and under mild conditions. In none of the described examples was it necessary to use a base catalyst. The reaction time was only 5–15 min. α-Amidoalkylation of benzotriazole sodium salt and sodium p-toluenesulfinate (entries 1–3, Table 3) was conducted at room temperature in CHCl3. Similar conditions were used for reaction with benzylamine (entries 4 and 5, Table 3). The obtained aminal derivatives 10d and 10e exhibited limited stability. Therefore, the use of excess amount of the nucleophile was required to make the alkaline environment safe for aminals.
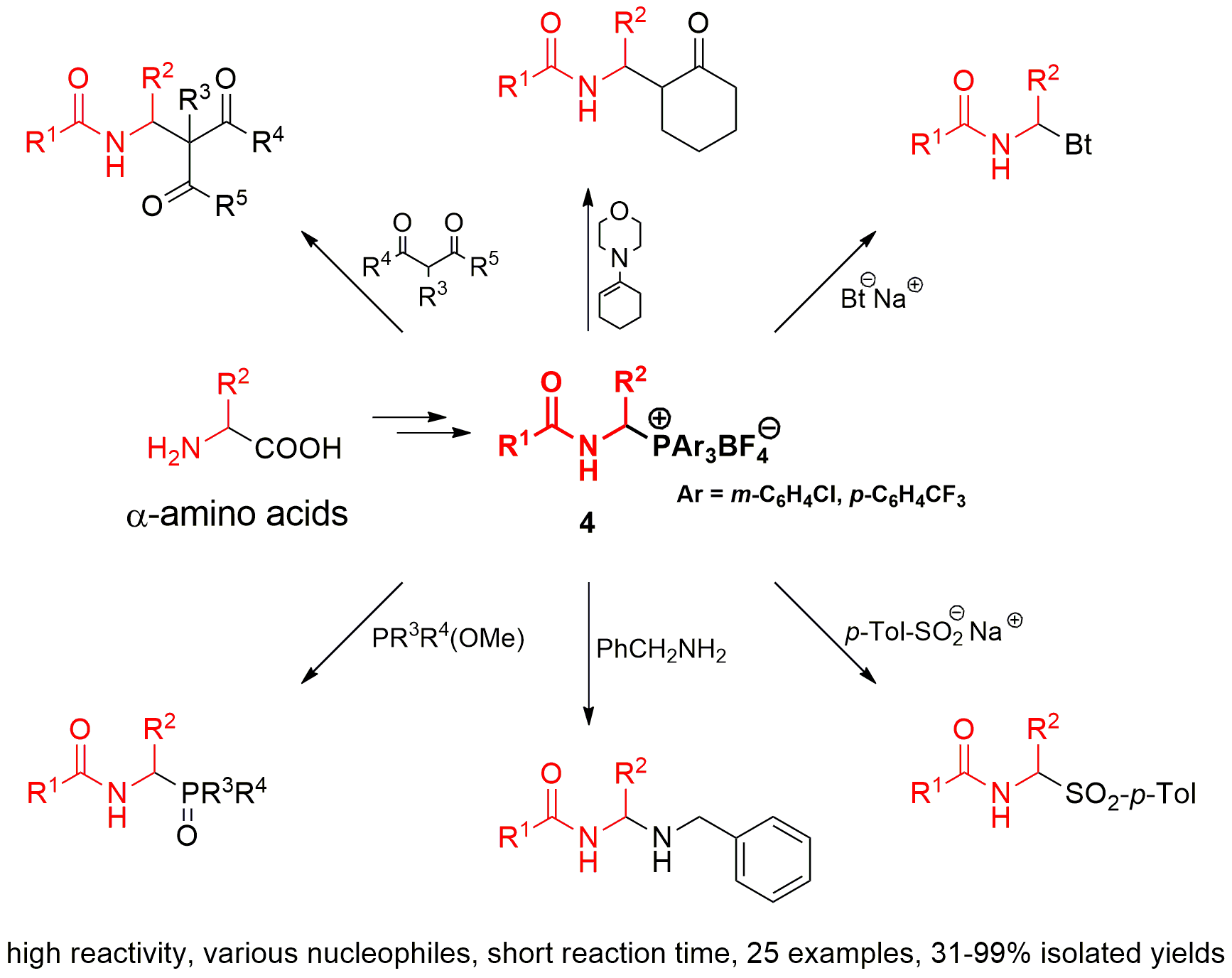
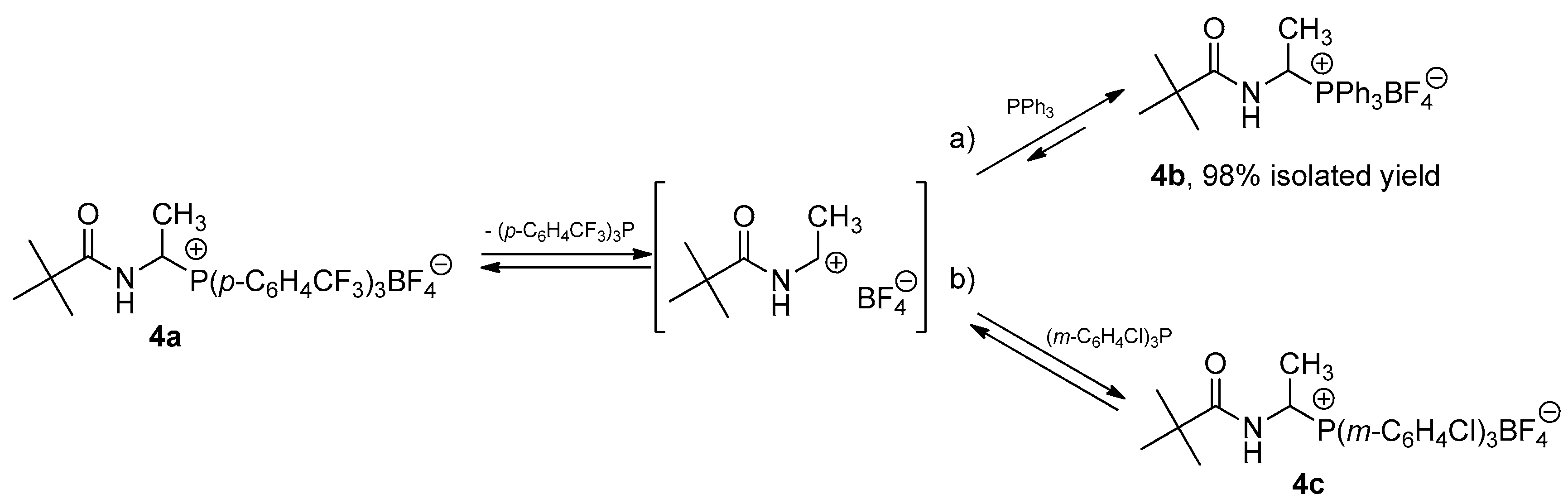
1-(N-Acylamino)alkyltriarylphosphonium salts 4 react also with triarylphosphines. For example, 1-(N-pivaloylamino)ethyltris(4-trifluoromethylphenyl)phosphonium tetrafluoroborate (4a) is completely transformed into 1-(N-pivaloylamino)ethyltriphenylphosphonium tetrafluoroborate (4b) during the reaction with triphenylphosphine. On the other hand, using tris(3-chlorophenyl)- phosphine as the nucleophile, we obtain a reaction mixture in which both 1-(N-pivaloylamino)ethyltris(4-trifluoromethylphenyl)phosphonium tetrafluoroborate (4a) and 1-(N-pivaloylamino)ethyltris(3-chlorophenyl)phosphonium tetrafluoroborate (4c) are present in a molar ratio of 1:3. A mixture of the same composition can also be obtained by the reaction of salt 4c with tris(4-trifluoromethylphenyl)phosphine. These observations may suggest the existence of some equilibrium, as described by the equations in Scheme 3. The equilibrium of the reaction is shifted toward more stable and less reactive phosphonium salts, which is evident for the reaction with triphenylphosphine (equation a, Scheme 3).
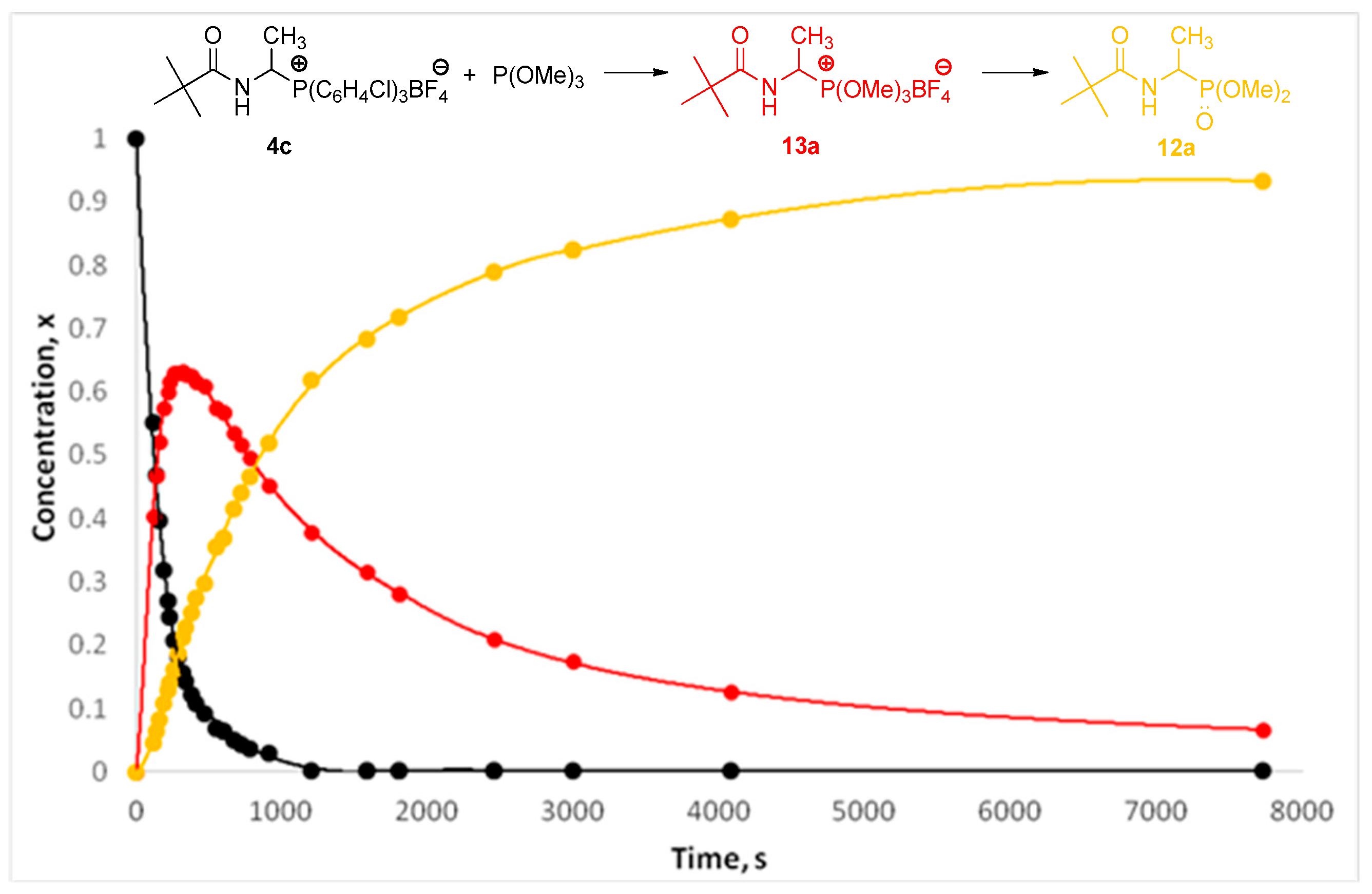
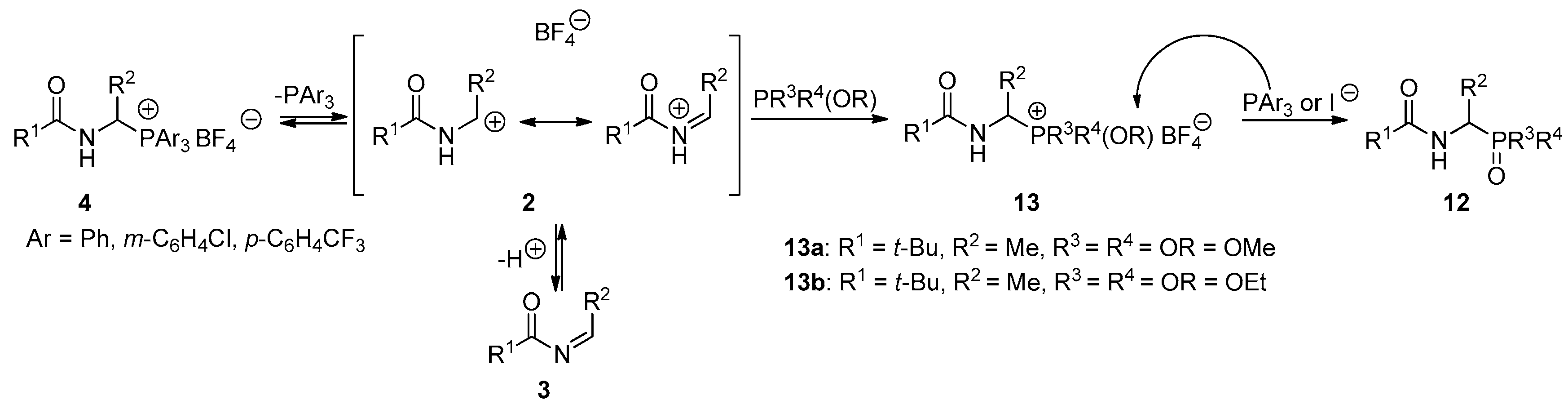
The results described above have encouraged us to extend the range of P-nucleophiles by trimethyl phosphite, dimethyl phenylphosphonite, and methyl diphenylphosphinite. It was expected that reactions with these types of phosphorus nucleophiles may occur quickly at room temperature in a non-catalytic environment, as for earlier tested nucleophiles. Surprisingly, the first experiments have shown that reactions are much slower. The analysis of reaction kinetics allowed us to explain these observations. In 2013, we proposed a plausible reaction mechanism wherein the N-acyliminium cation or N-acylimine, both generated from the 1-(N-acylamino)alkyltriphenylphosphonium salt 4 (Ar = Ph), reacts with phosphorus nucleophile to form alkoxyphosphonium salt 13—the characteristic intermediate of the Michaelis–Arbuzov reaction. The final step of the reaction is the dealkylation of the alkoxyphosphonium salt 13 and may occur directly with triphenylphosphine (see Scheme 4) [7]. Although we have not yet been able to isolate or even observe the formation of postulated intermediate product, we assume that 1-(N-acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength react with phosphorus nucleophiles in an analogous manner. To prove this, we performed the reaction of 1-(N-pivaloylamino)ethyltris(3-chlorophenyl)phosphonium tetrafluoroborate (4c) with trimethyl phosphite. The reaction progress was monitored using NMR spectroscopy. In the reaction mixture, besides substrates, two products were also detected. Changes in their concentration as a function of time were measured, as shown in Figure 1. The concentration of the first product quickly reaches its maximum and then rapidly decreases, while the concentration of the second one at the beginning of the reaction is low and then increases. The induction period, which is characteristic for the formation of the final product in the consecutive-type reaction, is very clearly visible. Detailed NMR (1H- and 31P-NMR) and HR-MS analysis confirmed that the first, fast-growing product was 1-(N-acylamino)alkyltrimethoxyphosphonium tetrafluoroborate (13a) —the postulated intermediate of the Michaelis–Arbuzov reaction. As a result of its demethylation, dimethyl 1-(N-pivaloylamino)ethanephosphonate (12a) is formed with a much slower reaction. It seems that the second step determines the overall rate of the process.
A plausible mechanism that explains the described kinetic facts is shown in Scheme 4 and is consistent with the mechanism proposed earlier in 2013. The only difference is that the generation of N-acyliminium cation from 1-(N-acylamino)alkyltriphenylphosphonium salt 4 (Ar = Ph) is more difficult. On the other hand, the highnucleophilicity of triphenylphosphine facilitates dealkylation, which makes the first step crucial for the course of the entire reaction. In the case of 1-(N-acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength 4 (R = m-C6H4Cl, p-C6H4CF3), the first step is easier, which facilitates the formation of alkoxyphosphonium salts 13. However, due to the lower nucleophilicity of phosphines with electron-withdrawing substituents [P(m-C6H4Cl)3, P(p-C6H4CF3)3], the rate of dealkylation drops. To overcome this drawback, we decided to add to the reaction mixture the substoichiometric amounts of methyltriphenylphosphonium iodide as the dealkylating agent (molar ratio of 1:0.25). This protocol allowed us to obtain a series of phosphorus analogs of α-amino acids 12 (N-acyl-α-aminoalkanephosphonates 12a–c, N-acyl-α-aminoalkanephosphinate (12d) and N-acyl-α-aminoalkylphosphine oxide (12e)) with good or very good yields. Usually, reactions occur efficiently at room temperature. Only in the reaction with dimethyl phenylphosphonite, it was necessary to raise the temperature to 60 °C. At room temperature, the reaction is very slow even after the addition of methyltriphenylphosphonium iodide (see Table 4).
Finally, we tried to isolate and fully characterize the intermediate of the Michaelis–Arbuzov reaction 13. To this end, we conducted the reaction of 1-(N-pivaloylamino)ethyltris(3-chlorophenyl)phosphonium tetrafluoroborate (4c) with trimethyl phosphite. Unfortunately, the expected 1-(N-pivaloylamino)ethyltrimethoxyphosphonium tetrafluoroborate (13a, Scheme 4) was too reactive and attempts to isolate the analytically pure sample failed. To increase the stability of the intermediate 13, we used triethyl phosphite as the phosphorus nucleophile. This allowed us to isolate 1-(N-pivaloylamino)ethyltriethoxyphosphonium tetrafluoroborate (13b) and determine its basic physicochemical properties.
3. Experimental Section
3.1. General Information
Melting points were determined in capillaries and were uncorrected. IR spectra were measured on an FT-IR spectrophotometer (ATR method). 1H- and 13C-NMR were recorded at operating frequencies of 400 and 100 MHz, respectively, using TMS as the resonance shift standard. 31P-NMR spectra were recorded at an operating frequency of 161.9 MHz without the resonance shift standard, with respect to H3PO4 as zero ppm. All chemical shifts (δ) are reported in ppm and coupling constants (J) in Hz. High-resolution mass spectrometry (HR-MS) analyses were performed on a Xevo G2 Q-TOF mass spectrometer (Waters, Milford, MA, USA) equipped with an ESI source operating in the positive ion mode. The accurate mass and composition of molecular ion adducts were calculated using the MassLynx software incorporated within the instrument.
1H, 13C, and 31P NMR spectra of all new compounds 6, 8, 10, 12, 13 as well as the summary table in which we compare conditions and yields for reactions of 1-(N-acylamino)alkyltriphenylphosphonium salts (former studies) and 1-(N-acylamino)alkyltriarylphosphonium salts (the current work) with selected nucleophiles, are placed in the Supplementary data.
3.2. Syntheses
3.2.1. Substrate Synthesis
The electrolysis was conducted in an undivided glass electrolyzer (85 cm3) equipped with a thermostatic jacket, a magnetic stirrer, a concentrically arranged, cylindrical Pt mesh anode (47 cm2) and cathode (44 cm2). To the thus prepared electrolyzer, N-acyl-α-amino acid (3.0 mmol), SiO2-Pip (200 mg), and methanol (30 cm3) were added. The electrochemical decarboxylative α-methoxylation was executed while stirring, at a constant current of 0.15 A, at 10 °C until a 3.75 F/mol charge had passed. Then, SiO2-Pip was filtered off, and methanol was evaporated under reduced pressure to obtain N-(1-methoxyalkyl)amide, which was used in the next reaction without further purification.
To a solution of triarylphosphine (1 mmol) in DCM (2 mL), a tetrafluoroboric acid diethyl ether complex (HBF4·Et2O, 136 µl, 161.9 mg, 1 mmol) was added at 0 °C. The reaction mixture was stirred for 2 h at room temperature. Thereafter, N-(1-methoxyalkyl)amide (1 mmol) was added and stirring was continued for 15 min. Evaporation of the solvent yielded the crude 1-(N-acylamino) alkyltriarylphosphonium salt 4, which was used in the next reaction without further purification.
3.2.2. Reactivity of 1-(N-acylamino)alkyltriarylphosphonium Salts 4 toward Carbon Nucleophiles
Reaction of 1-(N-acylamino)alkyltriarylphosphonium salts4with C-H acids—general procedure
A solution of a nucleophile (1.6 mmol), THF (0.5 mmol), and base LDA (2.0 M solution in THF, 0.1 cm3) was stirred under argon atmosphere. After a few minutes, this mixture was relegated to a solution of 1-(N-acylamino)alkyltriarylphosphonium salt (0.2 mmol) in THF (0.5 cm3) under argon atmosphere. The resulting reaction mixture was stirred for 15 min at room temperature, and then it was evaporated under reduced pressure. The product was separated using column chromatography [toluene/AcOEt 5:1 v/v].
Diethyl 1-(pivaloylamino)etylpropanedioate (6a) [10]. Colorless oil (38.5 mg, 67% yield). 1H-NMR (CDCl3) δ 6.80 (d, J = 7.5 Hz, 1H, NH), 4.74–4.62 (m, 1H, CαH), 4.29–4.21 (m, 2H, OCH2), 4.21–4.12 (m, 2H, OCH2), 3.57 (d, J = 3.4 Hz, 1H, CH), 1.32–1.25 (m, 9H, CH3 and 2 × OCH2CH3), 1.17 (s, 9H, t-Bu) ppm; 13C-NMR (CDCl3) δ 177.6 (NHC = O), 168.7 (C = O), 167.7 (C = O), 61.7 (OCH2), 61.5 (OCH2), 55.6 (CαH), 44.2 (CH), 38.6 (C(CH3)3), 27.4 (C(CH3)3), 19.0 (CH3), 14.0 (OCH2CH3) ppm; IR (ATR) 2977, 1731, 1647, 1510, 1175 cm−1.
Ethyl 2-acetyl-3-(pivaloylamino)butanoate (6b). A mixture of two disatereoisomers in a molar ratio of 1.4:1 (31.9 mg, 62% yield). 1H-NMR (CDCl3) δ 6.92 (d, J = 8.0 Hz, 1H, NH)a, 6.41 (d, J = 7.8 Hz, 1H, NH)a, 4.85–4.74 (m, 1H, CαH)a, 4.68–4.57 (m, 1H, CαH)a, 4.33–4.10 (m, 2H, OCH2)b, 3.72 (d, J = 4.9 Hz, 1H, CH)a, 3.65 (d, J = 3.6 Hz, 1H, CH)a, 2.26 (s, 3H, CH3CO)a, 2.25 (s, 3H, CH3CO)a, 1.35–1.20 (m, 6H, CH3 and OCH2CH3)b, 1.16 (s, 9H, t-Bu)a, 1.15 (s, 9H, t-Bu)a ppm; 13C-NMR (CDCl3) δ 203.4a (CH3C = O), 202.1a (CH3C = O), 178.0a (NHC = O), 177.7a (NHC = O), 169.5a (C = O), 168.2a (C = O), 63.4a (CαH), 62.2a (CαH), 61.6a (OCH2), 61.4a (OCH2), 44.3a (CH), 43.4a (CH), 38.6a (C(CH3)3), 38.6a (C(CH3)3), 30.4a (CH3CO), 29.0a (CH3CO), 27.4a (C(CH3)3), 27.3a (C(CH3)3), 19.7a (CH3), 18.7a (CH3), 14.1a (OCH2CH3), 14.1a (OCH2CH3) ppm; IR (ATR) 3318, 2971, 1730, 1635, 1534, 1301, 1190 cm−1. HRMS (TOF-ESI) calcd for C13H24NO4 [M + H]+ 258.1705, found 258.1701. a Separate signals from both diastereoisomers. b Overlapping signals of both diastereoisomers.
Ethyl 2-acetyl-5-methyl-3-(phenylacetylamino)hexanoate (6c). A mixture of two disatereoisomers in a molar ratio of 1.25:1 (55.4 mg, 83% yield). 1H-NMR (CDCl3) δ 7.39–7.19 (m, 5H, Ph)a, 6.43 (d, J = 9.7 Hz, 1H, NH)b, 6.02 (d, J = 9.5 Hz, 1H, NH)b, 4.77–4.68 (m, 1H, CαH)b, 4.63–4.54 (m, 1H, CαH)b, 4.21–4.07 (m, 2H, OCH2)a, 3.64 (d, J = 4.9 Hz, 1H, CH)b, 3.60 (d, J = 3.7 Hz, 1H, CH)b, 3.51 (s, 2H, PhCH2)a, 2.23 (s, 3H, CH3CO)b, 2.19 (s, 3H, CH3CO)b, 1.56–1.37 (m, 2H, CH2)a, 1.28–1.14 (m, 4H, CH and OCH2CH3)a, 0.95–0.80 (m, 6H, 2 × CH3)a ppm; 13C-NMR (CDCl3) δ 203.2b (CH3C = O), 202.2b (CH3C = O), 170.6b (NHC = O), 170.4b (NHC = O), 169.1b (C = O), 168.1b (C = O), aromatic carbons: 134.7b, 134.6b, 129.3b, 129.3b, 128.9b, 128.8b, 127.2b, 127.2b, 62.4b (CαH), 61.8b (CαH), 61.6b (OCH2), 61.3b (OCH2), 47.1b (PhCH2), 46.2b (PhCH2), 43.9b (CH), 43.8b (CH), 42.7b (CH2), 41.9b (CH2), 30.3b (CH3CO), 29.1b (CH3CO), 25.1b (CH), 25.1b (CH), 23.1b(CH3), 22.7b (CH3), 22.1b (CH3), 21.7b (CH3), 14.0b (OCH2CH3), 14.0b (OCH2CH3) ppm; IR (ATR) 3275, 2956, 1740, 1648, 1496, 1260, 1144 cm−1. HRMS (TOF-ESI) calcd for C19H28NO4 [M + H]+ 334.2018, found 334.2009. a Overlapping signals of both diastereoisomers. b Separate signals from both diastereoisomers.
Dimethyl 3-methyl-1-(phenylacetylamino)butylpropanedioate (6d). Colorless crystals (42.3 mg, 63% yield), mp 78–80 °C. 1H-NMR (CDCl3) δ 7.41–7.18 (m, 5H, Ph), 6.25 (d, J = 9.6 Hz, 1H, NH), 4.68–4.59 (m, 1H, CαH), 3.69 (s, 3H, OCH3), 3.62 (s, 3H, OCH3), 3.55 (d, J = 3.9 Hz, 1H, CH), 3.52 (s, 2H, PhCH2), 1.54–1.39 (m, 2H, CH2), 1.28–1.19 (m, 1H, CH), 0.90 (d, J = 6.5 Hz, 3H, CH3), 0.86 (d, J = 6.6 Hz, 3H, CH3) ppm; 13C-NMR (CDCl3) δ 170.3 (NHC = O), 168.6 (C = O), 168.0 (C = O), aromatic carbons: 134.8, 129.3, 128.8, 127.1, 54.6 (CαH), 52.6 (OCH3), 52.3 (OCH3), 46.8 (PhCH2), 43.9 (CH), 42.1 (CH2), 25.0 (CH), 22.9 (CH3), 21.9 (CH3) ppm; IR (ATR) 3283, 2954, 1736, 1648, 1454, 1263 cm−1. HRMS (TOF-ESI) calcd for C18H26NO5 [M + H]+ 336.1811, found 336.1805.
Diethyl 3-methyl-1-(phenylacetylamino)butylpropanedioate (6e). Colorless crystals (37.8 mg, 52% yield), mp 93.5–95.5 °C. 1H-NMR (CDCl3) δ 7.42–7.19 (m, 5H, Ph), 6.30 (d, J = 9.6 Hz, 1H, NH), 4.68–4.60 (m, 1H, CαH), 4.30–4.00 (m, 4H, 2 × OCH2), 3.52 (s, 2H, PhCH2), 3.52 (d, J = 2.8 Hz, 1H, CH), 1.57–1.37 (m, 2H, CH2), 1.36–1.06 (m, 7H, CH and 2 × OCH2CH3), 0.90 (d, J = 6.5 Hz, 3H, CH3), 0.86 (d, J = 6.6 Hz, 3H, CH3) ppm; 13C-NMR (CDCl3) δ 170.2 (NHC = O), 168.3 (C = O), 167.7 (C = O), aromatic carbons: 134.9, 129.3, 128.8, 127.1, 61.7 (OCH2), 61.4 (OCH2), 54.9 (CαH), 46.8 (PhCH2), 43.9 (CH), 42.2 (CH2), 25.0 (CH), 22.9 (CH3), 22.0 (CH3), 14.0 (OCH2CH3), 13.9 (OCH2CH3) ppm; IR (ATR) 3362, 2964, 1745, 1656, 1533, 1345, 1144 cm−1. HRMS (TOF-ESI) calcd for C20H30NO5 [M + H]+ 364.2124, found 364.2122.
Reaction of 1-(N-acylamino)alkyltriarylphosphonium salts4with 1-morpholinocyclohexene—general procedure
To a solution of 1-(N-acylamino)alkyltriarylphosphonium salt 4 (0.25 mmol) in MeCN (1 cm3), 1-morpholinocyclohexene (84.1 µL, 83.7 mg, 0.5 mmol) was added. After 1 h of stirring at room temperature, an aqueous solution of citric acid (20%) (1.125 cm3) was added. Stirring was continued for 45 min, and then a saturated solution of KHCO3 was added. Thereafter, the mixture was extracted with DCM (5 × 3 cm3), and the organic layer was combined and dried over MgSO4. Then, the solvent was evaporated under reduced pressure and the crude product was purified using column chromatography [toluene/EtOAc 2:1 v/v (8a), 5:1 (8b, 8c, 8e) or 10:1 v/v (8d)].
N-[1-(2-oxocyclohexyl)ethyl]pivalamide (8a). A mixture of two disatereoisomers in a molar ratio of 3.5:1 (35.5 mg, 63% yield). The major diastereoisomers was isolated using column chromatography [toluene/EtOAc 2:1] and crystallization from toluene. Colorless crystals, mp 124–126 °C. 1H-NMR (CDCl3) δ 6.51 (d, J = 8.1 Hz, 1H, NH), 4.19–4.10 (m, 1H, CαH), 2.56–2.48 (m, 1H, CH), 2.39–2.30 (m, 2H, CH2), 2.11–2.01 (m, 2H, CH2), 1.91–1.83 (m, 1H, CHH), 1.74–1.57 (m, 3H, CHH and CH2), 1.22 (d, J = 7.0 Hz, 3H, CH3), 1.17 (s, 9H, t-Bu) ppm; 13C-NMR (CDCl3) δ 214.2 (C = O), 178.0 (NHC = O), 55.4 (CαH), 45.6 (CH), 43.1 (CH2), 38.7 (C(CH3)3), 32.7 (CH2), 28.4 (CH2), 27.6 (C(CH3)3), 24.8 (CH2), 20.0 (CH3) ppm; IR (ATR) 3339, 2961, 2869, 1706, 1627, 1526, 1305, 1209, 1117 cm−1. HRMS (TOF-ESI) calcd for C13H24NO2 [M + H]+ 226.1807, found 226.1800.
N-[1-(2-oxocyclohexyl)-3-methylbutyl]phenylacetamide (8b). A mixture of two disatereoisomers in a molar ratio of 3.2:1 (46.7 mg, 62% yield). The major diastereoisomers was isolated using column chromatography [toluene/EtOAc 5:1] and crystallization from toluene. Colorless crystals, mp 115.5–117.5 °C. 1H-NMR (CDCl3) δ 7.42–7.09 (m, 5H, Ph), 6.13 (d, J = 9.7 Hz, 1H, NH), 4.10–3.99 (m, 1H, CαH), 3.51 (s, 2H, PhCH2), 2.49–2.40 (m, 1H, CH), 2.31–2.18 (m, 2H, CH2), 2.07–1.96 (m, 2H, CH2), 1.86–1.75 (m, 1H, CH), 1.65–1.37 (m, 5H, CH and 2 × CH2), 1.21–1.09 (m, 1H, CH), 0.86 (d, J = 6.5 Hz, 3H, CH3), 0.83 (d, J = 6.6 Hz, 3H, CH3) ppm; 13C-NMR (CDCl3) δ 213.5 (C = O), 170.7 (NHC = O), aromatic carbons: 135.3, 129.1, 128.8, 127.1, 54.8 (CαH), 48.7 (PhCH2), 44.2 (CH), 43.1 (CH2), 43.1 (CH2), 32.6 (CH2), 28.3 (CH2), 25.2 (CH), 25.0 (CH2), 23.1 (CH3), 22.0 (CH3) ppm; IR (ATR) 3273, 2954, 2865, 1702, 1633, 1552, 1455, 1315, 1138 cm−1. HRMS (TOF-ESI) calcd for C19H28NO2 [M + H]+ 302.2120, found 302.2115.
Benzyl N-[1-(2-oxocyclohexyl)-3-methylbutyl]carbamate (8c). Only one disatereoisomer was detected and isolated. Colorless oil (31.7 mg, 40% yield). 1H-NMR (CDCl3) δ 7.42–7.27 (m, 5H, Ph), 5.42 (d, J = 10.1 Hz, 1H, NH), 5.09 (d, J = 12.5 Hz, 1H, PhCHHO), 5.06 (d, J = 12.4 Hz, 1H, PhCHHO), 3.83–3.69 (m, 1H, CαH), 2.54–2.44 (m, 1H, CH), 2.38–2.23 (m, 2H, CH2), 2.15–1.99 (m, 2H, CH2), 1.93–1.82 (m, 1H, CHH), 1.74–1.55 (m, 5H, CHH and 2 × CH2), 1.28–1.16 (m, 1H, CH), 0.91 (d, J = 6.4 Hz, 3H, CH3), 0.89 (d, J = 6.5 Hz, 3H, CH3) ppm; 13C-NMR (CDCl3) δ 213.1 (C = O), 156.5 (NHC = O), aromatic carbons: 136.8, 128.4, 127.9, 127.9, 66.5 (PhCH2O), 54.7 (CαH), 50.8 (CH), 43.4 (CH2), 43.2 (CH2), 32.3 (CH2), 28.1 (CH2), 25.1 (CH2), 25.1 (CH), 23.2 (CH3), 21.9 (CH3) ppm; IR (ATR) 3439, 3333, 2952, 2866, 1699, 1499, 1213, 1052 cm−1. HRMS (TOF-ESI) calcd for C19H28NO3 [M + H]+ 318.2069, found 318.2061.
Benzyl N-[1-(2-oxocyclohexyl)-2-phenylethyl]carbamate (8d). A mixture of two disatereoisomers in a molar ratio of 13.4:1 (29.0 mg, 33% yield). The major diastereoisomers was isolated using column chromatography [toluene/EtOAc 10:1]. Colorless crystals, mp 99–101 °C. 1H-NMR (CDCl3) δ 7.42–7.06 (m, 10H, 2 × Ph), 5.65 (d, J = 10.0 Hz, 1H, NH), 5.07 (d, J = 12.3 Hz, 1H, PhCHHO), 5.00 (d, J = 12.4 Hz, 1H, PhCHHO), 3.95–3.87 (m, 1H, CαH), 2.99 (dd, J1 = 13.5, J2 = 7.3 Hz, 1H, PhCHH), 2.92 (dd, J1 = 13.5, J2 = 8.5 Hz, 1H, PhCHH), 2.51–2.41 (m, 1H, CH), 2.40–2.31 (m, 1H, CH), 2.29–2.17 (m, 1H, CH), 2.09–1.92 (m, 2H, CH2), 1.87–1.78 (m, 1H, CH), 1.77–1.51 (m, 3H, CH and CH2) ppm; 13C-NMR (CDCl3) δ 213.5 (C = O), 156.4 (NHC = O), aromatic carbons: 138.6, 136.7, 129.1, 128.5, 128.4, 127.9, 127.8, 126.4, 66.4 (PhCH2O), 54.2 (CαH), 51.9 (CH), 43.2 (CH2), 40.3 (PhCH2), 32.6 (CH2), 28.1 (CH2), 25.0 (CH2) ppm; IR (ATR) 3308, 2929, 1721, 1697, 1541, 1341, 1242, 1094, 1051 cm−1. HRMS (TOF-ESI) calcd for C22H26NO3 [M + H]+ 352.1913, found 352.1913.
Benzyl N-[1-(2-oxocyclohexyl)-2-tert-butoxyethyl]carbamate (8e). Only one disatereoisomer was detected and isolated. Colorless oil (26.9 mg, 31% yield). 1H-NMR (CDCl3) δ 7.38–7.28 (m, 5H, Ph), 5.57 (d, J = 9.7 Hz, 1H, NH), 5.11 (d, J = 12.3 Hz, 1H, PhCHHO), 5.06 (d, J = 12.3 Hz, 1H, PhCHHO), 3.89–3.79 (m, 1H, CαH), 3.44 (d, J = 7.1 Hz, 2H, CH2Ot-Bu), 2.90–2.82 (m, 1H, CH), 2.38–2.27 (m, 2H, CH2), 2.09–2.00 (m, 2H, CH2), 1.92–1.85 (m, 1H, CHH), 1.78–1.64 (m, 3H, CHH and CH2), 1.12 (s, 9H, t-Bu) ppm; 13C-NMR (CDCl3) δ 213.8 (C = O), 156.4 (NHC = O), aromatic carbons: 136.6, 128.5, 128.0, 73.1 (OC(CH3)3), 66.6 (PhCH2O), 62.1 (CH2Ot-Bu), 52.5 (CαH), 50.0 (CH), 42.8 (CH2), 31.9 (CH2), 28.1 (CH2), 27.5 (OC(CH3)3), 24.9 (CH2) ppm; IR (ATR) 3432, 2972, 2935, 2867, 1701, 1499, 1363, 1195, 1057 cm−1. HRMS (TOF-ESI) calcd for C20H30NO4 [M + H]+ 348.2175, found 348.2173.
3.2.3. Reactivity of 1-(N-Acylamino)alkyltriarylphosphonium Salts 4 toward Heteronucleophiles
Reaction of 1-(N-acylamino)alkyltriarylphosphonium salts4with benzotriazole sodium salt—general procedure
To a solution of 1-(N-acylamino)alkyltriarylphosphonium salt 4 (0.5 mmol) in CHCl3 (1 cm3), benzotriazole sodium salt (0.5 mmol) was added. The reaction mixture was stirred at room temperature for 15 min. Thereafter, it was filtered through a fluted filter and the filtrate was evaporated under reduced pressure. The residue was crystallized from toluene.
N-[1-(Benzotriazol-1-yl)ethyl]pivalamide (10a) [6]. Colorless crystals (121.9 mg, 99% yield), mp 142–144 °C. 1H-NMR (CDCl3) δ 8.06–8.02 (m, 1H, aromatic), 7.85–7.79 (m, 1H, aromatic), 7.54–7.48 (m, 1H, aromatic), 7.41–7.35 (m, 1H, aromatic), 6.88 (dq, J1 = 9.1, J2 = 6.7 Hz, 1H, CαH), 6.63 (d, J = 8.8 Hz, 1H, NH), 2.04 (d, J = 6.7 Hz, 3H, CH3), 1.15 (s, 9H, t-Bu) ppm; 13C-NMR (100 MHz, CDCl3) δ 178.2 (C = O), aromatic carbons: 145.5, 132.4, 127.7, 124.4, 119.5, 110.4, 58.7 (CαH), 38.7 (C(CH3)3), 27.2 (C(CH3)3), 20.7 (CH3) ppm; IR (ATR) 3346, 2969, 1668, 1512, 1193, 1152, 1065 cm−1.
Benzyl N-[1-(benzotriazol-1-yl)-2-phenylethyl]carbamate (10b) [6]. Colorless crystals (130.4 mg, 70% yield), mp 117.5–119.5 °C. 1H-NMR (CDCl3) δ 8.00 (d, J = 8.4 Hz, 1H, aromatic), 7.54 (d, J = 7.5 Hz, 1H, aromatic), 7.42–7.35 (m, 1H, aromatic), 7.33–7.27 (m, 4H, aromatic), 7.27–7.20 (m, 2H, aromatic), 7.19–7.12 (m, 3H, aromatic), 7.11–7.04 (m, 2H, aromatic), 6.71–6.58 (m, 1H, CαH), 6.02 (d, J = 8.9 Hz, 1H, NH), 5.10 (d, J = 12.4 Hz, 1H, PhCHHO), 4.97 (d, J = 12.2 Hz, 1H, PhCHHO), 3.80–3.68 (m, 1H, PhCHH), 3.63 (dd, J1 = 13.8, J2 = 6.6 Hz, 1H, PhCHH) ppm; 13C-NMR (CDCl3) δ 160.3 (C = O), aromatic carbons: 145.5, 136.0, 134.7, 131.9, 129.1, 128.7, 128.5, 128.4, 128.1, 127.7, 127.3, 124.1, 119.7, 109.7, 67.5 (PhCH2O), 65.8 (CαH), 41.1 (PHCH2) ppm; IR (ATR) 3177, 3008, 1712, 1548, 1280, 1261, 1244, 1195, 1046, 1022 cm−1.
Reaction of 1-(N-pivaloylamino)ethyltris(4-trifluoromethylphenyl)phosphonium tetrafluoroborate4awith sodium p-toluenesulfinate
To a solution of 1-(N-pivaloylamino)ethyltris(4-trifluoromethylphenyl)phosphonium tetrafluoroborate 4a (340.7 mg, 0.5 mmol) in CHCl3 (1 cm3), sodium p-toluenesulfinate (89.1 mg, 0.5 mmol) was added. The reaction mixture was stirred at room temperature for 15 min. Thereafter, it was filtered through a fluted filter and the filtrate was evaporated under reduced pressure. The residue was crystallized from toluene.
N-[1-(p-Toluenesulfonyl)ethyl]pivalamide (10c) [5]. Colorless crystals (124.7 mg, 88% yield), mp 143–145 °C. 1H-NMR (CDCl3) δ 7.77 (d, J = 8.3 Hz, 2H, aromatic), 7.33 (d, J = 7.9 Hz, 2H, aromatic), 5.98 (d, J = 10.2 Hz, 1H, NH), 5.41 (dq, J1 = 10.2, J2 = 7.0 Hz, 1H, CαH), 2.42 (s, 3H, CH3), 1.62 (d, J = 7.0 Hz, 3H, CH3), 1.01 (s, 9H, t-Bu) ppm; 13C-NMR (CDCl3) δ 176.9 (C = O), aromatic carbons: 145.1, 133.5, 129.6, 129.1, 64.3 (CαH), 38.7 (C(CH3)3), 27.2 (C(CH3)3), 21.6 (CH3), 13.2 (CH3) ppm; IR (ATR) 3372, 2973, 1677, 1516, 1308, 1287, 1135, 1083, 1016, 724 cm−1.
Reaction of 1-(N-acylamino)alkyltriarylphosphonium salts4with benzylamine—general procedure
To a stirred solution of 1-(N-acylamino)alkyltriarylphosphonium salt 4 (0.25 mmol) in DCM (1 cm3), benzylamine (109.4 µl, 110 mg, 1 mmol) was added dropwise. Stirring was continued for 5 min at room temperature. Thereafter, the mixture was evaporated under reduced pressure and dried. The residue was purified using column chromatography [DCM/MeOH/Et3N, 5:1:0.2 v/v/v].
N-[1-(Benzylamino)ethyl]pivalamide (10d). Yellow crystals (53.3 mg, 91% yield), mp 56.5–58.5 °C. 1H-NMR (CDCl3) δ 7.35–7.29 (m, 4H, aromatic), 7.26–7.21 (m, 1H, aromatic), 5.67 (d, J = 6.7 Hz, 1H, NH), 4.90 (qd, J1 = 6.2, J2 = 1.6 Hz, 1H, CαH), 3.77 (s, 2H, PhCH2), 1.92 (br s, 1H, NH), 1.32 (d, J = 6.2 Hz, 3H, CH3), 1.17 (s, 9H, t-Bu) ppm; 13C-NMR (CDCl3) δ 178.1 (C = O), aromatic carbons: 140.1, 128.4, 127.9, 126.9, 60.9 (CαH), 49.8 (PhCH2), 38.6 (C(CH3)3), 27.4 (C(CH3)3), 21.6 ppm (CH3); IR (ATR) 3328, 2971, 1622, 1527, 1475, 1144, 1097 cm−1. HRMS (TOF-ESI) calcd for C14H23N2O [M + H]+ 235.1810, found 235.1802.
N-[1-(Benzylamino)-3-methylbutyl]phenylacetamide (10e). Colorless crystals (42.7 mg, 55% yield), mp 93–95 °C. 1H-NMR (CDCl3) δ 7.59–6.96 (m, 10H, 2 × Ph), 5.33 (d, J = 8.3 Hz, 1H, NH), 4.82–4.75 (m, 1H, CαH), 3.68 (s, 2H, PhCH2NH), 3.54 (s, 2H, PhCH2), 1.66 (br s, 1H, NH), 1.64–1.53 (m, 1H, CH), 1.45–1.28 (m, 2H, CH2), 0.87 (d, J = 6.6 Hz, 3H, CH3), 0.84 (d, J = 6.6 Hz, 3H, CH3) ppm; 13C-NMR (CDCl3) δ 170.9 (C = O), aromatic carbons: 140.3, 135.0, 129.3, 129.0, 128.4, 128.0, 127.3, 126.9, 63.3 (CαH), 49.6 (PhCH2), 44.5 (PhCH2), 44.1 (CH2), 24.9 (CH), 22.6 (CH3), 22.5 (CH3) ppm; IR (ATR) 3306, 2962, 2946, 1632, 1518, 1144, 1009 cm−1. HRMS (TOF-ESI) calcd for C20H27N2O [M + H]+ 311.2123, found 311.2121.
Reaction of 1-(N-pivaloylamino)ethyltris(4-trifluoromethylphenyl)phosphonium tetrafluoroborate4awith triphenylphosphine
To a solution of 1-(N-pivaloylamino)ethyltris(4-trifluromethylphenyl)phosphonium tetrafluoroborate 4a (102.2 mg, 0.15 mmol) in DCM (1 cm3), triphenylphosphine (39.3 mg, 0.15 mmol) was added. The homogeneous mixture was allowed to react at room temperature for 5 min, and 1-(N-pivaloylamino)ethyltriphenylphosphonium tetrafluoroborate (4b) [9] was precipitated with Et2O, separated by decantation, and dried under reduced pressure.
1-(N-Pivaloylamino)ethyltriphenylphosphonium tetrafluoroborate (4b). Colorless crystals (70.2 mg, 98% yield), mp 160.5–162.5 °C. 1H-NMR (CDCl3) δ 7.89–7.64 (m, 16H, 3 × Ph and NH), 5.82–5.73 (m, 1H, CαH), 1.72 (dd, J1 = 17.8, J2 = 7.4 Hz, 3H, CH3), 0.91 (s, 9H, t-Bu) ppm; 13C-NMR (CDCl3) δ 179.7 (d, J = 2.3 Hz, C = O), aromatic carbons: 134.8 (d, J = 3.0 Hz), 134.5 (d, J = 9.2 Hz), 130.0 (d, J = 12.3 Hz), 118.4 (d, J = 82.5 Hz), 45.0 (d, J = 53.4 Hz, CαH), 38.5 (C(CH3)3), 26.8 (C(CH3)3), 17.4 (d, J = 4.6 Hz, CH3) ppm; 31P NMR (161.9 MHz, CDCl3) δ 29.2 ppm; IR (ATR) 3373, 1684, 1516, 1447, 1136, 1040 cm−1.
Reaction of 1-(N-acylamino)alkyltriarylphosphonium salts4with triarylphosphines—NMR scale
To a solution of 1-(N-acylamino)alkyltriarylphosphonium salt (0.025 mmol) in CDCl3 (0.65 cm3), the corresponding triarylphosphine (0.025 mmol) was added. Reactions were conducted in NMR tubes at 26 °C, and their course was monitored using NMR spectroscopy.
Reaction of 1-(N-acylamino)alkyltriarylphosphonium salts4with phosphorus nucleophiles in Michaelis–Arbuzov type reaction—general procedure
To a stirred solution of 1-(N-acylamino)alkyltriarylphosphonium salt 4 (0.25 mmol) and methyltriphenylphosphonium iodide (25.3 mg, 0.0625 mmol) in DCM (2 cm3), phosphorus nucleophile (0.375 mmol) was added, and the mixture was stirred for 3 h at room temperature (12a–c and 12e) or for 2 h at 60 °C (12d). The mixture was evaporated under reduced pressure and the residue was extracted with toluene (3 × 1 cm3) at 50 °C. The solvent was evaporated under reduced pressure, and the product was isolated using column chromatography [DCM/MeOH 20:1 v/v].
Dimethyl 1-(N-pivaloylamino)ethanephosphonate (12a) [7]. Colorless crystals (50.4 mg, 85% yield), mp 125.5–127.5 °C. 1H-NMR (CDCl3) δ 5.96 (d, J = 8.1 Hz, 1H, NH), 4.65–4.49 (m, 1H, CαH), 3.78 (d, J = 4.4 Hz, 3H, OCH3), 3.75 (d, J = 4.3 Hz, 3H, OCH3), 1.38 (dd, J1 = 16.8, J2 = 7.4 Hz, 3H, CH3), 1.21 (s, 9H, t-Bu) ppm; 13C-NMR (CDCl3) δ 177.6 (d, J = 5.1 Hz, C = O), 53.1 (d, J = 7.0 Hz, OCH3), 52.9 (d, J = 6.5 Hz, OCH3), 40.1 (d, J = 156.4 Hz, CαH), 38.6 (C(CH3)3), 27.3 (C(CH3)3), 15.5 (CH3) ppm; 31P-NMR (CDCl3) δ 28.5 ppm; IR (ATR) 3281, 2956, 1659, 1529, 1224, 1205, 1057, 1026, 1007 cm−1.
Dimethyl 1-(N-phenylcetylamino)-3-methylbutanephosphonate (12b) [4]. Colorless crystals (60.3 mg, 77% yield), mp 117–119 °C. 1H-NMR (CDCl3) δ 7.39–7.23 (m, 5H, Ph), 5.72 (d, J = 9.9 Hz, 1H, NH), 4.62–4.48 (m, 1H, CαH), 3.73 (d, J = 10.6 Hz, 3H, OCH3), 3.63 (d, J = 10.6 Hz, 3H, OCH3), 3.60 (s, 2H, PhCH2), 1.61–1.44 (m, 3H, CH2 and CH), 0.88 (d, J = 5.9 Hz, 6H, 2 × CH3) ppm; 13C-NMR (CDCl3) δ 170.5 (d, J = 4.3 Hz, C = O), aromatic carbons: 134.6, 129.2, 128.9, 127.4, 53.1 (d, J = 7.2 Hz, OCH3), 53.0 (d, J = 6.6 Hz, OCH3), 43.7 (PhCH2), 43.2 (d, J = 155.5 Hz, CαH), 38.1 (d, J = 1.8 Hz, CH2), 24.5 (d, J = 13.4 Hz, CH), 23.2 (CH3), 21.1 (CH3) ppm; 31P-NMR (CDCl3) δ 27.6 ppm; IR (ATR) 3243, 2959, 1673, 1540, 1217, 1027, 836, 742 cm−1.
Dimethyl 1-(N-benzyloxycarbonylamino)-2-tert-butoxyethanephosphonate (12c) [4]. Colorless crystals (54.8 mg, 61% yield), mp 59.5–61.5 °C. 1H-NMR (CDCl3) δ 7.41–7.28 (m, 5H, Ph), 5.29 (d, J = 8.8 Hz, 1H, NH), 5.13 (s, 2H, PhCH2O), 4.33–4.19 (m, 1H, CαH), 3.76 (d, J = 10.6 Hz, 3H, OCH3), 3.75 (d, J = 10.3 Hz, 3H, OCH3), 3.61 (dd, J1 = 9.4, J2 = 3.8 Hz, 1H, CHHOt-Bu), 3.54 (dd, J1 = 9.4, J2 = 3.8 Hz, 1H, CHHOt-Bu), 1.18 (s, 9H, t-Bu) ppm; 13C-NMR (CDCl3) δ 155.7 (C = O), aromatic carbons: 136.2, 128.5, 128.2, 128.1, 73.7 (OC(CH3)3), 67.2 (PhCH2O), 60.6 (CH2Ot-Bu), 53.3 (d, J = 6.0 Hz, OCH3), 52.6 (d, J = 5.8 Hz, OCH3), 48.3 (d, J = 156.0 Hz, CαH), 27.3 (OC(CH3)3) ppm; 31P-NMR (CDCl3) δ 26.1 ppm; IR (ATR) 3293, 2974, 1699, 1532, 1278, 1235, 1039, 1023, 756, 697 cm−1.
Methyl phenyl(1-pivaloylaminoethyl)phosphinate (12d). Colorless crystals (48.9 mg, 69% yield), mp 131–133 °C. 1H-NMR (CDCl3) δ 7.84–7.75 (m, 2H, aromatic), 7.59–7.43 (m, 3H, aromatic), 5.76 (d, J = 9.6 Hz, 1H, NH), 4.79–4.67 (m, 1H, CαH), 3.67 (d, J = 10.8 Hz, 3H, OCH3), 1.45 (dd, J1 = 14.6, J2 = 7.3 Hz, 3H, CH3), 0.92 (s, 9H, t-Bu) ppm; 13C-NMR (CDCl3) δ 177.3 (d, J = 4.7 Hz, C = O), aromatic carbons: 132.8 (d, J = 2.8 Hz), 132.4 (d, J = 9.4 Hz), 128.5 (d, J = 12.5 Hz), 127.5 (d, J = 124.2 Hz), 51.8 (d, J = 7.1 Hz, OCH3), 42.3 (d, J = 115.5 Hz, CαH), 38.5 (C(CH3)3), 27.1 (C(CH3)3), 14.4 (CH3) ppm; 31P-NMR (CDCl3) δ 42.9 ppm. IR (ATR) 3264, 2954, 1660, 1531, 1198, 1139, 1027, 799 cm−1; HRMS (TOF-ESI) calcd for C14H23NO3P [M + H]+ 284.1416, found 284.1405.
Diphenyl 1-(N-benzyloxycarbonylamino)-3-methylbutylphosphine oxide (12e) [8]. Colorless crystals (87.5 mg, 83% yield), mp 173.5–175.5 °C. 1H-NMR (CDCl3) δ 7.87–7.09 (m, 15H, 3 × Ph), 5.40 (d, J = 10.6 Hz, 1H, NH), 5.02 (d, J = 12.5 Hz, 1H, PhCHHO), 4.89 (d, J = 12.5 Hz, 1H, PhCHHO), 4.83–4.72 (m, 1H, CαH), 1.83–1.66 (m, 2H, CH2), 1.36–1.21 (m, 1H, CH), 0.90 (d, J = 6.5 Hz, 3H, CH3), 0.86 (d, J = 6.6 Hz, 3H, CH3) ppm; 13C-NMR (CDCl3) δ 156.1 (d, J = 4.5 Hz, C = O), aromatic carbons: 136.4, 132.0 (d, J = 2.8 Hz), 131.9 (d, J = 2.8 Hz), 131.5, 131.3, 131.1 (d, J = 9.2 Hz), 130.9 (d, J = 9.0 Hz), 128.8 (d, J = 11.4 Hz), 128.4 (d, J = 11.6 Hz), 128.4, 127.9, 127.6, 66.8 (PhCH2O), 48.1 (d, J = 79.5 Hz, CαH), 37.5 (d, J = 3.5 Hz, CH2), 24.5 (d, J = 11.0 Hz, CH), 23.4 (CH3), 21.0 (CH3) ppm; 31P-NMR (CDCl3) δ 33.5 ppm; IR (ATR) 3181, 2954, 1702, 1545, 1436, 1263, 1187, 1120 cm−1.
Reaction of 1-(N-pivaloylamino)ethyltris(3-chlorophenyl)phosphonium tetrafluoroborate4cwith trimethyl phosphite—measurement of changes in concentrations of the substrate4c, intermediate13aand product12aby NMR
To a solution of 1-(N-pivaloylamino)ethyltris(3-chlorophenyl)phosphonium tetrafluoroborate 4c (29.0 mg, 0.05 mmol) in CDCl3 (0.65 cm3), trimethyl phosphite (8.9 µL, 9.3 mg, 0.075 mmol) was added. Dimethyldiphenylsilane (5 mg) was used as the internal standard. The reaction mixture was placed directly into the NMR tube. Changes in the concentrations of substrate 4c, intermediate 13a, and product 12a were monitored using 1H-NMR spectroscopy.
Reaction of 1-(N-pivaloylamino)ethyltris(3-chlorophenyl)phosphonium tetrafluoroborate4cwith triethyl phosphite–synthesis of 1-(N-pivaloylamino)ethyltriethoxyphosphonium tetrafluoroborate13b
To a stirred solution of 1-(N-pivaloylamino)ethyltris(3-chlorophenyl)phosphonium tetrafluoroborate (4c, 145.2 mg, 0.25 mmol) in DCM (1 cm3), triethyl phosphite (64.6 µL, 62.3 mg, 0.375 mmol) was added. Stirring was continued for 30 min at room temperature. Thereafter the intermediate product 13b was precipitated with Et2O, separated by decantation, and dried under reduced pressure. In order to obtain the triethoxyphosphonium salt 13 with a higher purity, the reaction should be carried out under argon atmosphere.
1-(N-Pivaloylamino)ethyltriethoxyphosphonium tetrafluoroborate (13b). Colorless oil (47.6 mg, 50% yield). 1H-NMR (CDCl3) δ 7.53 (t, J = 8.2 Hz, 1H, NH), 4.69 (dq, J1 = 19.7, J2 = 7.0 Hz, 1H, CαH), 4.62–4.46 (m, 6H, 3 × OCH2), 1.56 (dd, J1 = 20.2, J2 = 7.3 Hz, 3H, CH3), 1.48 (td, J1 = 7.0, J2 = 0.9 Hz, 9H, 3 × OCH2CH3), 1.23 (s, 9H, t-Bu) ppm; 13C-NMR (CDCl3) δ 179.9 (C = O), 70.2 (d, J = 9.1 Hz, OCH2), 42.3 (d, J = 148.6 Hz, CαH), 38.5 (C(CH3)3), 27.1 (C(CH3)3), 15.9 (d, J = 6.0 Hz, OCH2CH3), 14.0 (CH3) ppm; 31P-NMR (CDCl3) δ 37.0 ppm; IR (ATR) 3375, 2973, 1664, 1515, 1030 cm−1. HRMS (TOF-ESI) calcd for C13H29NO4P [M]+ 294.1834, found 294.1829.
4. Conclusions
Modification of the phosphonium group by introducing electron-withdrawing substituents results in the weakening of the Cα-P+ bond and makes it susceptible to cleavage. This phenomenon is the cause of the high reactivity of 1-(N-acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength. As we have demonstrated, these types of phosphonium salts react smoothly, usually at room temperature with various types of nucleophiles such as 1-morpholinocyclohexene, 1,3-dicarbonyl compounds, benzotriazole sodium salt, p-toluenesulfinate sodium salt, benzylamine, triarylphosphines, and other P-nucleophiles. Only in the case of 1,3-dicarbonyl compounds, it was necessary to use a strong base to generate enolate anions. Reactions with 1-morpholinocyclohexene, benzotriazole sodium salt, p-toluenesulfinate sodium salt, benzylamine, and triarylphosphines do not require the use of any catalysts and occur quite fast (5–60 min). Other examined P-nucleophiles also react efficiently with 1-(N-acylamino)alkyltriarylphosphonium salts. However, the quickly formed intermediate, in the absence of any dealkylating agent, slowly transforms into a final product. Therefore, to facilitate the reaction, we used a substoichiometric amount of the dealkylating agent in the form of methyltriphenylphosphonium iodide.
The use of 1-(N-acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength allowed us to discover interesting mechanistic aspects of the examined reactions. Detection, isolation, and characterization of 1-(N-acylamino)alkyltrialkoxyphosphonium salt 13b—the reactive intermediate in the Michaelis–Arbuzov type reaction, were particularly important.
Futher studies on expanding the range of nucleophiles, which can be used in α-amidoalkylation by 1-(N-acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength, are in progress.
Supplementary Materials
The following are available online at https://www.mdpi.com/1420-3049/23/10/2453/s1, Supporting information includes a summary table (comparison of conditions and yields for reactions of 1-(N-acylamino)alkyltriphenylphosphonium salts (former studies) and 1-(N-acylamino)alkyltriarylphosphonium salts (the current work) with selected nucleophiles), 1H, 13C, and 31P NMR spectra of all new compounds 6, 8, 10, 12, 13. Supplementary data associated with this article can be found in the online version.
Author Contributions
Conceptualization, J.A.; Formal analysis, J.A., A.W., J.K. and K.E.; Investigation, J.A., A.W., J.K. and K.E.; Methodology, J.A. and A.W.; Supervision, J.A. and K.W.; Writing—original draft, J.A.; Writing—review & editing, J.A. and A.W.
Funding
This research received no external funding.
Acknowledgments
This work received financial support from the National Science Center, Poland (NCN) under Grant No. 2015/19/D/ST5/00733.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mazurkiewicz, R.; Październiok-Holewa, A.; Adamek, J.; Zielińska, K. α-Amidoalkylating agents: Structure, synthesis, reactivity and application. Adv. Heterocycl. Chem. 2014, 111, 43–94. [Google Scholar] [CrossRef]
- Adamek, J.; Węgrzyk, A.; Krawczyk, M.; Erfurt, K. Catalyst-free Friedel-Crafts reaction of 1-(N-acylamino)alkyltriarylphosphonium salts with electron-rich arenes. Tetrahedron 2018, 74, 2575–2583. [Google Scholar] [CrossRef]
- Adamek, J.; Mazurkiewicz, R.; Węgrzyk, A.; Erfurt, K. 1-Imidoalkylphosphonium salts with modulated Cα-P+ bond strength: Synthesis and application as new active α-imidoalkylating agents. Beilstein J. Org. Chem. 2017, 13, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, K.; Mazurkiewicz, R.; Szymańska, K.; Jarzębski, A.; Magiera, S.; Erfurt, K. Penicillin G acylase-mediated kinetic resolution of racemic 1-(N-acylamino)alkylphosphonic and 1-(N-acylamino)alkylphosphinic acids and their esters. J. Mol. Catal. B Enzym. 2016, 132, 31–40. [Google Scholar] [CrossRef]
- Adamek, J.; Mazurkiewicz, R.; Październiok-Holewa, A.; Grymel, M.; Kuźnik, A.; Zielińska, K. 1-(N-Acylamino)alkyl Sulfones from N-Acyl-α-amino Acids or N-Alkylamides. J. Org. Chem. 2014, 79, 2765–2770. [Google Scholar] [CrossRef] [PubMed]
- Adamek, J.; Mazurkiewicz, R.; Październiok-Holewa, A.; Kuźnik, A.; Grymel, M.; Zielińska, K.; Simka, W. N-[1-(Benzotriazol-1-yl)alkyl] amides from N-acyl-α-amino acids or N-alkylamides. Tetrahedron 2014, 70, 5725–5729. [Google Scholar] [CrossRef]
- Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Mazurkiewicz, R. Comparative Studies on the Amidoalkylating Properties of N-(1-Methoxyalkyl)Amides and 1-(N-Acylamino)Alkyltriphenylphosphonium Salts in the Michaelis–Arbuzov-Like Reaction: A New One-Pot Transformation of N-(1-Methoxyalkyl)Amides into Phosphonic or Phosphinic Analogs of N-Acyl-α-Amino Acids. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 967–980. [Google Scholar] [CrossRef]
- Październiok-Holewa, A.; Adamek, J.; Mazurkiewicz, R.; Zielińska, K. Amidoalkylating Properties of 1-(N-Acylamino)Alkyltriphenylphosphonium Salts. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 205–212. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Simka, W.; Gajos, A.; Szymura, K. α-Amidoalkylating Agents from N-Acyl-α-amino Acids: 1-(N-Acylamino)alkyltriphenylphosphonium Salts. J. Org. Chem. 2012, 77, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Październiok-Holewa, A.; Adamek, J.; Zielińska, K.; Piernikarczyk, K.; Mazurkiewicz, R. N-(1-acyloaminoalkyl)amidinium salts derived from DBU or related bases as reactive intermediates in α-amidoalkylation reactions. Arkivoc 2012, 314–329. [Google Scholar] [CrossRef]
- Zaugg, H.E. Recent Synthetic Methods Involving Intermolecular alpha-Amidoalkylation at Carbon. Synthesis 1970, 49–73. [Google Scholar] [CrossRef]
- Zaugh, H.E. α-Amidoalkylation at Carbon: Recent Advances–Part I. Synthesis 1984, 85–110. [Google Scholar] [CrossRef]
- Zaugg, H.E. α-Amidoalkylation at Carbon: Recent Advances–Part II. Synthesis 1984, 181–212. [Google Scholar] [CrossRef]
- Speckamp, W.N.; Hiemstra, H. Intramolecular reactions of N-acyliminium intermediates. Tetrahedron 1985, 41, 4367–4416. [Google Scholar] [CrossRef]
- Hiemstra, H.; Speckamp, W.N. N-Acyliminium Ions as Intermediates in Alkaloid Synthesis. Alkaloids 1988, 32, 271–339. [Google Scholar] [CrossRef]
- Hiemstra, H.; Speckamp, W.N. Comprehensive Organic Synthesis; Throst, B.M., Flemming, I., Eds.; Oxford: Pergamon, Turkey, 1991; Volume 2, pp. 1047–1082. [Google Scholar] [CrossRef]
- Kleinman, E.F. The Bimolecular Aliphatic Mannich and Related Reactions. In Comprehensive Organic Synthesis; Trost, B.M., Ed.; Pergamon Press: Oxford, UK, 1991; Volume 2, pp. 893–951. [Google Scholar]
- Katritzky, A.R.; Lan, X.; Yang, J.Z.; Denisko, O.V. Properties and Synthetic Utility of N-Substituted Benzotriazoles. Chem. Rev. 1998, 98, 409–548. [Google Scholar] [CrossRef] [PubMed]
- Speckamp, W.N.; Moolenaar, M.J. New developments in the chemistry of N-acyliminium ions and related intermediates. Tetrahedron 2000, 56, 3817–3856. [Google Scholar] [CrossRef]
- Mecozzi, T.; Petrini, M.; Profeta, R. Reactivity of chiral exocyclic N-acyliminium ions with aromatic derivatives. Tetrahedron Asymmetry 2003, 14, 1171–1178. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Zhang, H.C.; Cohen, J.H.; Turchi, I.J.; Maryanoff, C.A. Cyclizations of N-acyliminium ions. Chem. Rev. 2004, 104, 1431–1628. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Manju, K.; Singh, S.K.; Meher, N.K. Benzotriazole mediated amino-, amido-, alkoxy- and alkylthioalkylation. Tetrahedron 2005, 61, 2555–2581. [Google Scholar] [CrossRef]
- Petrini, M. α-Amido Sulfones as Stable Precursors of Reactive N-Acylimino Derivatives. Chem. Rev. 2005, 105, 3949–3977. [Google Scholar] [CrossRef] [PubMed]
- Ollevier, T.; Li, Z. The first catalytic Sakurai reaction of N-alkoxycarbonylamino sulfones with allyltrimethylsilane. Org. Biomol. Chem. 2006, 4, 4440–4443. [Google Scholar] [CrossRef] [PubMed]
- Ballini, R.; Palmieri, A.; Petrini, M.; Torregiani, E. Solventless Clay-Promoted Friedel−Crafts Reaction of Indoles with α-Amido Sulfones: Unexpected Synthesis of 3-(1-Arylsulfonylalkyl) Indoles. Org. Lett. 2006, 8, 4093–4096. [Google Scholar] [CrossRef] [PubMed]
- Marianacci, O.; Micheletti, G.; Bernardi, L.; Fini, F.; Fochi, M.; Pettersen, D.; Sgarzani, V.; Ricci, A. Organocatalytic Asymmetric Mannich Reactions with N-Boc and N-Cbz Protected α-Amido Sulfones. Chem. Eur. J. 2007, 13, 8338–8351. [Google Scholar] [CrossRef] [PubMed]
- Yazici, A.; Pyne, S.G. Intermolecular addition reactions of N-acyliminium ions (Part I). Synthesis 2009, 339–368. [Google Scholar] [CrossRef]
- Yazici, A.; Pyne, S.G. Intermolecular addition reactions of N-acyliminium ions (Part II). Synthesis 2009, 513–541. [Google Scholar] [CrossRef]
- Thirupathi, P.; Kim, S.S. InBr3: A Versatile Catalyst for the Different Types of Friedel−Crafts Reactions. J. Org. Chem. 2009, 74, 7755–7761. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Damodar, K.; Bhunia, N. A Simple and Efficient Access to α-Amino Phosphonates from N-Benzyloxycarbonylamino Sulfones Using Indium(III) Chloride. J. Org. Chem. 2009, 74, 5607–5609. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Estibalez, U.; Gómez-SanJuan, A.; García-Calvo, O.; Aranzamendi, E.; Lete, E.; Sotomayor, N. Strategies based on aryllithium and N-acyliminium ion cyclizations for the stereocontrolled synthesis of alkaloids and related systems. Eur. J. Org. Chem. 2011, 3610–3633. [Google Scholar] [CrossRef]
- Schneider, A.E.; Manolikakes, G. Bi(OTf)3-Catalyzed Multicomponent α-Amidoalkylation Reactions. J. Org. Chem. 2015, 80, 6193–6212. [Google Scholar] [CrossRef] [PubMed]
- Aranzamendi, E.; Arrasate, S.; Sotomayor, N.; González-Díaz, H.; Lete, E. Chiral Brønsted Acid Catalyzed Enantioselective α-Amidoalkylation Reactions: A Joint Experimental and Predictive Study. ChemistryOpen 2016, 5, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Touati, B.; El Bouakher, A.; Azizi, M.S.; Taillier, C.; Othman, R.B.; Trabelsi-Ayadi, M.; Antoniotti, S.; Dunach, E.; Dalla, V. Enolizable Carbonyls and N,O-Acetals: A Rational Approach for Room-Temperature Lewis Superacid-Catalyzed Direct α-Amidoalkylation of Ketones and Aldehydes. Chem. Eur. J. 2016, 22, 6012–6022. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, M.G.; Turova, O.V.; Zlotin, S.G. The progress in the chemistry of N-acyliminium ions and their use in stereoselective organic synthesis. Russ. Chem. Rev. 2017, 86, 1–17. [Google Scholar] [CrossRef]
- Aranzamendi, E.; Sotomayor, N.; González-Díaz, H.; Lete, E. Phenolic Activation in Chiral Brønsted Acid-Catalyzed Intramolecular α-Amidoalkylation Reactions for the Synthesis of Fused Isoquinolines. ACS Omega 2017, 2, 2706–2718. [Google Scholar] [CrossRef]
- Touati, B.; El Bouakher, A.; Azizi, M.S.; Taillier, C.; Othman, R.B.; Trabelsi-Ayadi, M.; Antoniotti, S.; Dunach, E.; Dalla, V. Atom-Economic Catalytic Direct Substitution of N,O-Acetals with Simple Ketones. Eur. J. Org. Chem. 2017, 4445–4460. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 6, 8, 10 and 12 are available from the authors. |
Scheme 1.
α-Amidoalkylation reactions—various synthetic routes.

Scheme 2.
1-(N-Acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength 4—preparation and synthetic application.
Scheme 2.
1-(N-Acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength 4—preparation and synthetic application.

Scheme 3.
1-(N-Acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength 4—reactions with triarylphosphines.
Scheme 3.
1-(N-Acylamino)alkyltriarylphosphonium salts with weakened Cα-P+ bond strength 4—reactions with triarylphosphines.

Scheme 4.
α-Amidoalkylation of P-nucleophiles by 1-(N-acylamino)alkyltriarylphosphonium salts 4—a plausible mechanism.
Scheme 4.
α-Amidoalkylation of P-nucleophiles by 1-(N-acylamino)alkyltriarylphosphonium salts 4—a plausible mechanism.

Figure 1.
Concentration of the substrate 4c, intermediate 13a, and product 12a as a function of time for the reaction of 1-(N-pivaloylamino)ethyltris(3-chlorophenyl)phosphonium tetrafluoroborate (4c) with trimethyl phosphite at 26 °C.
Figure 1.
Concentration of the substrate 4c, intermediate 13a, and product 12a as a function of time for the reaction of 1-(N-pivaloylamino)ethyltris(3-chlorophenyl)phosphonium tetrafluoroborate (4c) with trimethyl phosphite at 26 °C.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Conditions and yields for α-amidoalkylation of 1,3-dicarbonyl compounds by 1-(N-acylamino)alkyltriarylphosphonium salts 4.
Table 1.
Conditions and yields for α-amidoalkylation of 1,3-dicarbonyl compounds by 1-(N-acylamino)alkyltriarylphosphonium salts 4.

| Entry | Phosphonium Salt 4 | 1,3-Dicarbonyl Compound, 5 | Molar Ratio of 4:5:LDA | 6 | Yield, % | ||
|---|---|---|---|---|---|---|---|
| R1 | R2 | Ar | |||||
| 1 | t-Bu | Me | m-C6H4Cl | diethyl malonate | 1:2:1 | 6a | 40 |
| 2 | t-Bu | Me | m-C6H4Cl | diethyl malonate | 1:8:1 | 6a | 65 |
| 3 | t-Bu | Me | p-C6H4CF3 | diethyl malonate | 1:8:1 | 6a | 67 |
| 4 | t-Bu | Me | m-C6H4Cl | ethyl acetoacetate | 1:8:1 | 6b | 62 a |
| 5 | Bn | i-Bu | m-C6H4Cl | ethyl acetoacetate | 1:8:1 | 6c | 83 b |
| 6 | Bn | i-Bu | m-C6H4Cl | dimethyl malonate | 1:8:1 | 6d | 63 |
| 7 | Bn | i-Bu | m-C6H4Cl | diethyl malonate | 1:8:1 | 6e | 52 |
| 8 | BnO | Bn | m-C6H4Cl | ethyl acetoacetate | 1:8:1 | 6f | 30 c |
a A mixture of diastereoisomers in a molar ratio of 1.4:1. b A mixture of diastereoisomers in a molar ratio of 1.25:1. c Attempts to isolate an analytically pure sample failed.
Table 2.
Conditions and yields for α-amidoalkylation of 1-morpholinocyclohexene by 1-(N-acylamino)alkyltriarylphosphonium salts 4.
Table 2.
Conditions and yields for α-amidoalkylation of 1-morpholinocyclohexene by 1-(N-acylamino)alkyltriarylphosphonium salts 4.

| Entry | Phosphonium Salt 4 | 8 | Yield, % | ||
|---|---|---|---|---|---|
| R1 | R2 | Ar | |||
| 1 | t-Bu | Me | m-C6H4Cl | 8a | 63 a |
| 2 | Bn | i-Bu | m-C6H4Cl | 8b | 62 b |
| 3 | BnO | i-Bu | m-C6H4Cl | 8c | 40 c |
| 4 | BnO | Bn | m-C6H4Cl | 8d | 33 d |
| 5 | BnO | CH2Ot-Bu | p-C6H4CF3 | 8e | 31 c |
a A mixture of diastereoisomers in a molar ratio of 3.5:1. b A mixture of diastereoisomers in a molar ratio of 3.2:1. c Only one diastereoisomer was detected and isolated. d A mixture of diastereoisomers in a molar ratio of 13.4:1.
Table 3.
Conditions and yields for α-amidoalkylation of selected heteronucleophiles by 1-(N-acylamino)alkyltriarylphosphonium salts 4.
Table 3.
Conditions and yields for α-amidoalkylation of selected heteronucleophiles by 1-(N-acylamino)alkyltriarylphosphonium salts 4.

| Entry | Phosphonium Salt 4 | Nu−Na+/NuH | Temp., °C | Time, Min. | Molar Ratio of 4:9 | 10 | Yield, % | ||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | Ar | |||||||
| 1 | t-Bu | Me | p-C6H4CF3 | Bt−Na+ | 20 | 15 | 1:1 | 10a | 99 |
| 2 | BnO | Bn | m-C6H4Cl | Bt−Na+ | 20 | 15 | 1:1 | 10b | 70 |
| 3 | t-Bu | Me | p-C6H4CF3 | TolSO2−Na+ | 20 | 15 | 1:1 | 10c | 88 |
| 4 | t-Bu | Me | m-C6H4Cl | Bn-NH2 | 20 | 5 | 1:4 | 10d | 91 |
| 5 | Bn | i-Bu | p-C6H4CF3 | Bn-NH2 | 20 | 5 | 1:4 | 10e | 55 |
Table 4.
Conditions and yields for α-amidoalkylation of P-nucleophiles by 1-(N-acylamino)alkyltriarylphosphonium salts 4.
Table 4.
Conditions and yields for α-amidoalkylation of P-nucleophiles by 1-(N-acylamino)alkyltriarylphosphonium salts 4.

| Entry | Phosphonium Salt 4 | P-Nu 11 | Temp., °C | Time, h | Molar Ratio of 4:11 | 12 | Yield, % | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | Ar | R | R3 | R4 | ||||||
| 1 | t-Bu | Me | m-C6H4Cl | Me | OMe | OMe | 20 | 3 | 1:1.5 | 12a | 47 |
| 2 | t-Bu | Me | m-C6H4Cl | Me | OMe | OMe | 20 | 3 | 1:1.5 a | 12a | 85 |
| 3 | Bn | i-Bu | m-C6H4Cl | Me | OMe | OMe | 20 | 3 | 1:1.5 a | 12b | 77 |
| 4 | BnO | CH2Ot-Bu | p-C6H4CF3 | Me | OMe | OMe | 20 | 3 | 1:1.5 a | 12c | 61 |
| 5 | t-Bu | Me | m-C6H4Cl | Me | Ph | OMe | 60 | 2 | 1:1.5 a | 12d | 69 |
| 6 | BnO | i-Bu | p-C6H4CF3 | Me | Ph | Ph | 20 | 3 | 1:1.5 a | 12e | 83 |
a Substoichiometric amounts of methyltriphenylphosphonium iodide as a dealkylating agent (molar ratio of 1:0.25) was used.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Adamek, J.; Węgrzyk, A.; Kończewicz, J.; Walczak, K.; Erfurt, K. 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications. Molecules 2018, 23, 2453. https://doi.org/10.3390/molecules23102453
AMA Style
Adamek J, Węgrzyk A, Kończewicz J, Walczak K, Erfurt K. 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications. Molecules. 2018; 23(10):2453. https://doi.org/10.3390/molecules23102453
Chicago/Turabian StyleAdamek, Jakub, Anna Węgrzyk, Justyna Kończewicz, Krzysztof Walczak, and Karol Erfurt. 2018. "1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications" Molecules 23, no. 10: 2453. https://doi.org/10.3390/molecules23102453