Continuous Flow Alcoholysis of Dialkyl H-Phosphonates with Aliphatic Alcohols




Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, Hungary
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(7), 1618; https://doi.org/10.3390/molecules23071618
Submission received: 14 June 2018
/
Revised: 29 June 2018
/
Accepted: 1 July 2018
/
Published: 3 July 2018
(This article belongs to the Special Issue Organophosphorus Chemistry 2018)
Abstract
:The continuous flow alcoholysis of dialkyl H-phosphonates by aliphatic alcohols in the absence of a catalyst was elaborated using a microwave (MW) reactor equipped with a flow cell. By the precise control of the reaction conditions, the synthesis could be fine-tuned towards dialkyl H-phosphonates with two different and with two identical alkyl groups. In contrast to the “traditional” batch alcoholysis, flow approaches required shorter reaction times, and the products became available at a larger scale.
1. Introduction
Flow chemistry induced an inevitable revolution in the field of chemical transformations [1]. In modern chemistry, reactions have to be strictly controlled, which requires more and more precise synthetic techniques. In contrast to the “traditional” batch reactions, flow approaches create a significantly different processing environment, which enables a more efficient control of the reaction conditions (such as the reaction time or temperature). Due to this, flow protocols may be significantly better with respect to purity, selectivity and yields, as compared to batch approaches [2].
In microwave (MW) chemistry, the continuous flow technique represents a special importance. Although applications of the MW technique proved to be useful in many chemical transformations, the scale-up of MW-assisted reactions means a challenge due to the limited geometry of the MW devices [3,4,5,6]. One possibility to solve this problem is the use of continuous flow MW reactors, where the reaction mixture flows through an irradiated flow cell. The MW unit can be of a similar size used in batch mode. During the last decade, the advantages of the applications of continuous flow MW reactors were reported in certain transformations; however, in most cases, the usefulness of the occasional devices may be questionable, as non-professional MW reactors do not allow reproductions, and what is more important, the temperatures were not reported [7,8].
Esters represent a fundamental family among organic compounds. Esters of carboxylic acids may be important intermediates, solvents or products in organic chemistry [9].
The most common preparations of simple carboxylic esters comprise the acid-catalyzed reaction of a carboxylic acid with alcohol (Fischer esterification) and the reaction of an ester with alcohol (alcoholysis) [10]. Continuous flow Fischer esterifications may be carried out in systems containing a packed catalyst bed [11,12,13,14,15,16,17,18,19,20] or a heated coil [21,22], as well as in microreactors [23]. In a few cases, the esterifications were performed in continuous flow MW systems based on professional MW reactors [24,25,26] or in household MW ovens [27,28]. The continuous flow alcoholysis of carboxylic esters may also be performed in reactors equipped with a catalyst bed [29] or a heated coil [21,30]. The alcoholysis is also of great importance in biodiesel production, the area of which was recently summarized by Lee and co-workers [31].
Organophosphorus esters are also of great importance [32]. Dialkyl esters of phosphorous acid (dialkyl H-phosphonates) are widely applied building blocks in syntheses [33]. They are important starting materials of the Kabachnik–Fields condensations and the aza-Pudovik reactions [34] (towards α-aminophosphonates), the Pudovik reactions [35] (resulting in the formation of α-hydroxyphosphonates) and further organophosphorus transformations, such as the Hirao reaction [36] and the phospha-Michael addition [37]. Dialkyl H-phosphonates bearing different alkoxy groups on the phosphorus atom are valuable intermediates for P-chiral organophosphorus derivatives [38,39].
The industrial synthesis of dialkyl H-phosphonates is comprised of the reaction of phosphorus trichloride with alcohols (Scheme 1) [40,41,42,43,44,45]. Although this transformation is convenient, efficient and may be scaled up, it requires a solvent, and the liberating HCl decreases the atom efficiency. It is also a disadvantage that the synthesis of derivatives with different alkyl groups is not possible.
Another possibility for the preparation of dialkyl H-phosphonates is alcoholysis (Scheme 2), which provides the products in good yields [46,47,48,49,50,51,52,53,54]. The only by-product is the leaving alcohol. The relatively high temperature and the need for the high excess of the alcohol mean disadvantages.
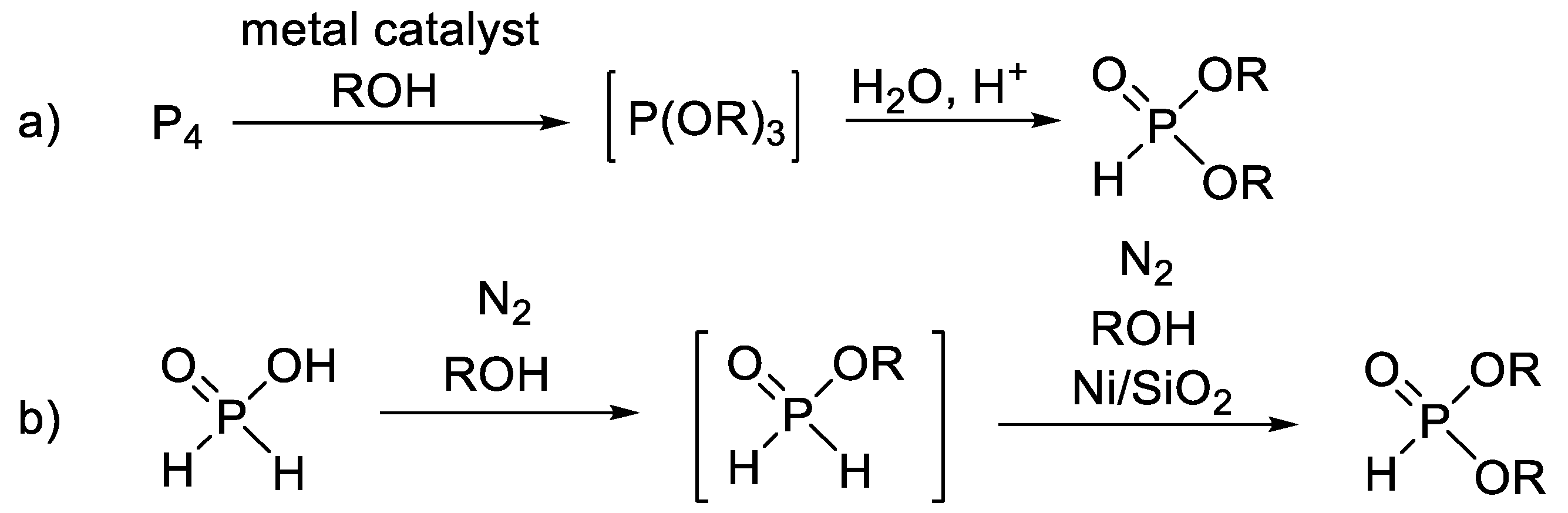
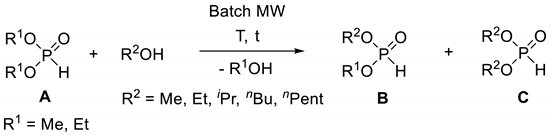
Besides the two main routes mentioned above, there are a few less important methods for the synthesis of dialkyl H-phosphonates. Such protocols are the oxidative reaction of elemental phosphorus with alcohols (Scheme 3a) [55,56,57,58,59,60] and the Ni-catalyzed oxidation of hypophosphorous esters (Scheme 3b) [61].
The synthesis of dialkyl-H-phosphonates bearing different alkyl groups is not easy and requires special methods (Table 1).
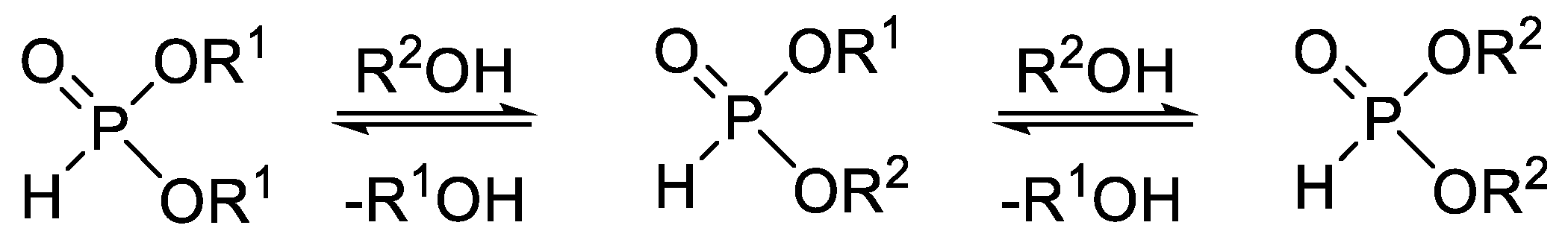
Phosphites with different alkyl groups may be synthesized by the partial alcoholysis of dialkyl H-phosphonates (Table 1, Entry 1). The reaction conditions must be controlled strictly to avoid complete transesterification.
Dialkyl H-phosphonates with two different alkyl groups may be obtained by the controlled O–C cleavage of dialkyl H-phosphonates (Table 2, Entries 2 and 3). In the first step of the first method, a phosphite-ammonium salt is prepared, which is then alkylated with alkyl iodides (Table 2, Entry 2), or with alcohols in the presence of pivaloyl chloride and pyridine (Table 2 entry 3) to furnish the target compounds selectively. A serious drawback of these transformations is the low atom efficiency.
Dialkyl H-phosphonates bearing different alkoxy groups can also be obtained in the reaction of alkyl dichlorophosphites and alcohols. The primarily formed trialkyl phosphite is cleaved by a part of the hydrochloric acid formed (Table 2, Entry 4).
From these synthetic methods, the alcoholysis may be flow compatible. This solvolytic reaction may be carried out without any catalyst by fine-tuning the reaction conditions, to afford mixed or fully-transesterified dialkyl phosphites as the predominant component.
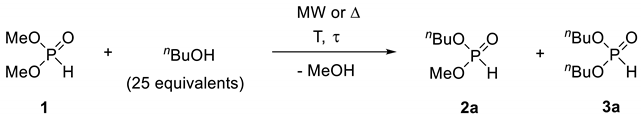
The alcoholysis of dialkyl H-phosphonates in a batch MW reactor was investigated by us previously (Table 2) [52]. The reaction of dialkyl H-phosphonates (A) with aliphatic alcohols under mild conditions (lower alcohol excess and lower temperature) afforded the derivatives with different alkyl groups (B) as the main products, while in the case of applying higher excess of alcohol and higher temperature, the fully-transesterified dialkyl H-phosphonates (C) predominated in the mixture.
In this paper, we aimed at elaborating an MW-assisted continuous flow method for the alcoholysis of dialkyl H-phosphonates. Our purpose was to find the optimum conditions for the formation of dialkyl H-phosphonates bearing different alkyl groups and for the fully-transesterified phosphites in a flow MW reactor. Furthermore, we wished to prepare new dialkyl H-phosphonates with two different alkyl groups, which may be valuable building blocks of chiral organophosphorus compounds.
2. Results and Discussion
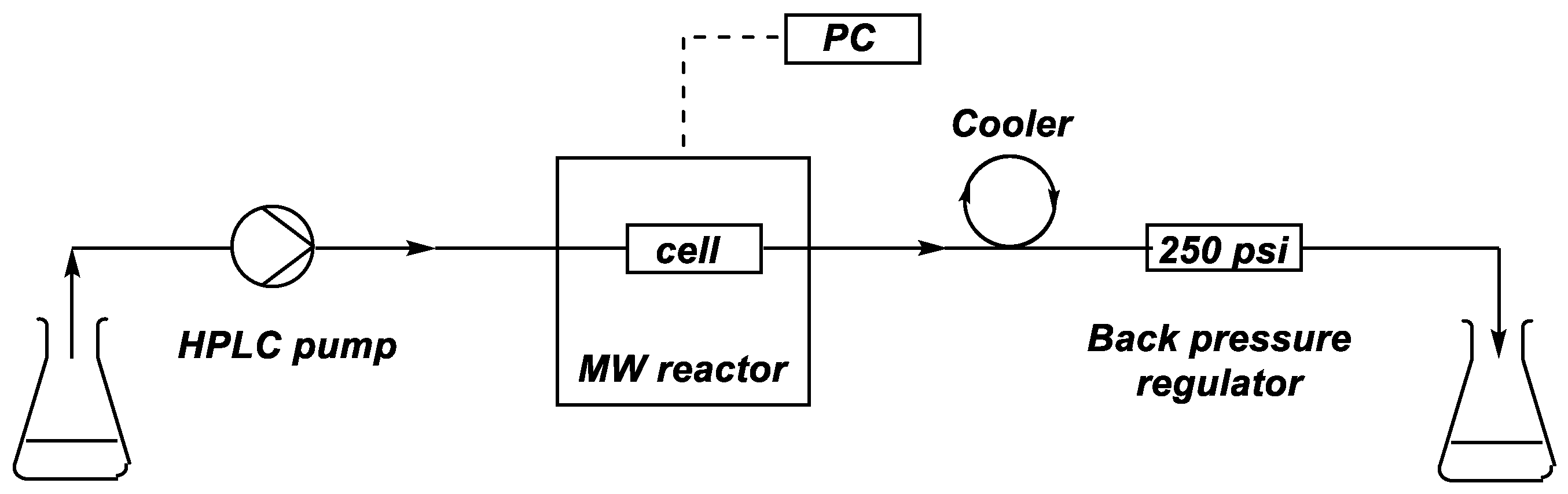
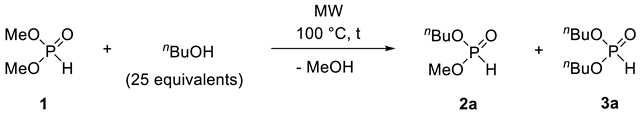
In the first case, the alcoholysis of dimethyl H-phosphonate (DMP) (1) with n-butanol was investigated in the absence of any catalyst, in a CEM® (Matthews, NC, USA) MW reactor equipped with a commercially available CEM® continuous flow cell (Figure 1 and Figure 2). The mixture of DMP (1) and a 25-fold excess of n-butanol was fed into the reactor by an HPLC pump at a flow rate of 0.1–1.4 mL/min (corresponding to residence times of 60–5 min, respectively). The temperature was monitored and controlled by an IR sensor. The mixture leaving the reactor was cooled down using a spiral-like cooler and was passed through a back pressure regulator operating at 250 psi. Consecutive fractions of the leaving mixture were analyzed by GC measurements in order to determine the composition and to identify the stationary operation.
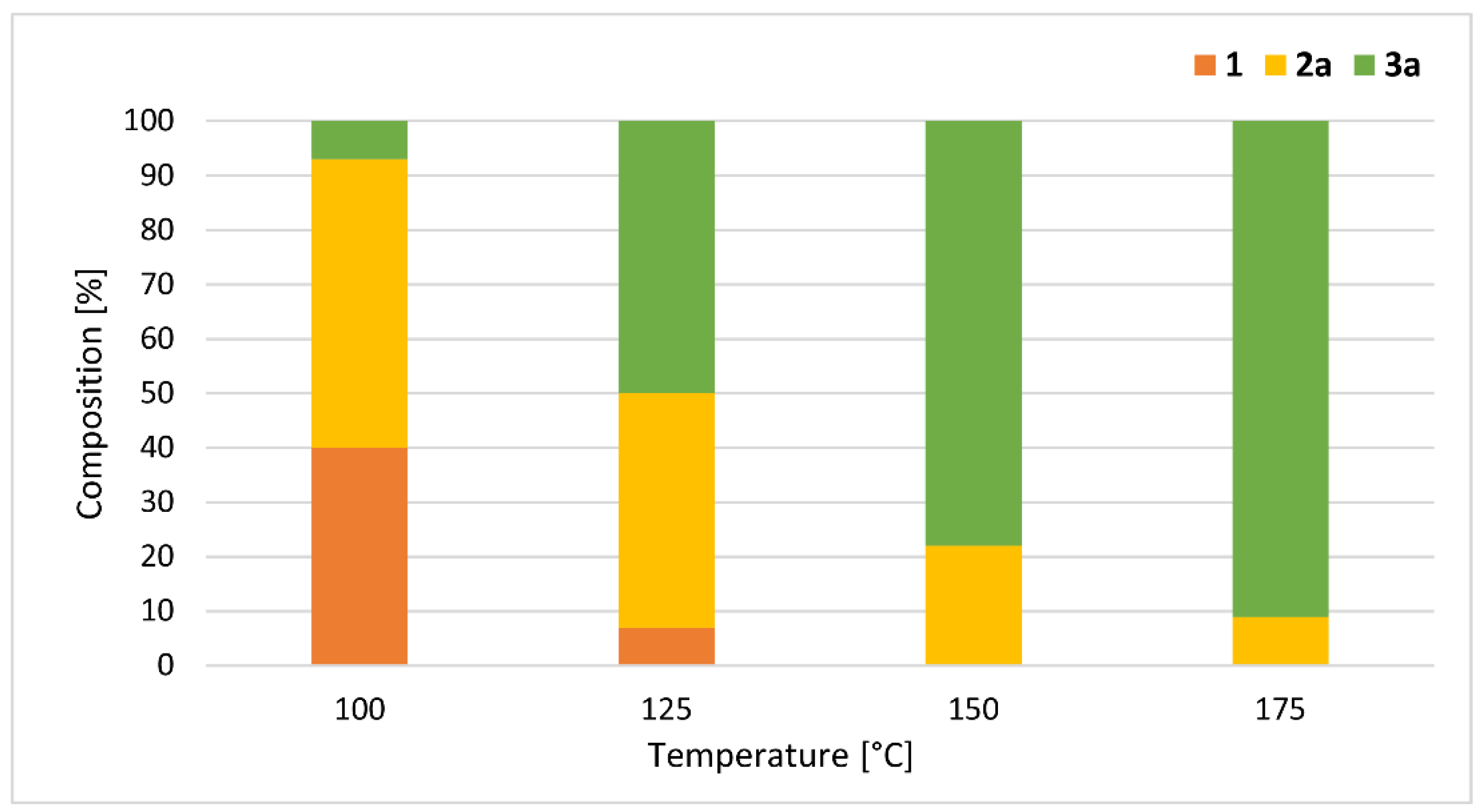
The reaction conditions and the results are summarized in Table 3. To find the optimum conditions for the formation of n-butyl methyl H-phosphonate (nBMP) (2a) and n-dibutyl H-phosphonate (DnBP) (3a), the alcoholysis was carried out applying different temperatures and residence times. Performing the reaction at 100 °C, at a flow rate of 1.4 mL/min (with a residence time of 5 min), the conversion was only 26%, and the proportion of nBMP (2a) was 25%, along with 1% of DnBP (3a) (Table 3, Entry 1). Using a flow rate of 0.45 mL/min (with a residence time of 15 min), the starting DMP (1) was still the main component of the leaving mixture; however, the proportion of nBMP (2a) was 44% (Table 3, Entry 2). Increasing the residence time to 30 min (by decreasing the flow rate to 0.25 mL/min), the ratio of mixed H-phosphonate (2a) was 9% higher (53%) (Table 3, Entry 3). Applying a longer residence time of 45 or 60 min (flow rate of 0.15 or 0.10 mL/min), the proportion of phosphite 2a was somewhat lower (50% or 47%), and the ratio of DnBP (3a) increased to 26% or 35%, respectively (Table 3, Entries 4 and 5). The maximum proportion of nBMP (2a) (53%) was obtained applying a reaction time of 30 min at 100 °C (Table 3, Entry 3). Increasing or decreasing the residence time at the same temperature resulted in lower ratio of product 2a (Table 3, Entries 1, 2 and 4, 5, respectively). In a comparative thermal experiment at 100 °C at a residence time of 30 min, the conversion was only 15% (Table 3, Entry 6). Next, the effect of the reaction temperature was investigated at a residence time of 30 min (Table 3, Entries 6–8) (Figure 3). At 125 °C, the mixture was comprised of 7% of DMP (1), 43% of nBMP (2a) and 50% of DnBP (3a) (Table 3, Entry 7). Increasing the temperature to 150 °C, the conversion was complete, and the fully-transesterified product (3a) predominated in a proportion of 78% (Table 3, Entry 8). Performing the alcoholysis at 175 °C, the ratio of DnBP (3a) was 91% (Table 3, Entry 9). Carrying out the reaction at a longer residence time (45 min) and/or higher excess of the nBuOH (50 equiv), the composition did not change. Applying common heating at 175 °C, the reaction was not complete, and the ratio of product 3a was 8% lower, than under MW conditions (Table 3, Entry 10). It can be observed that the difference between MW-assisted and conventionally-heated experiments was significant at 100 °C, while significantly smaller at 175 °C. After column chromatography, nBMP (2a) was obtained in a yield of 48%, while DnBP (3a) was isolated in a yield of 88% (Table 3, Entries 3 and 9).
Comparative batch experiments were also carried out in the DMP (1)—nBuOH model reaction at 100 °C. The alcohol excess (25 equiv) and the reaction time range studied (5–60 min) were the same as in the flow approaches. The batch results are listed in Table 4. Similarly to the flow experiments, the composition was highly reaction time-dependent.
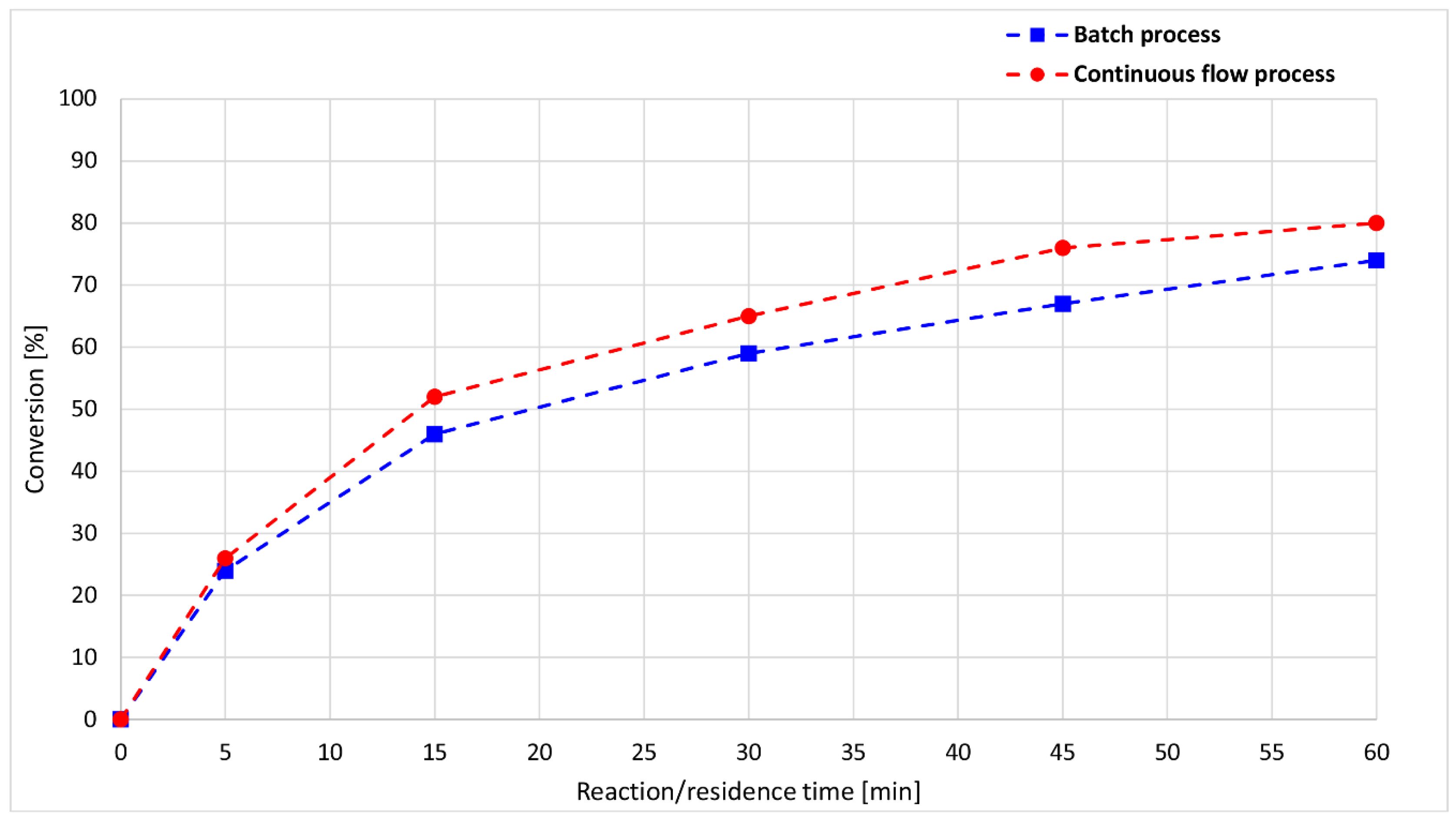
Comparing the conversions of the flow and batch processes, the two plots show a similar shape; however, after a reaction time of 15 min, the batch conversions were 5–10% lower than the flow results (Figure 4).
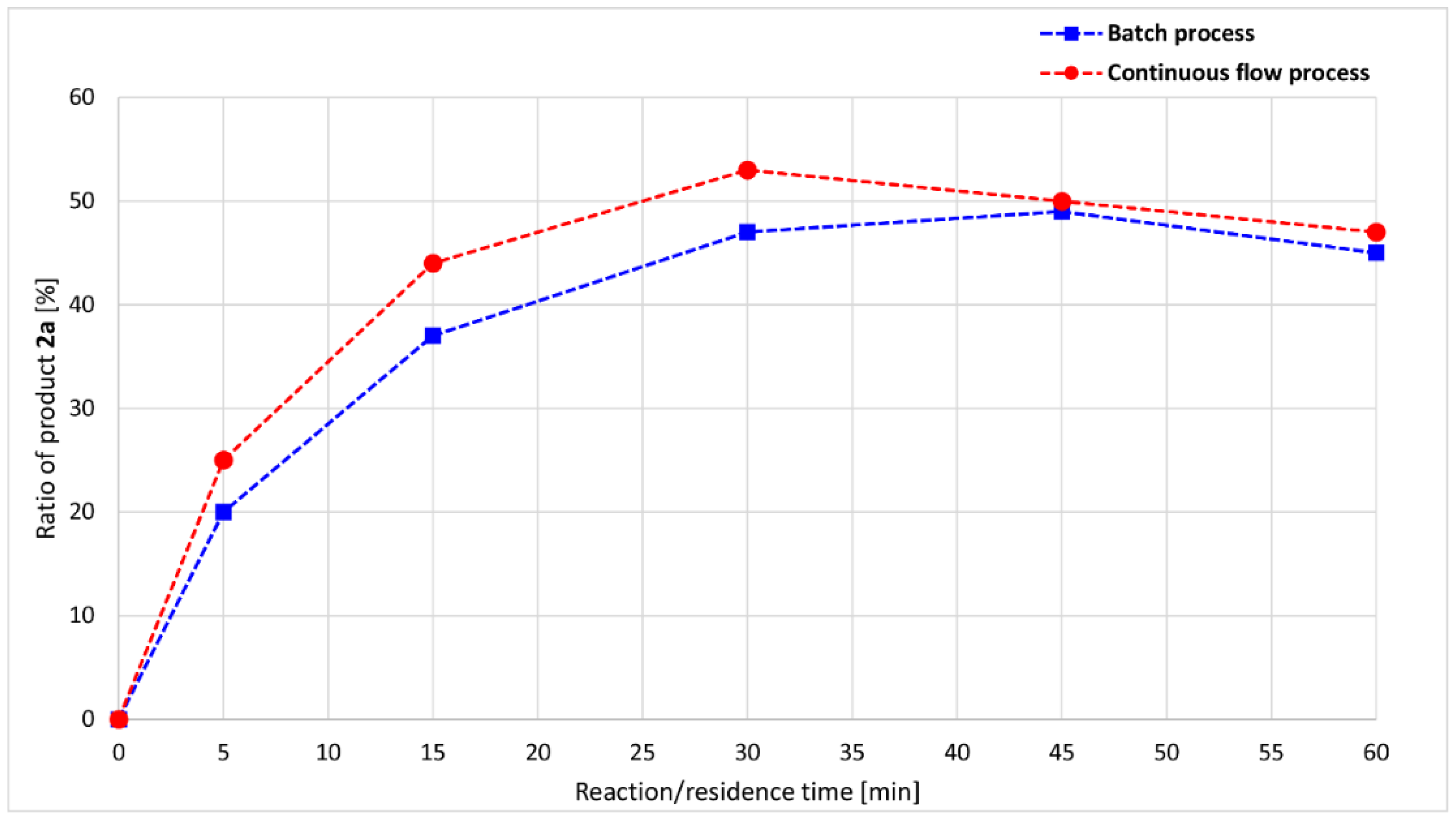
A more significant difference may be observed if the ratio of product 2a is examined separately (Figure 5). Maximum points are present on both curves: at 30 min and 53% in the flow experiments and at 45 min 49% in the case of the batch approaches. Based on the comparison, the flow reaction reaches the optimum point of phosphite 2a under a shorter time, with a higher ratio.
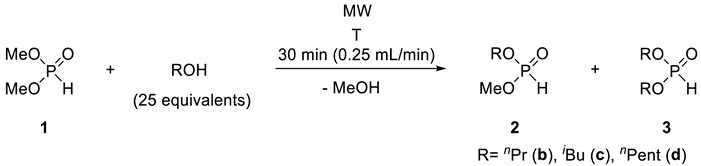
Based on the experiences above, the alcoholysis of DMP (1) was also carried out with n-propanol, i-butanol, as well as n-pentanol at 100 °C and 175 °C with a residence time of 30 min. In all cases, a mixture containing DMP (1) and 25 equivalents of the corresponding alcohol was passed through the continuous flow system. The data shown in Table 4 are concerned to the stationary operation. In the reaction of DMP (1) with nPrOH at 100 °C, the leaving mixture contained 45% of unreacted DMP (1), 46% of n-propyl methyl H-phosphonate (PrMP) (2b) and 9% of n-dipropyl H-phosphonate (DPrP) (3b) (Table 5, Entry 1). At a higher temperature of 175 °C, the fully-transesterified product (3b) was the major component (90%) (Table 5, Entry 2). The outcome of the alcoholysis of DMP (1) with iBuOH and nPentOH was similar (Table 5, Entries 3–6) However, in the case of the experiments carried out at 100 °C, the ratio of i-butyl methyl H-phosphonate (iBMP) (2c) and n-pentyl methyl H-phosphonate (PeMP) (2d) was above 50% (Table 5, Entries 3 and 5). After column chromatography, the corresponding mixed phosphonates (2b, 2c and 2d) were obtained in yields of 42–51% (Table 5, Entries 1, 3 and 5), and the fully-transesterified products (3b, 3c and 3d) were isolated in yields of 85–88% (Table 5, Entries 2, 4 and 6).
The next model was the continuous flow alcoholysis of diethyl H-phosphonate (DEP) (4) (Table 6). Based on the experiences from the transesterification of DMP (1), the reactions were carried out using 25 equivalents of the alcohols at a flow rate of 0.25 mL/min (or at a residence time of 30 min). First, the alcoholysis of DEP (4) by nBuOH was investigated. Performing the reaction at 100 °C, the unreacted DEP (4) was the main component along with 34% of n-butyl ethyl H-phosphonate (nBEP) (5a) and 8% of DBP (3a) (Table 6, Entry 1). At a higher temperature of 125 °C, the ratio of nBEP (5a) was 42%, and after column chromatography, compound 5a could be isolated in a yield of 38% (Table 6, Entry 2). Increasing the temperature to 150 °C and 175 °C, the proportion of 5a was decreased, while that of DBP (3a) increased (Table 6, Entries 3 and 4). In the latter case, the alcoholysis was complete, and the DBP (3a) was obtained in a yield of 85% (Table 6, Entry 4). The transesterification of DEP (4) by nPrOH at 125 °C took place similarly as the alcoholysis by nBuOH, and the n-propyl ethyl H-phosphonate (PrEP) (5b) was obtained in a yield of 36% (Table 6, Entries 2 and 5). At 175 °C, the conversion was not complete, and the resulting mixture contained only 74% of DPrP (3b) besides 4% of unreacted DEP (4) and 22% of PrEP (5b) (Table 6, Entry 6). To obtain a higher proportion of product 3b, the temperature was increased to 200 °C. Hence, phosphite 3b was formed in 84% (Table 6, Entry 7). In the case of iBuOH, the tendency was similar, and the highest amount of mixed phosphonate (5c) was formed at 125 °C, while at 175 and 200 °C, the fully-transesterified product (3c) predominated (Table 6, Entries 8–10). Using PentOH at 125 °C, the pentyl ethyl H-phosphonate (PeEP) (5d) was the main component in the departing mixture, from which it was isolated in a yield of 40% (Table 6, Entry 11). For the preparation of dipentyl H-phosphonate (DPeP) (3d), the application of 175 °C was enough, and compound 3d was obtained in a yield of 80% (Table 6, Entry 12).
The catalyst-free alcoholysis of dialkyl H-phosphonates with various aliphatic alcohols was carried out efficiently in a continuous flow MW reactor. Applying a residence time of 30 min at different temperatures, the alcoholysis could be fine-tuned towards the dialkyl H-phosphonates with two different or with two identical alkyl groups. In the case of the reaction studied, the dimethyl phosphite proved to be the more reactive starting material. After almost complete conversions, the (RO)2P(O)H species formed by transesterification were obtained in yields above 85%. The ratio of (RO)(R’O)P(O)H derivatives with two different alkyl groups was around 50% in all mixtures, affording the target compounds in a yield range of ca. 40–50%. By the method developed, a series of mixed dialkyl H-phosphonates, which are valuable building blocks for the preparation of chiral organophosphorus derivatives, was synthesized.
3. Materials and Methods
3.1. General
GC measurements were performed on an HP5890 Series 2 GC-FID chromatograph, using a 15 m × 0.18 mm Restek, Rtx-5 column with a film layer of 0.20 μm. The temperature of the column was initially held at 40 °C for 1 min, followed by programming at 25 °C/min up to 300 °C and a final period at 300 °C (isothermal) for 10 min. The temperature of the injector was 290 °C and of the FID detector was 300 °C. The carrier gas was N2.
GC-MS measurements were performed on an Agilent 6890 N-GC-5973 N-MSD chromatograph, using a 30 m × 0.25 mm Restek, Rtx-5SILMS column with a film layer of 0.25 μm. The initial temperature of the column was 45 °C for 1 min, followed by programming at 10 °C/min. up to 310 °C and a final period at 310 °C (isothermal) for 17 min. The temperature of the injector was 250 °C. The carrier gas was He, and the operation mode was splitless.
High resolution mass spectrometric measurements were performed using a TripleTOF 5600+ mass spectrometer in positive electrospray mode.
The 13C- and 1H-NMR spectra were obtained in CDCl3 solution on a Bruker DRX-500 spectrometer operating at 125.7 and 500.1 MHz, respectively. The 13C and 1H chemical shifts are referred to TMS. 31P-NMR spectra were obtained on a Bruker AV-300 spectrometer at 121.5 MHz. Chemical shifts are downfield relative to 85% H3PO4.
3.2. Equipment
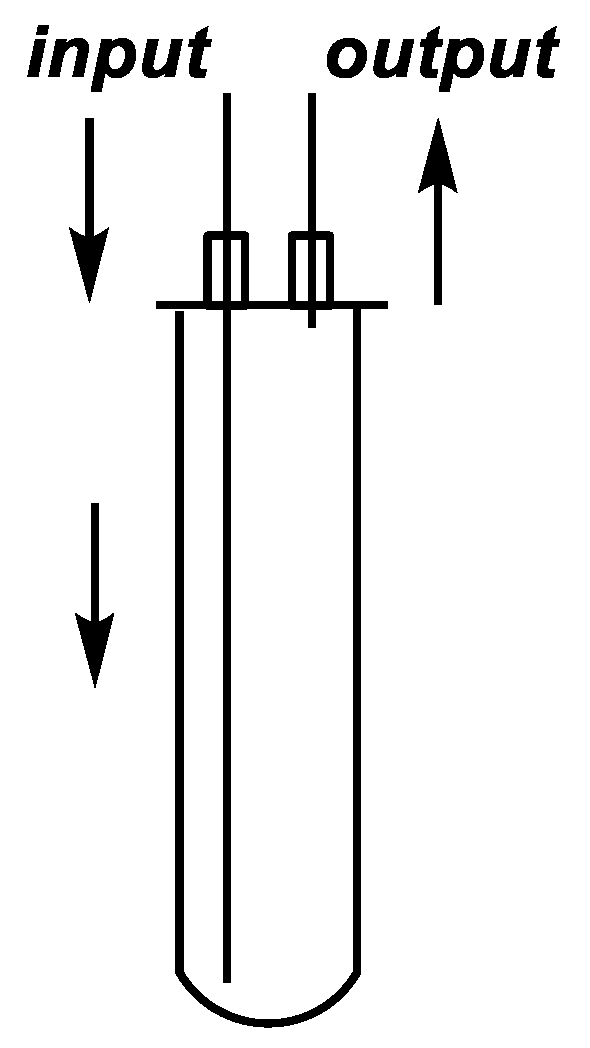
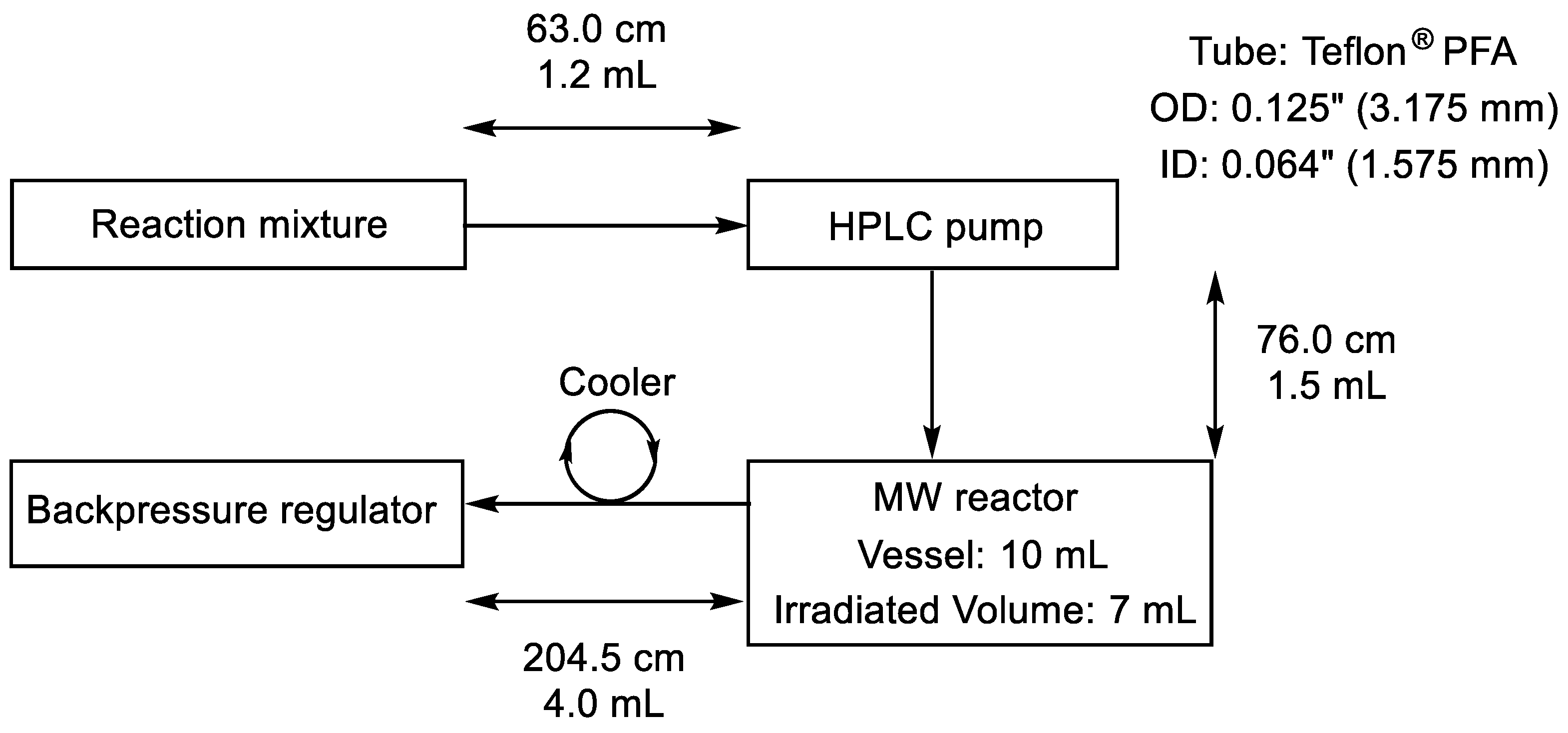
The continuous flow reactions were performed in a self-developed continuous flow system comprising a 300-W CEM® Discover-focused microwave reactor equipped with a CEM® 10-mL Flow Cell Accessory continuous flow unit (irradiated volume 7 mL), a Gilson 305 HPLC pump, an HPLC backpressure regulator with a 250-psi (17.2 bar) cartridge and a cooler. Teflon® (Wilmington, DE, USA) PFA tubes with an outside diameter of 0.125” (3.175 mm) and an inside diameter of 0.064” (1.575 mm) were used. The exact lengths and volumes of each tube part are shown in Figure 6. All of the tubes, screws and ferrules applied were fully compatible with a regular HPLC system.
3.3. General Procedure for the Continuous Flow Alcoholysis of Dialkyl H-Phosphonates
A mixture of 25.0 mmol of the dialkyl H-phosphonate (2.7 mL of dimethyl H-phosphonate, 3.9 mL of diethyl H-phosphonate) and 25 equivalents, 0.625 mol of alcohol (47 mL of n-propanol, 57 mL of n-butanol, 58 mL of i-butanol, 68 mL of n-pentanol) was homogenized by stirring for 5 minutes at 25 °C. The reactor was flushed with 20 mL of the mixture with a flow rate of 10 mL/min at 25 °C and 17 bar. Next, under the same pressure, the flow rate was set to the desired value, and the vessel was irradiated with a power of 45–300 W for 3–5 min, until the temperature reached the desired value, then the power was controlled automatically by the software of the MW reactor. The flow rate and the necessary power at stationary operation can be seen in Table 3, Table 5 and Table 6. The operation was regarded as the steady state on the basis of the results of the GC measurements. The products were separated by column chromatography using silica gel as the absorbent and ethyl acetate as the eluent. Yields were calculated on the basis of the weights obtained after separation and evaporation taking into consideration the quantity of the dialkyl H-phosphonate fed in during a given time. The 31P-NMR and mass data for the dialkyl H-phosphonates are listed in Table 7.
3.4. General Procedure for the Comparative Batch Experiments
A mixture of 0.5 mmol (0.05 mL) of dimethyl H-phosphonate and 25 equivalents, 12.5 mmol (1.1 mL) of n-butanol was heated at 100 °C in a vial in the MW reactor for 5–60 min as shown in Table 4. The volatile components were removed in vacuum, and the residual oil was analyzed by GC.
Methyl propyl H-phosphonate (2b): Yield: 42%; 31P-NMR and HRMS (see Table 7); 13C-NMR (CDCl3) δ 10.0 (CH3CH2), 23.8 (J = 6.2, CH2), 52.0 (J = 5.8, CH3O), 67.1 (J = 6.0, CH2O); 1H-NMR (CDCl3) δ 0.98 (t, JHH = 7.4, 3H, CH3CH2), 1.65–1.82 (m, 2H, CH2), 3.78, (d, JPH = 11.9, 3H, CH3O) 3.97–4.12 (m, 2H, CH2O), 6.79 (d, JPH = 695.4, 1H, PH).
i-Butyl methyl H-phosphonate (2c): Yield: 46%; 31P-NMR and HRMS (see Table 7); 13C-NMR (CDCl3) δ 18.6 (J = 3.1, CH3CH), 29.2 (J = 6.4, CH), 51.9 (J = 5.7, CH3O), 71.8 (J = 6.3, CH2O); 1H-NMR (CDCl3) δ 0.89 (d, JHH = 6.8, 6H, CH3CH), 1.84–1.96 (m, 1H, CH), 3.71 (d, JPH = 11.9, 3H, CH3O) 3.74–3.83 (m, 2H, CH2O), 6.72 (d, JPH = 695.8, 1H, PH).
Methyl pentyl H-phosphonate (2d): Yield: 50%; 31P-NMR and HRMS (see Table 7); 13C-NMR (CDCl3) δ 13.9 (CH3CH2), 22.2 (CH3CH2), 27.6 (CH3CH2CH2), 30.1 (J = 6.2, CH2CH2O), 51.9 (J = 5.7, CH3O), 67.0 (J = 6.1, CH2O); 1H-NMR (CDCl3) δ 0.91 (t, J = 6.9, 3H, CH3CH2), 1.29–1.44 (m, 4H, CH2CH2CH3), 1.63–1.78 (m, 2H, CH2CH2O), 3.78, (d, JPH = 11.9, 3H, CH3O) 3.99–4.17 (m, 2H, CH2O), 6.79 (d, JPH = 695.2, 1H, PH).
Ethyl propyl H-phosphonate (5b): Yield: 30%; 31P-NMR and HRMS (see Table 7); 13C-NMR (CDCl3) δ 10.0 (CH3CH2CH2), 16.3 (J = 6.1, CH3CH2O), 23.8 (J = 6.4, CH3CH2CH2), 61.8 (J = 5.8, CH2CH2O), 67.3 (J = 6.0, CH3CH2O); 1H NMR (CDCl3) δ 0.96 (t, JHH = 7.4, 3H, CH3CH2CH2), 1.35 (t, JHH = 7.1, 3H, CH3CH2O), 1.63–1.78 (m, 2H, CH3CH2CH2), 3.94–4.07, (m, 2H, CH2CH2O) 4.07–4.21 (m, 2H, CH3CH2O), 6.80 (d, JPH = 692.4, 1H, PH).
i-Butyl ethyl H-phosphonate (5c): Yield: 36%; 31P-NMR and HRMS (see Table 7); 13C-NMR (CDCl3) δ 16.3 (J = 6.2, CH3CH2), 18.7 (J = 1.7, CH3CH), 29.1 (J = 6.5, CH), 61.8 (J = 5.7, CH3CH2O), 71.6 (J = 6.2, CHCH2O); 1H NMR (CDCl3) δ 0.96 (d, JHH = 6.7, 6H, CH3CH), 1.37 (t, JHH = 7.1, 3H, CH3CH2), 1.89–2.06 (m, 1H, CH), 3.77–3.91, (m, 2H, CHCH2O) 4.09–4.22 (m, 2H, CH3CH2O), 6.82 (d, JPH = 692.7, 1H, PH).
4. Conclusions
In summary, a MW-assisted continuous flow method was developed for the catalyst-free alcoholysis of dialkyl H-phosphonates. By the optimization of the reaction parameters, the alcoholysis was fine-tuned towards dialkyl H-phosphonates with two different or with two identical alkyl groups. The selectivity of the reaction was similar in the flow and batch approaches; however, the continuous protocol offered several advantages. The dialkyl H-phosphonates with two different alkyl groups can be obtained at a shorter reaction time and in a higher ratio. Furthermore, a somewhat higher productivity (0.8–1.0 g/h) can be attained by the continuous flow method. Five new dialkyl H-phosphonates with different alkyl groups were isolated and characterized, which may be valuable building blocks in the synthesis of chiral organophosphorus compounds.
Author Contributions
E.B., Á.T. and G.K. conceived of and designed the experiments. Á.T. and N.T. performed the experiments. E.B. and G.K. contributed reagents/materials/analysis tools. E.B., Á.T. and G.K. wrote the paper.
Funding
The project was supported by the Hungarian Research Development and Innovation Fund (FK123961 and K119202), by the National Research, Development and Innovation Fund of Hungary in the frame of the FIEK_16-1-2016-0007 (Higher Education and Industrial Cooperation Center) project and by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00278/17/7) (E.B.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s guide to flow chemistry. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef] [PubMed]
- Glasnov, T. Continuous-Flow Chemistry in the Research Laboratory; Springer International Publishing: Basel, Switzerland, 2016; ISBN 978-3-319-32194-3. [Google Scholar]
- Bálint, E.; Keglevich, G. The Spread of the Application of the Microwave Technique in Organic Synthesis. In Milestones in Microwave Chemistry; Keglevich, G., Ed.; Springer: Basel, Switzerland, 2016; pp. 1–10. ISBN 978-3-319-30632-2. [Google Scholar] [Green Version]
- de la Hoz, A.; Loupy, A. (Eds.) Microwaves in Organic Synthesis, 3rd ed.; Wiley: Weinheim, Germany, 2012; ISBN 978-3-527-65131-3. [Google Scholar]
- Moseley, J.D. Microwave heating as a tool for process chemistry. In Microwave Heating as a Tool for Sustainable Chemistry; Leadbeater, N., Ed.; CRC Press: New York, NY, USA, 2010; pp. 105–147. ISBN 9781138111981. [Google Scholar]
- Kappe, C.O.; Stadler, A.; Dallinger, D. Microwaves in Organic and Medicinal Chemistry, 2nd ed.; Wiley: Weinheim, Germany, 2012; Volume 52, ISBN 978-3-527-33185-7. [Google Scholar]
- Estela, L.; Pouxb, M.; Benamaraa, N.; Polaerta, I. Continuous flow-microwave reactor: Where are we? Chem. Eng. Process. 2016, 113, 56–64. [Google Scholar] [CrossRef]
- Baxendale, I.; Hayward, J.; Ley, S. Microwave reactions under continuous flow conditions. Comb. Chem. High Throughput Screen. 2007, 10, 802–836. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, W.; Bolt, H.M. Esters, Organic. In Ullmann’s Encyclopedia of Industrial Chemistry, 7th ed.; Wiley: Weinheim, Germany, 2005; pp. 245–265. ISBN 978-3-527-30673-2. [Google Scholar]
- Otera, J.; Nishikido, J. (Eds.) Esterification: Methods, Reactions, and Applications, 2nd ed.; Wiley: Weinheim, Germany, 2010; ISBN 978-3-527-32289-3. [Google Scholar]
- Woodcock, L.L.; Wiles, C.; Greenway, G.M.; Watts, P.; Wells, A.; Eyley, S. Enzymatic synthesis of a series of alkyl esters using novozyme 435 in a packed-bed, miniaturized, continuous flow reactor. Biocatal. Biotransform. 2008, 26, 501–507. [Google Scholar] [CrossRef]
- Junior, I.I.; Flores, M.C.; Sutili, F.K.; Leite, S.G.F.; Miranda, L.S.D.M.; Leal, I.C.R.; de Souza, R.O.M.A. Fatty acids residue from palm oil refining process as feedstock for lipase catalyzed monoacylglicerol production under batch and continuous flow conditions. J. Mol. Catal. B Enzym. 2012, 77, 53–58. [Google Scholar] [CrossRef]
- Sutili, F.K.; Ruela, H.S.; Leite, S.G.F.; Miranda, L.S.D.M.; Leal, I.C.R.; de Souza, R.O.M.A. Lipase-catalyzed esterification of steric hindered fructose derivative by continuous flow and batch conditions. J. Mol. Catal. B Enzym. 2013, 85–86, 37–42. [Google Scholar] [CrossRef]
- Patil, N.G.; Benaskar, F.; Rebrov, E.V.; Meuldijk, J.; Hulshof, L.A.; Hessel, V.; Schouten, J.C. Scale-up of Microwave Assisted Flow Synthesis by Transient Processing through Monomode Cavities in Series. Org. Process Res. Dev. 2014, 18, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Sutili, F.K.; Ruela, H.S.; Nogueira, D.D.O.; Leal, I.C.R.; Miranda, L.S.D.M.; de Souza, R.O.M.A. Enhanced production of fructose ester by biocatalyzed continuous flow process. Sustain. Chem. Process. 2015, 3, 6. [Google Scholar] [CrossRef]
- Okuno, Y.; Isomura, S.; Sugamata, A.; Tamahori, K.; Fukuhara, A.; Kashiwagi, M.; Kitagawa, Y.; Kasai, E.; Takeda, K. Convenient and Simple Esterification in Continuous-Flow Systems using g-DMAP. ChemSusChem 2015, 8, 3587–3589. [Google Scholar] [CrossRef] [PubMed]
- Koreniuk, A.; Maresz, K.; Odrozek, K.; Jarzebski, A.B.; Mrowiec-Bialon, J. Highly effective continuous-flow monolithic silica microreactors for acid catalyzed processes. Appl. Catal. A 2015, 489, 203–208. [Google Scholar] [CrossRef]
- Baek, H.; Minakawa, M.; Yamada, Y.M.A.; Han, J.W.; Uozumi, Y. In-Water and Neat Batch and Continuous-Flow Direct Esterifcation and Transesterifcation by a Porous Polymeric Acid Catalyst. Sci. Rep. 2016, 6, 25925. [Google Scholar] [CrossRef] [PubMed]
- Furuta, A.; Fukuyama, T.; Ryu, I. Efficient Flow Fischer Esterification of Carboxylic Acids with Alcohols Using Sulfonic Acid-Functionalized Silica as Supported Catalyst. Bull. Chem. Soc. Jpn. 2017, 90, 607–612. [Google Scholar] [CrossRef]
- Iemhoff, A.; Sherwood, J.; McElroy, C.R.; Hunt, A.J. Towards sustainable kinetic resolution, a combination of bio-catalysis, flow chemistry and bio-based solvents. Green Chem. 2018, 20, 136–140. [Google Scholar] [CrossRef]
- Razzaq, T.; Glasnov, T.N.; Kappe, C.O. Continuous-Flow Microreactor Chemistry under High-Temperature/Pressure Conditions. Eur. J. Org. Chem. 2009, 1321–1325. [Google Scholar] [CrossRef]
- Archambault, C.M.; Leadbeater, N.E. A benchtop NMR spectrometer as a tool for monitoring mesoscale continuous-flow organic synthesis: Equipment interface and assessment in four organic transformations. RSC Adv. 2016, 6, 101171–101177. [Google Scholar] [CrossRef]
- Gumel, A.M.; Annuar, M.S.M. Thermomyces lanuginosus lipase-catalyzed synthesis of natural flavor esters in a continuous flow microreactor. 3 Biotech 2016, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Tajti, Á.; Tóth, N.; Bálint, E.; Keglevich, G. Esterification of benzoic acid in a continuous flow microwave reactor. J. Flow Chem. 2017, 8, 11–19. [Google Scholar] [CrossRef]
- Krull, M.; Moschhaeuser, R. Continuous Method for Producing Esters of Aromatic Carboxylic Acids. U.S. Patent 0088918, 12 April 2012. [Google Scholar]
- Cablewski, T.; Faux, A.F.; Strauss, C.R. Development and Application of a Continuous Microwave Reactor for Organic Synthesis. J. Org. Chem. 1994, 59, 3408–3412. [Google Scholar] [CrossRef]
- Chen, S.-T.; Chiou, S.-H.; Wang, K.-T. Preparative scale organic synthesis using a kitchen microwave oven. J. Chem. Soc. Chem. Commun. 1990, 807–809. [Google Scholar] [CrossRef]
- Pipus, G.; Plazl, I.; Koloini, T. Esterification of benzoic acid in microwave tubular flow reactor. Chem. Eng. J. 2000, 76, 239–245. [Google Scholar] [CrossRef]
- Asadi, M.; Hooper, J.F.; Lupton, D.W. Biodiesel synthesis using integrated acid and base catalysis in continuous flow. Tetrahedron 2016, 72, 3729–3733. [Google Scholar] [CrossRef]
- Adeyemi, A.; Bergman, J.; Branalt, J.; Savmarker, J.; Larhed, M. Continuous Flow Synthesis under High-Temperature/High-Pressure Conditions Using a Resistively Heated Flow Reactor. Org. Process Res. Dev. 2017, 21, 947–955. [Google Scholar] [CrossRef]
- Tran, D.-T.; Chang, J.-S.; Lee, D.-J. Recent insights into continuous-flow biodiesel production via catalytic and non-catalytic transesterification processes. Appl. Energy 2017, 185, 376–409. [Google Scholar] [CrossRef]
- Corbridge, D.E.C. Phosphorus: Chemistry, Biochemistry and Technology, 6th ed.; CRC Press: New York, NY, USA, 2013; ISBN 978-1-439-84088-7. [Google Scholar]
- Troev, K.D. Chemistry and Application of H-Phosphonates; Elsevier: Amsterdam, The Netherlands, 2006; ISBN 978-0-080-47649-0. [Google Scholar]
- Bálint, E.; Tajti, Á.; Tripolszky, A. Synthesis of α-aminophosphonates by the Kabachnik–Fields reaction and by the Pudovik reaction. In Organophosphorus Chemistry; Keglevich, G., Ed.; Walter de Gruyter GmbH: Berlin, Germany, 2018; pp. 108–147. ISBN 978-3-11-053453-5. [Google Scholar]
- Rádai, Z.; Kiss, N.Z.; Keglevich, G. Synthesis of α-hydroxyphosphonates, an important class of bioactive compounds. In Organophosphorus Chemistry; Keglevich, G., Ed.; Walter de Gruyter GmbH: Berlin, Germany, 2018; pp. 91–107. ISBN 978-3-11-053453-5. [Google Scholar]
- Henyecz, R.; Keglevich, G. P-C couplings by the Hiaro reaction. In Organophosphorus Chemistry; Keglevich, G., Ed.; Walter de Gruyter GmbH: Berlin, Germany, 2018; pp. 158–178. ISBN 978-3-11-053453-5. [Google Scholar]
- Enders, D.; Saint-Dizier, A.; Lannou, M.-I.; Lenzen, A. The phospha-Michael addition in organic synthesis. Eur. J. Org. Chem. 2006, 29–49. [Google Scholar] [CrossRef]
- Tajti, Á.; Bálint, E.; Keglevich, G. Synthesis of ethyl octyl α-aminophosphonate derivatives. Curr. Org. Synth. 2016, 13, 638–675. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Kalocsai, D.; Mátravölgyi, B.; Konstantin, K.; Czugler, M.; Keglevich, G. Synthesis and utilization of optically active α-aminophosphonate derivatives by Kabachnik-Fields reaction. Tetrahedron 2017, 73, 5659–5667. [Google Scholar] [CrossRef]
- Gerrard, W. The interaction of n-butyl alcohol and the chlorides and oxychloride of phosphorus in the absence and in the presence of pyridine. J. Chem. Soc. 1940, 1466–1469. [Google Scholar] [CrossRef]
- Hardy, E.E.; Anniston, A.; Kosolapoff, G.M. Halogenated Compounds and Process for Making Same. U.S. Patent 2409039, 8 October 1946. [Google Scholar]
- Foss, O. Di-O-alkylmonothiophosphates and the corresponding pseudohalogens. Acta Chem. Scand. 1947, 1, 8–31. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.H.; Chadwick, D.H.; Kaufman, S. Continuous process for preparing dialkyl phosphites. Ind. Eng. Chem. Res. 1957, 49, 1871–1873. [Google Scholar] [CrossRef]
- Mitschke, K.-H. Method for the Combined Production of Diethyl Phosphite and Ethyl Chloride. Application Number WO200424742 A1, 25 March 2004. [Google Scholar]
- Kendall, A.J.; Salazar, C.A.; Martino, P.F.; Tyler, D.R. Direct conversion of phosphonates to phosphine oxides: An improved synthetic route to phosphines including the first synthesis of methyl JohnPhos. Organometallics 2014, 33, 6171–6178. [Google Scholar] [CrossRef]
- Kosolapoff, G.M. Preparation of some mixed dialkyl phosphites. J. Am. Chem. Soc. 1951, 73, 4989. [Google Scholar] [CrossRef]
- Fields, E. The synthesis of esters of substituted amino phosphonic acids. J. Am. Chem. Soc. 1952, 74, 1528–1531. [Google Scholar] [CrossRef]
- Oswald, A.A. Synthesis of cyclic phosphorus acid esters by transesterification. Can. J. Chem. 1959, 37, 1499–1504. [Google Scholar] [CrossRef]
- Kuskov, V.K.; Gradis, G.K. Reaction of diethyl phosphite with sodium alcoholates. Dokl. Akad. Nauk SSSR 1953, 92, 323–324. [Google Scholar]
- Froneman, M.; Modro, T.A. The titanium-mediated transesterification of phosphorus esters. Tetrahedron Lett. 1988, 27, 3327–3330. [Google Scholar] [CrossRef]
- Aitken, R.A.; Collett, C.J.; Mesher, S.T.E. Convenient preparation of long-chain dialkyl phosphates: Synthesis of dialkyl phosphates. Synthesis 2012, 44, 2515–2518. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Drahos, L.; Ilia, G.; Keglevich, G. Alcoholysis of dialkyl phosphites under microwave conditions. Curr. Org. Chem. 2013, 17, 555–562. [Google Scholar] [CrossRef]
- Zarrougui, R.; Raouafi, N.; Lemordant, D. New series of green cyclic ammonium-based room temperature ionic liquids with alkylphosphite-containing anion: Synthesis and physicochemical characterization. J. Chem. Eng. Data 2014, 59, 1193–1201. [Google Scholar] [CrossRef]
- Salin, A.V.; Il’in, A.V.; Shamsutdinova, F.G.; Fatkhutdinov, A.R.; Galkin, V.I.; Islamov, D.R.; Kataeva, O.N. Phosphine-catalyzed addition of P(O)-H compounds to ethyl phenylpropiolate. Tetrahedron Lett. 2015, 56, 6282–6286. [Google Scholar] [CrossRef]
- Ernsberger, M.L.; Hill, J.W. Preparation of Organic Phosphorus Compounds, and in Particular, of Dialkyl Phosphites. U.S. Patent 2661364, 1 December 1953. [Google Scholar]
- Budnikova, Y.G.; Kargin, Y.M. Electrosynthesis of aliphatic esters of phosphorus acids from white phosphorus in alcohol solutions, involving radical cations of phenothiazine and triarylamine. Russ. J. Gen. Chem. 1995, 65, 504–507. [Google Scholar]
- Abdreimova, R.R.; Akbayeva, D.N.; Polimbetova, G.S.; Caminade, A.-M.; Majoral, J.-P. Chlorine free synthesis of organophosphorus compounds based on the functionalization of white phosphorus (P4). Phosphorus Sulfur Silicon Relat. Elem. 2000, 156, 239–254. [Google Scholar] [CrossRef]
- Trofimov, B.; Timokhin, B.; Gusarova, N.; Kazantseva, M. Golubin, Cu-catalyzed oxidative phosphorylation of alkanols with white phosphorus and H2O2. Phosphorus Sulfur Silicon Relat. Elem. 2002, 177, 2385–2390. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Kafiyatullina, A.G.; Sinyashin, O.G.; Abdreimova, R.R. Electrochemical synthesis of phosphorus esters from white phosphorus in the presence of copper complexes and ethanol. Russ. Chem. Bull. 2003, 52, 929–938. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Yakhvarov, D.G.; Sinyashin, O.G. Electrocatalytic eco-efficient functionalization of white phosphorus. J. Organomet. Chem. 2005, 690, 2416–2425. [Google Scholar] [CrossRef]
- Fisher, H.C.; Prost, L.; Montchamp, J.L. Organophosphorus chemistry without PCl3: A bridge from hypophosphorous acid to H-phosphonate diesters. Eur. J. Org. Chem. 2013, 7973–7978. [Google Scholar] [CrossRef]
- Kluba, M.; Zwierzak, A. Alkylation of tetra-n-butylaminium alkyl hydrogen phosphites. A new route to mixed dialkyl phosphites. Synthesis 1978, 2, 134–137. [Google Scholar] [CrossRef]
- Ilia, G.; Kurunczi, L. Synthesis of mixed alkylphosphites and alkylphosphates. Phosphorus Sulfur Silicon Relat. Elem. 2003, 178, 1513–1519. [Google Scholar] [CrossRef]
- Berchel, M.; Haddad, J.; Le Corre, S.S.; Haelters, J.-P.; Jaffrès, P.-A. Synthesis of lipid-based unsymmetrical O,O-dialkylphosphites. Tetrahedron Lett. 2015, 56, 2345–2348. [Google Scholar] [CrossRef]
- Guin, J.; Wang, Q.; van Gemmeren, M.; List, B. The catalytic asymmetric Abramov reaction. Angew. Chem. Int. Ed. 2014, 53, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Schreeve, J.M. Rapid and high yield oxidation of phosphine, phosphite and phosphinite compounds to phosphine oxides, phosphates and phosphinates using hypofluorous acid–acetonitrile complex. J. Fluorine Chem. 2005, 126, 1054–1056. [Google Scholar] [CrossRef]
- Santschi, N.; Togni, A. Electrophilic Trifluoromethylation of S-Hydrogen Phosphorothioates. J. Org. Chem. 2011, 76, 4189–4193. [Google Scholar] [CrossRef] [PubMed]
- Kers, A.; Kers, I.; Stawinski, J.; Sobkowski, M.; Kraszewski, A. Studies on Aryl H-Phosphonates; Part 2: A General Method for the Preparation of Alkyl H-Phosphonate Monoesters. Synthesis 1995, 427–430. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 2a–d, 3a–d, 5a–d are available from the authors. |
Scheme 1.
Synthesis of dialkyl H-phosphonates from phosphorus trichloride.

Scheme 2.
Synthesis of dialkyl H-phosphonates by alcoholysis.

Scheme 3.
Miscellaneous synthesis of dialkyl H-phosphonates.
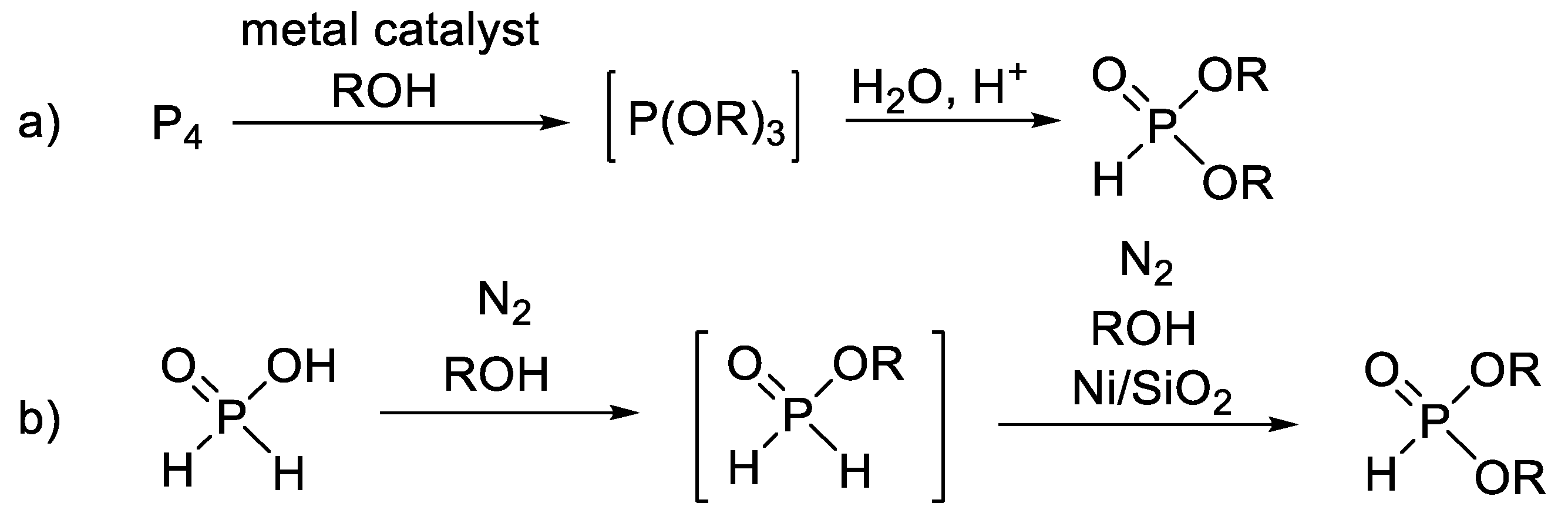
Figure 1.
Schematic drawing of the continuous flow system developed.
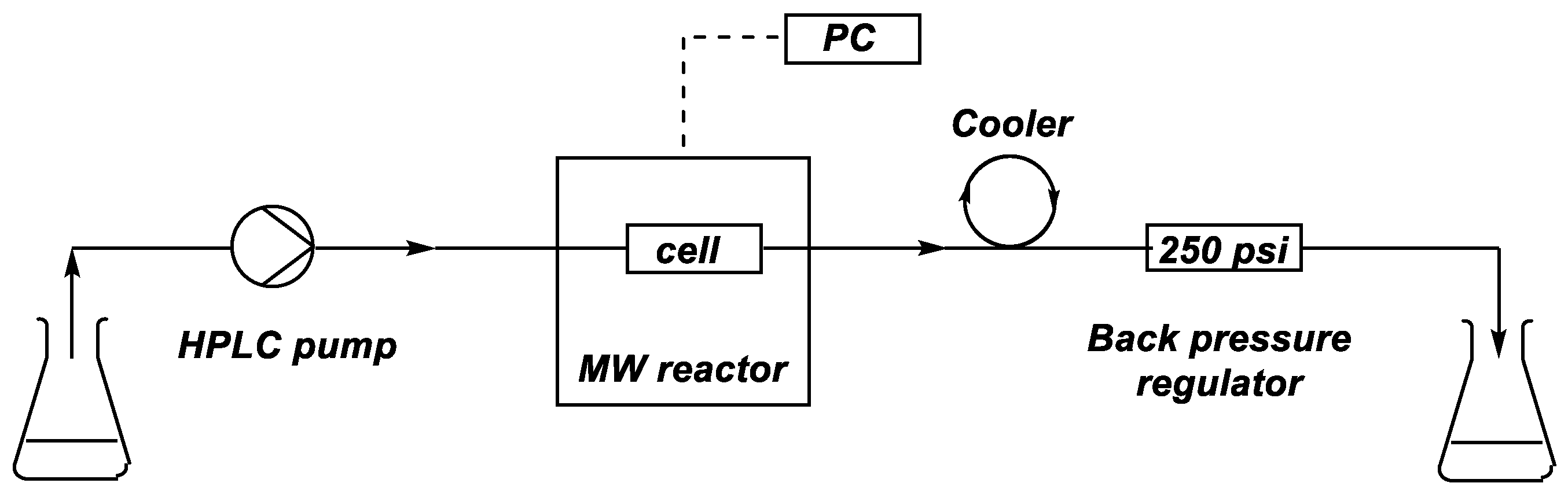
Figure 2.
Sketch of the continuous flow cell.
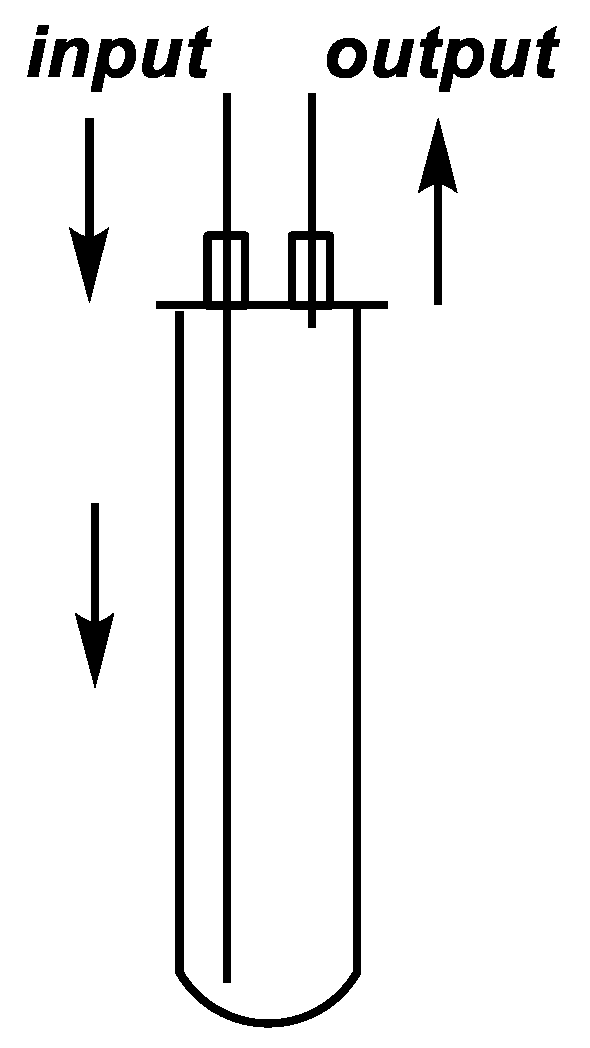
Figure 3.
Composition of the reaction mixtures of the MW-assisted alcoholysis of DMP with nBuOH at different temperatures at a residence time of 30 min (Table 3 entries 3 and 7–9).
Figure 3.
Composition of the reaction mixtures of the MW-assisted alcoholysis of DMP with nBuOH at different temperatures at a residence time of 30 min (Table 3 entries 3 and 7–9).
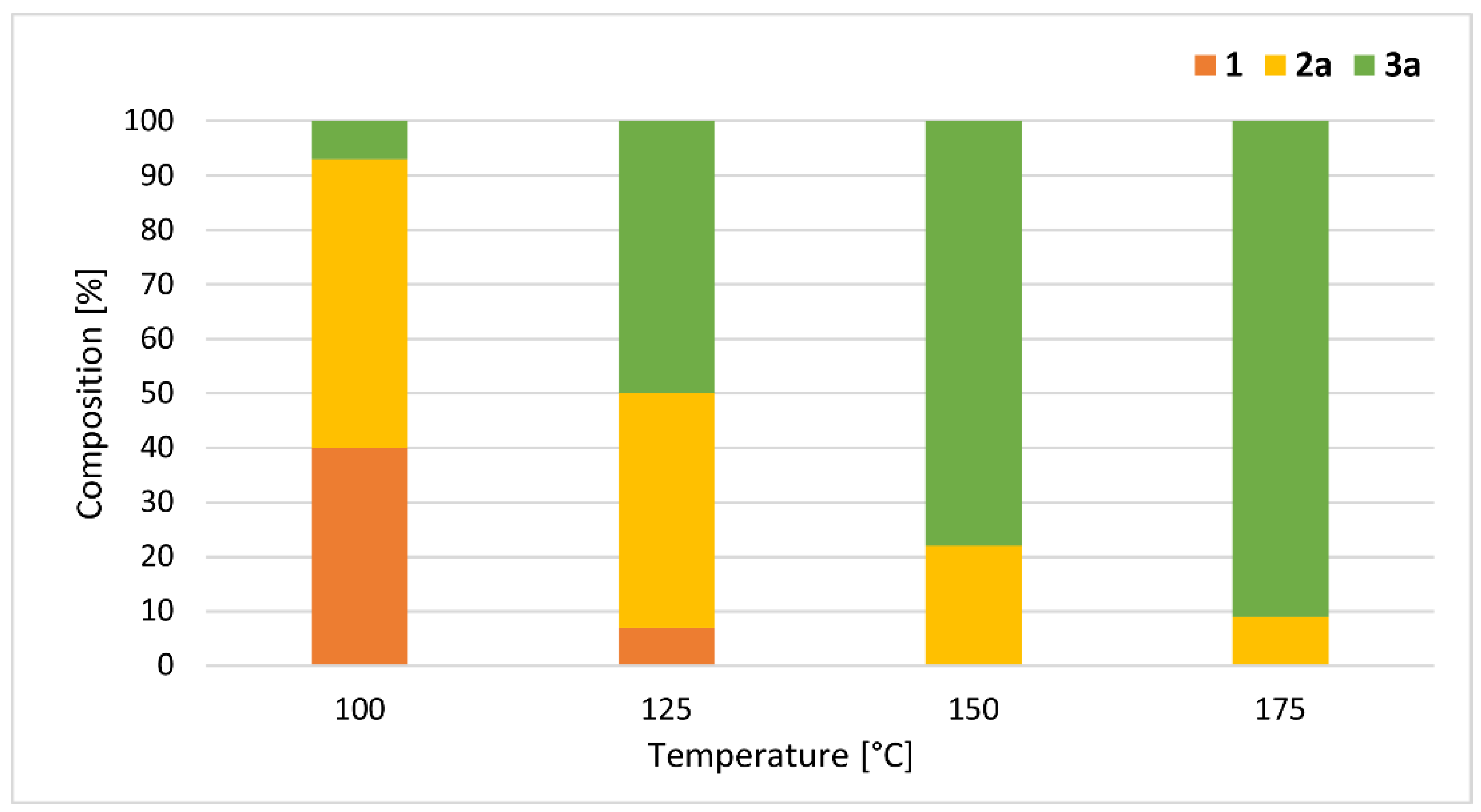
Figure 4.
Reaction/residence time-dependence of the conversion of the alcoholysis of DMP with nBuOH at 100 °C.
Figure 4.
Reaction/residence time-dependence of the conversion of the alcoholysis of DMP with nBuOH at 100 °C.

Figure 5.
Reaction/residence time-dependence of the product 2a ratio in the alcoholysis of DMP with nBuOH at 100 °C.
Figure 5.
Reaction/residence time-dependence of the product 2a ratio in the alcoholysis of DMP with nBuOH at 100 °C.
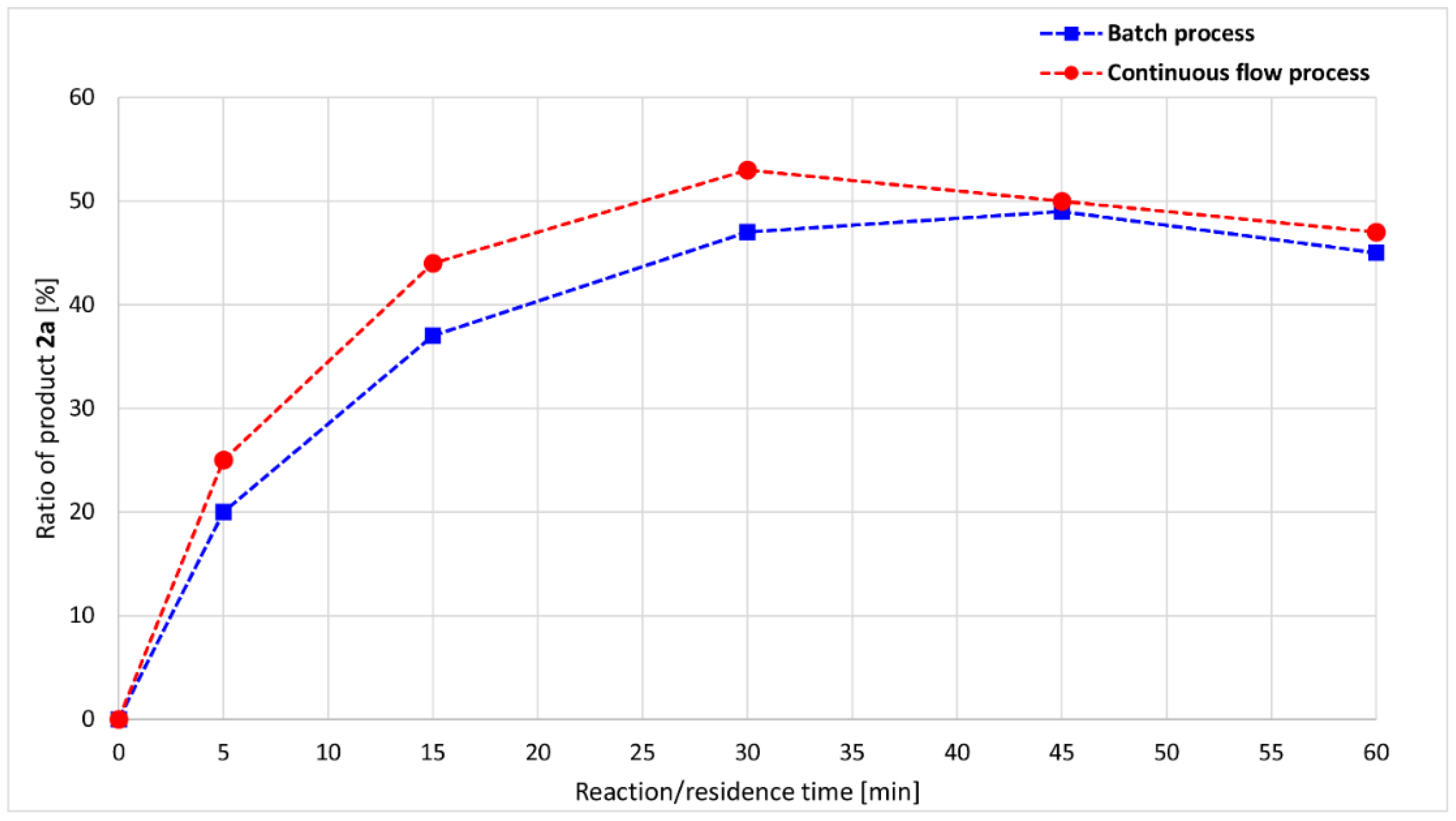
Figure 6.
Design parameters of the continuous flow system.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
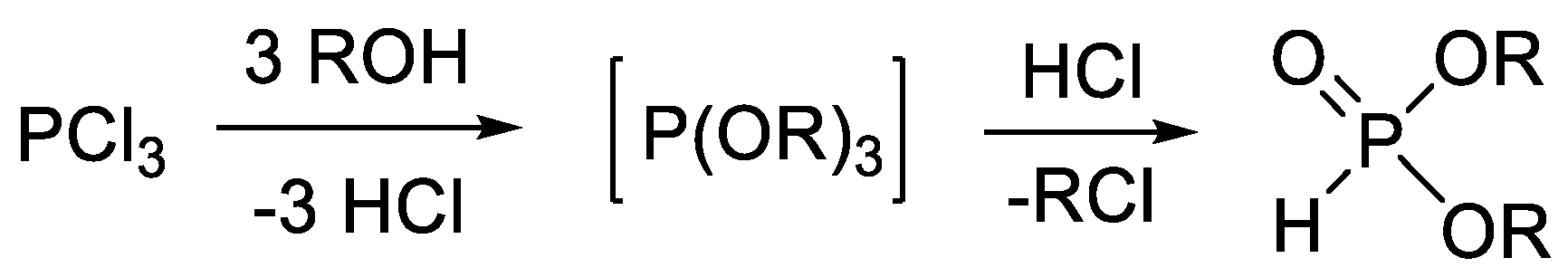{kind=link}
{kind=link}
{kind=link}
Table 1.
Synthetic methods for the preparation of dialkyl H-phosphonates bearing two different alkyl groups.
Table 1.
Synthetic methods for the preparation of dialkyl H-phosphonates bearing two different alkyl groups.
| Entry | Reaction | Average Yield (%) | Flow Compatible | Ref. |
|---|---|---|---|---|
| 1 | 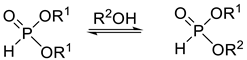 | 30–50 | + | [46,50,52] |
| 2 | 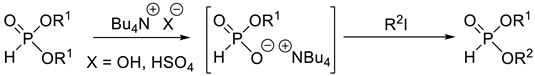 | 50–70 | − | [62,63] |
| 3 | 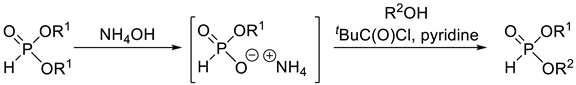 | 50–85 | − | [64] |
| 4 | 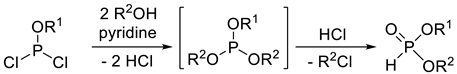 | 30–60 | − | [65] |
Table 2.
Alcoholysis of dialkyl H-phosphonates in a batch microwave (MW) reactor [33].
Table 2.
Alcoholysis of dialkyl H-phosphonates in a batch microwave (MW) reactor [33].
| Entry | R1 | R2 | R2OH (equiv) | T (°C) | t (min) | Composition (%) a | ||
|---|---|---|---|---|---|---|---|---|
| A | B | C | ||||||
| 1 | Me | Et | 25 | 100 | 120 | 22 | 56 | 22 |
| 2 | Me | Et | 50 | 175 | 40 | 0 | 4 | 96 |
| 3 | Me | nBu | 25 | 125 | 60 | 0 | 40 | 60 |
| 4 | Me | nBu | 50 | 150 | 60 | 0 | 0 | 96 |
| 5 | Et | Me | 25 | 125 | 120 | 49 | 38 | 13 |
| 6 | Et | Me | 50 | 175 | 40 | 0 | 21 | 79 |
| 7 | Et | iPr | 25 | 125 | 60 | 26 | 57 | 17 |
| 8 | Et | iPr | 50 | 175 | 40 | 0 | 6 | 94 |
| 9 | Et | nBu | 25 | 125 | 60 | 25 | 54 | 21 |
| 10 | Et | nBu | 50 | 175 | 40 | 0 | 2 | 98 |
| 11 | Et | nPent | 25 | 125 | 60 | 3 | 52 | 45 |
| 12 | Et | nPent | 50 | 175 | 40 | 0 | 8 | 92 |
a Analyzed by GC. Bold numbers indicate the target compounds in the reactions.
Table 3.
Continuous flow alcoholysis of DMP with nBuOH.
| Entry | Mode of Heating | Power (W) | T (°C) a | Flow Rate | τ (min) | Conversion (%) | Composition (%) c | Yield (%) d | ||
|---|---|---|---|---|---|---|---|---|---|---|
| (mL/min) b | 1 | 2a | 3a | |||||||
| 1 | MW | 22 | 100 | 1.4 | 5 | 26 | 74 | 25 | 1 | - |
| 2 | MW | 14 | 100 | 0.45 | 15 | 52 | 48 | 44 | 8 | - |
| 3 | MW | 10 | 100 | 0.25 | 30 | 65 | 35 | 53 | 12 | 48 (2a) |
| 4 | MW | 8 | 100 | 0.15 | 45 | 76 | 24 | 50 | 26 | - |
| 5 | MW | 5 | 100 | 0.10 | 60 | 83 | 18 | 47 | 35 | - |
| 6 | Δ | - | 100 | 0.25 | 30 | 15 | 85 | 15 | 0 | - |
| 7 | MW | 18 | 125 | 0.25 | 30 | 93 | 7 | 43 | 50 | - |
| 8 | MW | 38 | 150 | 0.25 | 30 | 100 | 0 | 22 | 78 | - |
| 9 | MW | 59 | 175 | 0.25 | 30 | 100 | 0 | 9 | 91e,f | 88 (3a) |
| 10 | Δ | - | 175 | 0.25 | 30 | 98 | 2 | 15 | 83 | - |
a The pressure was 17 bar; b Based on the HPLC pump; c the mixtures from the stationary operation were analyzed by GC; d after column chromatography; e No change on longer residence time (45 min); f No change on higher excess (50 equiv) of nBuOH. Bold numbers indicate the target compounds in the reactions.
Table 4.
Comparative experiments for the alcoholysis of dimethyl H-phosphonate (DMP) with nBuOH in a batch MW reactor.
Table 4.
Comparative experiments for the alcoholysis of dimethyl H-phosphonate (DMP) with nBuOH in a batch MW reactor.
| Entry | t (min) | Conversion (%) | Composition (%) a | ||
|---|---|---|---|---|---|
| 1 | 2a | 3a | |||
| 1 | 5 | 24 | 76 | 20 | 4 |
| 2 | 15 | 46 | 54 | 37 | 9 |
| 3 | 30 | 59 | 41 | 47 | 12 |
| 4 | 45 | 67 | 33 | 49 | 18 |
| 5 | 60 | 73 | 26 | 45 | 29 |
a Based on GC.
Table 5.
Continuous flow alcoholysis of DMP with various alcohols.
| Entry | R | Power (W) | T (°C) a | Composition (%) b | Yield (%) c | ||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | |||||
| 1 | nPr | 10 | 100 | 45 | 46 | 9 | 42 (2b) |
| 2 | 57 | 175 | 0 | 10 | 90 | 85 (3b) | |
| 3 | iBu | 10 | 100 | 38 | 51 | 11 | 46 (2c) |
| 4 | 71 | 175 | 0 | 10 | 90 | 86 (3c) | |
| 5 | nPent | 10 | 100 | 32 | 55 | 13 | 51 (2d) |
| 6 | 55 | 175 | 0 | 8 | 92 | 88 (3d) | |
a The pressure was 17 bar; b the mixtures from the stationary operation were analyzed by GC; c after column chromatography. Bold numbers indicate the target compounds in the reactions.
Table 6.
Continuous flow alcoholysis of DEP with alcohols.

| Entry | R | Power (W) | T (°C) a | Composition (%) b | Yield (%) c | ||
|---|---|---|---|---|---|---|---|
| 4 | 5 | 3 | |||||
| 1 | nBu | 10 | 100 | 58 | 34 | 8 | - |
| 2 | 17 | 125 | 44 | 42 | 14 | 38 (5a) | |
| 3 | 38 | 150 | 7 | 32 | 61 | - | |
| 4 | 57 | 175 | 0 | 11 | 89 d,e | 85 (3a) | |
| 5 | nPr | 18 | 125 | 51 | 41 | 8 | 36 (5b) |
| 6 | 54 | 175 | 4 | 22 | 74 | – | |
| 7 | 83 | 200 | 2 | 14 | 84 d,e | 78 (3b) | |
| 8 | iBu | 21 | 125 | 21 | 40 | 39 | 36 (5c) |
| 9 | 63 | 175 | 3 | 26 | 71 | – | |
| 10 | 105 | 200 | 0 | 16 | 84 d,e | 77 (3c) | |
| 11 | nPent | 10 | 125 | 31 | 44 | 25 | 40 (5d) |
| 12 | 39 | 175 | 2 | 13 | 85 d,e | 80 (3d) | |
a The pressure was 17 bar; b the mixtures from the stationary operation were analyzed by GC; c after column chromatography; d no change upon longer residence time (45 min); e no change upon higher excess (50 equiv) of nBuOH. Bold numbers indicate the target compounds in the reactions.
Table 7.
31P-NMR and mass data for the dialkyl H-phosphonates prepared.
| Compound | δP (CDCl3) | δP [Ref] | [M + H]+found | [M + H]+requires |
|---|---|---|---|---|
| 2a | 9.3 (695.0) | 9.3 [52] | 153.0684 | 153.0681 |
| 2b | 9.3 (695.4) | ‒ | 139.0522 | 139.0523 |
| 2c | 6.7 (695.7) | ‒ | 153.0676 | 153.0681 |
| 2d | 6.8 (695.2) | ‒ | 167.0833 | 167.0837 |
| 3a | 7.8 | 7.9 [66] | 195.1153 | 195.1150 |
| 3b | 7.9 | 7.8 [67] | 167.0833 | 167.0837 |
| 3c | 8.1 | 8.0 [67] | 195.1145 | 195.1150 |
| 3d | 7.8 | 8.1 [68] | 223.1462 | 223.1463 |
| 5a | 7.6 (693.0) | 7.6 [52] | 167.0840 | 167.0837 |
| 5b | 8.6 (692.4) | ‒ | 153.0673 | 153.0681 |
| 5c | 7.8 (692.8) | ‒ | 167.0832 | 167.0837 |
| 5d | 7.7 (692.1) | 7.7 [52] | 181.0992 | 181.0994 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bálint, E.; Tajti, Á.; Tóth, N.; Keglevich, G. Continuous Flow Alcoholysis of Dialkyl H-Phosphonates with Aliphatic Alcohols. Molecules 2018, 23, 1618. https://doi.org/10.3390/molecules23071618
AMA Style
Bálint E, Tajti Á, Tóth N, Keglevich G. Continuous Flow Alcoholysis of Dialkyl H-Phosphonates with Aliphatic Alcohols. Molecules. 2018; 23(7):1618. https://doi.org/10.3390/molecules23071618
Chicago/Turabian StyleBálint, Erika, Ádám Tajti, Nóra Tóth, and György Keglevich. 2018. "Continuous Flow Alcoholysis of Dialkyl H-Phosphonates with Aliphatic Alcohols" Molecules 23, no. 7: 1618. https://doi.org/10.3390/molecules23071618