Compressional-Puffing Pretreatment Enhances Neuroprotective Effects of Fucoidans from the Brown Seaweed Sargassum hemiphyllum on 6-Hydroxydopamine-Induced Apoptosis in SH-SY5Y Cells



Abstract
:1. Introduction
2. Results and Discussion
2.1. Effects of Compressional-Puffing Parameters on the Degree of Moisture Loss of Puffed Algal Samples and Extraction Yields of Fucoidan
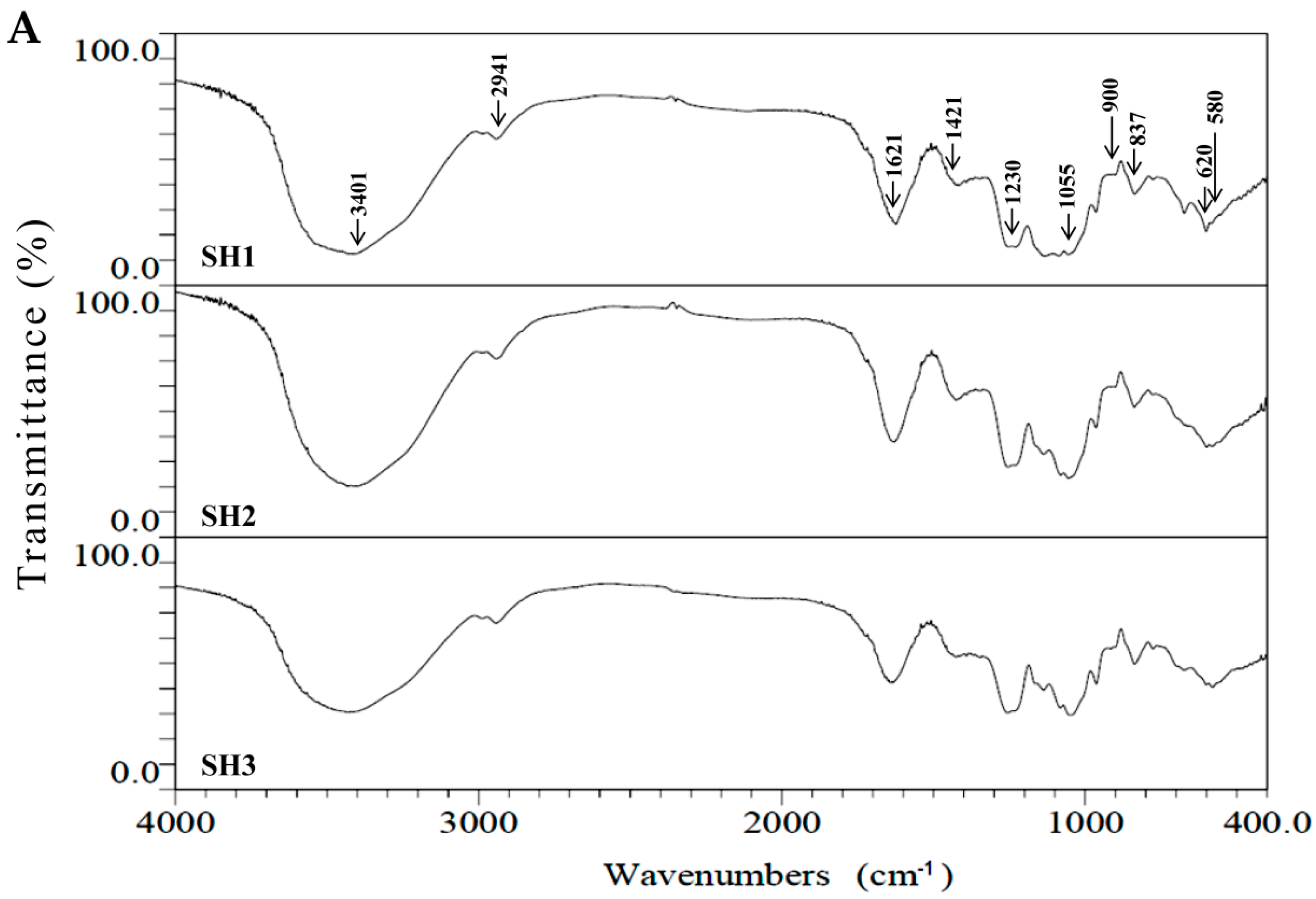
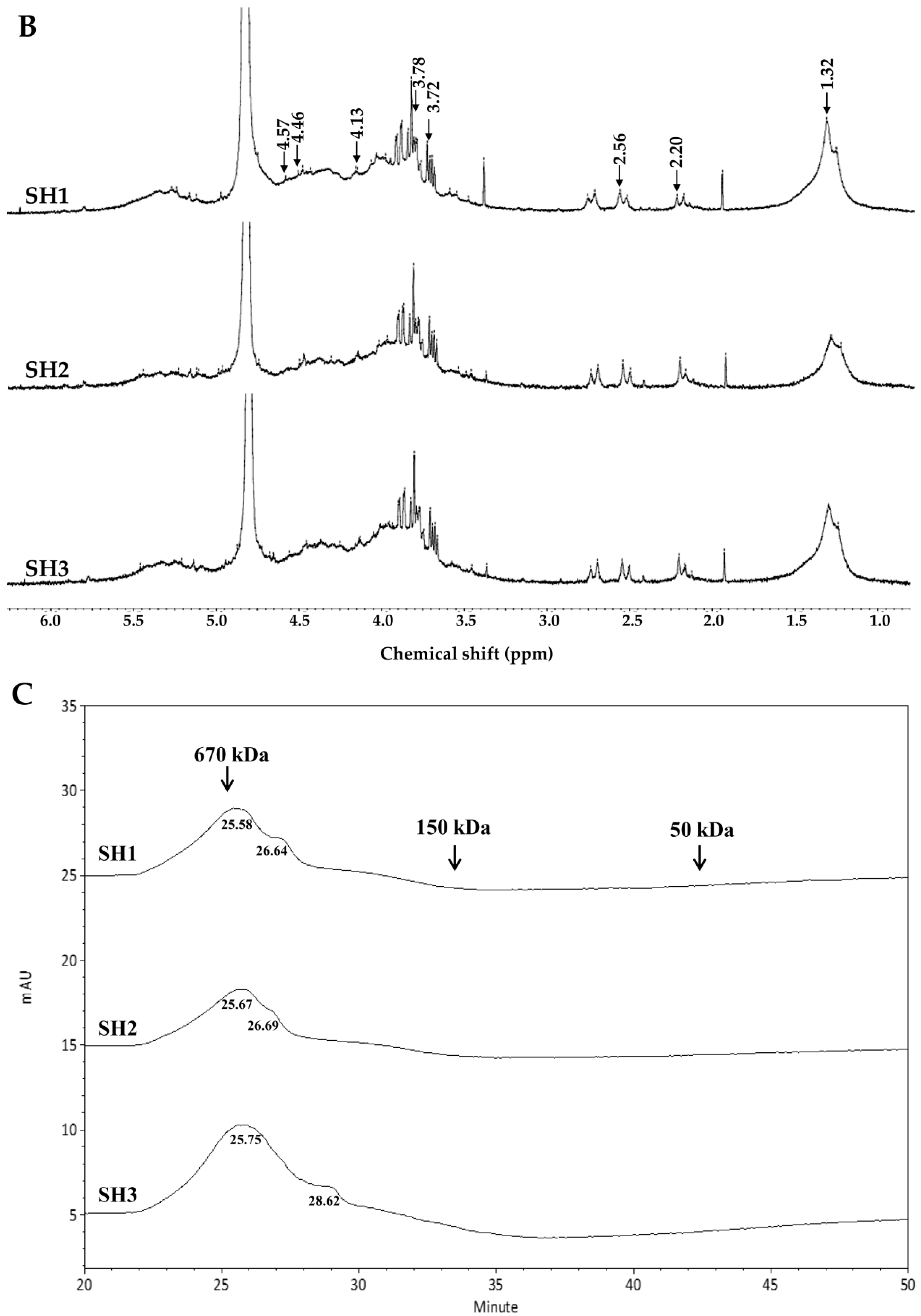
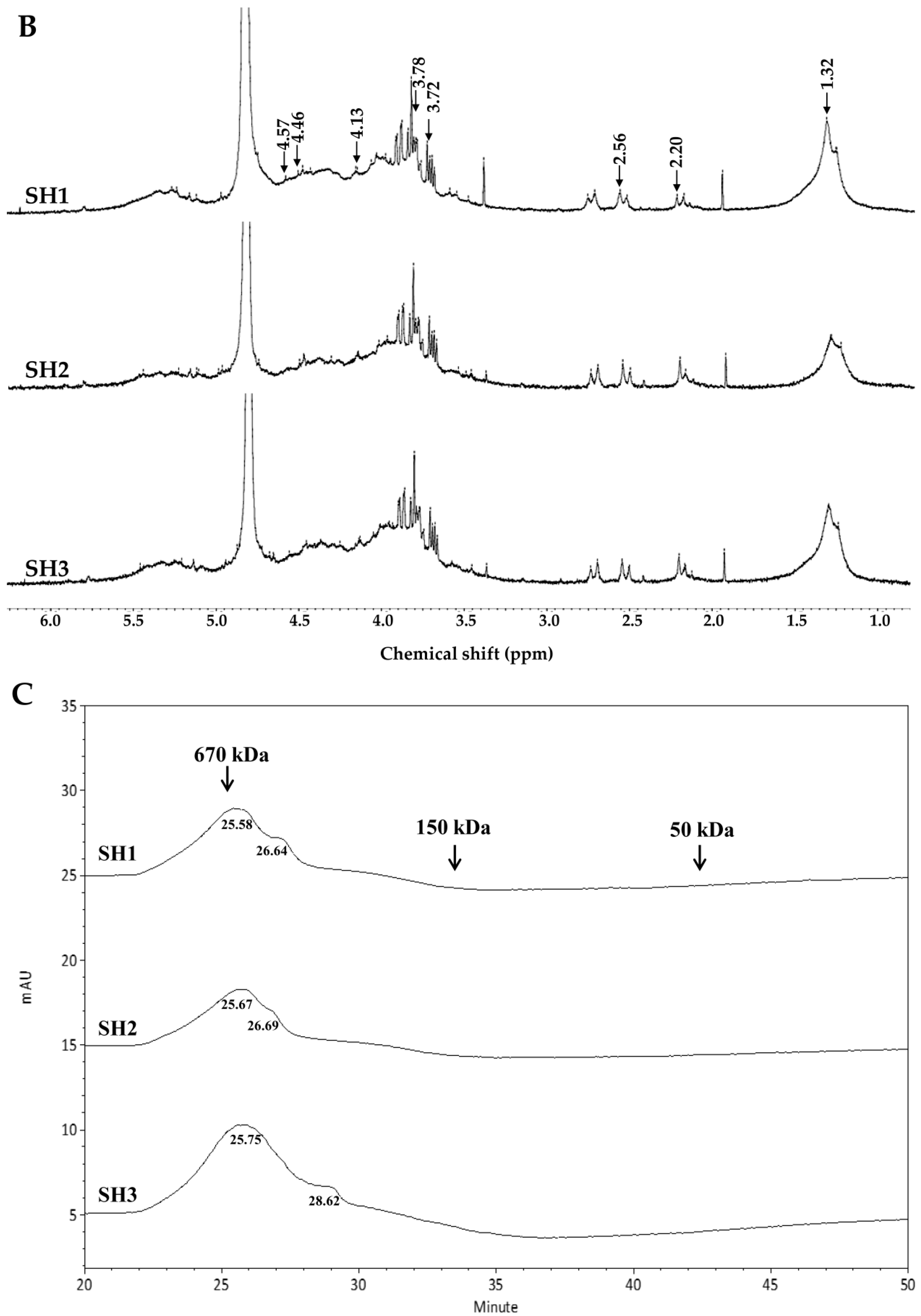
2.2. Compositional and Physicochemical Analyses of Fucoidans for SH1, SH2, and SH3
2.3. Antioxidant Activities for SH1, SH2, and SH3
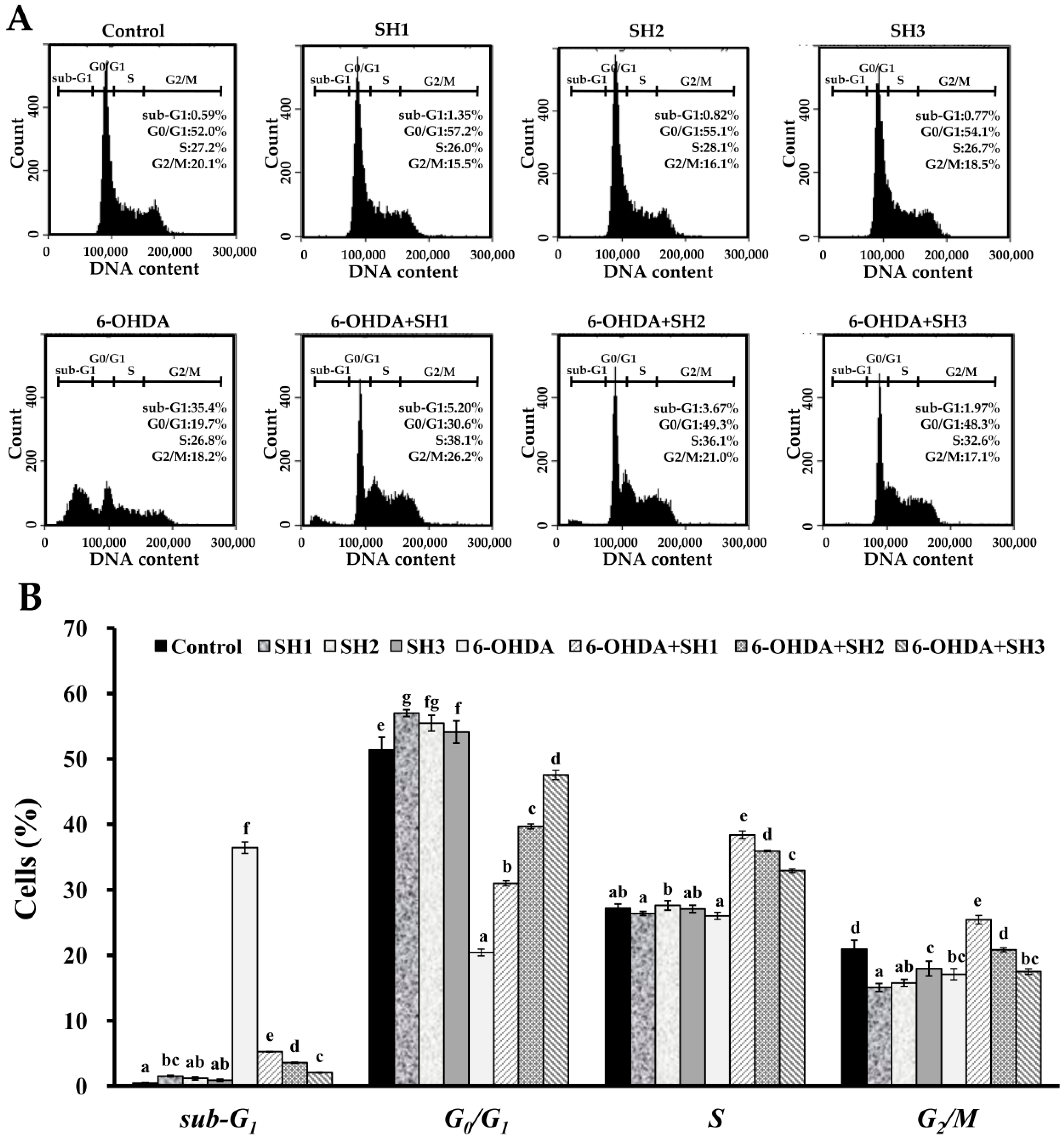
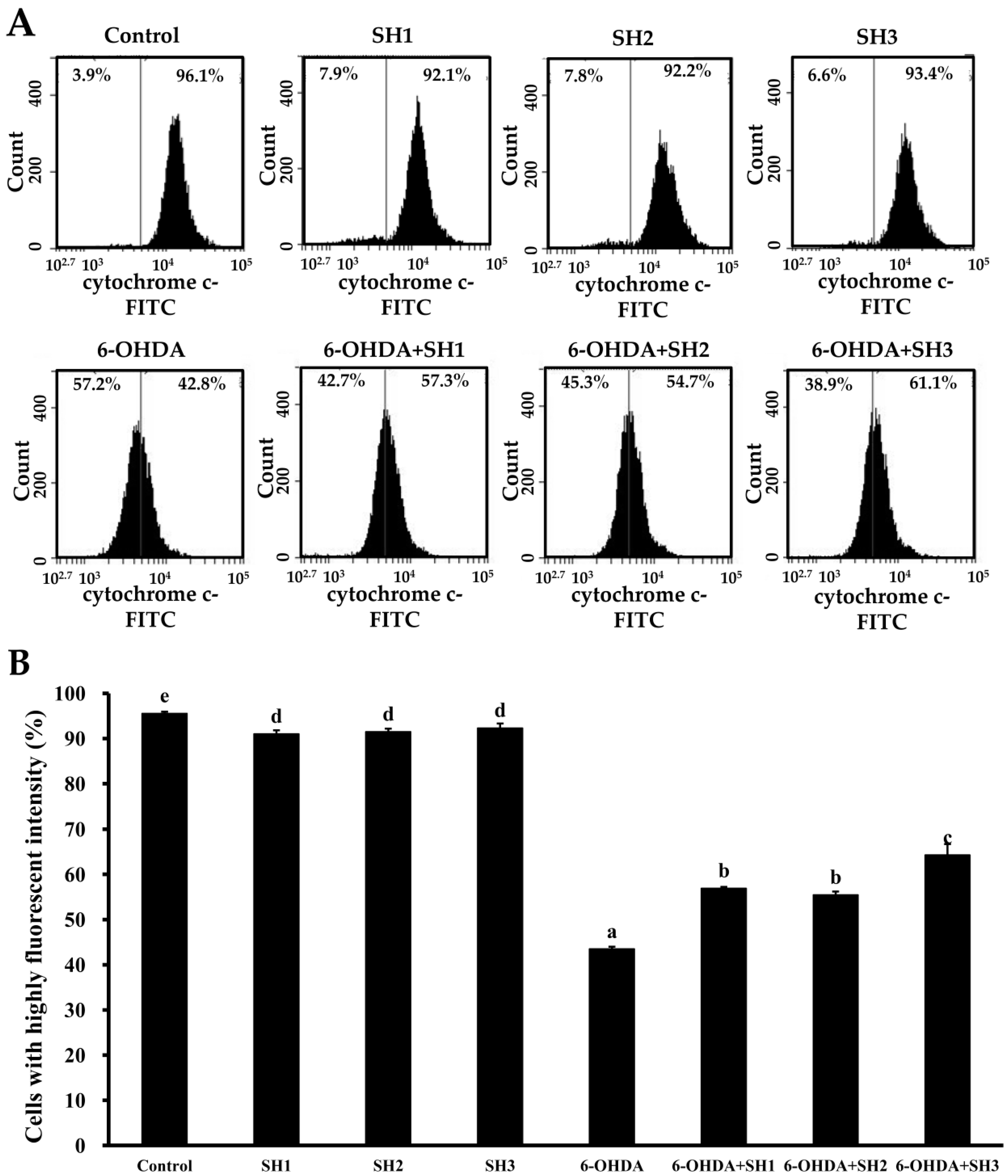
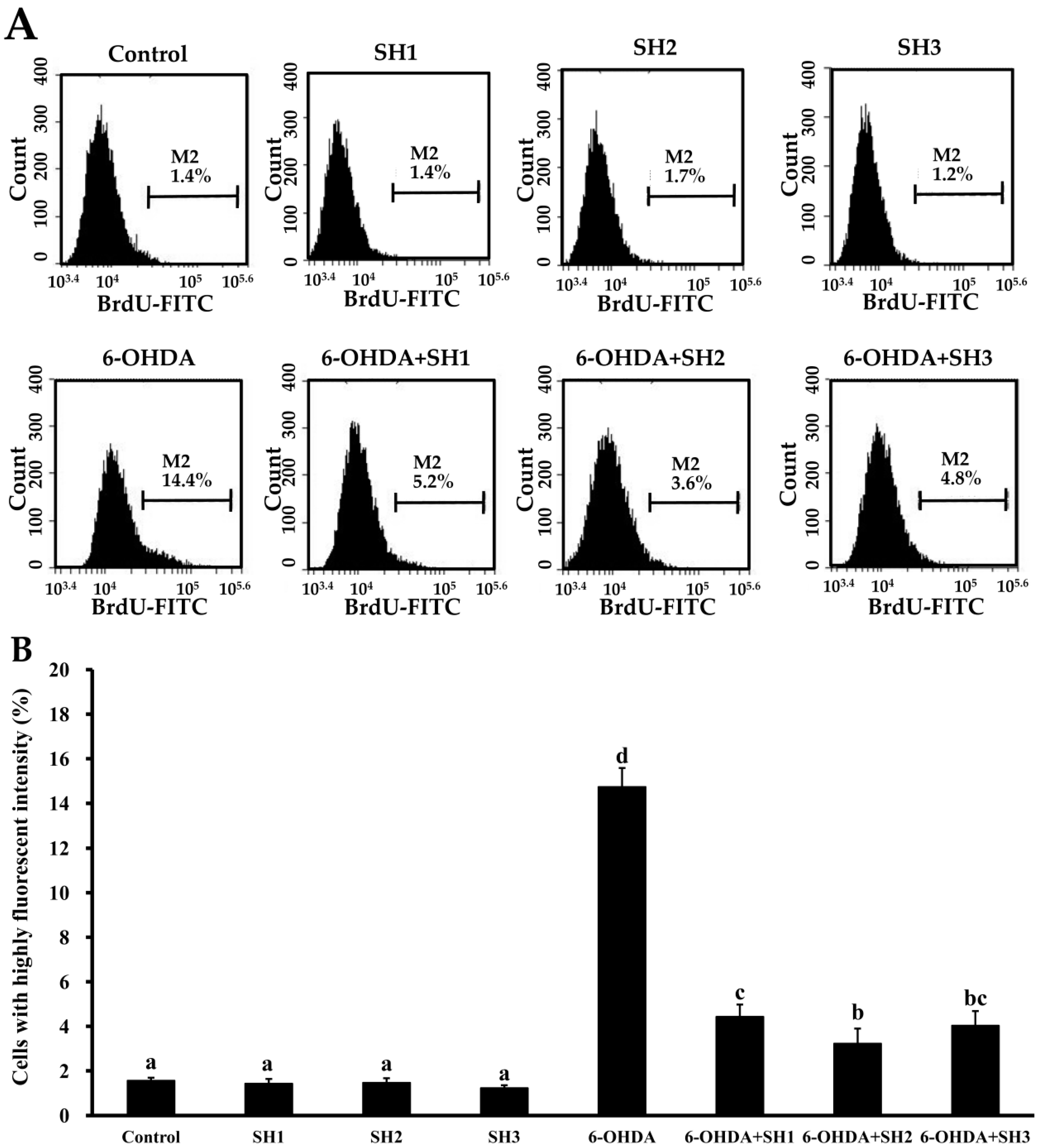
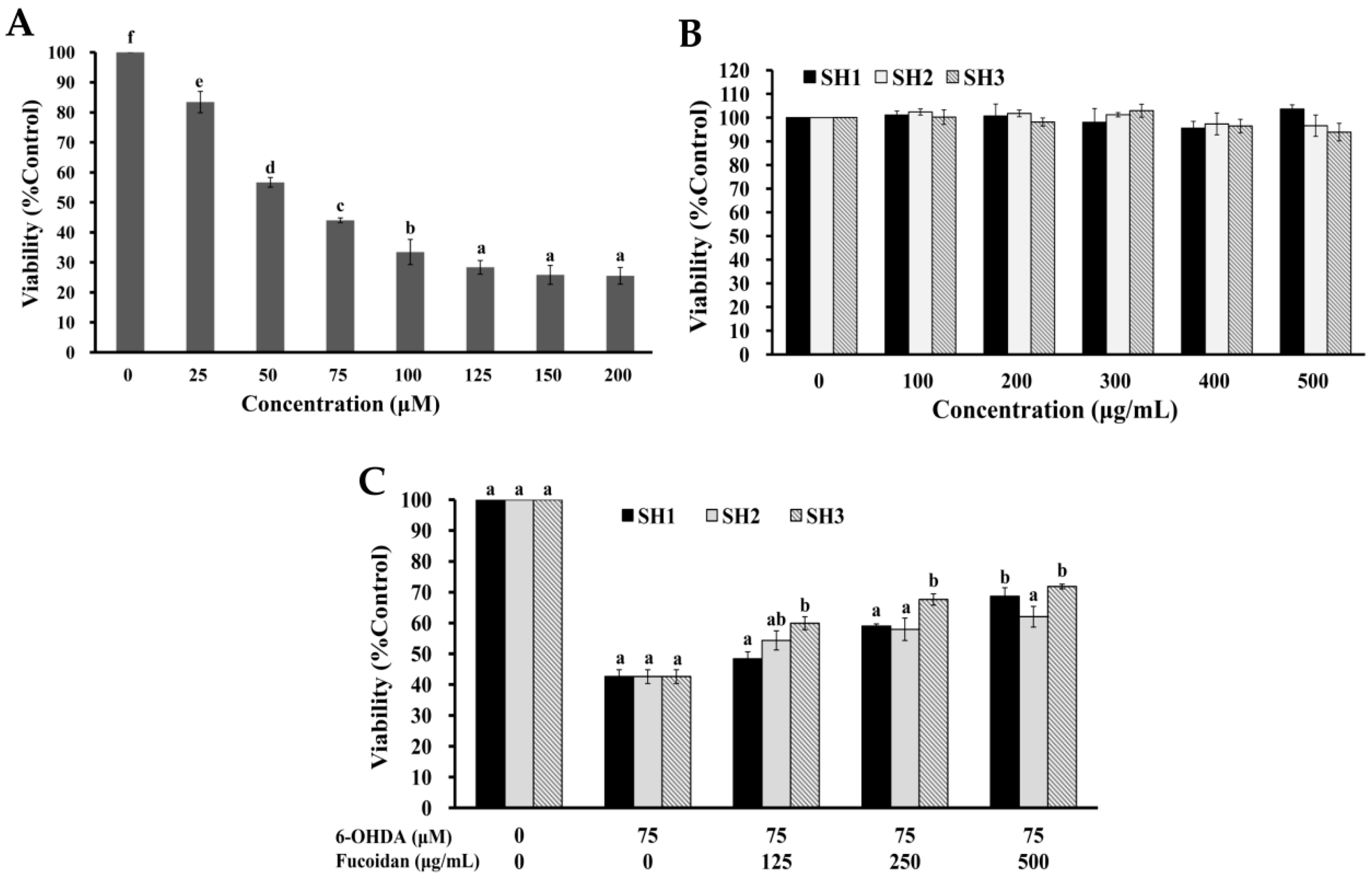
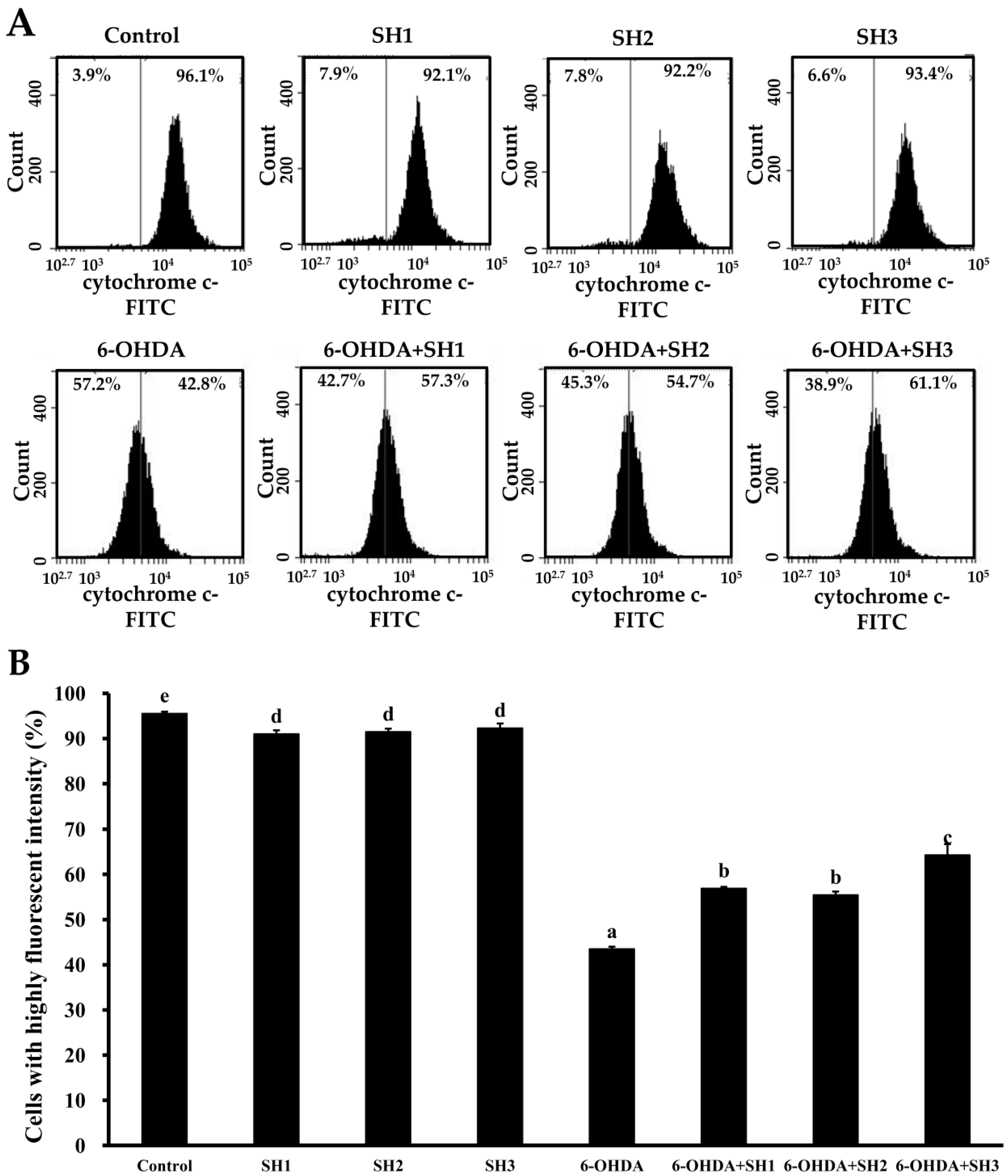
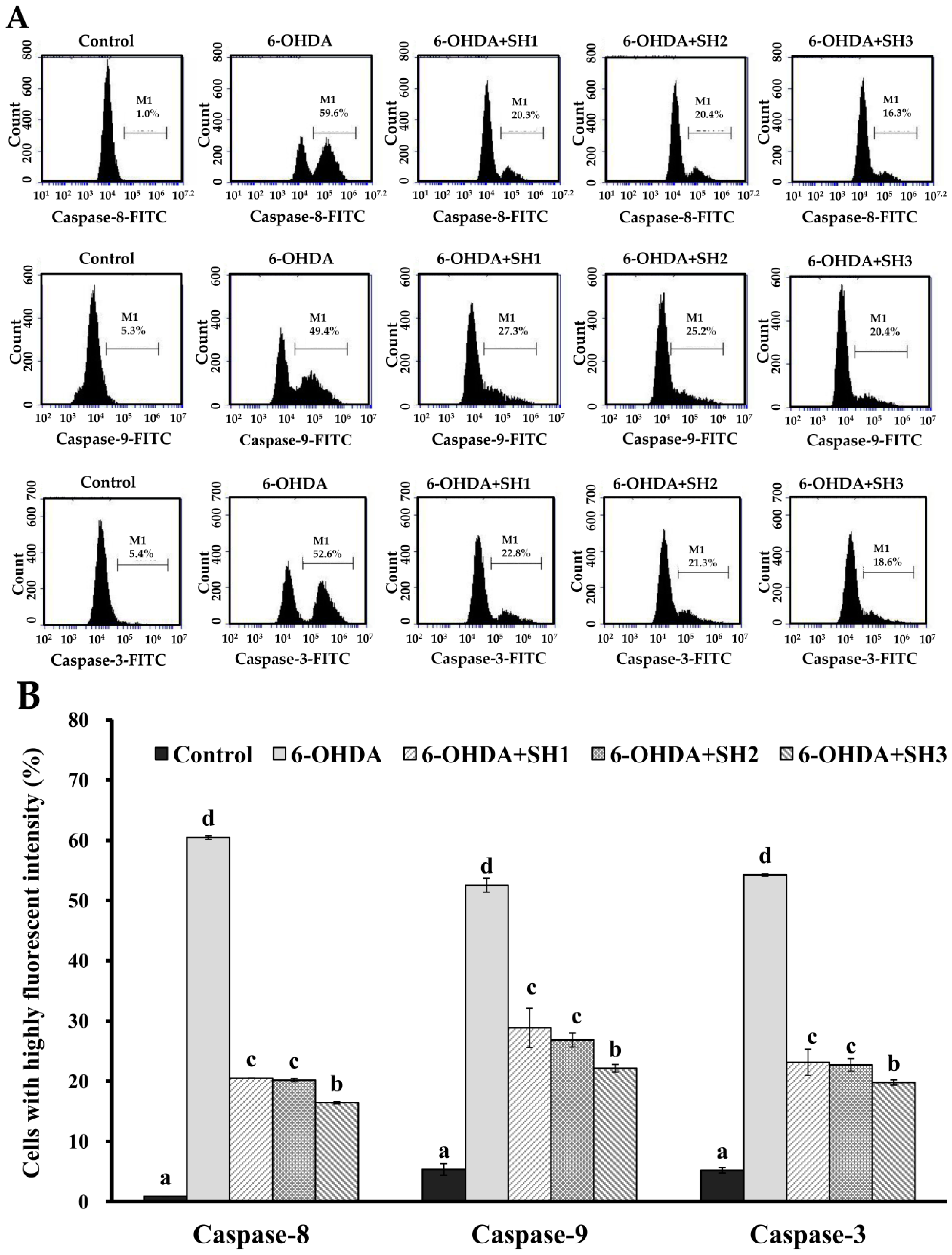
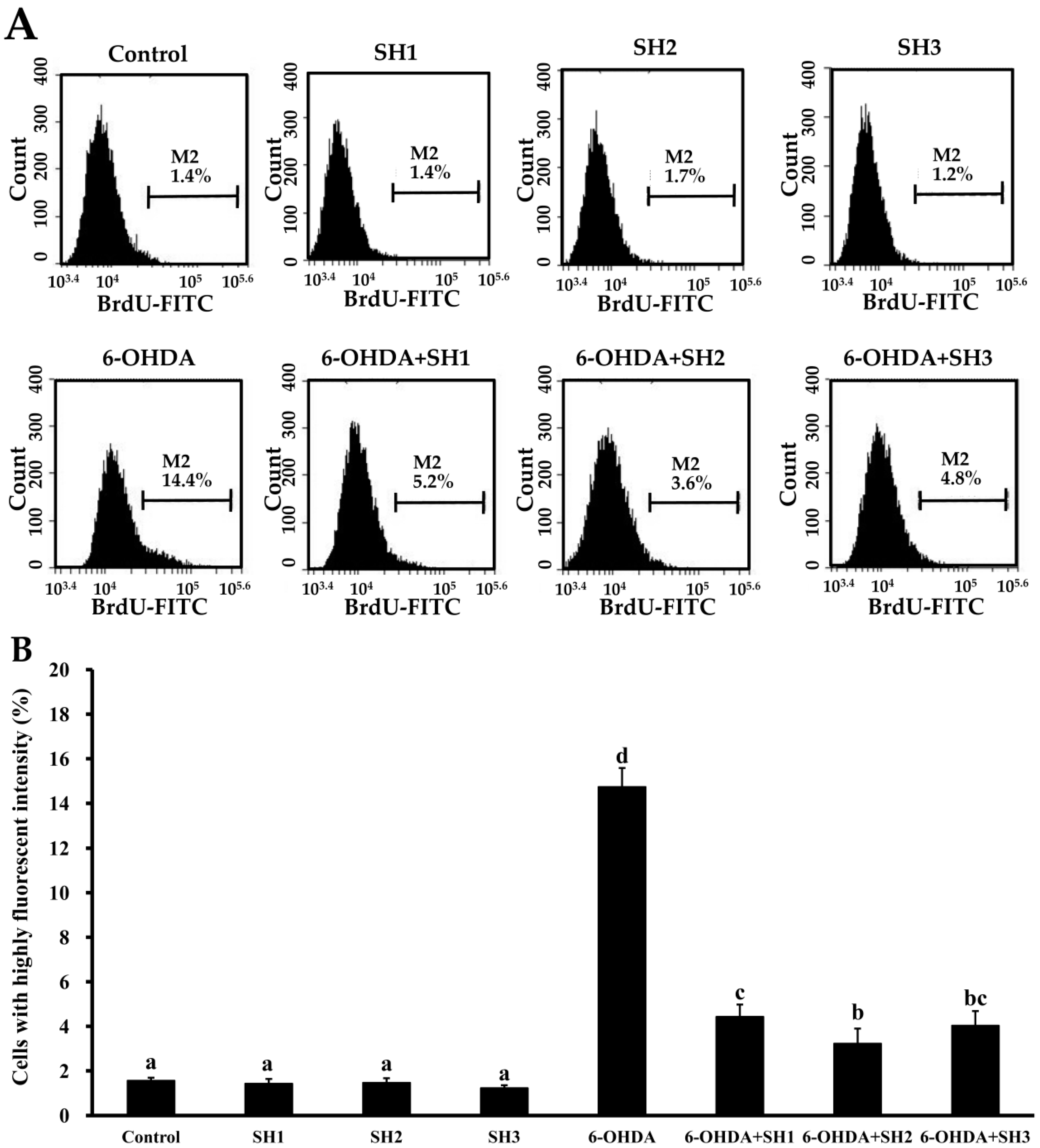
2.4. Effects of SH1, SH2, and SH3 on 6-OHDA-Induced Apoptosis in SH-SY5Y Cells
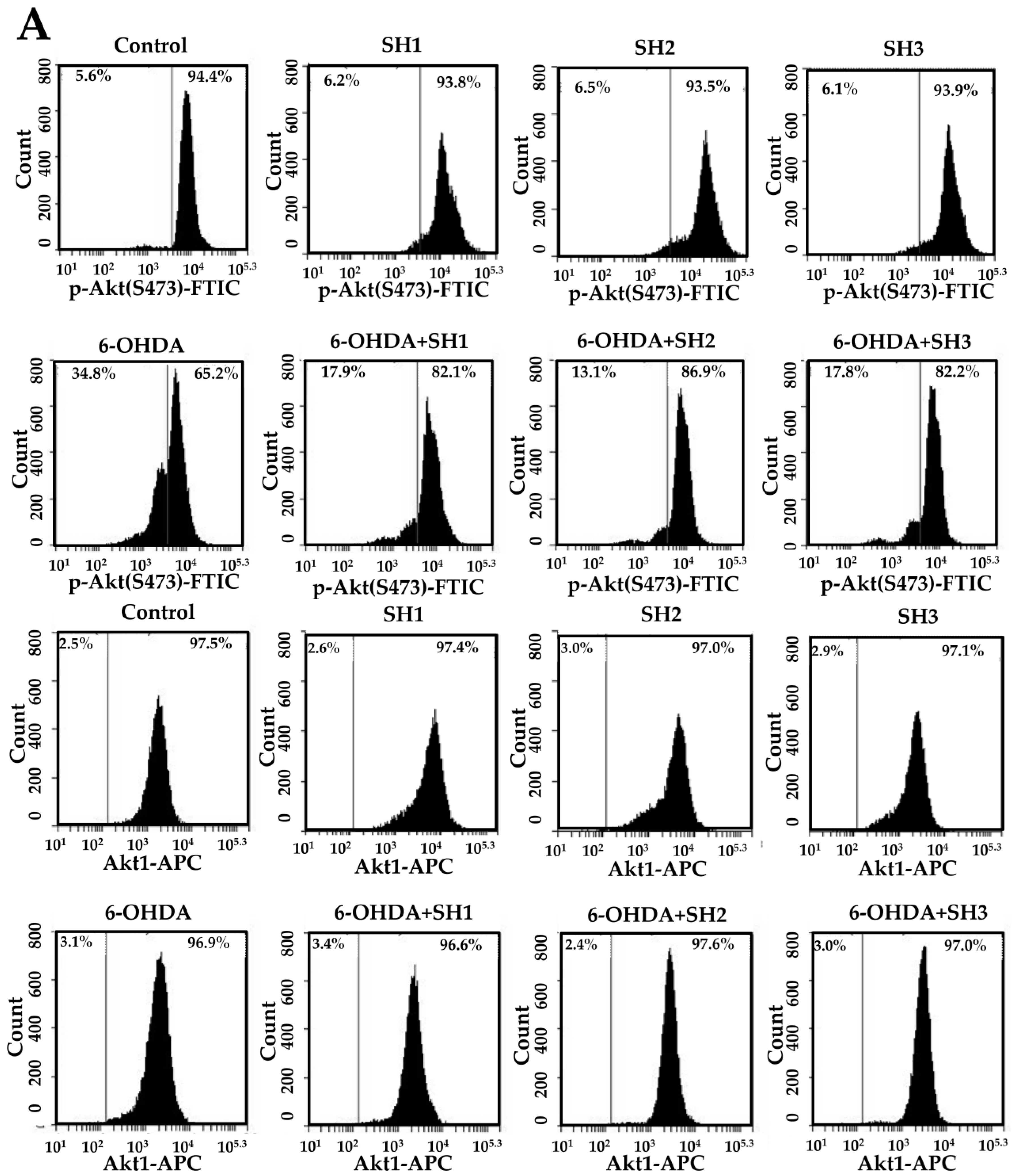
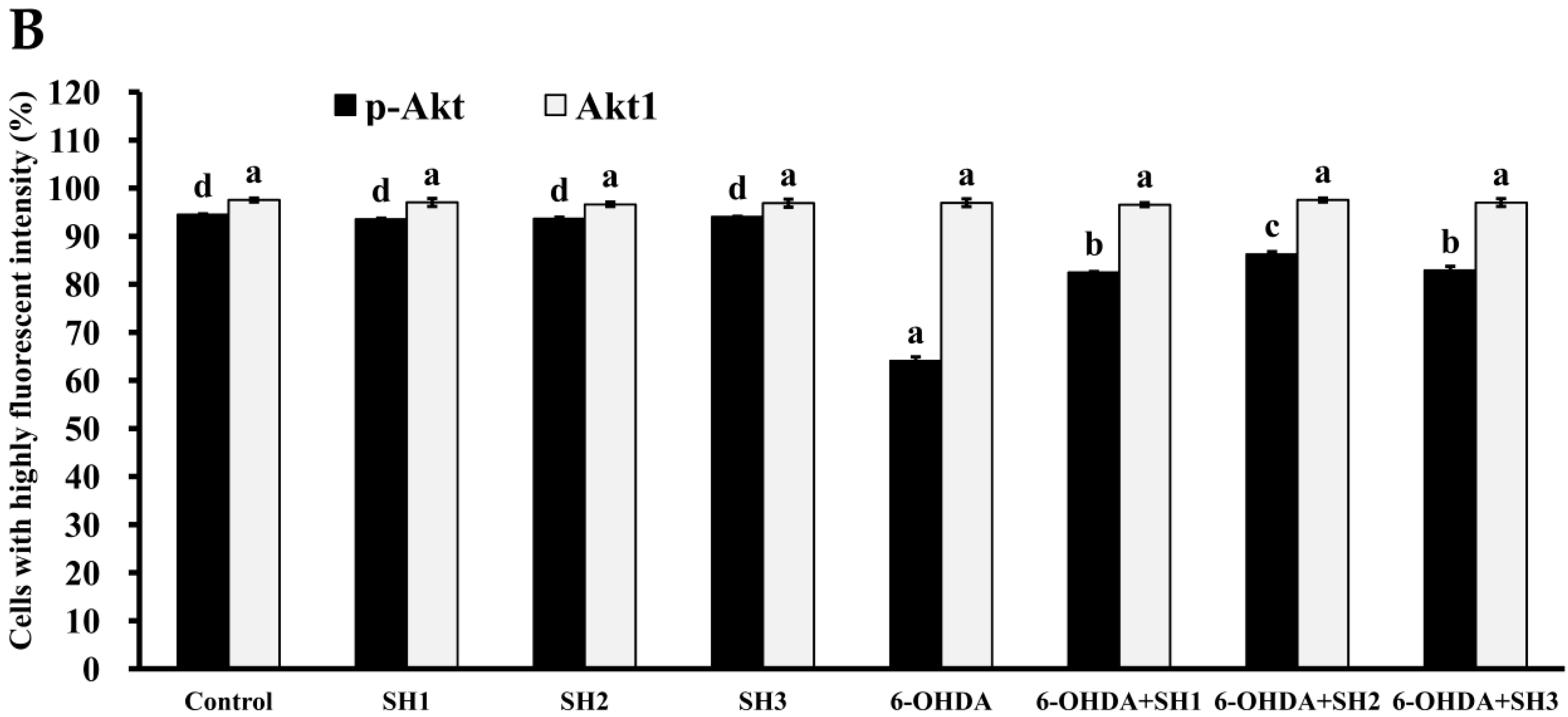
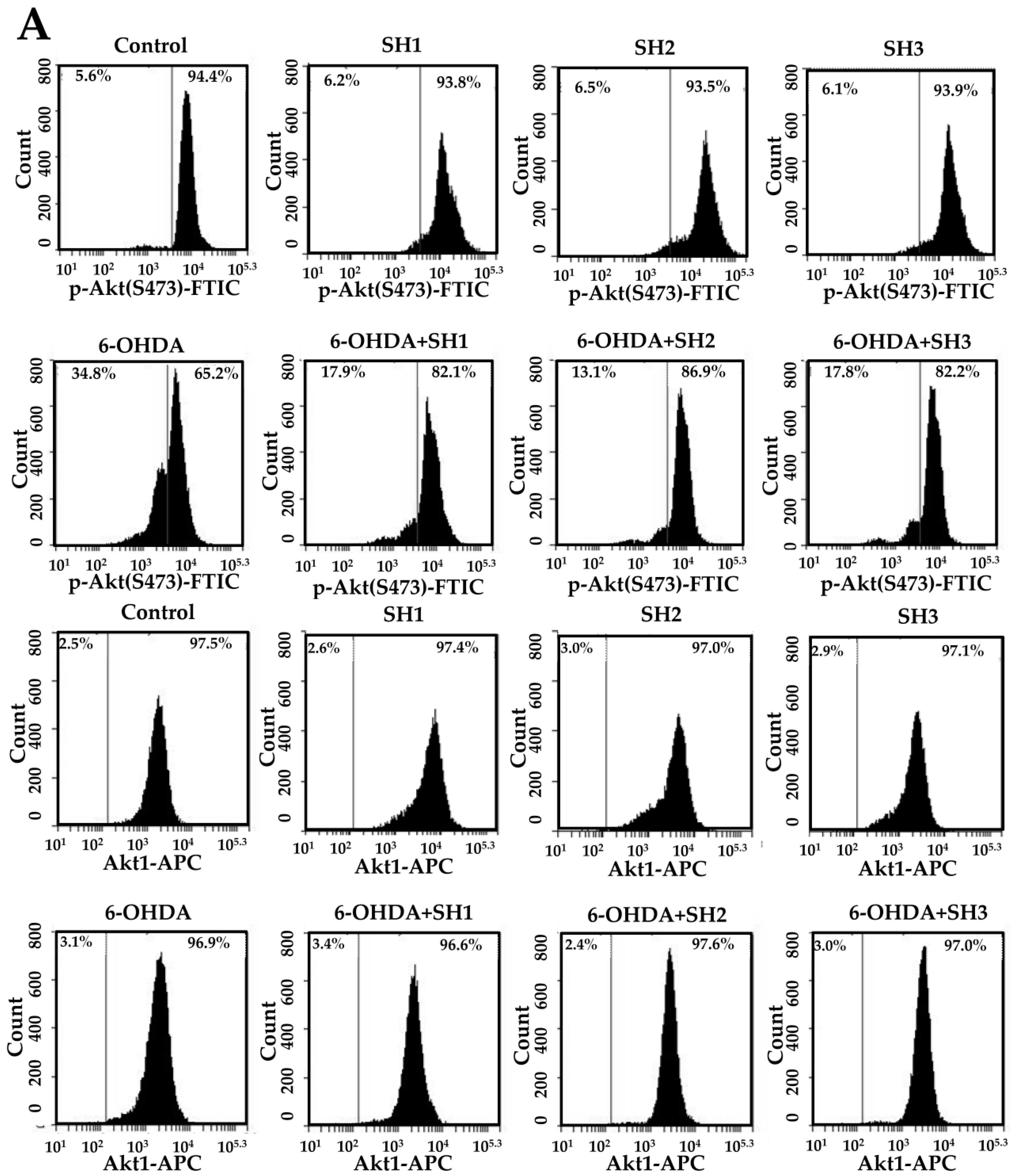
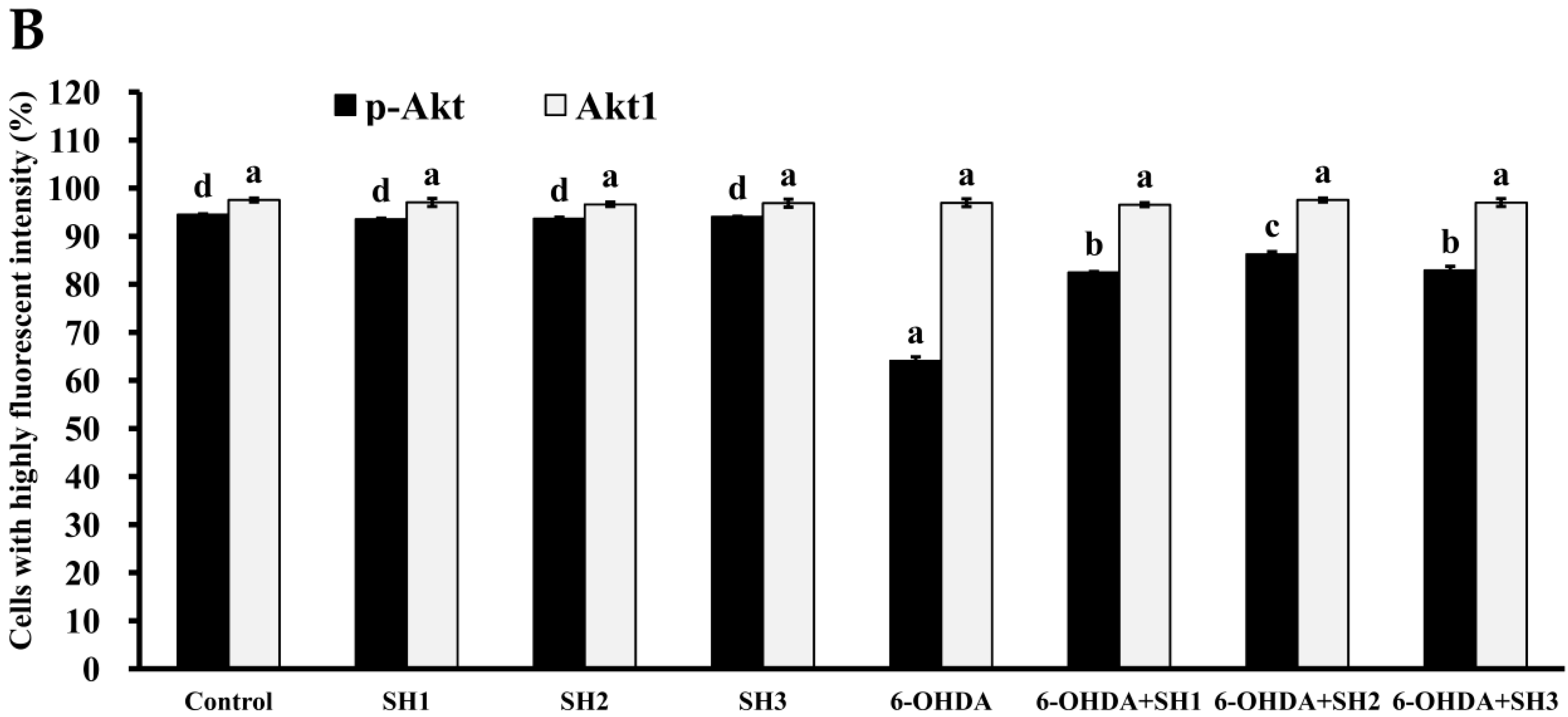
2.5. Phosphorylation of Akt Is Involved in the Neuroprotective Effects of SH1, SH2, and SH3 on 6-OHDA-Induced Apoptosis in SH-SY5Y Cells
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Compressional-Puffing Procedure
3.3. Water Extraction Procedure
3.4. Chemical Methods
3.5. Analysis of Monosaccharide Composition
3.6. NMR Spectroscopy
3.7. Molecular Weight Analysis
3.8. FTIR Spectroscopy
3.9. DPPH Radical Scavenging Activity
3.10. ABTS Radical Cation Scavenging Activity
3.11. FRAP Assay
3.12. Cell Culture
3.13. Cell Viability Test
3.14. Cell Cycle Analysis
3.15. Cytochrome c Release Analysis
3.16. Activated Caspase-8, -9, and -3 Analyses
3.17. Quantitation of DNA Fragmentation by TUNEL Assay
3.18. Phosphorylated Akt Analysis
3.19. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reed, T.T. Lipid peroxidation and neurodegenerative disease. Free Radic. Biol. Med. 2011, 51, 1302–1319. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R.; Carney, J.M. Oxidative alterations in Alzheimer’s disease. Brain Pathol. 1999, 9, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Imbs, T.I.; Skriptsova, A.V.; Zvyagintseva, T.N. Antioxidant activity of fucose-containing sulfated polysaccharides obtained from Fucus evanescens by different extraction methods. J. Appl. Phycol. 2015, 27, 545–553. [Google Scholar] [CrossRef]
- Ale, M.T.; Maruyama, H.; Tamauchi, H.; Mikkelsen, J.; Meyer, A.S. Fucoidan from Sargassum sp. and Fucus vesiculosus reduces cell viability of lung carcinoma and melanoma cells in vitro and activates natural killer cells in mice in vivo. Int. J. Biol. Macromol. 2011, 49, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Ale, M.T.; Mikkelsen, J.D.; Meyer, A.S. Important determinants for fucoidan bioactivity: A critical review of structure-function relations and extraction methods for fucose-containing sulfated polysaccharides from brown seaweeds. Mar. Drugs 2011, 9, 2106–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.Y.; Wu, S.J.; Yang, W.N.; Kuan, A.W.; Chen, C.Y. Antioxidant activities of crude extracts of fucoidan extracted from Sargassum glaucescens by a compressional-puffing-hydrothermal extraction process. Food Chem. 2016, 197, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.N.; Chen, P.W.; Huang, C.Y. Compositional characteristics and in vitro evaluations of antioxidant and neuroprotective properties of crude extracts of fucoidan prepared from compressional puffing-pretreated Sargassum crassifolium. Mar. Drugs 2017, 15, 183. [Google Scholar] [CrossRef] [PubMed]
- Chiang, P.S.; Lee, D.J.; Whiteley, C.G.; Huang, C.Y. Antioxidant phenolic compounds from Pinus morrisconicola using compressional-puffing pretreatment and water-ethanol extraction: Optimization of extraction parameters. J. Taiwan Inst. Chem. Eng. 2017, 70, 7–14. [Google Scholar] [CrossRef]
- Chiang, P.S.; Lee, D.J.; Whiteley, C.G.; Huang, C.Y. Extracting antioxidant phenolic compounds from compressional-puffing pretreated Pinus morrisonicola: Effects of operational parameters, kinetics and characterization. J. Taiwan Inst. Chem. Eng. 2017, 75, 70–76. [Google Scholar] [CrossRef]
- Sinha, S.; Astani, A.; Ghosh, T.; Schnitzler, P.; Ray, B. Polysaccharides from Sargassum tenerrimum: Structural features, chemical modification and anti-viral activity. Phytochemistry 2010, 71, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lu, F.; Wei, X.J.; Zhao, R.X. Fucoidan: Structure and bioactivity. Molecules 2008, 13, 1671–1695. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Cui, N.; Bo, Z.X.; Xiang, F.X. Structural determinant and its underlying molecular mechanism of STPC2 related to anti-angiogenic activity. Mar. Drugs 2017, 15, 48. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Q.B.; Zhang, Z.S.; Song, H.F.; Li, P.C. Potential antioxidant and anticoagulant capacity of low molecular weight fucoidan fractions extracted from Laminaria japonica. Int. J. Biol. Macromol. 2010, 46, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Movasaghi, Z.; Rehman, S.; ur Rehman, D.I. Fourier transform infrared (FTIR) spectroscopy of biological tissues. Appl. Spectrosc. Rev. 2008, 43, 134–179. [Google Scholar] [CrossRef]
- Shao, P.; Pei, Y.P.; Fang, Z.X.; Sun, P.L. Effects of partial desulfation on antioxidant and inhibition of DLD cancer cell of Ulva fasciata polysaccharide. Int. J. Biol. Macromol. 2014, 65, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, S.; Vinosha, M.; Marudhupandi, T.; Rajasekar, P.; Prabhu, N.M. Isolation of fucoidan from Sargassum polycystum brown algae: Structural characterization, in vitro antioxidant and anticancer activity. Int. J. Biol. Macromol. 2017, 102, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Synytsya, A.; Bleha, R.; Synytsya, A.; Pohl, R.; Hayashi, K.; Yoshinaga, K.; Nakano, T.; Hayashi, T. Mekabu fucoidan: Structural complexity and defensive effects against avian influenza A viruses. Carbohydr. Polym. 2014, 111, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Bilan, M.I.; Grachev, A.A.; Shashkov, A.S.; Nifantiev, N.E.; Usov, A.I. Structure of a fucoidan from the brown seaweed Fucus serratus L. Carbohydr. Res. 2006, 341, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Tako, M.; Nakada, T.; Hongou, F. Chemical characterization of fucoidan from commercially cultured Nemacystus decipiens (Itomozuku). Biosci. Biotechnol. Biochem. 1999, 63, 1813–1815. [Google Scholar] [CrossRef] [PubMed]
- Jégou, C.; Kervarec, N.; Cérantola, S.; Bihannic, I.; Stiger-Pouvreau, V. NMR use to quantify phlorotannins: The case of Cystoseira tamariscifolia, a phloroglucinol-producing brown macroalga in Brittany (France). Talanta 2015, 135, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ermakova, S.; Sokolova, R.; Kim, S.M.; Um, B.H.; Isakov, V.; Zvyagintseva, T. Fucoidans from brown seaweeds Sargassum hornery, Eclonia cava, Costaria costata: Structural characteristics and anticancer activity. Appl. Biochem. Biotechnol. 2011, 164, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Lean, C.; Doran, S.; Somorjai, R.L.; Malycha, P.; Clarke, D.; Himmelreich, U.; Bourne, R.; Dolenko, B.; Nikulin, A.E.; Mountford, C. Determination of grade and receptor status from the primary breast lesion by magnetic resonance spectroscopy. Technol. Cancer Res. Treat. 2004, 3, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Mlynárik, V.; Cudalbu, C.; Xin, L.; Gruetter, R. 1H NMR spectroscopy of rat brain in vivo at 14.1 Tesla: Improvements in quantification of the neurochemical profile. J. Magn. Reson. 2008, 194, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Bilan, M.I.; Grachev, A.A.; Ustuzhanina, N.E.; Shashkov, A.S.; Nifantiev, N.E.; Usov, A.I. A highly regular fraction of a fucoidan from the brown seaweed Fucus distichus L. Carbohydr. Res. 2004, 339, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Synytsya, A.; Kim, W.J.; Kim, S.M.; Pohl, R.; Synytsya, A.; Kvasnička, F.; Čopíková, J.; Park, Y. Structure and antitumour activity of fucoidan isolated from sporophyll of Korean brown seaweed Undaria pinnatifida. Carbohydr. Polym. 2010, 81, 41–48. [Google Scholar] [CrossRef]
- Mandemakers, W.; Morais, V.A.; De Strooper, B. A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J. Cell Sci. 2007, 120, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lazaro, M.; Bonekamp, N.A.; Galindo, M.F.; Jordán, J.; Schrader, M. 6-Hydroxydopamine (6-OHDA) induces Drp1-dependent mitochondrial fragmentation in SH-SY5Y cells. Free Radic. Biol. Med. 2008, 44, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Noh, Y.H.; Kim, Y.S.; Kim, K.Y.; Chung, Y.H.; Lee, W.B.; Kim, S.S. Baicalein attenuates 6-hydroxydopamine-induced neurotoxicity in SH-SY5Y cells. Eur. J. Cell Biol. 2005, 84, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Woodgate, A.; MacGibbon, G.; Walton, M.; Dragunow, M. The toxicity of 6-hydroxydopamine on PC12 and P19 cells. Mol. Brain Res. 1999, 69, 84–92. [Google Scholar] [CrossRef]
- Bové, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-induced models of Parkinson’s disease. NeuroRX 2005, 2, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Lopes, F.M.; Schröder, R.; da Frota Júnior, M.L.C.; Zanotto-Filho, A.; Müller, C.B.; Pires, A.S.; Meurer, R.T.; Colpo, G.D.; Gelain, D.P.; Kapczinski, F.; et al. Comparison between proliferative and neuron-like SH-SY5Y cells as an in vitro model for Parkinson disease studies. Brain Res. 2010, 1337, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.T.; Lau, W.K.W.; Yu, M.S.; Lai, C.S.W.; Yeung, S.C.; So, K.F.; Chang, R.C.C. Effects of all-trans-retinoic acid on human SH-SY5Y neuroblastoma as in vitro model in neurotoxicity research. Neurotoxicology 2009, 30, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Jordán, J.; Galindo, M.F.; Tornero, D.; González-García, C.; Ceña, V. Bcl-XL blocks mitochondrial multiple conductance channel activation and inhibits 6-OHDA-induced death in SH-SY5Y cells. J. Neurochem. 2004, 89, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Wu, T.C.; Hsieh, S.L.; Tsai, Y.H.; Yeh, C.W.; Huang, C.Y. Antioxidant activity and growth inhibition of human colon cancer cells by crude and purified fucoidan preparations extracted from Sargassum cristaefolium. J. Food Drug Anal. 2015, 23, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Campos, C.B.; Paim, B.A.; Cosso, R.G.; Castilho, R.F.; Rottenberg, H.; Vercesi, A.E. Method for monitoring of mitochondrial cytochrome c release during cell death: Immunodetection of cytochrome c by flow cytometry after selective permeabilization of the plasma membrane. Cytom. A 2006, 69, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.; Aubert, M. Flow cytometric detection of activated caspase-3. In Apoptosis and Cancer: Methods and Protocols, 2008th ed.; Mor, G., Alvero, A., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2008; pp. 47–56. [Google Scholar]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Li, P.; Widlak, P.; Zou, H.; Luo, X.; Garrard, W.T.; Wang, X.D. The 40-kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc. Natl. Acad. Sci. USA 1998, 95, 8461–8466. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, R.; Ganjalikhani-Hakemi, M.; Esmaeili, M.; Abolhassani, H.; Vaeli, S.; Rezaei, A.; Sharifi, Z.; Azizi, G.; Rezaei, N.; Aghamohammadi, A. Impaired Akt phosphorylation in B-cells of patients with common variable immunodeficiency. Clin. Immunol. 2017, 175, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Nakaso, K.; Ito, S.; Nakashima, K. Caffeine activates the PI3K/Akt pathway and prevents apoptotic cell death in a Parkinson’s disease model of SH-SY5Y cells. Neurosci. Lett. 2008, 432, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, M.N. The determination of methylpentoses. Analyst 1955, 80, 268–276. [Google Scholar] [CrossRef]
- Filisetti-Cozzi, T.M.; Carpita, N.C. Measurement of uronic acids without interference from neutral sugars. Anal. Biochem. 1991, 197, 157–162. [Google Scholar] [CrossRef]
- Honya, M.; Kinoshita, T.; Ishikawa, M.; Mori, H.; Nisizawa, K. Monthly determination of alginate, M/G ratio, mannitol, and minerals in cultivated Laminaria japonica. Bull. Jpn. Soc. Sci. Fish. 1993, 59, 295. [Google Scholar] [CrossRef]
- Huang, C.Y.; Kuo, J.M.; Wu, S.J.; Tsai, H.T. Isolation and characterization of fish scale collagen from tilapia (Oreochromis sp.) by a novel extrusion-hydro-extraction process. Food Chem. 2016, 190, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.F.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “Antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables of Compressional-Puffing | SH1 | SH2 | SH3 | |
|---|---|---|---|---|
| Mechanical compression | Pressure (kg/cm2) | 0 | 5 | 5 |
| Number of compression times | 0 | 3 | 3 | |
| Puffing | Temperature (°C) | 0 | 140 | 180 |
| Pressure (kg/cm2) | 0 | 1.7 | 10.0 | |
| Reaction time | Time (s) | 0 | 10 | 10 |
| Degree of moisture loss of puffed algal samples | SH1 1 | SH2 1 | SH3 1 | |
| Degree of moisture loss (%) | 0.00 ± 0.00 a | 16.21 ± 1.17 b | 29.56 ± 2.21 c | |
| Variables of water extraction | SH1 | SH2 | SH3 | |
| Extraction temperature | Temperature (°C) | 85 | 85 | 85 |
| Extraction time | Time (h) | 1 | 1 | 1 |
| Extraction yields of fucoidan | SH1 1 | SH2 1 | SH3 1 | |
| Extraction yield (%) | 1.51 ± 0.09 a | 1.93 ± 0.28 ab | 2.06 ± 0.14 b | |
| Chemical Composition | SH1 2 | SH2 2 | SH3 2 |
| Total sugar (%) | 53.67 ± 2.59 a | 58.83 ± 1.61 b | 68.83 ± 1.91 c |
| Uronic acid (%) | 5.66 ± 0.08 a | 7.04 ± 0.22 c | 6.12 ± 0.08 b |
| Fucose (%) | 24.53 ± 1.15 a | 22.76 ± 0.38 a | 23.87 ± 1.76 a |
| Sulfate (%) | 18.87 ± 1.65 b | 15.58 ± 0.16 a | 24.16 ± 2.80 c |
| Sulfate/fucose (molar ratio) 1 | 1.32 ± 0.11 a | 1.17 ± 0.01 a | 1.74 ± 0.20 b |
| Protein (%) | 2.28 ± 0.05 c | 2.05 ± 0.12 b | 1.83 ± 0.07 a |
| Polyphenols (%) | 1.25 ± 0.08 c | 1.06 ± 0.06 b | 0.71 ± 0.06 a |
| Monosaccharide compositions (molar ratio) | SH1 | SH2 | SH3 |
| Fucose | 1.00 | 1.00 | 1.00 |
| Mannose | 0.32 | 0.27 | 0.28 |
| Rhamnose | 0.15 | 0.15 | 0.14 |
| Glucose | 0.09 | 0.07 | 0.06 |
| Glucuronic acid | 0.21 | 0.20 | 0.20 |
| Galactose | 0.40 | 0.35 | 0.35 |
| Xylose | ND 3 | 0.02 | 0.02 |
| Antioxidant Activity | SH1 2 | SH2 2 | SH3 2 |
|---|---|---|---|
| DPPH/IC50 (mg/mL) 1 | 1.72 ± 0.07 a | 2.42 ± 0.15 b | 2.58 ± 0.03 b |
| ABTS/IC50 (mg/mL) 1 | 0.17 ± 0.01 a | 0.26 ± 0.00 b | 0.34 ± 0.00 c |
| FRAP/vitamin C equivalent (μmol vitamin C/g extract, dry basis) | 18.36 ± 0.11 c | 15.81 ± 0.24 b | 13.10 ± 0.06 a |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.-Y.; Kuo, C.-H.; Chen, P.-W. Compressional-Puffing Pretreatment Enhances Neuroprotective Effects of Fucoidans from the Brown Seaweed Sargassum hemiphyllum on 6-Hydroxydopamine-Induced Apoptosis in SH-SY5Y Cells. Molecules 2018, 23, 78. https://doi.org/10.3390/molecules23010078
Huang C-Y, Kuo C-H, Chen P-W. Compressional-Puffing Pretreatment Enhances Neuroprotective Effects of Fucoidans from the Brown Seaweed Sargassum hemiphyllum on 6-Hydroxydopamine-Induced Apoptosis in SH-SY5Y Cells. Molecules. 2018; 23(1):78. https://doi.org/10.3390/molecules23010078
Chicago/Turabian StyleHuang, Chun-Yung, Chia-Hung Kuo, and Po-Wei Chen. 2018. "Compressional-Puffing Pretreatment Enhances Neuroprotective Effects of Fucoidans from the Brown Seaweed Sargassum hemiphyllum on 6-Hydroxydopamine-Induced Apoptosis in SH-SY5Y Cells" Molecules 23, no. 1: 78. https://doi.org/10.3390/molecules23010078