Analysis of Chemical Variations between Crude and Salt-Processed Anemarrhenae rhizoma Using Ultra-High-Performance Liquid Chromatography–Mass Spectrometry Methods

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
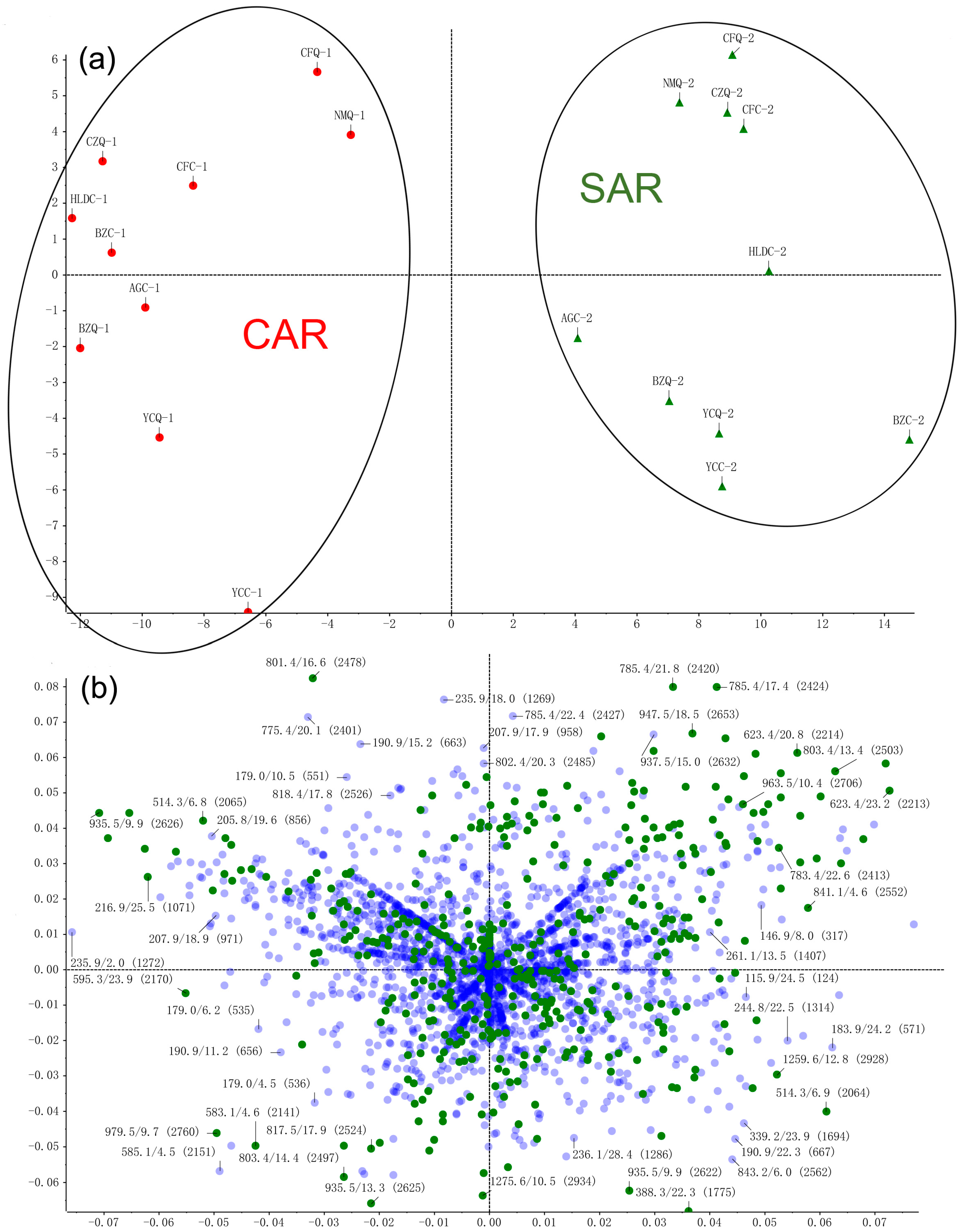
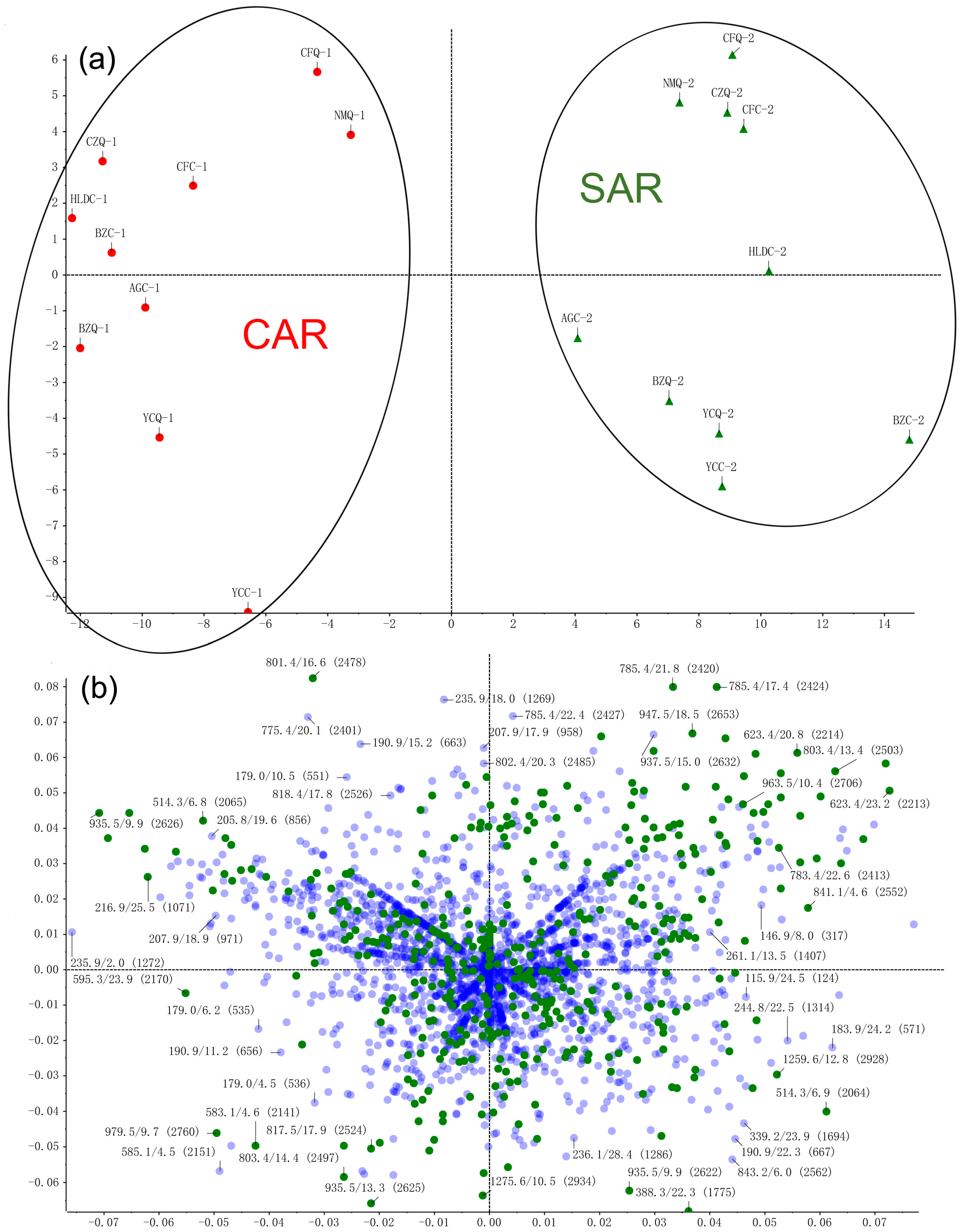
2.1. Multivariate Data Analyses and Identification of Characteristic Chemical Compositions Differentiation of CAR and SAR
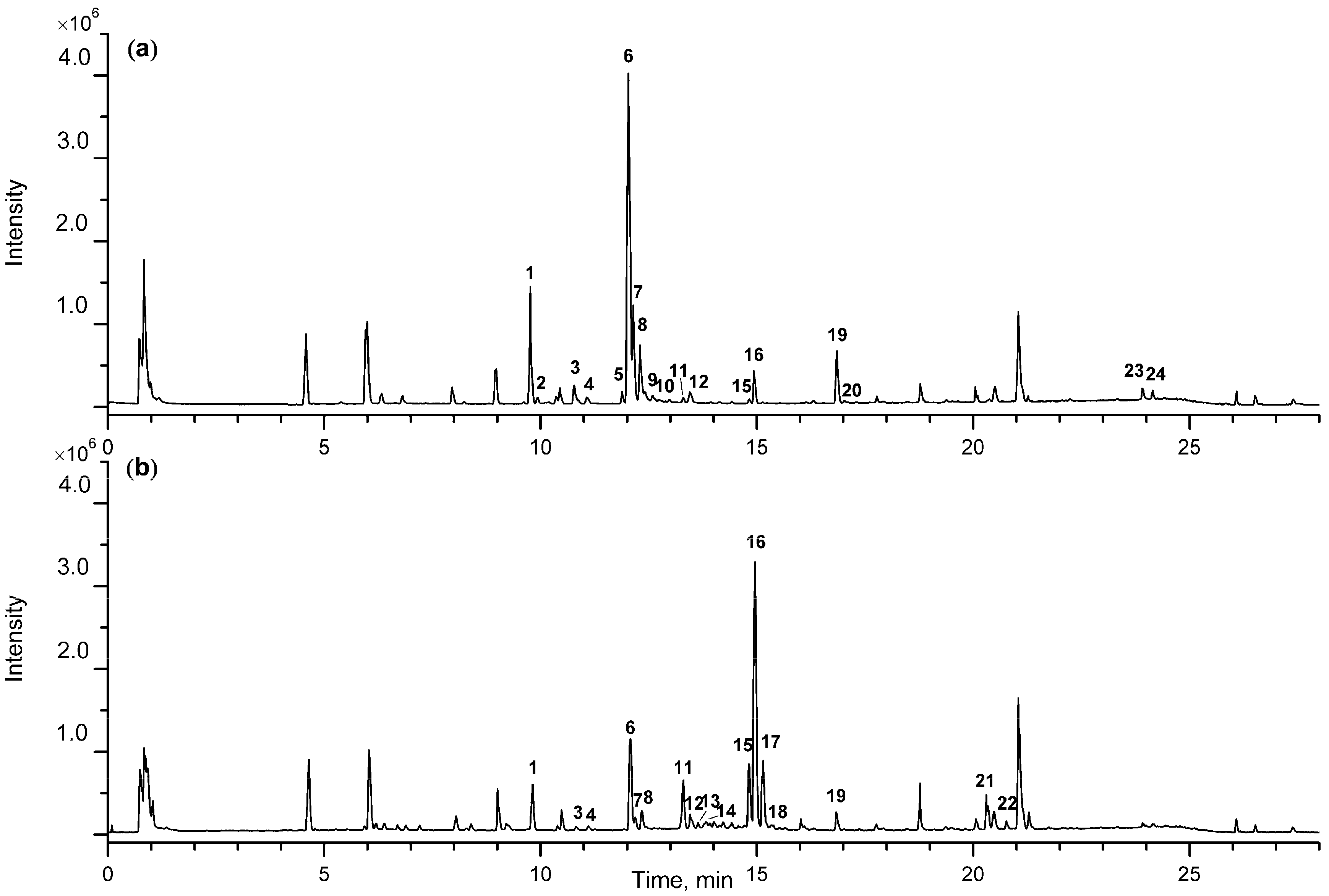
2.2. Determination of the Seven Main Components in CAR and SAR
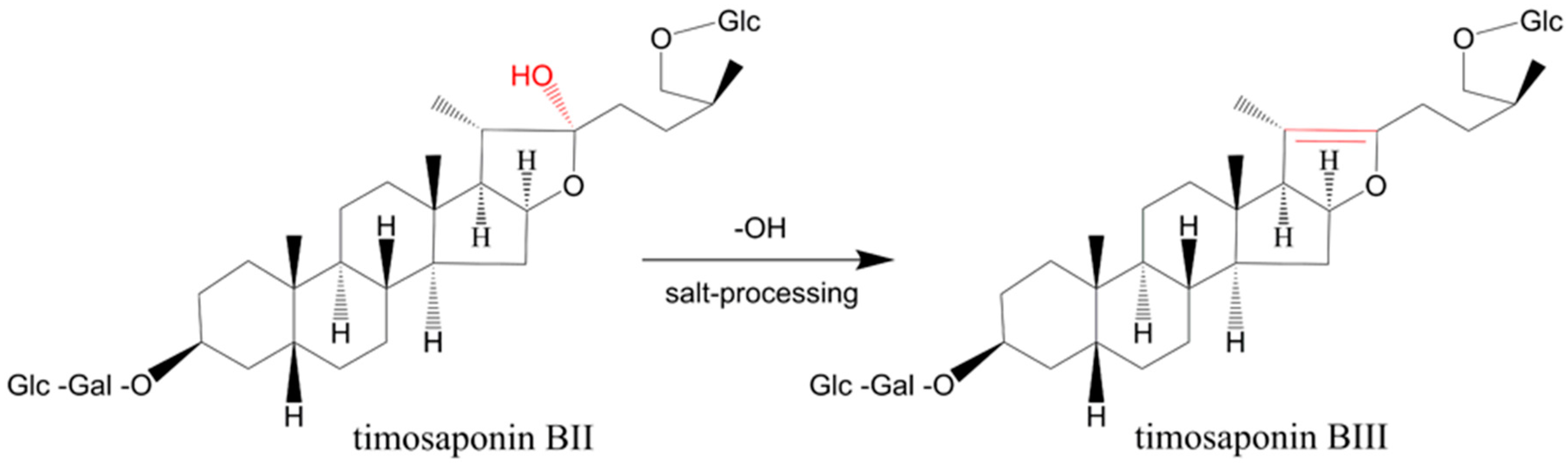
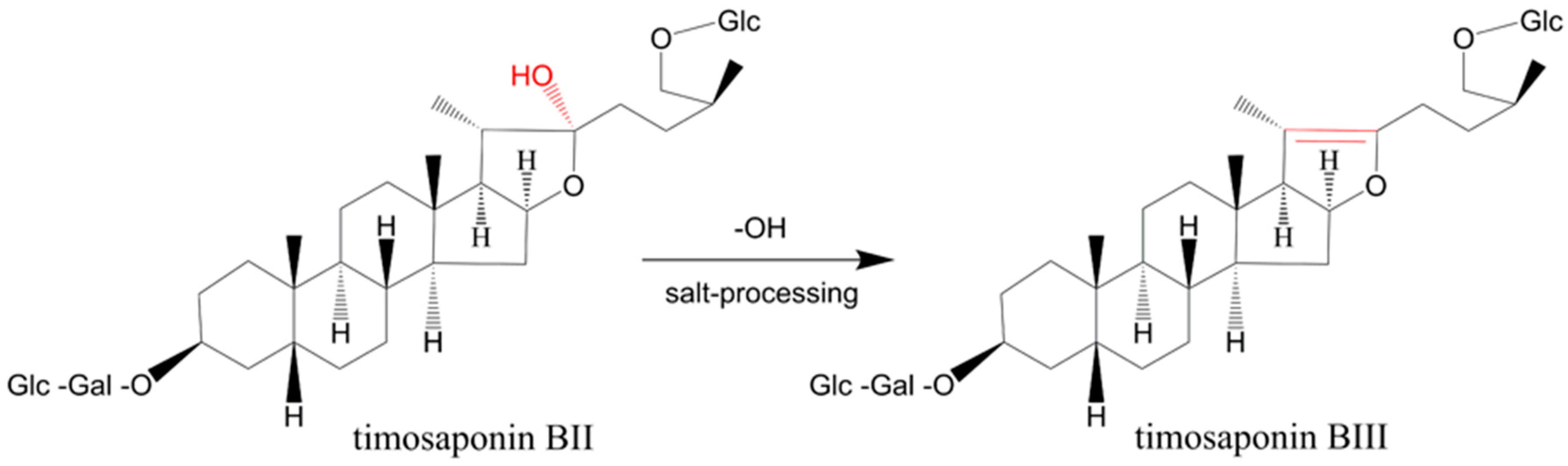
2.3. Compounds Changed upon Processing
3. Materials and Methods
3.1. Materials and Reagents
3.2. Preparation of Standard Solutions
3.3. Sample Preparation
3.4. UHPLC–QTOF-MS Analysis
3.4.1. Instrument and Chromatographic Conditions
3.4.2. Data Analysis
3.5. Quantification of Seven Compounds by UHPLC-MS
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jin, S.Y.; Wang, Q. Studies on Processing of Chinese Medicinal Yinpian and Its Clinical Application; Chemical Industry Press: Beijing, China, 2004; pp. 12–13. [Google Scholar]
- Ye, D.J.; Yuan, S.T. Dictionary of Chinese Herbal Processing Science; Shanghai Science and Technology Press: Shanghai, China, 2005; p. 102. [Google Scholar]
- Lu, T.L.; Hu, C.J. Chinese Medicine Processing; China Medical Science Press: Beijing, China, 2014; p. 3. [Google Scholar]
- Waltenberger, B.; Mocan, A.; Smejkal, K.; Heiss, E.H.; Atanasov, A.G. Natural Products to Counteract the Epidemic of Cardiovascular and Metabolic Disorders. Molecules 2016, 21, 807. [Google Scholar] [CrossRef] [PubMed]
- Eid, H.M.; Wright, M.L.; Anil, K.N.; Qawasmeh, A.; Hassan, S.; Mocan, A.; Nabavi, S.M.; Rastrelli, L.; Atanasov, A.G.; Haddad, P.S. Significance of Microbiota in Obesity and Metabolic Diseases and the Modulatory Potential by Medicinal Plant and Food Ingredients. Front. Pharmacol. 2017, 8, 387. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, L.; Xiao, Y.Q.; Yao, J.Q.; Li, P.Y.; Yu, D.R.; Ma, Y.L. Global metabolite profiling and diagnostic ion filtering strategy by LC-QTOF MS for rapid identification of raw and processed pieces of Rheum palmatum L. Food Chem. 2016, 192, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xu, H.; Zhao, B.; Li, S.; Li, T.; Xu, X.; Zhang, T.; Lin, R.; Li, J.; Li, X. The Difference of Chemical Components and Biological Activities of the Crude Products and the Salt-Processed Product from Semen Cuscutae. Evid. Based Complement. Altern. Med. 2016, 2016, 8656740. [Google Scholar] [CrossRef] [PubMed]
- State Pharmacopoeia Commission. Chinese Pharmacopoeia; Chinese Medical Science and Technology Press: Beijing, China, 2015; pp. 212–213. [Google Scholar]
- Peng, Y.; Zhao, L.; Lin, D.; Liu, Y.; Zhang, M.; Song, S. Determination of the chemical constituents of the different processed products of Anemarrhena asphodeloides Rhizomes by high-performance liquid chromatography quadrupole time-of-flight mass spectrometry. Biomed. Chromatogr. 2016, 30, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Gao, H.; Jiang, Y.; Zhang, F.; Jia, T. Influences of Anemarrhenae rhizoma and one processed with salt-water on Na+-K+-ATP enzyme in erythrocyte membrane in rats with hyperthyroidism and yin deficiency. Chin. J. Exp. Tradit. Med. Formulae 2011, 17, 184–186. [Google Scholar]
- Wu, Y.; Song, Z.; Gao, H.; Liu, T. Study on hypoglycemic effects of Rhizoma anemarrhenae before and after processed with salt-water and its mechanism. Chin. J. Hosp. Pharm. 2014, 34, 1977–1980. [Google Scholar]
- Zhao, Y.; Kang, L.; Yu, H.; Zhang, J.; Xiong, C.; Pang, X.; Gao, Y.; Liu, C.; Ma, B. Structure Characterization and Identification of Steroidal Saponins from the Rhizomes of Anemarrhena asphodeloides by Ultra Performance Liquid Chromatography and Hybrid Quadrupole Time-of-Flight Mass Spectrometry. Int. J. Mass Spectrom. 2013, 341, 7–17. [Google Scholar] [CrossRef]
- Shvets, S.A.; Kintia, P.K.; Naibi, M.A. Steroidal glycosides from Petunia hybrida L. seeds and their biological activity. Adv. Exp. Med. Biol. 1996, 404, 251–262. [Google Scholar] [PubMed]
- Wang, Y.; Dan, Y.; Yang, D.; Hu, Y.; Zhang, L.; Zhang, C.; Zhu, H.; Cui, Z.; Li, M.; Liu, Y. The genus Anemarrhena Bunge: A review on ethnopharmacology, phytochemistry and pharmacology. J. Ethnopharmacol. 2014, 153, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yuan, J.; Kang, L.; Pang, X.; Yan, R.; Zhao, Y.; Zhang, J.; Sun, X.; Ma, B. An Efficient Approach to Identify Different Chemical Markers between Fibrous Root and Rhizome of Anemarrhena asphodeloides by Ultra High-Performance Liquid Chromatography Quadrupole Time-of-Flight Tandem Mass Spectrometry with Multivariate Statistical Analysis. J. Pharm. Biomed. Anal. 2016, 129, 105–116. [Google Scholar] [PubMed]
- Meng, Z.Y.; Zhang, J.Y.; Xu, S.X.; Sugahara, K. Steroidal saponins from Anemarrhena asphodeloides and their effects on superoxide generation. Planta Med. 1999, 65, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Trinh, H.T.; Jung, K.; Han, S.J.; Kim, D.H. Inhibitory effects of steroidal timosaponins isolated from the rhizomes of Anemarrhena asphodeloides against passive cutaneous anaphylaxis reaction and pruritus. Immunopharmacol. Immunotoxicol. 2010, 32, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Nagase, S.; Ichinose, K. New steroidal saponins from the rhizomes of Anemarrhena asphodeloides Bunge (Liliaceae). Chem. Pharm. Bull. 1994, 42, 2342–2345. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, C.; Lozano-Sanchez, J.; Rodriguez-Perez, C.; Segura-Carretero, A.; Fernandez-Gutierrez, A. Comprehensive, untargeted, and qualitative RP-HPLC-ESI-QTOF/MS2 metabolite profiling of green asparagus (Asparagus officinalis). J. Food Compos. Anal. 2016, 46, 78–87. [Google Scholar] [CrossRef]
- Kite, G.C.; Porter, E.A.; Simmonds, M.S. Chromatographic behaviour of steroidal saponins studied by high-performance liquid chromatography-mass spectrometry. J. Chromatogr. A 2007, 1148, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Li, L.J.; Abliz, Z.; Yang, Y.C.; Shi, J.G. Structural characterization of steroidal saponins by electrospray ionization and fast-atom bombardment tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2002, 16, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound timosaponin N is available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak | tR | Formula | Negative Ion Mode of ESI-MS (m/z) | Positive Ion Mode of ESI-MS (m/z) | Identification | t-Value | p-Value | Changing Direction c | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Precursor Ion | Selective Ion | MS2 Fragmentation | Precursor Ion | Selective Ion | MS2 Fragmentation | |||||||
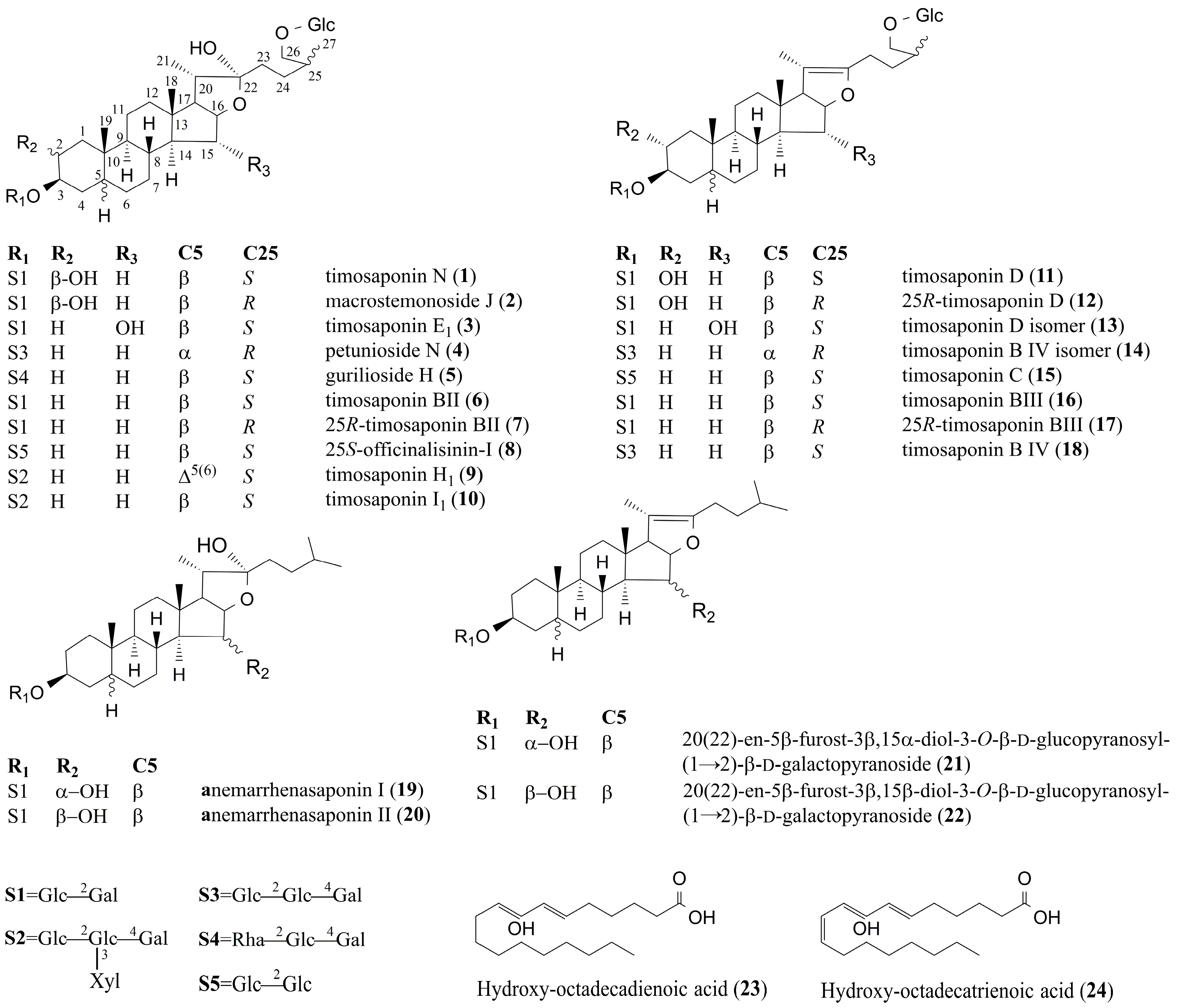
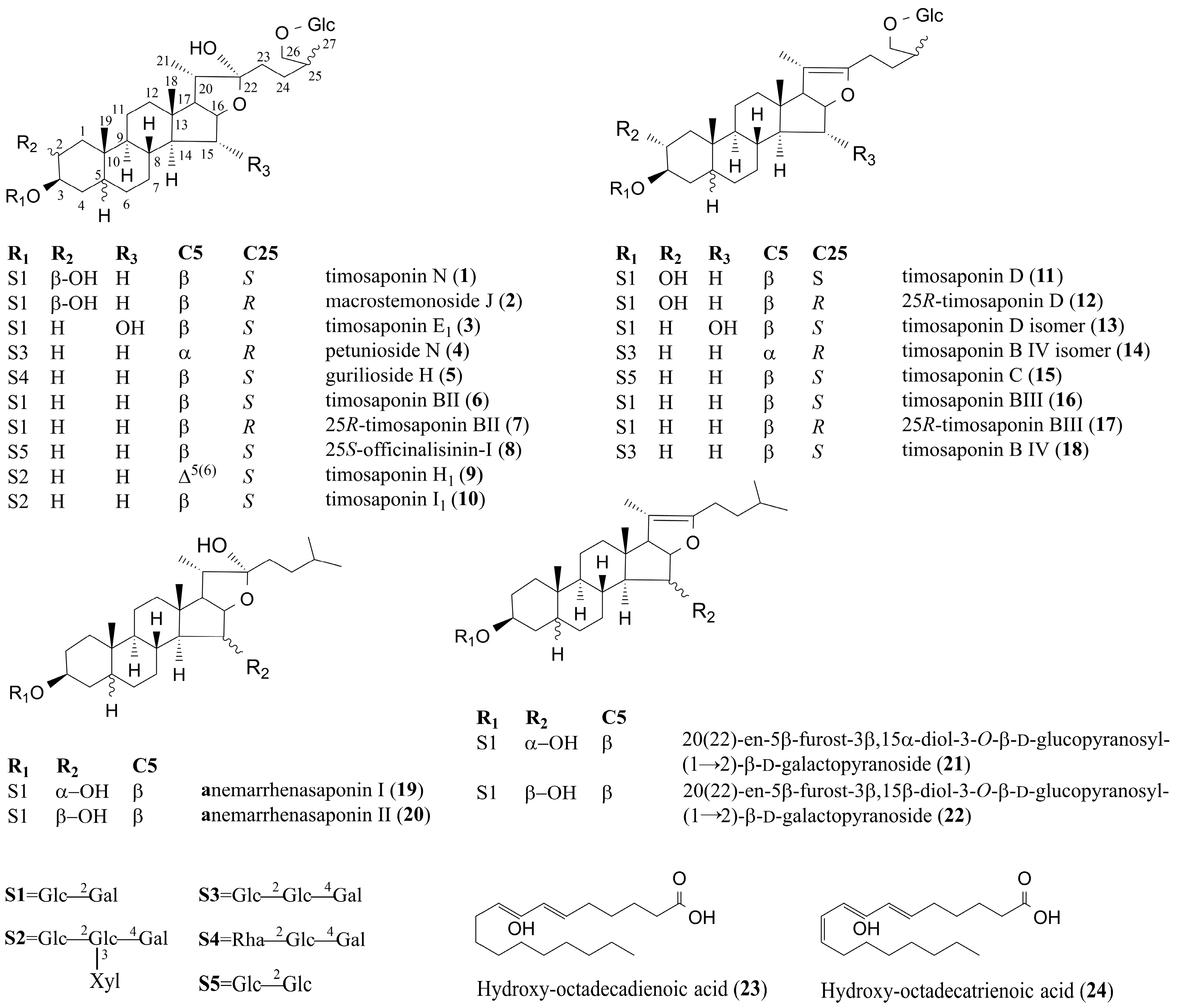
| 1 | 9.81 | C45H76O20 | 981.4892 | [M + HCOO]− | 935, 773, 611 | 919.4911 | [M − H2O + H]+ | 757, 595, 433, 415, 289, 271, 253 | Timosaponin N a | 2.77 | 0.01271 | ↓ * |
| 2 | 9.97 | C45H76O20 | 981.4925 | [M + HCOO]− | 935, 773, 611 | 919.4916 | [M − H2O + H]+ | 757, 595, 433, 415, 289, 271, 253 | Macrostemonoside J [12] | 2.06 | 0.03379 | ↓ * |
| 3 | 10.78 | C45H76O20 | 981.4914 | [M + HCOO]− | 935, 773, 611 | 919.4918 | [M − H2O + H]+ | 757, 595, 433, 415, 289, 271, 253 | Timosaponin E1 a | 3.25 | 0.00447 | ↓ ** |
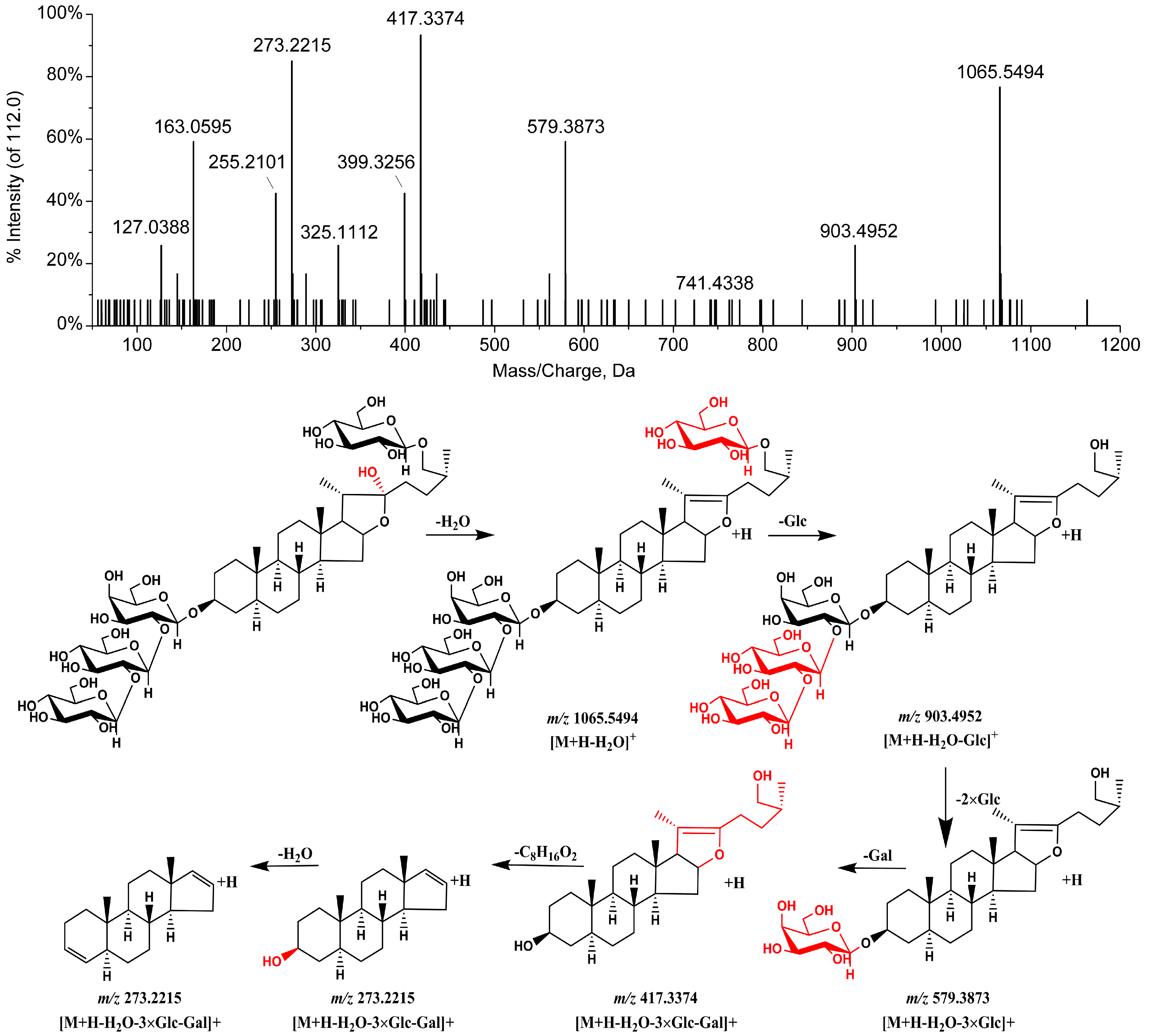
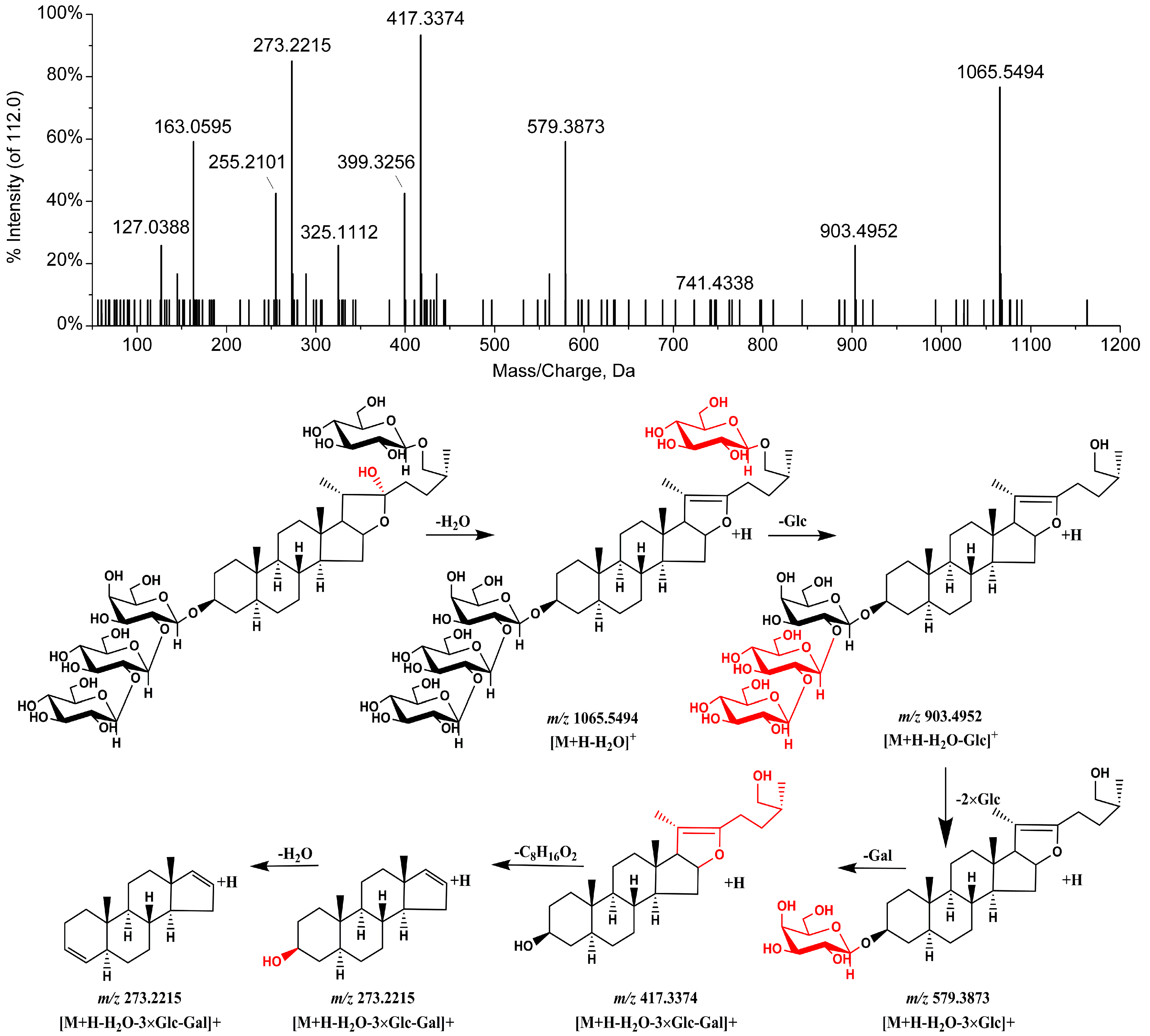
| 4 | 11.08 | C51H86O24 | 1127.5433 | [M + HCOO]− | 1081, 919, 757, 595 | 1065.5482 | [M − H2O + H]+ | 903, 741, 579, 417, 399, 273, 255 | Petunioside N [13] | 2.21 | 0.03999 | ↓ * |
| 5 | 11.89 | C51H86O23 | 1111.5528 | [M + HCOO]− | 1065, 919, 757, 595 | 1049.5289 | [M − H2O + H]+ | 903, 741, 579, 417, 399, 273, 255 | Curilioside H [14] | 2.92 | 0.00921 | ↓ ** |
| 6 | 12.03 | C45H76O19 | 965.5029 | [M + HCOO]− | 919, 757, 595 | 903.4982 | [M − H2O + H]+ | 741, 579, 417, 399, 273, 255 | Timosaponin BII a | 4.10 | 0.00067 | ↓ ** |
| 7 | 12.14 | C45H76O19 | 965.4945 | [M + HCOO]− | 919, 757, 595 | 903.4931 | [M − H2O + H]+ | 741, 579, 417, 399, 273, 255 | 25R-timosaponin BII [12,15] | 4.17 | 0.00058 | ↓ ** |
| 8 | 12.30 | C45H76O19 | 965.4943 | [M + HCOO]− | 919, 757, 595 | 903.4940 | [M − H2O + H]+ | 741, 579, 417, 399, 273, 255 | 25S-officinalisinin-I [12,15] | 2.48 | 0.03575 | ↓ * |
| 9 | 12.60 | C56H92O28 | 1257.5733 | [M + HCOO]− | 1211, 1079, 1049, 917, 755, 593 | 1195.4256 | [M − H2O + H]+ | 1033, 901, 739, 577, 433, 415, 271, 253 | Timosaponin H1 [12,16] | 2.73 | 0.01360 | ↓ * |
| 10 | 12.75 | C56H94O28 | 1259.5853 | [M + HCOO]− | 1213, 1081, 919, 757, 595 | 1197.4558 | [M − H2O + H]+ | 1065, 903, 741, 579, 435, 417, 273, 255 | Timosaponin I1 [12,16] | 2,17 | 0.04392 | ↓ * |
| 11 | 13.30 | C45H74O19 | 963.4798 | [M + HCOO]− | 917, 755, 593 | 919.4893 | [M + H]+ | 757, 595, 433, 415, 289, 271, 253 | Timosaponin D [12,17] | −5.82 | 1.6189 × 10−5 | ↑ ** |
| 12 | 13.46 | C45H74O19 | 963.4806 | [M + HCOO]− | 917, 755, 593 | 919.4901 | [M + H]+ | 757, 595, 433, 415, 289, 271, 253 | 25R-timosaponin D [12] | 3.53 | 0.00241 | ↑ ** |
| 13 | 13.64 | C45H74O19 | 963.4793 | [M + HCOO]− | 917, 755, 593 | 919.4889 | [M + H]+ | 757, 595, 433, 415, 289, 271, 253 | Timosaponin D isomer [12] | −2.02 | 0.03806 | ↑ * |
| 14 | 13.83 | C51H84O23 | 1109.5370 | [M + HCOO]− | 1063, 901, 739 | 1065.5008 | [M + H]+ | 903, 741, 579, 417, 273, 255 | (25R)-26-O-β-d-glucopyranosyl-5α-furostane-20(22)-en-3β, 26-diol-3-O-β-d-glucopyranosyl-(1→2)-β-d-glucopyranosyl-(1→4)-β-d-galactopyranoside [12,14] | −2.82 | 0.01144 | ↑ * |
| 15 | 14.82 | C45H74O18 | 947.4865 | [M+HCOO]− | 901, 739, 721, 577 | 903.4984 | [M + H]+ | 741, 579, 417, 273, 255 | Timosaponin C [12,14] | −5.24 | 5.4915 × 10−5 | ↑ ** |
| 16 | 14.95 | C45H74O18 | 947.4851 | [M + HCOO]− | 901, 739, 577 | 903.4941 | [M + H]+ | 741, 579, 417, 273, 255 | Timosaponin BIII a | −9.15 | 3.4202 × 10−8 | ↑ ** |
| 17 | 15.14 | C45H74O18 | 947.4854 | [M + HCOO]− | 901, 739, 577 | 903.4958 | [M + H]+ | 741, 579, 417, 273, 255 | 25R-timosaponin BIII [12,14] | −11.53 | 9.5454 ×10−10 | ↑ ** |
| 18 | 15.33 | C51H84O23 | 1109.5577 | [M + HCOO]− | 1063, 901, 739 | 1065.5078 | [M + H]+ | 903, 741, 579, 417, 273, 255 | Timosaponin B IV [12,14] | −4.44 | 0.00032 | ↑ ** |
| 19 | 16.85 | C39H66O14 | 803.4412 | [M + HCOO]− | 757, 595, 433 | 741.4397 | [M − H2O + H]+ | 579, 417, 399, 289, 271, 253 | Anemarrhenasaponin I a | 3.23 | 0.00464 | ↓ ** |
| 20 | 17.03 | C39H66O14 | 803.4415 | [M + HCOO]− | 757, 595, 433 | 741.4398 | [M − H2O + H]+ | 579, 417, 399, 289, 271, 253 | Anemarrhenasaponin II [18] | 2.12 | 0.04854 | ↓ * |
| 21 | 20.32 | C39H64O13 | 785.4321 | [M + HCOO]− | 739, 577 | 741.4308 | [M + H]+ | 579, 417, 399, 289, 271, 253 | 20(22)-en-5β-furost-3β,15α-diol-3-O-β-d-glucopyranosyl-(1→2)-β-d-galactopyranoside b | −4.71 | 0.00017 | ↑ ** |
| 22 | 20.77 | C39H64O13 | 785.4323 | [M + HCOO]− | 739, 577 | 741.4303 | [M + H]+ | 579, 417, 399, 289, 271, 253 | 20(22)-en-5β-furost-3β,15β-diol-3-O-β-d-glucopyranosyl-(1→2)-β-d-galactopyranoside b | −2.12 | 0.04033 | ↑ * |
| 23 | 23.92 | C18H32O3 | 295.2280 | [M − H]− | 277, 195, 171 | Hydroxy-octadecadienoic acid [19] | 4.33 | 0.00040 | ↓ ** | |||
| 24 | 24.15 | C18H30O3 | 293.2124 | [M − H]− | Hydroxy-octadecatrienoic acid [19] | 2.39 | 0.02800 | ↓ * | ||||
| Compounds | Linear Regression | r2 | Linear Range (μg/mL) | LOD (ng/mL) | LOQ (ng/mL) |
|---|---|---|---|---|---|
| Timosaponin N | y = 68,743x + 27,585 | 0.9997 | 2.0–16.0 | 2.8 | 9.8 |
| Timosaponin E1 | y = 100,376x + 13,353 | 0.9997 | 0.1–6.0 | 3.6 | 11.3 |
| Timosaponin BII | y = 13,964x + 8444 | 0.9999 | 20.0–120.0 | 4.8 | 19.0 |
| Timosaponin BIII | y = 53,259x + 21,310 | 0.9998 | 3.0–60.0 | 2.7 | 8.6 |
| Anemarrhenasaponin I | y = 71,064x + 29,916 | 0.9999 | 2.0–16.0 | 3.0 | 10.0 |
| Timosaponin AII | y = 58,520x + 17,831 | 0.9998 | 2.0–16.0 | 2.0 | 7.4 |
| Timosaponin AIII | y = 38,170x + 32,791 | 0.9998 | 3.0–60.0 | 1.0 | 3.5 |
| No. | Timosaponin N | Timosaponin E1 | Timosaponin BII | Timosaponin BIII | Anemarrhenasaponin I | Timosaponin AII | Timosaponin AIII | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CAR | SAR | CAR | SAR | CAR | SAR | CAR | SAR | CAR | SAR | CAR | SAR | CAR | SAR | |
| 1 | 11.03 ± 0.28 | 5.20 ± 0.13 ** | 12.57 ± 0.03 | 7.45 ± 0.02 ** | 81.58 ± 1.38 | 72.44 ± 0.61 ** | 1.44 ± 0.18 | 11.18 ± 0.57 ** | 9.16 ± 0.23 | 7.92 ± 0.10 ** | 9.09 ± 0.23 | 9.00 ± 0.23 | 37.82 ± 0.96 | 38.33 ± 0.97 |
| 2 | 13.44 ± 0.34 | 6.82 ± 0.17 ** | 15.78 ± 0.04 | 10.79 ± 0.03 * | 83.13 ± 1.41 | 72.80 ± 0.62 ** | 2.02 ± 0.26 | 10.53 ± 0.53 ** | 7.54 ± 0.19 | 6.07 ± 0.08 * | 5.63 ± 0.14 | 5.41 ± 0.14 | 26.77 ± 0.68 | 25.79 ± 0.65 |
| 3 | 12.59 ± 0.32 | 5.26 ± 0.13 ** | 19.70 ± 0.05 | 6.64 ± 0.02 ** | 91.21 ± 1.54 | 63.05 ± 0.53 ** | 1.73 ± 0.22 | 13.04 ± 0.66 ** | 7.15 ± 0.18 | 3.95 ± 0.05 ** | 2.91 ± 0.07 | 4.06 ± 0.10 | 19.94 ± 0.38 | 21.88 ± 0.56 |
| 4 | 14.47 ± 0.37 | 5.00 ± 0.13 ** | 15.98 ± 0.04 | 8.89 ± 0.02 ** | 84.31 ± 1.43 | 71.33 ± 0.60 ** | 0.63 ± 0.18 | 12.01 ± 0.61 ** | 6.54 ± 0.17 | 5.86 ± 0.07 * | 2.26 ± 0.06 | 2.34 ± 0.06 | 13.49 ± 0.34 | 13.46 ± 0.34 |
| 5 | 11.53 ± 0.29 | 5.65 ± 0.14 ** | 6.53 ± 0.02 | 4.07 ± 0.01 * | 82.76 ± 1.40 | 72.01 ± 0.61 ** | 6.39 ± 0.15 | 11.58 ± 0.59 ** | 8.24 ± 0.21 | 7.30 ± 0.09 * | 4.23 ± 0.11 | 4.41 ± 0.11 | 35.56 ± 0.90 | 36.99 ± 0.94 |
| 6 | 9.21 ± 0.23 | 4.11 ± 0.10 ** | 19.37 ± 0.05 | 6.07 ± 0.02 ** | 91.01 ± 1.54 | 60.15 ± 0.51 ** | 1.30 ± 0.17 | 11.92 ± 0.61 ** | 9.78 ± 0.25 | 5.98 ± 0.08 ** | 6.15 ± 0.16 | 7.04 ± 0.18 | 32.63 ± 0.65 | 32.05 ± 0.81 |
| 7 | 12.58 ± 0.35 | 5.55 ± 0.14 ** | 8.47 ± 0.02 | 4.65 ± 0.01 ** | 71.68 ± 1.21 | 55.24 ± 0.47 ** | 3.32 ± 0.42 | 5.94 ± 0.81 ** | 7.12 ± 0.18 | 5.51 ± 0.07 * | 8.16 ± 0.21 | 9.82 ± 0.25 | 36.65 ± 0.78 | 39.88 ± 1.01 |
| 8 | 6.89 ± 0.17 | 2.60 ± 0.07 ** | 1.26 ± 0.00 | 0.52 ± 0.01 * | 85.47 ± 1.45 | 68.21 ± 0.58 ** | 1.22 ± 0.15 | 14.20 ± 0.72 ** | 11.69 ± 0.30 | 8.34 ± 0.11 ** | - | - | 6.32 ± 0.12 | 7.22 ± 0.11 |
| 9 | 10.13 ± 0.26 | 4.87 ± 0.12 ** | 13.84 ± 0.04 | 10.15 ± 0.03 * | 98.30 ± 1.66 | 92.92 ± 0.79 * | 1.06 ± 0.13 | 8.07 ± 0.41 ** | 6.31 ± 0.16 | 4.94 ± 0.08 * | 2.93 ± 0.07 | 3.51 ± 0.09 | 17.71 ± 0.40 | 18.11 ± 0.46 |
| 10 | 9.06 ± 0.23 | 4.10 ± 0.10 ** | 4.91 ± 0.01 | 2.56 ± 0.01 ** | 73.79 ± 1.25 | 55.96 ± 0.47 ** | 5.99 ± 0.15 | 9.44 ± 0.48 ** | 4.61 ± 0.12 | 3.21 ± 0.04 * | 4.52 ± 0.11 | 4.68 ± 0.12 | 41.02 ± 1.04 | 41.82 ± 1.06 |
| No. | Place of Collection | Collection Time | Growing Condition |
|---|---|---|---|
| 1 | Changzhi, Shanxi | 2014.09 | Cultivated |
| 2 | Anguo, Hebei | 2014.10 | Cultivated |
| 3 | Bozhou, Anhui | 2014.08 | Cultivated |
| 4 | Huludao, Liaoning | 2014.11 | wild |
| 5 | Chifeng, Neimenggu | 2014.12 | wild |
| 6 | Changzhi, Shanxi | 2015.03 | Cultivated |
| 7 | Anguo, Hebei | 2015.04 | Cultivated |
| 8 | Datong, Shanxi | 2015.03 | Cultivated |
| 9 | Bozhou, Anhui | 2015.04 | Cultivated |
| 10 | Chifeng, Neimenggu | 2015.05 | wild |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, D.; Su, X.; Huang, Z.; Su, L.; Li, L.; Lu, T. Analysis of Chemical Variations between Crude and Salt-Processed Anemarrhenae rhizoma Using Ultra-High-Performance Liquid Chromatography–Mass Spectrometry Methods. Molecules 2018, 23, 23. https://doi.org/10.3390/molecules23010023
Ji D, Su X, Huang Z, Su L, Li L, Lu T. Analysis of Chemical Variations between Crude and Salt-Processed Anemarrhenae rhizoma Using Ultra-High-Performance Liquid Chromatography–Mass Spectrometry Methods. Molecules. 2018; 23(1):23. https://doi.org/10.3390/molecules23010023
Chicago/Turabian StyleJi, De, Xiaonan Su, Ziyan Huang, Lialin Su, Lin Li, and Tulin Lu. 2018. "Analysis of Chemical Variations between Crude and Salt-Processed Anemarrhenae rhizoma Using Ultra-High-Performance Liquid Chromatography–Mass Spectrometry Methods" Molecules 23, no. 1: 23. https://doi.org/10.3390/molecules23010023