Identification of Novel Bisbenzimidazole Derivatives as Anticancer Vacuolar (H+)-ATPase Inhibitors †


Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
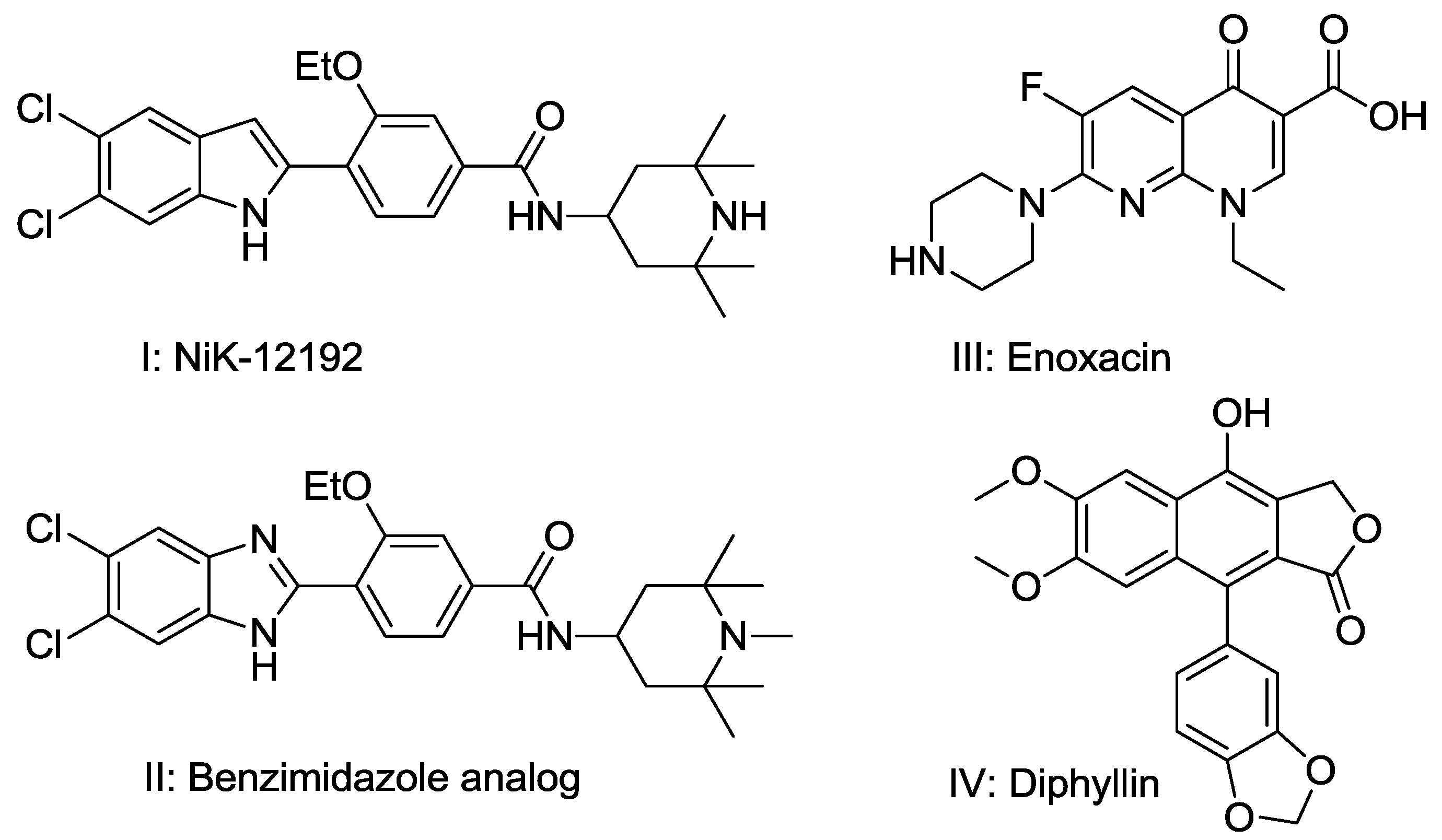
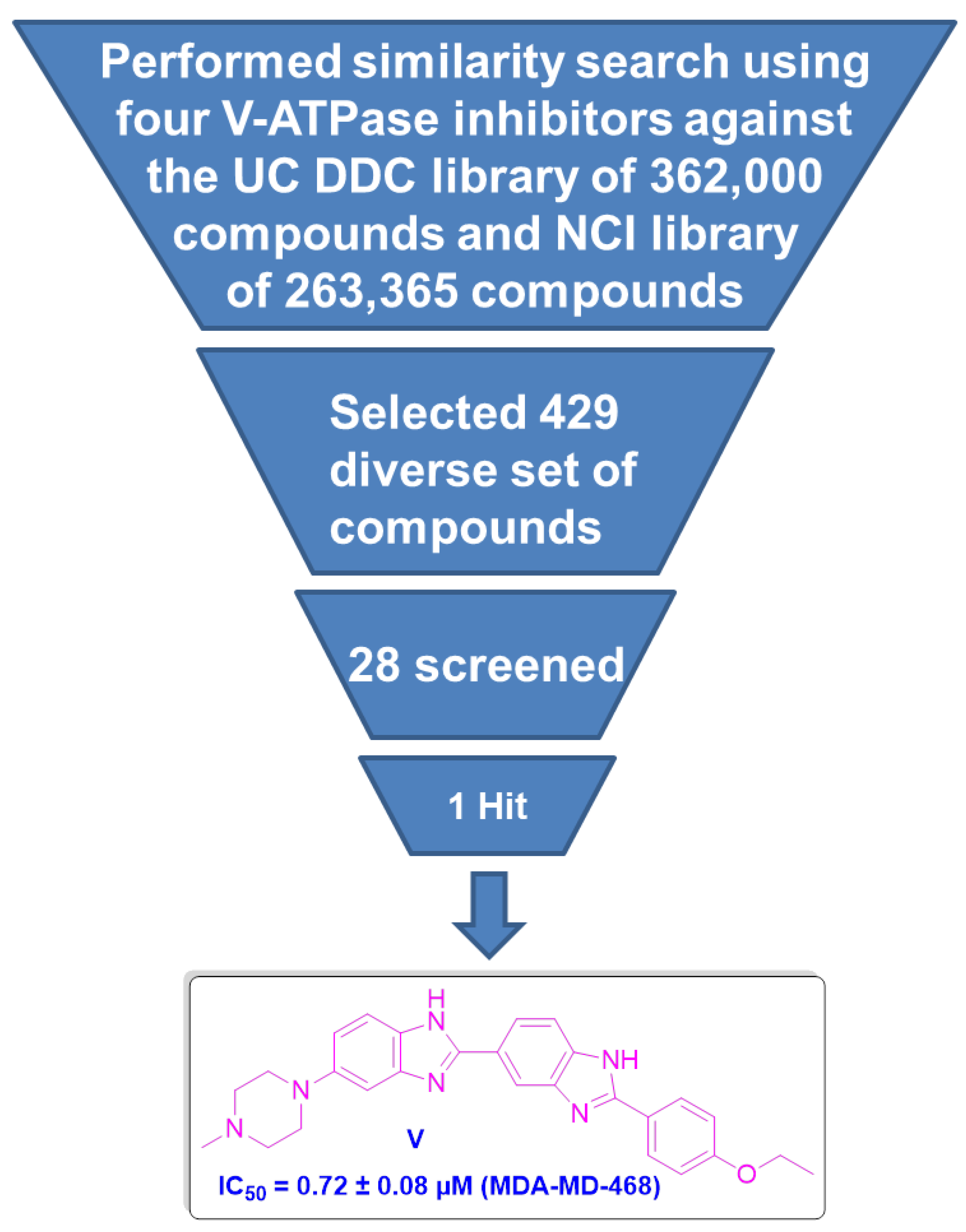
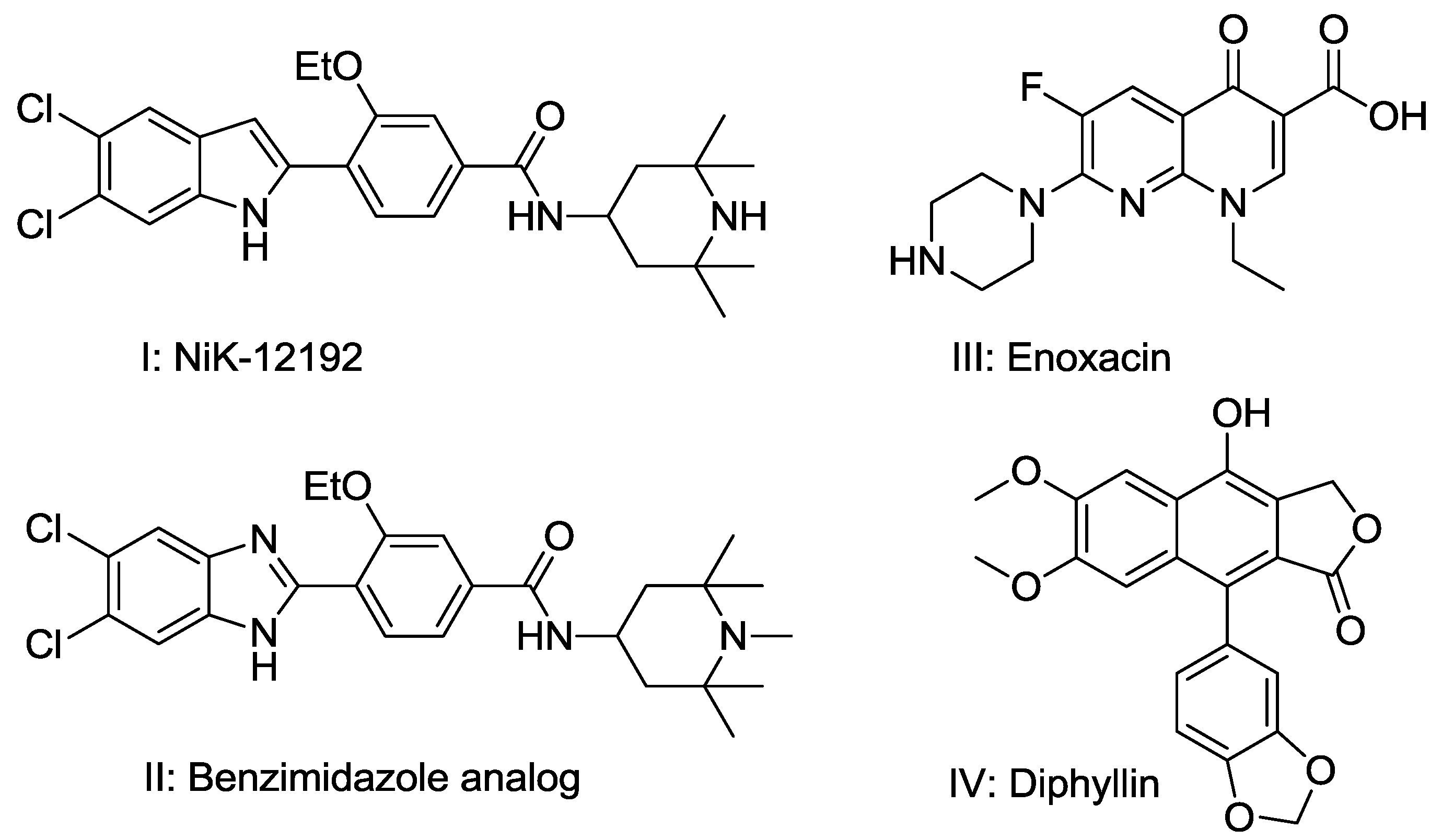
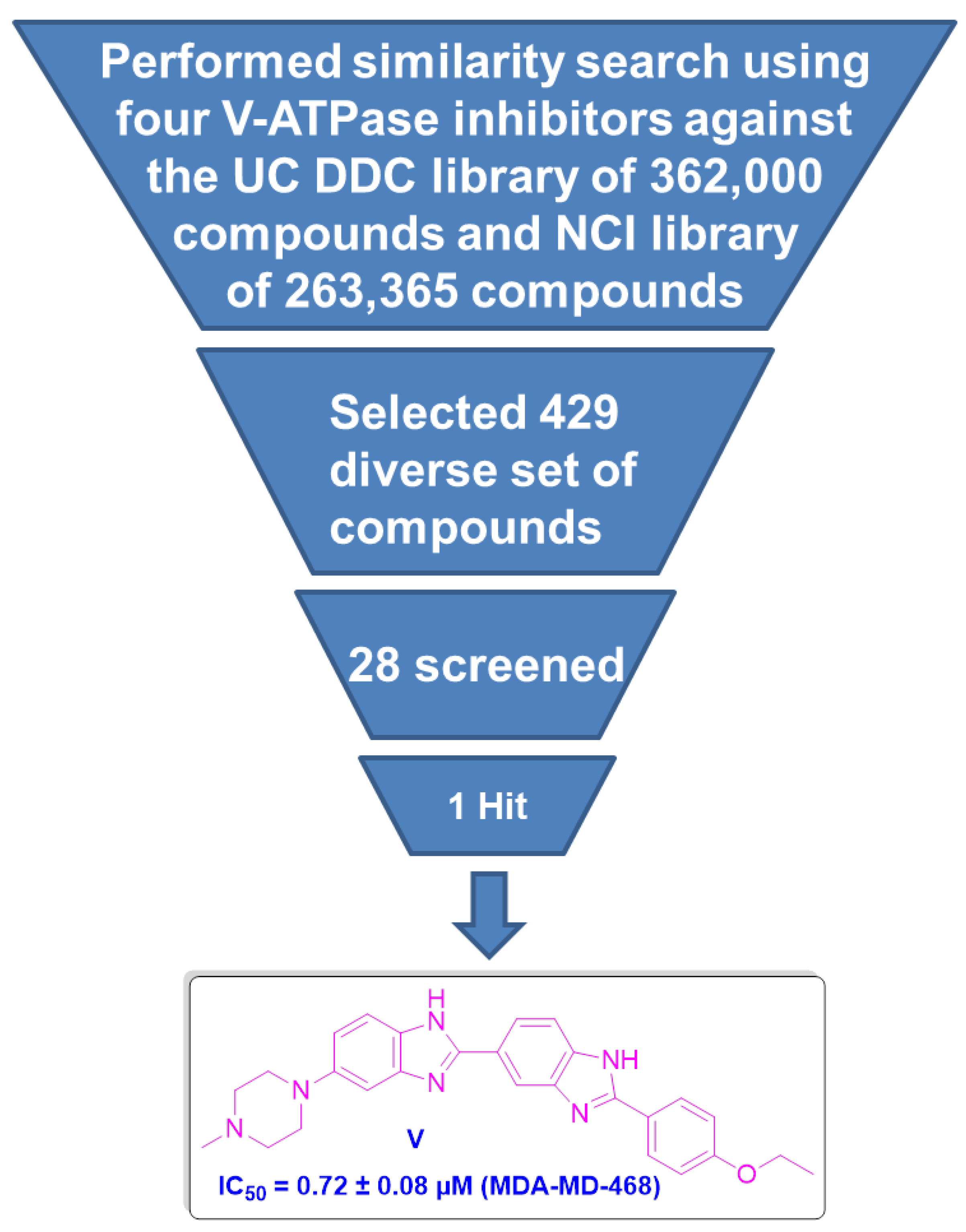
2.1.1. Similarity Search
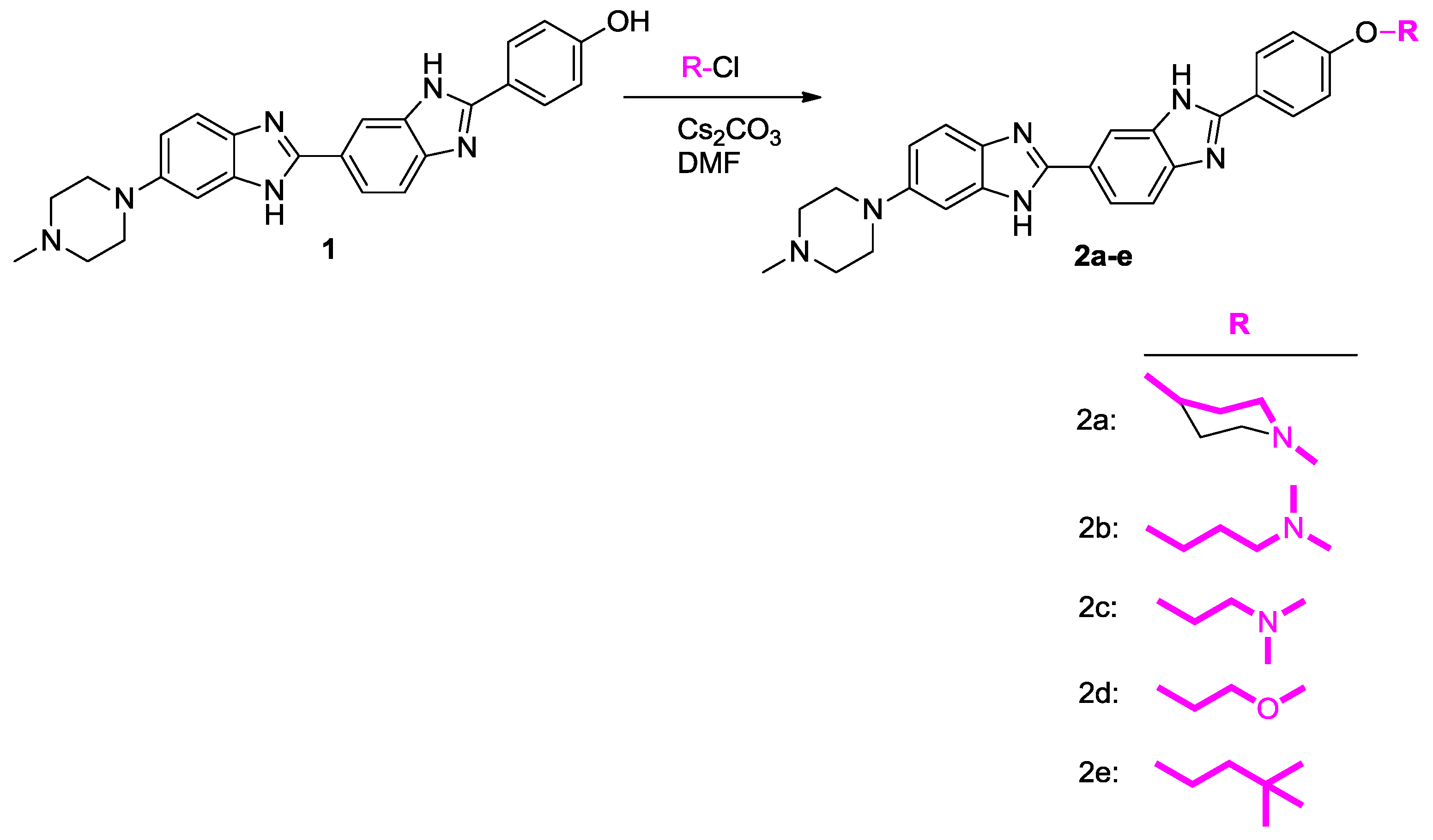
2.1.2. Chemical Synthesis
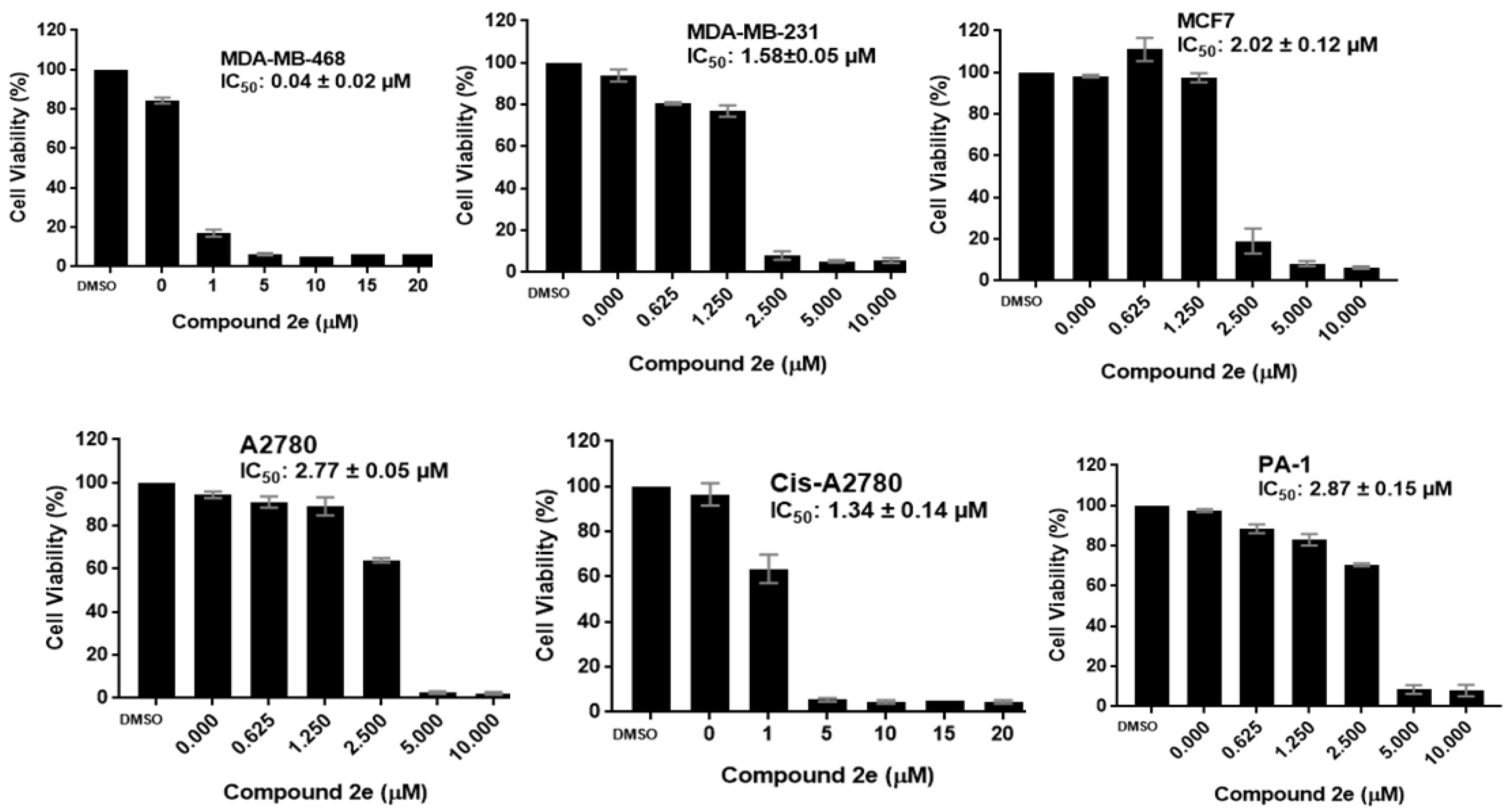
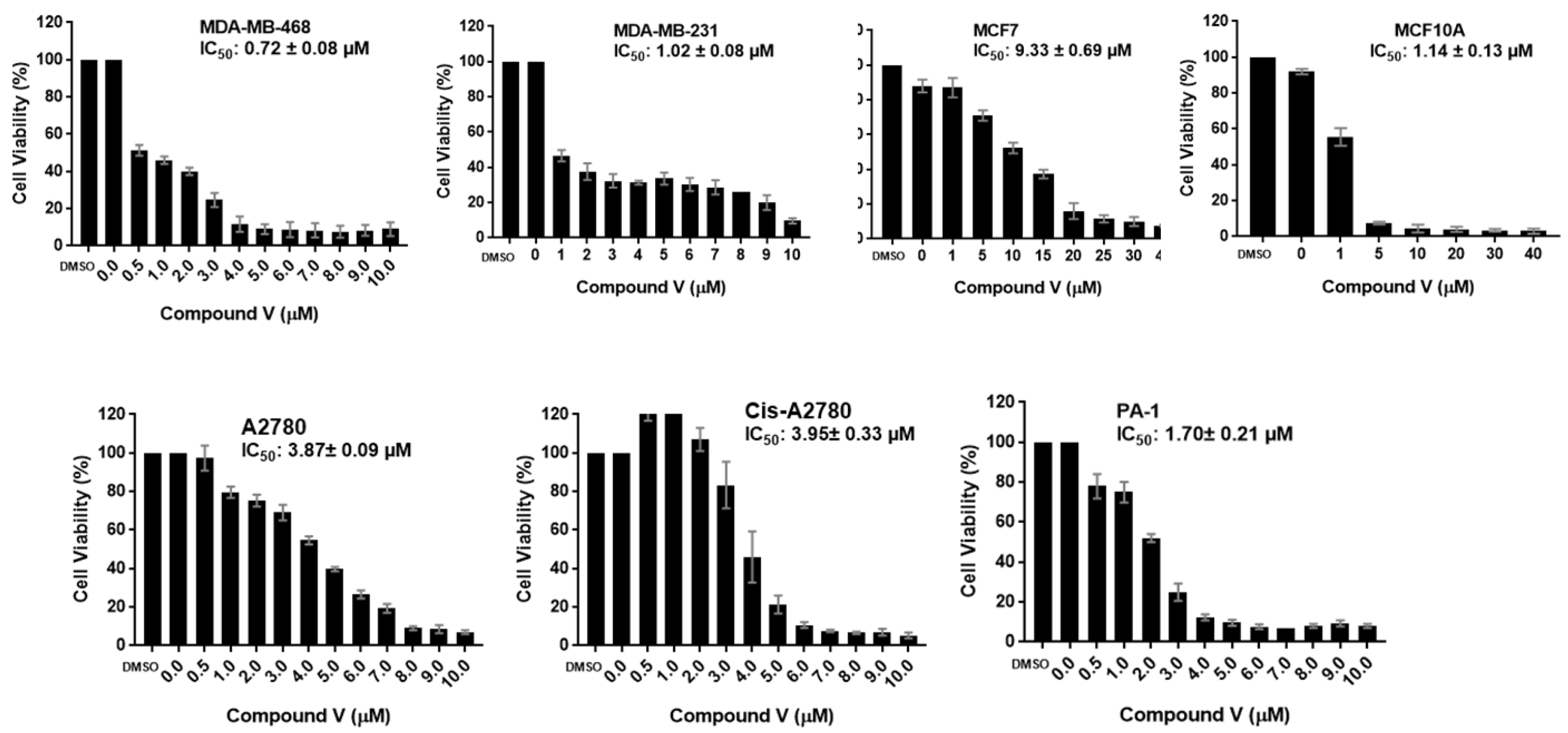
2.2. Biology
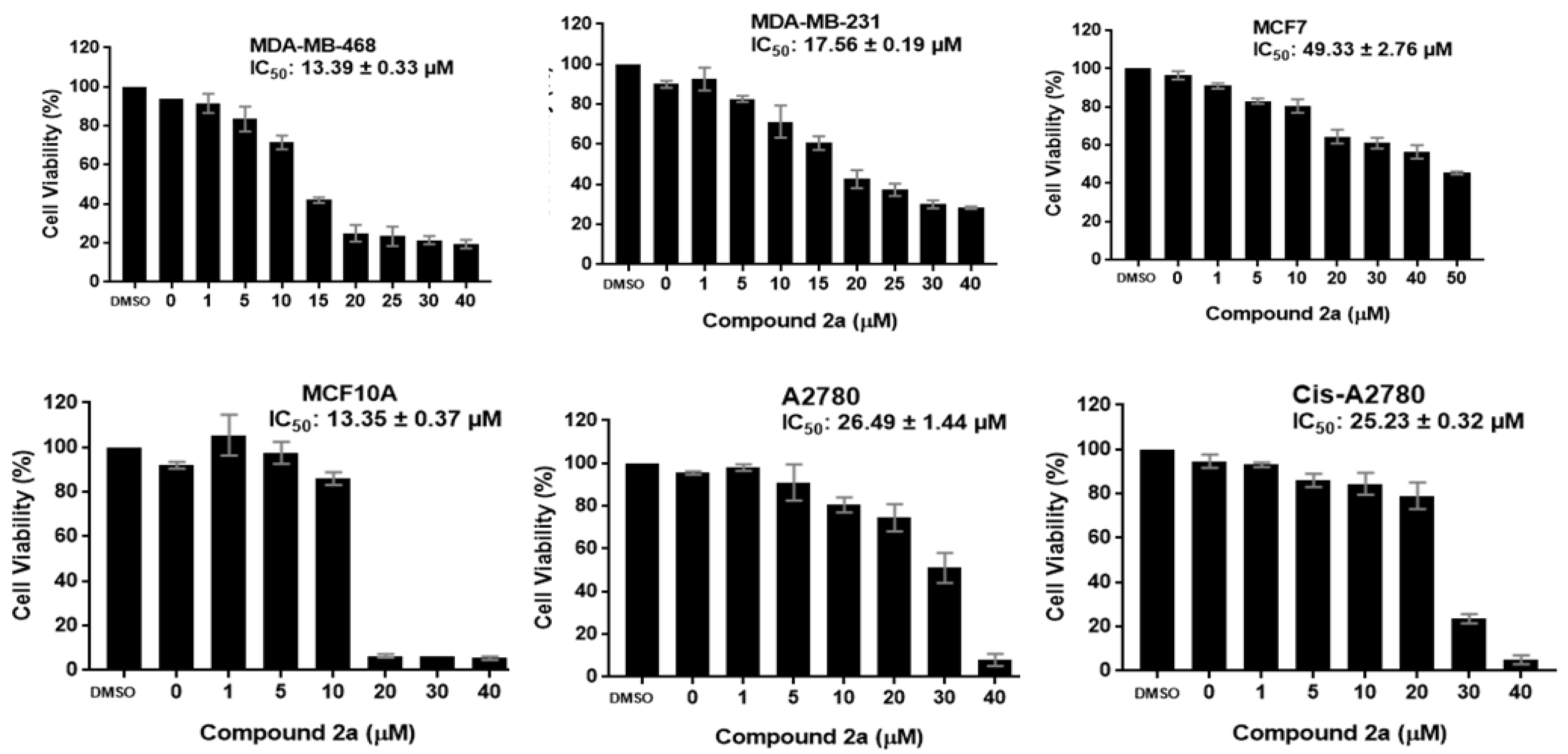
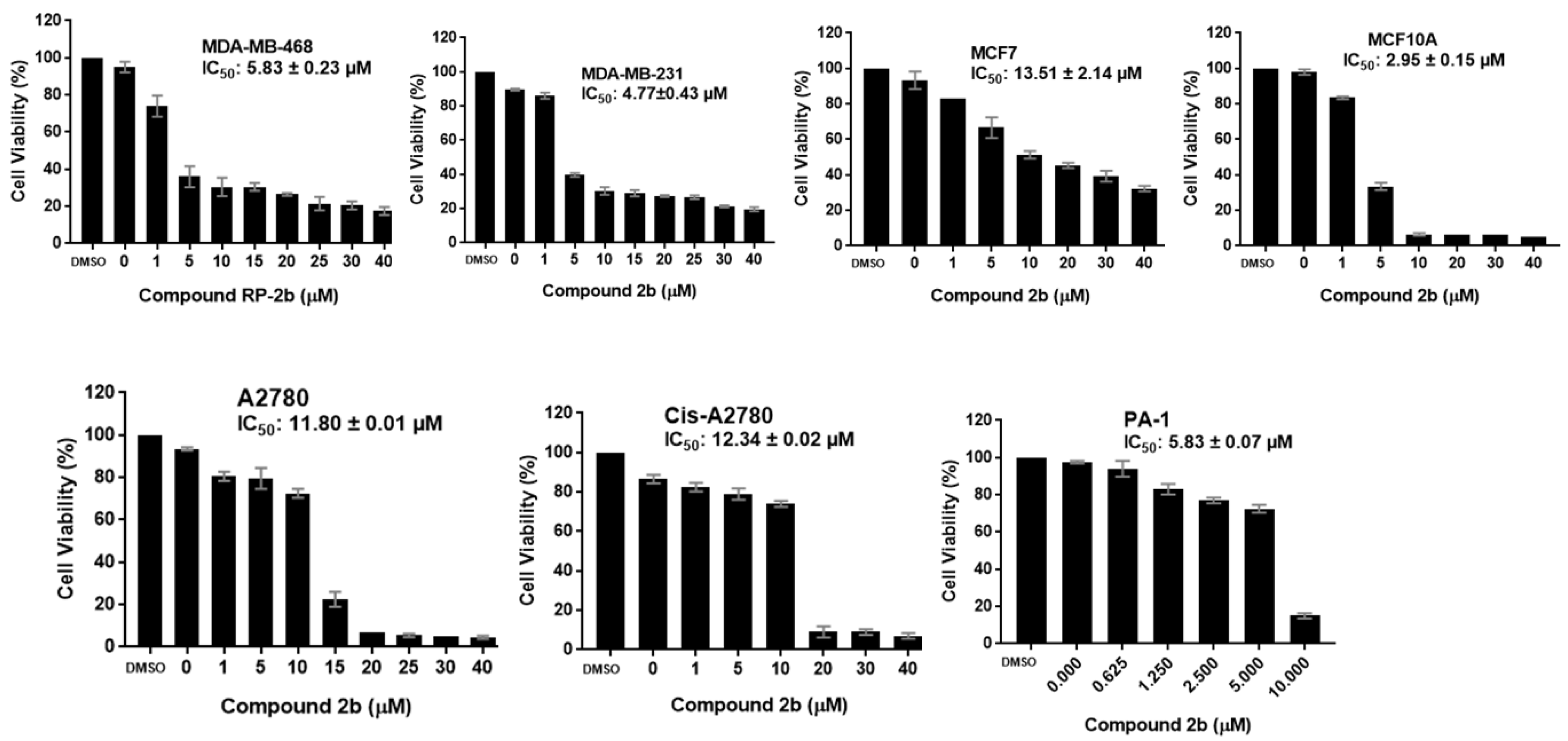
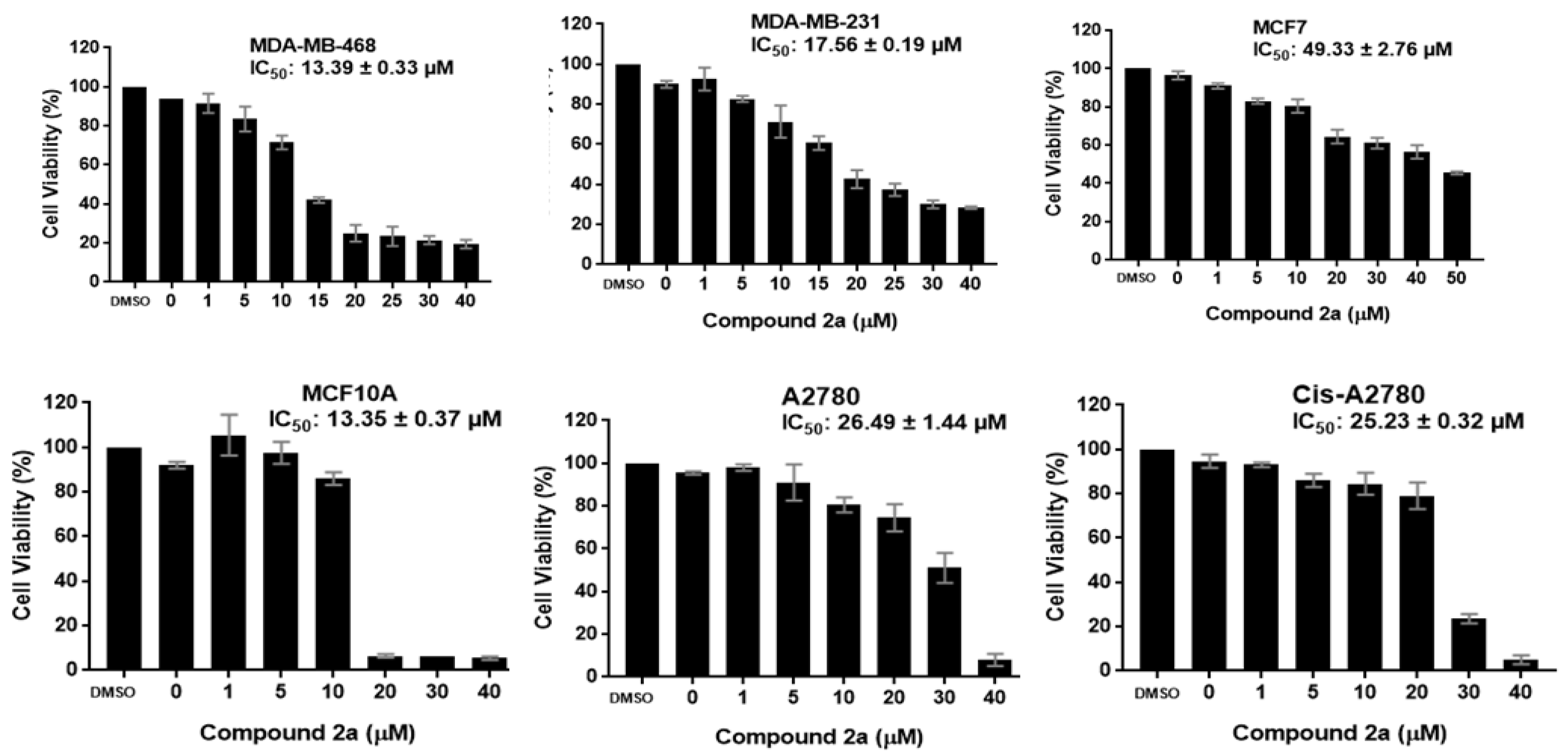
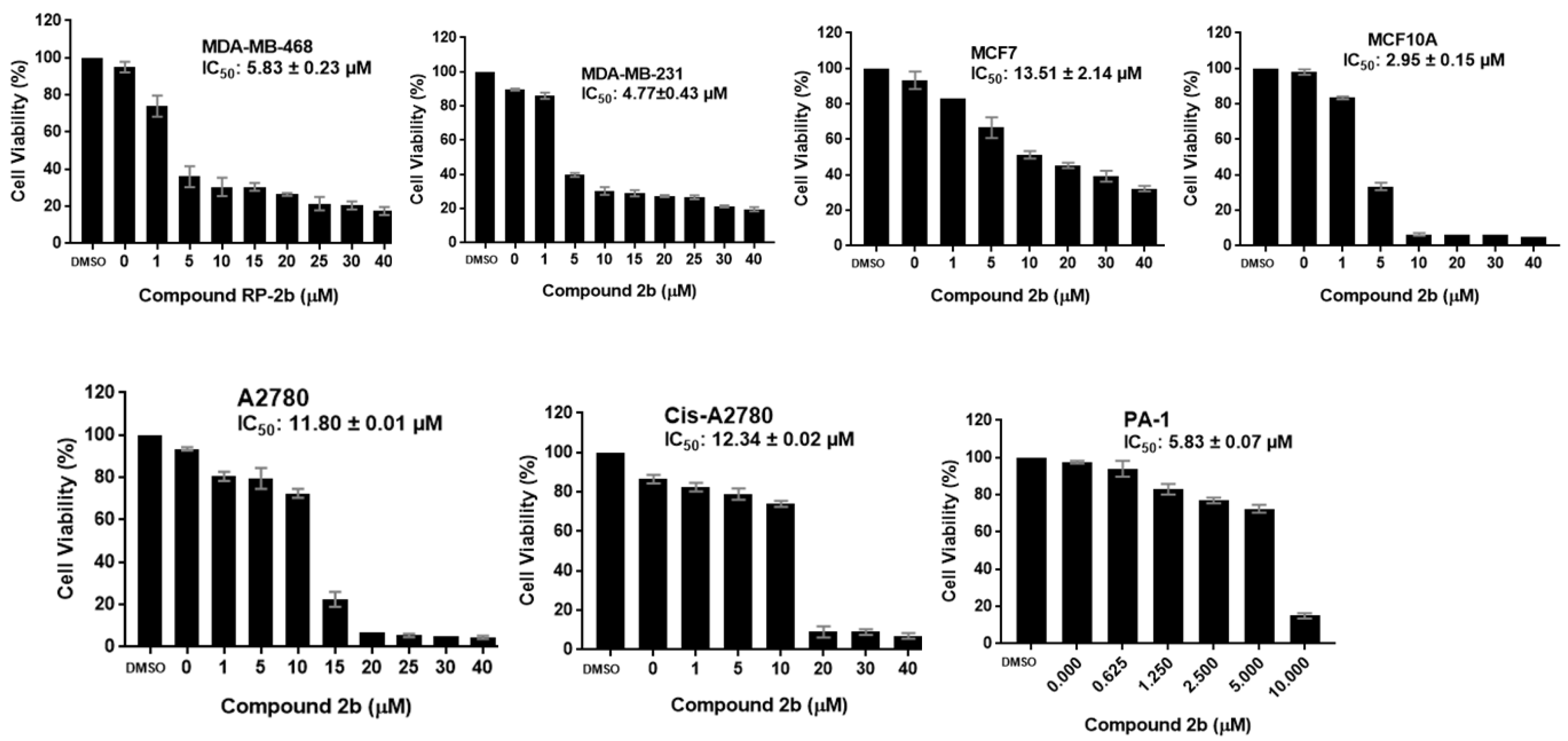
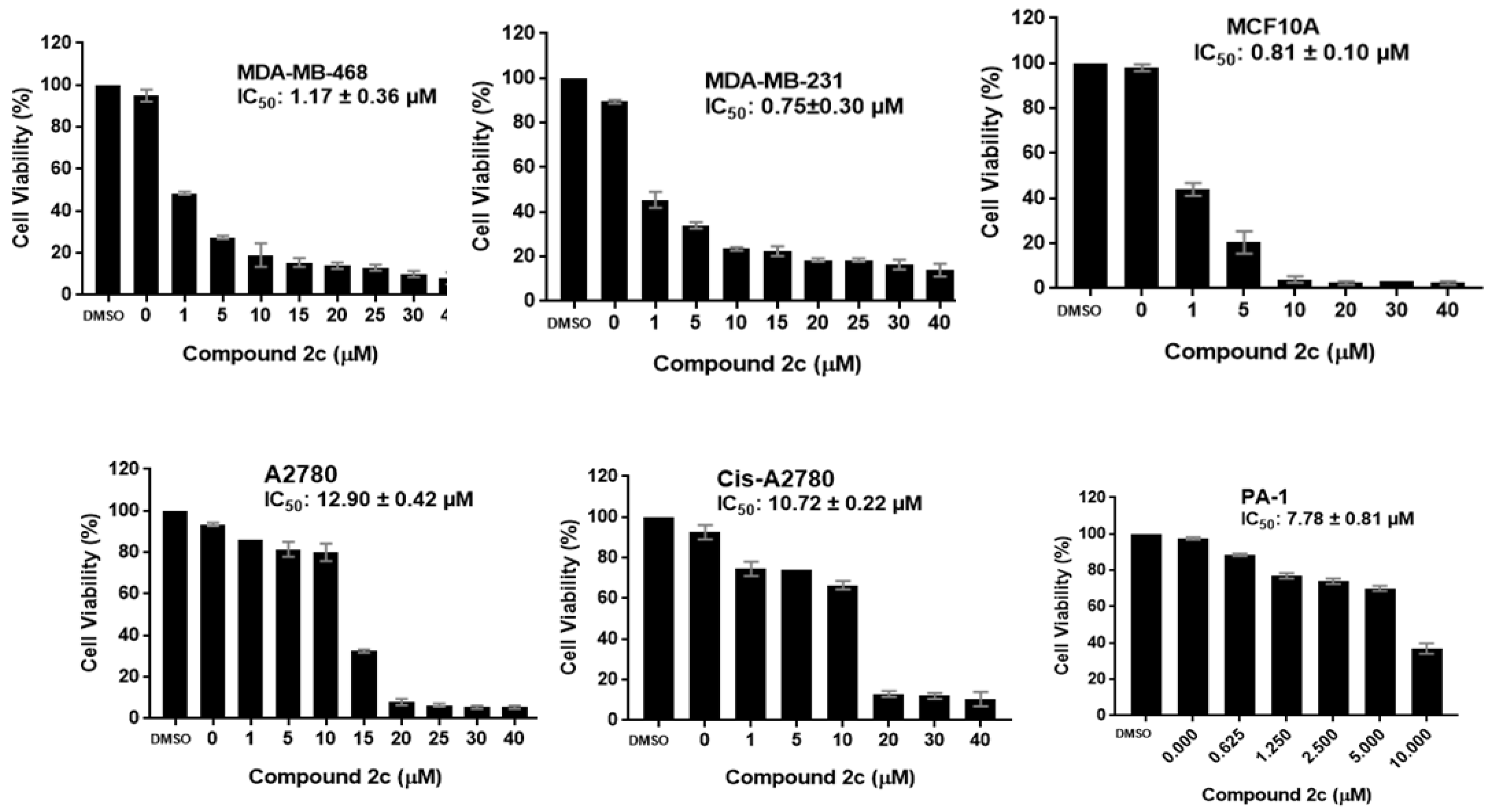
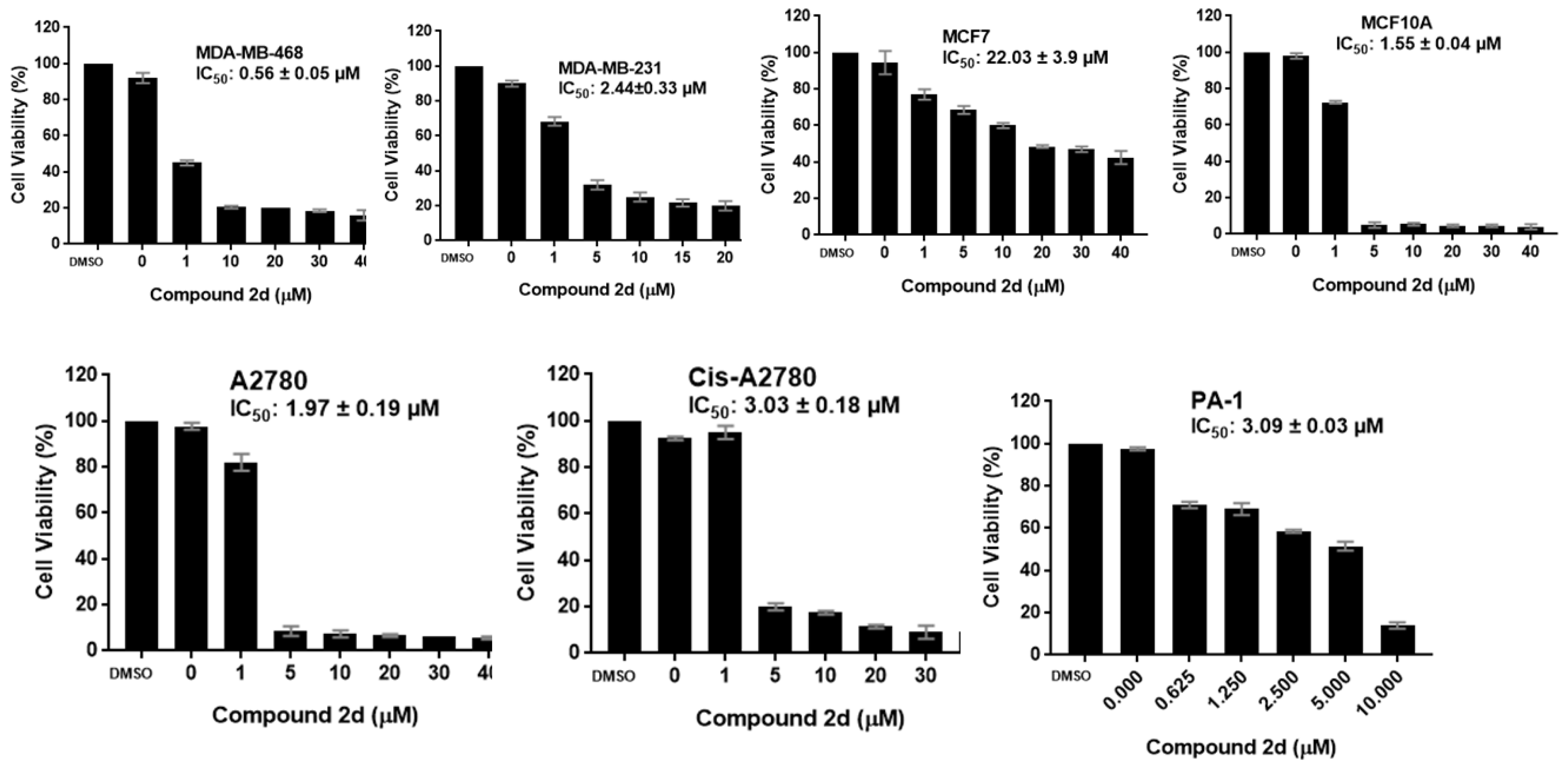
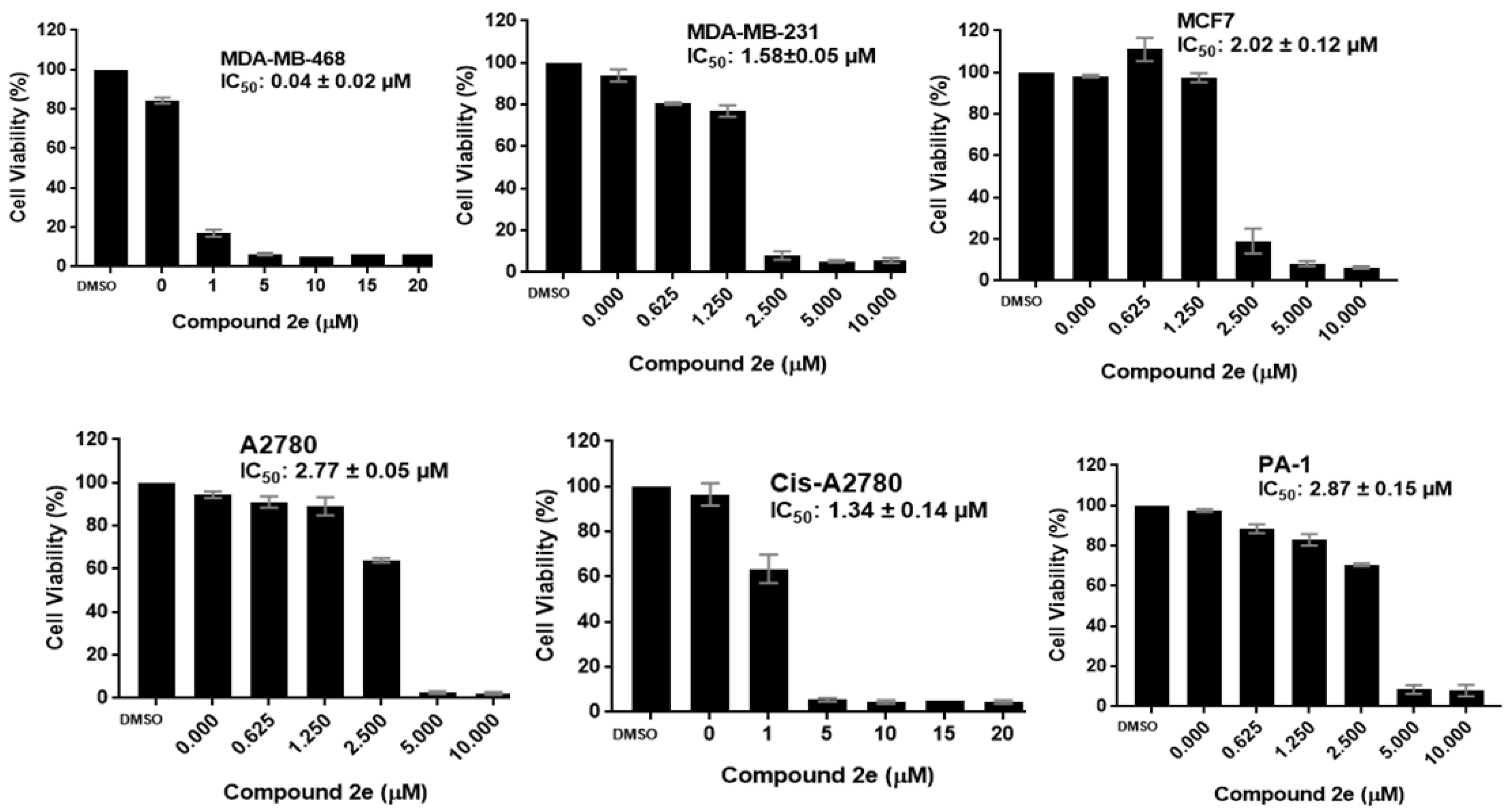
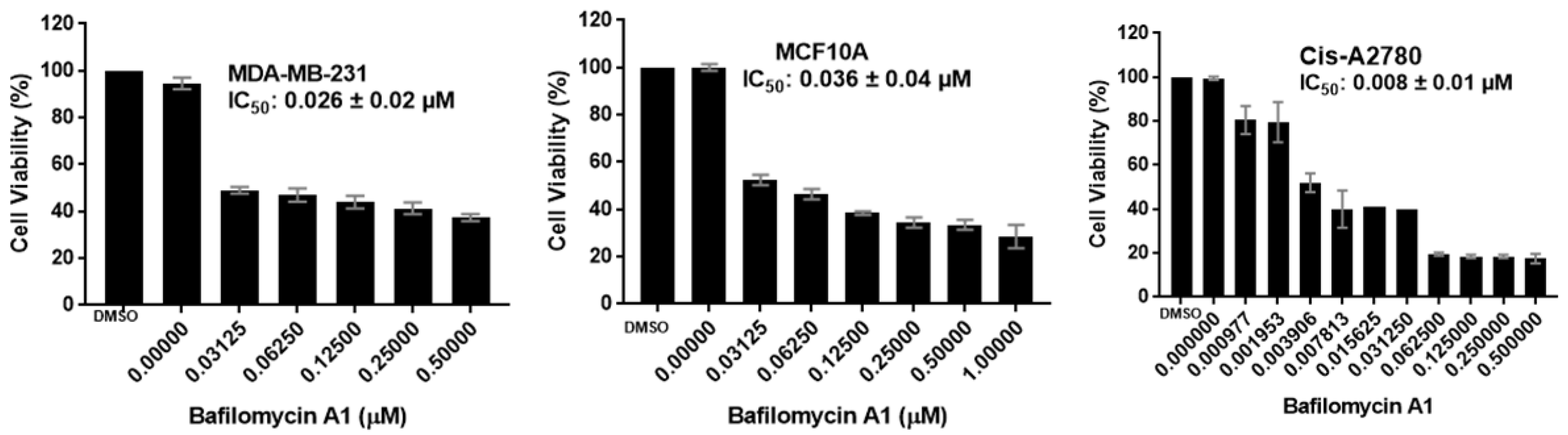
2.2.1. Antiproliferative Activity
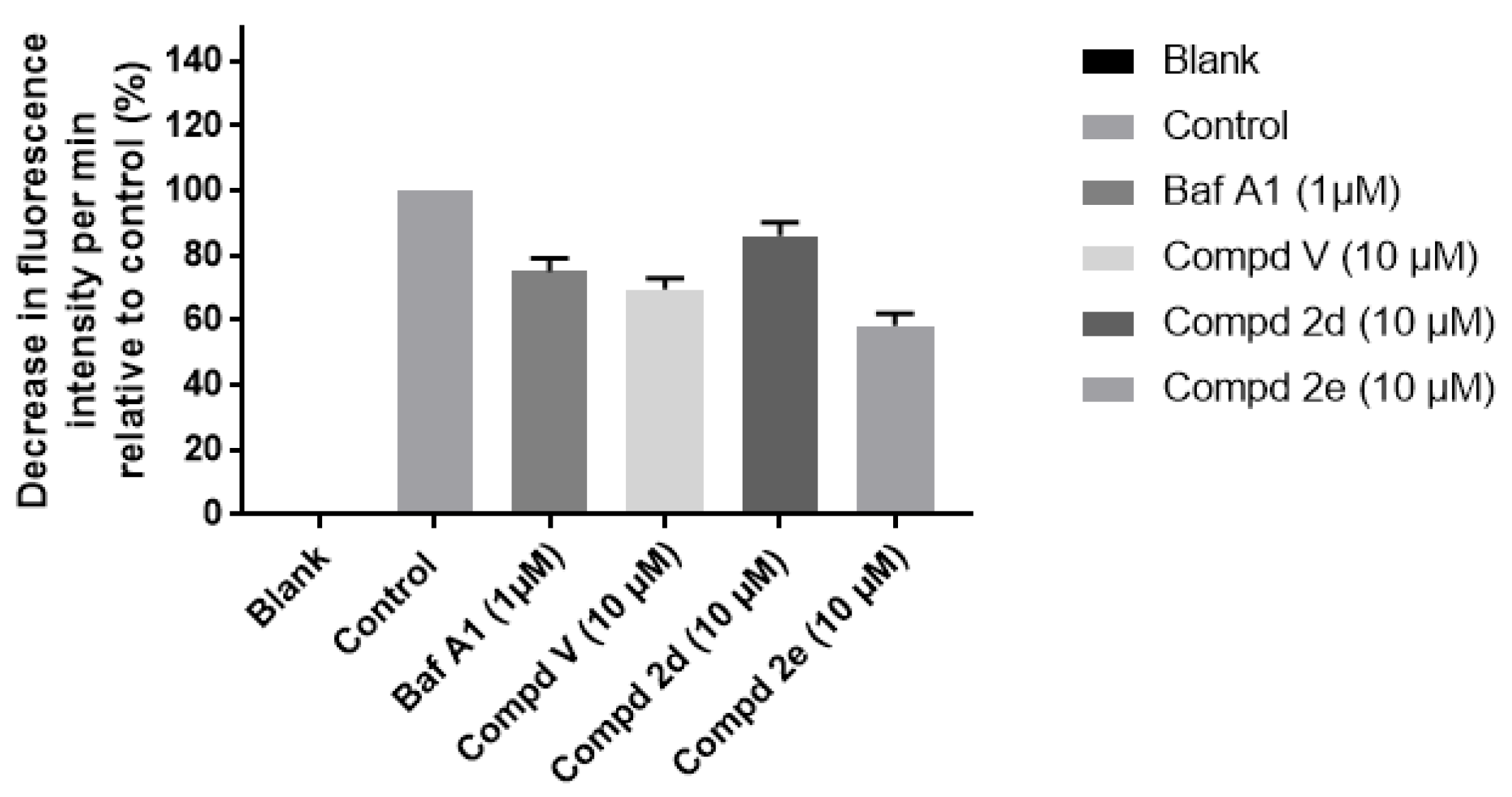
2.2.2. Proton (H+) Pump Activity
3. Experimental Section
3.1. Chemical General Information
3.2. Synthesis
3.3. Antiproliferative Activity
3.3.1. Cell Lines and Cell Culture
3.3.2. Cell Viability Assay
3.3.3. Measurement of the Proton (H+) Pump Activity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cotter, K.; Stransky, L.; McGuire, C.; Forgac, M. Recent Insights into the Structure, Regulation, and Function of the V-ATPases. Trends Biochem. Sci. 2015, 40, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Nishi, T.; Forgac, M. The vacuolar (H+)-ATPases-nature’s most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 2002, 3, 94–103. [Google Scholar] [CrossRef] [PubMed]
- McGuire, C.; Stransky, L.; Cotter, K.; Forgac, M. Regulation of V-ATPase activity. Front Biosci (Landmark Ed). 2017, 22, 609–622. [Google Scholar] [PubMed]
- Yokoyama, K.; Imamura, H. Rotation, structure, and classification of prokaryotic V-ATPase. J. Bioenerg. Biomembr. 2005, 37, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cipriano, D.J.; Forgac, M. Arrangement of subunits in the proteolipid ring of the V-ATPase. J. Biol. Chem. 2007, 282, 34058–34065. [Google Scholar] [CrossRef] [PubMed]
- Fais, S.; De Milito, A.; You, H.; Qin, W. Targeting vacuolar H+-ATPases as a new strategy against cancer. Cancer Res. 2007, 67, 10627–10630. [Google Scholar] [CrossRef] [PubMed]
- Sennoune, S.R.; Luo, D.; Martínez-Zaguilán, R. Plasmalemmal vacuolar-type H+-ATPase in cancer biology. Cell Biochem. Biophys. 2004, 40, 185–206. [Google Scholar] [CrossRef]
- Rofstad, E.K.; Mathiesen, B.; Kindem, K.; Galappathi, K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006, 66, 6699–6707. [Google Scholar] [CrossRef] [PubMed]
- Sennoune, S.R.; Bakunts, K.; Martínez, G.M.; Chua-Tuan, J.L.; Kebir, Y.; Attaya, M.N.; Martínez-Zaguilán, R. Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: Distribution and functional activity. Am. J. Physiol. Cell Physiol. 2004, 286, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Torigoe, T.; Izumi, H.; Ise, T.; Murakami, T.; Uramoto, H.; Ishiguchi, H.; Yoshida, Y.; Tanabe, M.; Nomoto, M.; Kohno, K. Vacuolar H+-ATPase: Functional mechanisms and potential as a target for cancer chemotherapy. Anticancer Drugs. 2002, 13, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Bowman, E.J.; Graham, L.A.; Stevens, T.H.; Bowman, B.J. The bafilomycin/concanamycin binding site in subunit c of the V-ATPases from Neurospora crassa and Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 33131–33138. [Google Scholar] [CrossRef] [PubMed]
- Scheidt, K.A.; Bannister, T.D.; Tasaka, A.; Wendt, M.D.; Savall, B.M.; Fegley, G.J.; Roush, W.R. Total synthesis of (−)-bafilomycin A(1). J. Am. Chem. Soc. 2002, 124, 6981–6990. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Park, J.W.; Kim, M.S.; Park, S.K.; Johnson, R.S.; Chun, Y.S. Bafilomycin induces the p21-mediated growth inhibition of cancer cells under hypoxic conditions by expressing hypoxia-inducible factor-1α. Mol. Pharmacol. 2006, 70, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Katayama, K.; Togawa, T.; Kimura, T.; Yamaguchi, A. Effects of bafilomycin A1, a vacuolar type H+-ATPase inhibitor, on the thermosensitivity of a human pancreatic cancer cell line. Int. J. Hyperth. 2006, 22, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Huss, M.; Ingenhorst, G.; König, S.; Gassel, M.; Dröse, S.; Zeeck, A.; Altendorf, K.; Wieczorek, H. Concanamycin A, the specific inhibitor of V-ATPases, binds to the Vo subunit c. J. Biol. Chem. 2002, 277, 40544–40548. [Google Scholar] [CrossRef] [PubMed]
- Hert, J.; Willett, P.; Wilton, D.J.; Acklin, P.; Azzaoui, K.; Jacoby, E.; Schuffenhauer, A. Comparison of topological descriptors for similarity-based virtual screening using multiple bioactive reference structures. Org. Biomol. Chem. 2004, 2, 3256–3266. [Google Scholar] [CrossRef] [PubMed]
- López-Ramos, M.; Perruccio, F. HPPD: Ligand-and target-based virtual screening on a herbicide target. J. Chem. Inf. Model. 2010, 50, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Sciabola, S.; Carosati, E.; Cucurull-Sanchez, L.; Baroni, M.; Mannhold, R. Novel TOPP descriptors in 3D-QSAR analysis of apoptosis inducing 4-aryl-4H-chromenes: Comparison versus other 2D- and 3D-descriptors. Bioorg. Med. Chem. 2007, 15, 6450–6462. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.; Loura, L.; Koehorst, R.B.; Dixon, N.; Kee, T.P.; Hemminga, M.A.; Prieto, M. Interaction of the indole class of vacuolar H+-ATPase inhibitors with lipid bilayers. Biochemistry 2006, 45, 5271–5279. [Google Scholar] [CrossRef] [PubMed]
- Petrangolini, G.; Supino, R.; Pratesi, G.; Dal Bo, L.; Tortoreto, M.; Croce, A.C.; Misiano, P.; Belfiore, P.; Farina, C.; Zunino, F. Effect of a novel vacuolar-H+-ATPase inhibitor on cell and tumor response to camptothecins. J. Pharmacol. Exp. Ther. 2006, 318, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Ostrov, D.A.; Magis, A.T.; Wronski, T.J.; Chan, E.K.; Toro, E.J.; Donatelli, R.E.; Sajek, K.; Haroun, I.N.; Nagib, M.I.; Piedrahita, A.; et al. Identification of enoxacin as an inhibitor of osteoclast formation and bone resorption by structure-based virtual screening. J. Med. Chem. 2009, 52, 5144–5151. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.G.; Henriksen, K.; Neutzsky-Wulff, A.V.; Dziegiel, M.H.; Karsdal, M.A. Diphyllin, a novel and naturally potent V-ATPase inhibitor, abrogates acidification of the osteoclastic resorption lacunae and bone resorption. J. Bone Miner. Res. 2007, 22, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Pamarthy, S.; Jaiswal, M.K.; Kulshreshtha, A.; Katara, G.K.; Gilman-Sachs, A.; Beaman, K.D. The Vacuolar ATPase a2-subunit regulates Notch signaling in triple-negative breast cancer cells. Oncotarget 2015, 6, 34206–34220. [Google Scholar] [PubMed]
- Ibrahim, S.A.; Katara, G.K.; Kulshrestha, A.; Jaiswal, M.K.; Amin, M.A.; Beaman, K.D. Breast cancer associated a2 isoform vacuolar ATPase immunomodulates neutrophils: Potential role in tumor progression. Oncotarget 2015, 6, 33033–33045. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.; Katara, G.K.; Ginter, J.; Pamarthy, S.; Ibrahim, S.A.; Jaiswal, M.K.; Sandulescu, C.; Periakaruppan, R.; Dolan, J.; Gilman-Sachs, A.; et al. Selective inhibition of tumor cell associated Vacuolar-ATPase ‘a2’ isoform overcomes cisplatin resistance in ovarian cancer cells. Mol. Oncol. 2016, 10, 789–805. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.; Katara, G.K.; Ibrahim, S.; Pamarthy, S.; Jaiswal, M.K.; Gilman-Sachs, A.; Beaman, K.D. Vacuolar ATPase ‘a2’ isoform exhibits distinct cell surface accumulation and modulates matrix metalloproteinase activity in ovarian cancer. Oncotarget 2015, 6, 3797–3810. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wang, Y.; Wang, Z.; Wang, Y.; Zheng, H. Inhibiting autophagy increases epirubicin’s cytotoxicity in breast cancer cells. Cancer Sci. 2016, 107, 1610–1621. [Google Scholar] [CrossRef] [PubMed]
- Kandala, P.K.; Srivastava, S.K. Regulation of macroautophagy in ovarian cancer cells in vitro and in vivo by controlling glucose regulatory protein 78 and AMPK. Oncotarget 2012, 3, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Afsar, T.; Trembley, J.H.; Salomon, C.E.; Razak, S.; Khan, M.R.; Ahmed, K. Growth inhibition and apoptosis in cancer cells induced by polyphenolic compounds of Acacia hydaspica: Involvement of multiple signal transduction pathways. Sci. Rep. 2016, 6, 23077. [Google Scholar] [CrossRef] [PubMed]
- Lohberger, B.; Steinecker-Frohnwieser, B.; Stuendl, N.; Kaltenegger, H.; Leithner, A.; Rinner, B. The proteasome inhibitor bortezomib affects chondrosarcoma cells via the mitochondria-caspase dependent pathway and enhances death receptor expression and autophagy. PLoS ONE 2016, 11, e0168193. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Citro, G.; Fais, S. Proton pump inhibitors as anti vacuolar-ATPases drugs: A novel anticancer strategy. J. Exp. Clin. Cancer Res. 2010, 29, 44. [Google Scholar] [CrossRef] [PubMed]
- De Milito, A.; Canese, R.; Marino, M.L.; Borghi, M.; Iero, M.; Villa, A.; Venturi, G.; Lozupone, F.; Iessi, E.; Logozzi, M.; et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int. J. Cancer 2010, 127, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; Spugnini, E.P.; Assaraf, Y.G.; Azzarito, T.; Rauch, C.; Fais, S. Microenvironment acidity as a major determinant of tumor chemoresistance: Proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist. Updates 2015, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.; Fais, S.; Spugnini, E.P.; Harguindey, S.; Abu Izneid, T.; Scacco, L.; Williams, P.; Allegrucci, C.; Rauch, C.; Omran, Z. Proton pump inhibitors for the treatment of cancer in companion animals. J. Exp. Clin. Cancer Res. 2015, 34, 93. [Google Scholar] [CrossRef] [PubMed]
- Babichev, Y.; Tamir, A.; Park, M.; Muallem, S.; Isakov, N. Cloning, expression and functional characterization of the putative regeneration and tolerance factor (RTF/TJ6) as a functional vacuolar ATPase proton pump regulatory subunit with a conserved sequence of immunoreceptor tyrosine-based activation motif. Int. Immunol. 2005, 17, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Lundholm, L.; Hååg, P.; Zong, D.; Juntti, T.; Mörk, B.; Lewensohn, R.; Viktorsson, K. Resistance to DNA-damaging treatment in non-small cell lung cancer tumor-initiating cells involves reduced DNA-PK/ATM activation and diminished cell cycle arrest. Cell Death Dis. 2013, 4, e478. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | IC50 ± SD (μM) | ||||||
|---|---|---|---|---|---|---|---|
| MDA-MB-468 | MDA-MB-231 | MCF7 | MCF10A | A2780 | Cis-A2780 | PA-1 | |
| V | 0.72 ± 0.08 | 1.02 ± 0.08 | 9.33 ± 0.69 | 1.14 ± 0.13 | 3.87 ± 0.09 | 3.95 ± 0.33 | 1.70 ± 0.21 |
| 2a | 13.39 ± 0.33 | 17.56 ± 0.19 | 49.33 ± 2.76 | 13.35 ± 0.37 | 26.49 ± 1.44 | 25.23 ± 0.32 | ND |
| 2b | 5.83 ± 0.23 | 4.77 ± 0.43 | 13.51 ± 2.14 | 2.95± 0.15 | 11.80 ± 0.01 | 12.34 ± 0.02 | 5.83 ± 0.07 |
| 2c | 1.17 ± 0.36 | 0.75 ± 0.30 | ND | 0.81 ± 0.10 | 12.90 ± 0.42 | 10.72 ± 0.22 | 7.78 ± 0.81 |
| 2d | 0.56 ± 0.05 | 2.44 ± 0.33 | 22.03 ± 3.9 | 1.55 ± 0.04 | 1.97 ± 0.19 | 3.03 ± 0.18 | 3.09 ± 0.03 |
| 2e | 0.04 ± 0.02 | 1.58 ± 0.05 | 2.02 ± 0.12 | 1.62 ± 0.14 | 2.77 ± 0.05 | 1.34 ± 0.14 | 2.87 ± 0.15 |
| Baf A1 | ND | 0.026 ± 0.02 | ND | 0.036 ± 0.04 | ND | 0.008 ± 0.01 | ND |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, R.; Kulshrestha, A.; Tikoo, A.; Fleetwood, S.; Katara, G.; Kolli, B.; Seibel, W.; Gilman-Sachs, A.; Patil, S.A.; Beaman, K.D. Identification of Novel Bisbenzimidazole Derivatives as Anticancer Vacuolar (H+)-ATPase Inhibitors. Molecules 2017, 22, 1559. https://doi.org/10.3390/molecules22091559
Patil R, Kulshrestha A, Tikoo A, Fleetwood S, Katara G, Kolli B, Seibel W, Gilman-Sachs A, Patil SA, Beaman KD. Identification of Novel Bisbenzimidazole Derivatives as Anticancer Vacuolar (H+)-ATPase Inhibitors. Molecules. 2017; 22(9):1559. https://doi.org/10.3390/molecules22091559
Chicago/Turabian StylePatil, Renukadevi, Arpita Kulshrestha, Anjali Tikoo, Sara Fleetwood, Gajendra Katara, Bala Kolli, William Seibel, Alice Gilman-Sachs, Shivaputra A. Patil, and Kenneth D. Beaman. 2017. "Identification of Novel Bisbenzimidazole Derivatives as Anticancer Vacuolar (H+)-ATPase Inhibitors" Molecules 22, no. 9: 1559. https://doi.org/10.3390/molecules22091559