The Application of Quantitative 1H-NMR for the Determination of Orlistat in Tablets

Abstract
:1. Introduction
2. Results and Discussion
2.1. Selection of Deuterated Solvent
2.2. Determination of Relaxation Time
2.3. Validation
2.3.1. Specificity and Selectivity
2.3.2. Linearity and Range
2.3.3. Accuracy
2.3.4. Precision
2.3.5. Stability
2.3.6. Limit of Detection (LOD) and Limit of Quantification (LOQ)
2.3.7. Robustness
2.4. Assay of Orlistat in Tablets
3. Materials and Methods
3.1. Materials
3.2. Instruments
3.3. Test Solutions Preparation
3.3.1. Standard Solution Preparation for qNMR
3.3.2. Tablet Powder Solution Preparation for qNMR
3.3.3. Sample Solution Preparation for HPLC
3.4. Data Acquisition and Processing
3.5. Content Calculation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jungnickel, J.L.; Forbes, J.W. Quantitative measurement of hydrogen types by integrated nuclear magnetic resonance intensities. Anal. Chem. 1963, 35, 938–942. [Google Scholar] [CrossRef]
- Hollis, D.P. Quantitative analysis of aspirin, phenacetin and caffeine mixtures by nuclear magnetic resonance spectrometry. Anal. Chem. 1963, 35, 1682–1684. [Google Scholar] [CrossRef]
- Simmler, C.; Napolitano, J.G.; McAlpine, J.B.; Chen, S.N.; Pauli, G.F. Universal quantitative NMR analysis of complex natural samples. Curr. Opin. Biotechnol. 2014, 25, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nakaie, S.; Kinoshita, M.; Ihara, T.; Kinugasa, S.; Nomura, A.; Meada, T. Practical guide for accurate quantitative solution state NMR analysis. Metrologia 2004, 41, 213–218. [Google Scholar] [CrossRef]
- Farrant, R.D.; Hollerton, J.C.; Lynn, S.M.; Provera, S.; Sidebottom, P.J.; Upton, R.J. NMR quantification using an artificial signal. Magn. Reson. Chem. 2010, 48, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Pauli, G.F.; Godecke, T.; Jaki, B.U.; Lankin, D.C. Quantitative 1H-NMR. Development and potential of an analytical method: An update. J. Nat. Prod. 2012, 75, 834–851. [Google Scholar] [CrossRef] [PubMed]
- Pinciroli, V.; Biancardi, R.; Visentin, G.; Rizzo, V. The well-characterized synthetic molecule: A role for quantitative 1H-NMR. Org. Process. Res. Dev. 2004, 8, 381–384. [Google Scholar] [CrossRef]
- Sahu, A.; Narayanam, M.; Kurmi, M.; Ladumor, M.K.; Singh, S. Quantitation of memantine hydrochloride bulk drug and its tablet formulation using proton nuclear magnetic resonance spectrometry. Magn. Reson. Chem. 2016, 54, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.R.; Du, L.P.; Su, F.; Parekh, H.S.; Su, W.K. The application of quantitative NMR for the facile, rapid and reliable determination of clindamycin phosphate in a conventional tablet formulation. Magn. Reson. Chem. 2014, 52, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.Y.; Qiu, H.; Guo, W.; Wang, D.M.; Zhou, X.N.; Xue, D.; Zhang, J.L.; Wu, S.; Wang, Y.H. Quantitative 1H-NMR method for the determination of tadalafil in bulk drugs and its tablets. Molecules 2015, 20, 12114–12124. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.Y.; Yang, K.M.; Yang, L.; Miao, Z.X.; Wang, Y.H.; Zhu, H.B. A 1H-NMR-based metabonomic investigation of time-related metabolic trajectories of the plasma, urine and liver extracts of Hyperlipidemic Hamsters. PLoS ONE 2013, 8, e66786. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.A.; Mossa, H.A. Method validation and determination of levofloxacin, metronidazole and sulfamethoxazole in an aqueous pharmaceutical, urine and blood plasma samples using quantitative nuclear magnetic resonance spectrometry. Talanta 2012, 88, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Garrido, R.; Puyada, A.; Fernandez, A.; Gonzalez, M.; Ramirez, U.; Cardoso, F.; Valdes, Y.; Gonzalez, D.; Fernandez, V.; Verez, V.; et al. Quantitative proton nuclear magnetic resonance evaluation and total assignment of the capsular polysaccharide Neisseria meningitides serogroup X. J. Pharm. Biomed. Anal. 2012, 70, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Staneva, J.; Denkova, P.; Todorova, M.; Evstatieva, L. Quantitative analysis of sesquiterpene lactones in extract of Arnica montana L. by 1H-NMR spectroscopy. J. Pharm. Biomed. Anal. 2011, 54, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Goger, N.G.; Parlatan, H.K.; Basan, H.; Berkkan, A.; Ozden, T. Quantitative determination of azathioprine in tablets by 1H-NMR spectroscopy. J. Pharm. Biomed. Anal. 1999, 21, 685–689. [Google Scholar] [CrossRef]
- Zoppi, A.; Linares, M.; Longhi, M. Quantitative analysis of enalapril by 1H-NMR spectroscopy in tablets. J. Pharm. Biomed. Anal. 2005, 37, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, Z.Q.; Wang, M.; Bai, X.D.; Liu, M.T.; Chen, Y.; Liu, J. Content determination of orlistat tablet by RP-HPLC method. Prog. Mod. Biomed. 2010, 10, 4360–4362. [Google Scholar]
- Xiao, S.; Zhu, X.L.; Chen, B.; Yao, S.Z. Determination of orlistat in Capsules by HPLC/MS. Chin. J. Pharm. Anal. 2005, 25, 1055–1057. [Google Scholar]
- Xu, Y.H.; Peng, W.; Tang, B.X.; Wang, D.G.; Liu, D.F. Purification of Orlistat by preparative HPLC and its determination by UPLC. Chin. J. Pharm. 2012, 43, 690–692. [Google Scholar]
- Ma, W.; Fu, L.; Wang, H.B.; Chen, W.; Wang, X.J.; Yu, J.; Tang, Y.Z. Determination of illegal drug orlistat added in the weight-loss functional foods by Microwave Assisted Extraction/High Performance Liquid Chromatography tandem Mass Spectrometry. J. Instrum. Anal. 2009, 28, 1045–1048. [Google Scholar]
- Holzgrabe, U. Quantitative NMR spectroscopy in pharmaceutical applications. Prog. Nucl. Magn. Reson. Spectrosc. 2010, 57, 229–240. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds Orlistat and Phloroglucinol Anhydrous are available from the authors. |
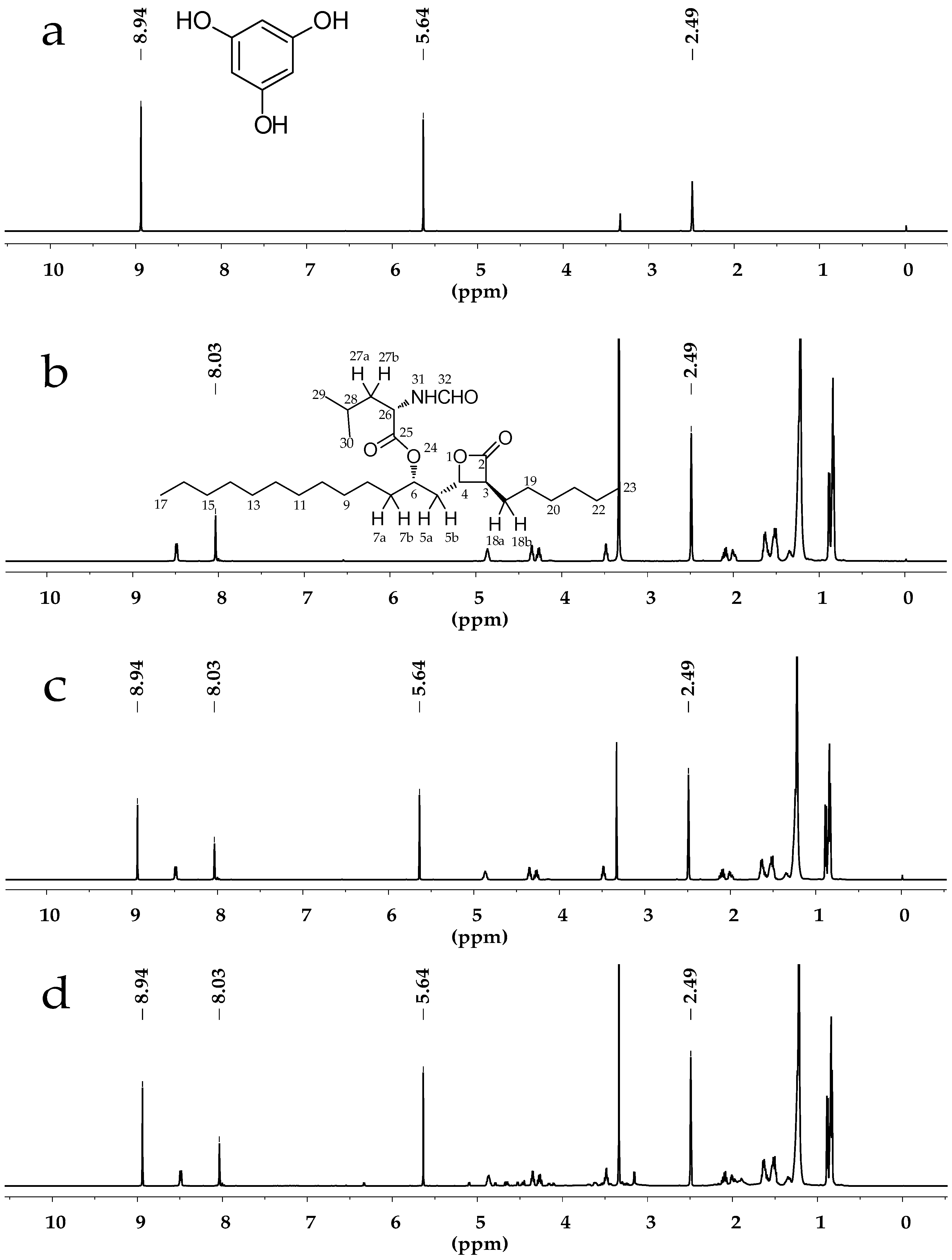

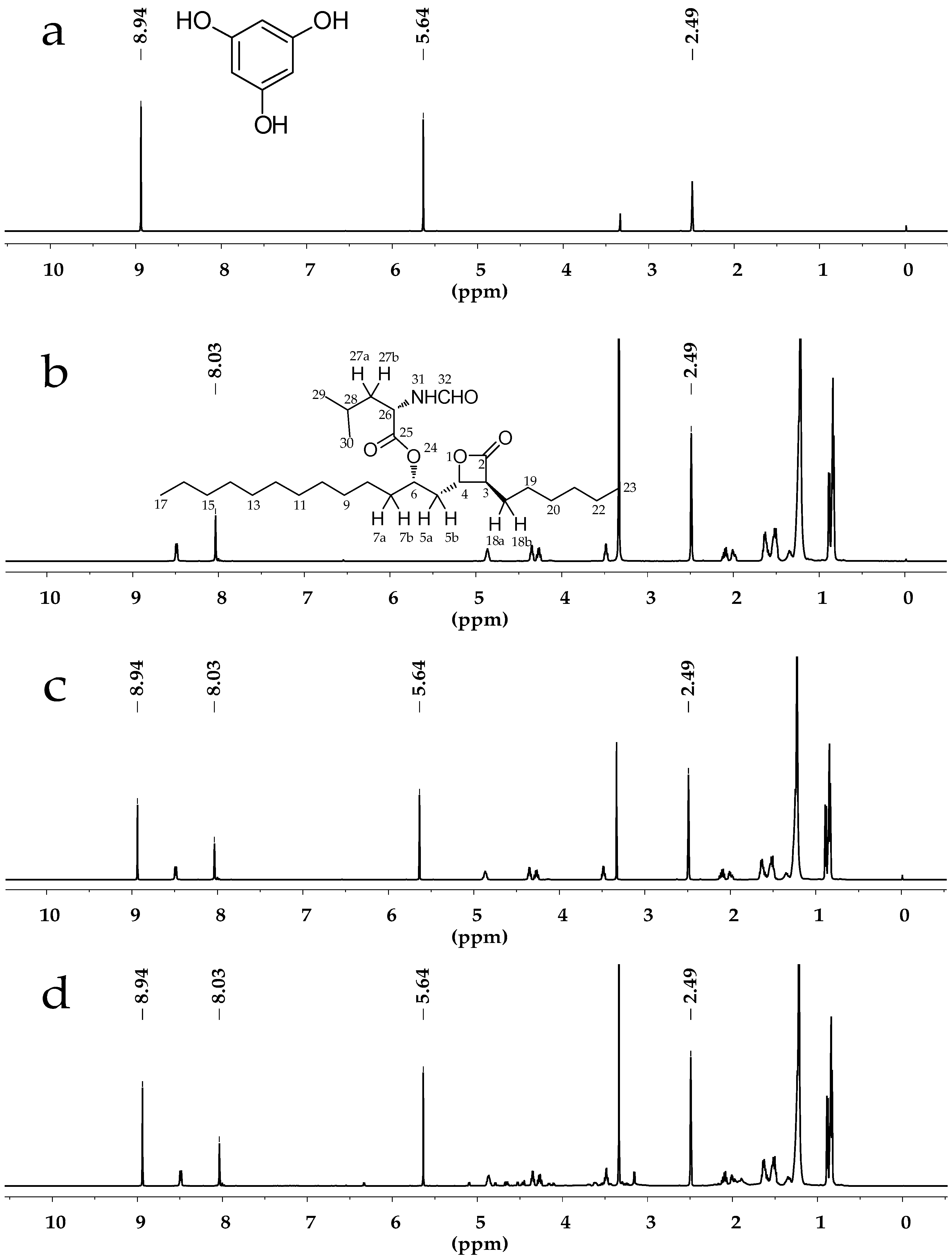{kind=link}
| No. | Orlistat | Recovery (%) | ||||
|---|---|---|---|---|---|---|
| m0 (mg) | ms (mg) | mx (mg) | ||||
| δ*5.64 | δ*8.94 | δ*5.64 | δ*8.94 | |||
| 1 | 2.302 | 1.795 | 4.096 | 4.091 | 99.94 | 99.68 |
| 2 | 2.238 | 1.943 | 4.136 | 4.136 | 97.71 | 97.71 |
| 3 | 2.295 | 1.875 | 4.170 | 4.154 | 100.03 | 99.19 |
| 4 | 2.259 | 2.197 | 4.442 | 4.442 | 99.35 | 99.39 |
| 5 | 2.311 | 2.271 | 4.556 | 4.527 | 98.85 | 97.57 |
| 6 | 2.248 | 2.295 | 4.507 | 4.497 | 98.43 | 97.97 |
| 7 | 2.099 | 2.768 | 4.873 | 4.896 | 100.20 | 101.02 |
| 8 | 2.229 | 2.853 | 5.136 | 5.114 | 101.89 | 101.13 |
| 9 | 2.213 | 2.821 | 5.093 | 5.073 | 102.07 | 101.36 |
| Average value | / | / | / | / | 99.83 | 99.45 |
| RSD% | / | / | / | / | 1.46 | 1.50 |
| No. | mstd (mg) | mx (mg) | Px (%) | ||
|---|---|---|---|---|---|
| δ*5.64 | δ*8.94 | ||||
| Repeatability | 1 | 1.119 | 6.042 | 98.91 | 99.16 |
| 2 | 1.068 | 6.024 | 99.09 | 98.14 | |
| 3 | 1.072 | 6.050 | 98.83 | 97.87 | |
| 4 | 1.122 | 9.343 | 98.64 | 99.25 | |
| 5 | 1.092 | 9.060 | 98.96 | 98.53 | |
| 6 | 1.067 | 8.833 | 98.11 | 97.10 | |
| 7 | 1.078 | 11.164 | 98.49 | 97.83 | |
| 8 | 1.055 | 12.930 | 99.08 | 98.16 | |
| 9 | 1.118 | 11.200 | 98.65 | 97.82 | |
| Average value | / | / | 98.75 | 98.21 | |
| RSD % | / | / | 0.32 | 0.70 | |
| Inter-day precision | 1 | / | / | 100.40 | 99.65 |
| 2 | 100.46 | 99.68 | |||
| 3 | 100.43 | 99.62 | |||
| 4 | 100.35 | 99.61 | |||
| 5 | 100.20 | 99.32 | |||
| 6 | 100.10 | 99.49 | |||
| Average value | / | / | 100.32 | 99.56 | |
| RSD % | / | / | 0.14 | 0.14 | |
| Time (h) | δ*5.64 | δ*8.94 | ||
|---|---|---|---|---|
| Assay (%) | Diff (%) | Assay (%) | Diff (%) | |
| 0 | 100.30 | / | 99.53 | / |
| 6 | 100.14 | 0.16 | 99.37 | 0.16 |
| 12 | 100.07 | 0.23 | 99.26 | 0.28 |
| 24 | 100.01 | 0.29 | 99.23 | 0.31 |
| 48 | 99.90 | 0.40 | 99.24 | 0.30 |
| 72 | 99.74 | 0.60 | 99.12 | 0.41 |
| Average value | 100.03 | / | 99.30 | / |
| RSD % | 0.19 | / | 0.14 | / |
| Parameters (Target Value) | Change | δ*5.64 | δ*8.94 | ||
|---|---|---|---|---|---|
| Assay (%) | Diff (%) | Assay (%) | Diff (%) | ||
| Number of scans (32) | 16 | 100.11 | 0.35 | 99.39 | 0.28 |
| 48 | 100.05 | 0.41 | 99.33 | 0.34 | |
| Relaxation delay (32 s) | 24 | 100.08 | 0.38 | 99.36 | 0.31 |
| 40 | 99.98 | 0.47 | 99.37 | 0.30 | |
| Acquisition time (3.277 s) | 2.277 | 100.13 | 0.33 | 99.55 | 0.12 |
| 4.277 | 100.21 | 0.25 | 99.51 | 0.16 | |
| Data points (64 K) | 32 | 100.03 | 0.43 | 99.43 | 0.25 |
| 128 | 99.93 | 0.53 | 99.44 | 0.23 | |
| Spectral width (20 ppm) | 15 | 99.89 | 0.57 | 99.35 | 0.32 |
| 25 | 99.91 | 0.55 | 99.36 | 0.31 | |
| P1 (8.00 μsec) | 7.00 | 99.95 | 0.51 | 99.30 | 0.35 |
| 9.00 | 99.96 | 0.50 | 99.40 | 0.27 | |
| Batch No. | qNMR (n = 3) | HPLC | |||
|---|---|---|---|---|---|
| δ | % Label Claim | RSD% | % Label Claim | RSD% | |
| 15082101 | 5.64 | 100.65 | 0.38 | 99.71 | 0.61 |
| 8.94 | 99.99 | 0.34 | |||
| 21606151 | 5.64 | 98.58 | 1.11 | 96.75 | 0.85 |
| 8.94 | 99.03 | 0.86 | |||
| 21605111 | 5.64 | 98.75 | 0.33 | 97.87 | 1.20 |
| 8.94 | 97.42 | 0.51 | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, S.; Jin, M.; Zhou, X.; Ni, J.; Jin, X.; Liu, H.; Wang, Y. The Application of Quantitative 1H-NMR for the Determination of Orlistat in Tablets. Molecules 2017, 22, 1517. https://doi.org/10.3390/molecules22091517
Sun S, Jin M, Zhou X, Ni J, Jin X, Liu H, Wang Y. The Application of Quantitative 1H-NMR for the Determination of Orlistat in Tablets. Molecules. 2017; 22(9):1517. https://doi.org/10.3390/molecules22091517
Chicago/Turabian StyleSun, Shanshan, Mengxia Jin, Xia Zhou, Jinghua Ni, Xiangju Jin, Hongyue Liu, and Yinghong Wang. 2017. "The Application of Quantitative 1H-NMR for the Determination of Orlistat in Tablets" Molecules 22, no. 9: 1517. https://doi.org/10.3390/molecules22091517
APA StyleSun, S., Jin, M., Zhou, X., Ni, J., Jin, X., Liu, H., & Wang, Y. (2017). The Application of Quantitative 1H-NMR for the Determination of Orlistat in Tablets. Molecules, 22(9), 1517. https://doi.org/10.3390/molecules22091517