Abstract
Deep eutectic solvents, as green and environmentally friendly media, were utilized in the synthesis of novel coumarinyl Schiff bases. Novel derivatives were synthesized from 2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide and corresponding aldehyde in choline chloride:malonic acid (1:1) based deep eutectic solvent. In these reactions, deep eutectic solvent acted as a solvent and catalyst as well. Novel Schiff bases were synthesized in high yields (65–75%) with no need for further purification, and their structures were confirmed by mass spectra, 1H and 13C NMR. Furthermore, their antioxidant activity was determined and compared to antioxidant activity of previously synthesized derivatives, thus investigating their structure–activity relationship utilizing quantitative structure-activity relationship QSAR studies. Calculation of molecular descriptors has been performed by DRAGON software. The best QSAR model (Rtr = 0.636; Rext = 0.709) obtained with three descriptors (MATS3m, Mor22u, Hy) implies that the pairs of atoms higher mass at the path length 3, three-dimensional arrangement of atoms at scattering parameter s = 21 Å−1, and higher number of hydrophilic groups (-OH, -NH) enhanced antioxidant activity. Electrostatic potential surface of the most active compounds showed possible regions for donation of electrons to 1,1-diphenyl-2-picryhydrazyl (DPPH) radicals.
1. Introduction
Coumarins are a class of compounds widely distributed in the plant kingdom [1], but lots of synthetic studies have been done on them in the last few decades. Most of the synthetic modifications aim at the synthesis of biologically active derivatives with more potent specific biological activity and their potential application in pharmacy, cosmetic industry or medicine. Different antioxidants have been synthesized and investigated by many researchers in this regard. We have been investigating the antioxidant activity of different synthetic coumarin derivatives for years and showed that different synthetic modifications on the basic coumarin core can increase its antioxidant activity [2,3,4,5]. For this purpose, a series of Schiff bases were synthesized [2] conventionally; however, we have noticed their formation when deep eutectic solvents (DESs) are applied as well. DESs have proven to be a convenient media for many synthetic routes and are often characterized as environmentally friendly [6,7,8]. Their application in organic synthesis and extraction, as well as an extensive analysis of their properties, were described in some good reviews published in the last few years [6,7,8,9,10,11].
As the emphasis is put on green chemistry approaches on a daily basis, “green solvents” are the focus of many researchers, since the most waste that is produced in the synthesis of different compounds are actually waste solvents [12]. DESs are an emerging class of new, non-toxic, biodegradable solvents, which have already found their application in organic synthesis, and they are becoming more prominent every day, since they have many advantages compared to conventional solvents. Capua et al. [12] synthesized 2-aminoimidazoles in one-pot protocol in high yields and with very simple post synthetic workup, in shorter times than with conventional organic solvents like tetrahydrofuran (THF), and this procedure resulted in no by-products, while DES was recovered and recycled. Massolo et al. [13] employed DESs in enantioselective transformations, also obtaining products at high yields with the possibility of DESs being recycled and reused, and the same advantages of DESs were noticed in the synthesis of imines and hydrobenzamides [14] and enantioselective aldol reaction [15]. DESs have proven to be effective in umpolung sulfenylation reactions [16], applicable in material science in epoxy resin polymerization [17], and as catalysts in CO2 fixation epichlorohydrin [18].
Due to DESs’ numerous advantages compared to organic solvents, the aim of this study was to synthesize novel coumarinyl Schiff bases utilizing choline chloride based DES as reaction media and catalyst as well. The fact that there was no need for a specific catalyst, like acetic acid, which is often used in these kinds of reactions, was noticed. Not only did we get a novel compounds, but we also applied an environmentally friendly approach, utilizing a solvent made of biodegradable components, with low toxicity and vapor pressure, which can be easily recycled and reused [19]. The purpose of the study was also to determine their antioxidant activity and compare it to antioxidant activities of previously synthesized similar Schiff bases [2,5].
Many theoretical studies have showed that biological activity of natural and synthetic antioxidants is related to their structure [20]. Different quantitative structure–activity relationship (QSAR) techniques have been employed to correlate the antioxidant capacity with various structural features of coumarin derivatives that affect the antioxidant activity and predict the same for the untested and future molecules, such as multiple linear regression [21,22], genetic function approximation genetic partial least squares [23,24], and artificial neural networks [25]. Wide ranges of molecular descriptors, mathematical representations of a molecule, have been used for QSAR modeling of antioxidant activities of a series of coumarin derivatives. Thus, physicochemical properties such as hydrophilic factor [24,25] and lipophilicity [21]; energy based descriptors, such as bond dissociation enthalpy [26]; two-dimensional (2D) descriptors Moran autocorrelation and BCUT (Burden - CAS - University of Texas eigen values) descriptors and three-dimensional (3D) descriptors GATEWAY descriptors [27] showed the most significant influences on the QSAR modeling of antioxidant activity of coumarines. Accurate QSAR models have been generated using quantum-chemistry descriptors, such as HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbital), the difference of the energies HOMO-LUMO, and hardness [24,25,28,29].
QSAR models obtained for series of coumarin derivatives that were modeled for their antioxidant activity based on their ability to inhibit DPPH free radical, revealed that compounds with a higher degree of branching, oxygen atom-bearing fragment and tertiary carbon as substituent lie in the higher activity range [23]. QSAR study of 3-carboxycoumarin derivatives has implied an importance of the presence of hydroxyl groups on the ring structure and a higher number of hydrophilic groups for enhanced DPPH radical scavenging activity [25]. In this work, QSAR study was performed on the set of synthesized coumarin derivatives in order to signify the importance of structural and chemical attributes for their antioxidant activity.
2. Results
2.1. Synthesis
Synthesis of Schiff bases is often performed by a reaction of amine with aldehyde, usually with the addition of catalysts such as acetic acid. Here, we describe a synthesis of Schiff bases from 2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide and different aldehydes utilizing choline chloride:malonic acid DES. DES was easily prepared from choline chloride and malonic acid (1:1), simply by heating their mixture at 80 °C until clear liquid was formed and used, as such, without any further purification, in synthesis of desired Schiff bases. First, we performed reactions with some commercial aldehydes, 4-methoxybenzaldehyde and 4-dimethylaminobenzaldehyde, to obtain Schiff bases 10 and 29, which have already been synthesized conventionally in our previous research [2,5]. The yields of compounds obtained conventionally (60% for compound 10 and 86% for compound 29 [5]) compared to those obtained in DES (44% for compound 10 and 70% for compound 29) are not much higher, which justified the use of this environmentally acceptable approach in further synthesis of novel Schiff bases. Therefore, further synthesis was performed with some new aldehydes to obtain compounds 30–36 (Scheme 1). Pyrazole based aldehydes utilized for a synthesis of Schiff bases 30–35 were synthesized in a reaction of substituted acetophenones with different hydrazines and subsequent formylation to yield 1H-pyrazole-4-carbaldehyde. For synthesis of Schiff bases, choline chloride:malonic acid (1:1) DES was used as reaction media. The synthetic approach (Scheme 1) in the synthesis of mentioned Schiff bases in DES is rather simple. When DES is prepared, an equimolar ratio of 2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide and aldehyde was added to the solvent, stirred at 70 °C and, upon completion of the reaction, water was added and a solid product separated. DESs were found to be very effective in this kind of synthesis, both, as solvents and catalysts, since reaction times were not longer than four hours, and purity of final compounds was satisfying as well as the yields. The mechanism of their action in accordance with Yadav et al. [30] is shown in Scheme 2.
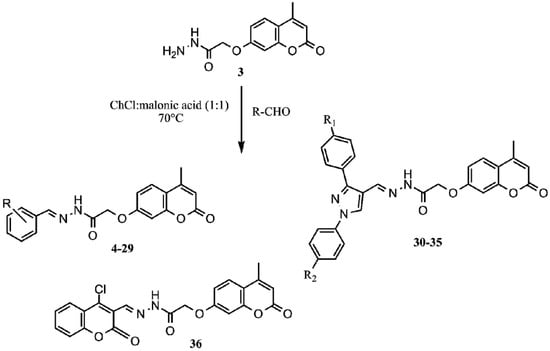
Scheme 1.
Synthesis of novel coumarinyl Schiff bases in deep eutectic solvent (DES).
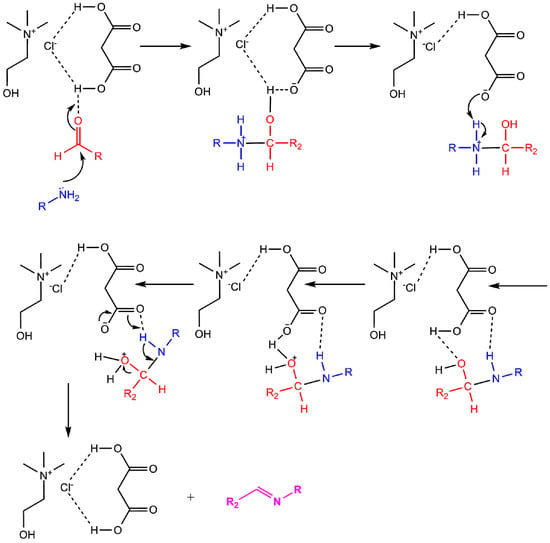
Scheme 2.
Proposed mechanism for synthesis of Schiff bases in choline chloride:malonic acid (1:1) DES.
Schiff bases were obtained in high yields (44–95%) and characterized by 1H- and 13C-NMR, as well as mass spectrometry. Typical shifts for coumarin C3 proton (6.20–6.22 ppm) and C4-methyl group (2.40 ppm) were noticed. The synthesized coumarin derivatives show characteristic peaks for aromatic protons, coumarin aromatic protons, protons of aromatic rings derived from corresponding aldehyde and pyrazole proton. All molecular masses were also in accordance with molecular ions obtained by mass spectrometry. The structures of synthesized compounds (30–36) are presented in Table 1. The 1H- and 13C-NMR spectra of synthesized compounds are presented in supplementary file 1 (File S1).

Table 1.
Structures of analysed compounds with their antioxidant activities (%DPPH) *.
2.2. Antioxidant Activity
Novel compounds (30–36), together with Schiff bases synthesized in our previous work [2,5], were investigated for their antioxidant activity expressed as % DPPH scavenging activity.
Data presented in Table 1 show that substituents on the phenyl ring have a great influence on antioxidant activity. Compounds 1–3 were used as basic structures in antioxidant activity determination to demonstrate how modification of a starting compound can have a great influence on antioxidant activity. Compared to coumarin's basic structure (1), substitution in position 4 with methyl and in position 7 with hydroxy group enhances antioxidant activity (2). Substitution of 7-OH group with -CO-NH-NH2 group in 2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (3) additionally increased antioxidant activity.
Novel compounds, together with Schiff bases synthesized in our previous work [2,5] were investigated for their antioxidant activity expressed as % DPPH scavenging activity. Structural details of all studied molecules and % DPPH are shown in Table 1. Data presented in Table 1 show that substituents on the phenyl ring have a great influence on antioxidant activity. Compounds with two hydroxyl groups in ortho position (11,14) exhibited the best antioxidant activity, although hydroxyl groups at the 2,3-position (11) compared to the 3,4-position (14) showed enhanced antioxidant activity. Catehol structure or 3,4-diOH substitution of phenyl ring allows oxygen atoms to cause electron delocalization and stabilisation of phenoxyl radical [2,5,31]. Novel compounds (30–35) did not show significant antioxidant activity. Replacement of aromatic ring by pyrazole ring in these compounds reduced antioxidant power compared to compounds (4–29).
2.3. QSAR Models
The best QSAR model for antioxidant activity of 36 coumarinyl Schiff bases was obtained by DRAGON descriptors:
log % DPPH = 0.696 + 1.474 (0.488) Mor22u + 1.948 (0.398) MATS3m + 0.387 (0.398) Hy
N(training) = 29; N(test) = 7 (6,12,19,24,28,33,34)
N(training) = 29; N(test) = 7 (6,12,19,24,28,33,34)
Statistical parameters of obtained models are given in Table 2. The variables in Equation (1) are listed in order of relative importance by their standardized regression coefficients (β, in brackets). Table 3 presents a correlation matrix of descriptors included in model (1) and proves that descriptors are not mutually correlated (correlation coefficient, R ≤ 0.7). Low collinearity is also verified by the low values of Kxx and ΔK (≥0.05) [32] (Table 2). The molecular descriptor values; experimental and calculated log %DPPH by model (1) have been tabulated in supplementary file 2 (Table S2). A scatter plot of experimentally obtained antioxidant activity versus calculated by model (1) is shown in Figure 1.
Table 2.
The model statistical results for the QSAR for antioxidant activity.
Table 3.
Correlation matrix for the descriptors included in model 1.
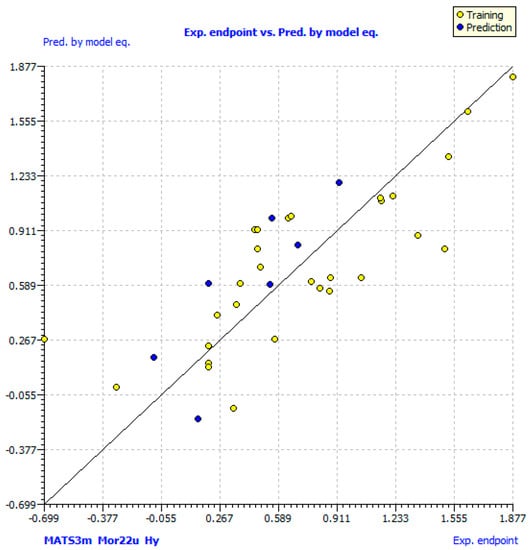
Figure 1.
Observed versus predicted log % DPPH for the coumarinyl Schiff bases calculated by model (1).
According the statistical results presented in Table 2, model (1) satisfactoriness threshold for fitting validation parameters: R2 > 0.60 and value of R2adj is close to the value of R2. The leave-one out (LOO) validation highlights that the model is stable, not obtained by chance, since Q2LOO > 0.5; R2yscr, and Q2yscr < 0.2, as R2yscr > Q2yscr [33,34]. Moreover, the root-mean-square error (RMSE) values for the training and validation sets are similar. The chosen models demonstrate a satisfactory stability in external validation: R2ext ≥ 0.60; small difference between CCCtr and CCCext; small difference between RMSEtr and RMSEex, and between mean absolute error of the training set (MAEtr) and mean absolute error of the external validation set (MAEex) [35]. Parameter considers the actual difference between the experimental and the predicted values, serving as more accurate measures for rating of model predictivity. Value of confirms high prediction ability for both internal and external validation sets [36]. Williams plot (Figure 2) was used in order to define the chemical domain of applicability for which a given QSAR model makes reliable predictions [37]. Inspection of the Williams plot revealed no compounds out of hat value of leverage or warning leverage (HAT, h* = 0.414), which means that all predicted data for compounds belonging to the chemical domain and have reliable prediction. Williams plot identified one outlier (compound 9), which has standardized residual predictions greater than 2.5 standard deviation units. Outlying behaviour of compound 9 was expected since it has demonstrated lowest antioxidant activity (Table 1). The methoxy group in phenyl ring, especially in position 3 (compound 9), inactivates the ring, which negatively influences the ability of the compound to form a stable radical upon scavenging DPPH radicals [38].
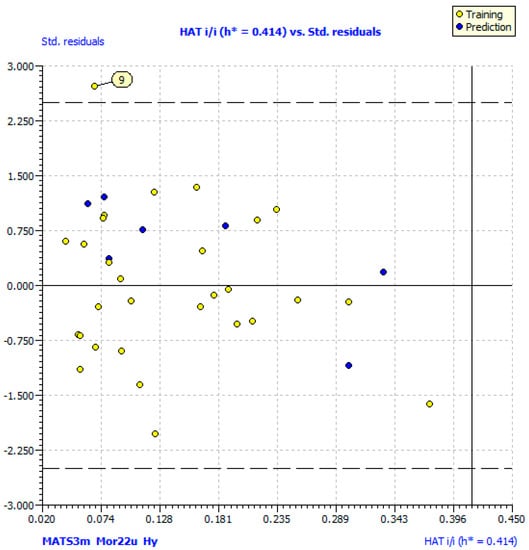
Figure 2.
Williams plot of the applicability domain of the the QSAR model for antioxidant activity calculated by model (1).
After removal of the compound 9 from the training set, subsequent re-analysis produced a following improved QSAR model:
log % DPPH = 0.733 + 1.331 (0.495) Mor22u + 1.744 (0.401) MATS3m + 0.375 (0.375) Hy
N(training) = 28; N(test) = 7 (6,12,19,24,28,33,34) (β in brackets)
N(training) = 28; N(test) = 7 (6,12,19,24,28,33,34) (β in brackets)
Calculated values of log %DPPH by model (2) have been tabulated in Table S2. Since compound 9 belonged to the training set, the obtained model gives the better statistics in terms of improvement in fitting and internal criteria. Exclusion of outliers generated the model with higher values of R2, R2adj, F, CCCtr, CCCcv, Q2LOO and lower values of s, RMSEtr, MAEtr RMSEcv, and MAEcv. The obtained model in external validation showed only improvement in R2ext, which demonstrated that the removal of outliers can improve the fitting, but not the predictivity of a model.
The largest values of standardized regression coefficients in Equations (1) and (2) has 3D-MoRSE (Molecular Representation of Structures based on Electronic diffraction) descriptor Mor22u. According the positive coefficients of Mor22u in Equations (1) and (2), compounds with significant antioxidant ability have enhanced positive values of this descriptor (Table S2). Descriptor Mor22u denotes unweighted descriptors with scattering parameter s = 21 Å−1. Since it is unweighted, the descriptor has no discriminative ability precisely and treats each atom equally. Though each 3D-MoRSE descriptor reflects the three-dimensional arrangement of the atoms in molecules, their final values are derived mostly from short distances [39]. Since mainly QSAR study covers structurally similar sets of compounds, 3D-MoRSE descriptors in a model can be interpreted using just several pairs of neighbor atoms. Specifically, descriptor Mor22u has the possibility to distinguish the difference between bond lengths of any kinds of atoms at least 0.03 Å [39]. The relative discriminative power of descriptor Mor22u can be observed at molecules 11–15, which have two hydroxyl groups on phenyl rings (Table 1 and Table S2). This descriptor is extremely sensitive to the difference in di-OH substitution of the phenyl ring. The value of Mor22u for the most active compound 11 (log %DPPH = 1.817), which has two hydroxyl groups in position 2,3 is 0.3. Substitution of two OH groups in compound 12 at the position 2,4 decreased values of Mor22u (0.216) and antioxidant activity (log %DPPH = 0.924). Our recent study has also evinced sensitivity of the 3D-MoRSE descriptor Mor19e to the changes of substituents and their position on the 3,4-ethylenedioxythiophene central unit [40].
The 2D autocorrelation molecular descriptor, MATS3m, corresponds to the Moran autocorrelation descriptor –lag 3/weighted by atomic masses. The given descriptor describes how atomic mass is distributed along a topological molecular structure, precisely indicating dependence of one atom on value of mass through a topological structure of compounds [41]. Its positive regression coefficient in models (1) and (2) suggests that the increased number of pairs of atoms higher atomic mass at the path length 3 characterized compounds with enhanced antioxidant activity. The descriptor is sensitive to kind and position of substituents on the phenyl ring (Table 1 and Table S2). The best differences can be observed between compounds 11 and 14, as well as 12 and 15. Compounds bearing 2,3-dihydroxyphenyl (11) and 3,4-dihydroxyphenyl moieties (14) on benzene rings have two oxygen atoms at path length 3. Consequently, these compounds have higher values of MATS3m than compounds with 2,4-dihydroxyphenyl (12) and 3,5-dihydroxyphenyl (15) moieties, and exhibit higher antioxidant activity. Hence, descriptor MATS3m has ability to distinguish the structure with two hydroxyl groups in ortho position at the phenolic ring from structure with two hydroxyl groups in meta position, the structural requirements relevant for the effective radical scavenging of phenolic compounds.
The third variable in models (1) and (2) is hydrophilic index (Hy). Hydrophilic index takes into account a total number of hydrogen atoms attached to oxygen, sulfur and nitrogen atoms, and the number of carbon atoms in relation of number of non-hydrogen atoms [42]. Positive regression coefficients of Hy in both models indicate that higher number of -OH and -NH groups are favorable for antioxidant activity. This is expected since DPPH radicals become stable molecules by accepting hydrogen radicals from organic substances [43]. Since the hydrophilic factor can predict the ability of molecules to donate a hydrogen atom, it is an important descriptor in DPPH radical scavenging. Hydrophilic index counts only the number of hydrogen donating groups, and it is independent from their positions in molecules; therefore, its relative importance in models (1) and (2) is minimal. Hydrophilic index has been found in previous QSAR studies of the DPPH radical scavenging activity of coumarin derivatives. Results of those studies showed that incorporation of hydroxyl groups in coumarin molecules enhanced hydrophilic index, which increased DPPH radical scavenging effects [24,25].
2.4. Electrostatic Potential (ESP) Surface
Electrostatic potential (ESP) surface provides a visualization of total charge distribution of the molecule and relative polarity of the molecule [44]. Figure 3 presents an ESP mapped density surface of the most active molecule (11) and the least active molecule (9), for comparison. The red region shows the greatest increase in electron density or region of negative ESP. The light blue shows about zero, while the white color positive electron density. The negative regions (red color) correspond to the aggregation of electron density. The positive regions, which increase in order blue < violet < white color, indicate positive ESP that corresponds to the repulsion of the proton by the atomic nuclei or nucleophilic reactivity [45].
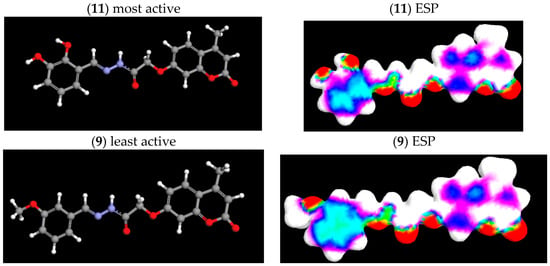

Figure 3.
Three-dimensional optimized structure (MM+, AM1) and electrostatic potential (ESP) surface maps of the most active compound (11) and the least active compound (9).
ESP maps of compounds (9) and (11) shows that the greatest negative electrostatic potential is located mainly over the oxygen and nitrogen from imino groups (>C=N-) with minimum value of −0.0409 au. A weakly positive region (from 0.0045 to 0.0318 au) is localized mostly on the carbon atoms of phenyl rings. Highly positive electron density is located on the hydrogen atoms. Observing ESP maps can help in the explanation of the role of substitution in antioxidant activity. Comparing the ESP maps of molecules, it could be observed that positive charge is more spread over the phenyl ring of inactive molecule (9). Negative charge of oxygen atoms from the 3-OCH3 group stabilizes positive charge, decreasing the ability for electron transfer to the DPPH radical. On the contrary, a smaller positive region in the phenyl ring of the most active molecule (11) allows easy electron transfer from negative charges located on two oxygen atoms from 2,3-OH groups.
3. Materials and Methods
3.1. Chemistry
All the chemicals were of p.a. purity and purchased from commercial suppliers. Melting points were determined on a capillary melting point apparatus (Electrothermal Engineering Ltd., Rochford, UK) and are uncorrected. Thin-layer chromatography was performed with fluorescent silica gel plates F254 (Merck, Darmstadt, Germany), under UV (254 and 365 nm) light, with benzene–acetone–acetic acid (8:1:1, v/v) as a solvent. The mass spectra were recorded on liquid chromatography tandem mass spectrometry (LC/MS/MS) API 2000 (Applied Biosystems/MDS SCIEX, Foster City, CA, USA). NMR spectra were recorded on a Bruker Avance 600 MHz NMR Spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany) at 293 K in dimethylsulfoxide-d6 (DMSO-d6). The absorbance was measured on UV visible spectrophotometer Helios γ (ThermoSpectronic, Cambridge, UK).
3.2. Preparation of DES
DES was prepared as described previously [7]. Briefly, choline chloride and malonic acid were mixed together in molar ratio 1:1 and heated up to 80 °C until a clear liquid was obtained. This DES was used as such in synthesis of desired compounds as described in Section 3.3.
3.3. Synthesis of Schiff Bases in DES Choline Chloride:Malonic Acid (1:1)
An equimolar ratio of 2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide and corresponding aldehyde was mixed in DES and stirred at 70 °C until the completion of reaction, which was monitored by thin-layer chromatography (TLC). Upon addition of water, a crude product was precipitated and washed with ethanol to obtain a pure Schiff base.
3.4. Characterization of Compounds
(E)-N′-(4-Methoxybenzylidene)-2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (10). Yield 44%; m.p. = 263 °C; 1H-NMR (ppm): 2.40 (s, 3H, CH3), 3.80 (s, 3H, OCH3), 4.79–5.27 (s, 2H, CH2), 6.22 (s, H, CH), 6.95–7.70 (7H, arom), 7.98 (s, H, CH), 11.53 (s, H, NH); 13C-NMR (ppm): 18.1, 55.3, 65.2, 101.5, 111.2, 112.3, 113.3, 114.2, 114.5, 126.3, 128.6, 128.8, 143.8, 147.9, 153.3, 154.5, 160.1, 160.8, 161.3, 163.3, 168.2; MS m/z: 365.20 [M − H]+, (Mr = 366.37).
(E)-N′-(4-(Dimethylamino)benzylidene)-2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (29). Yield 70%; m.p. = 262 °C; Rf = 0.4; 1H-NMR (ppm): 2.40 (s, 3H, CH3), 2.97 (s, 6H, 2(CH3)), 4.76–5.25 (s, 2H, CH2), 6.22 (s, H, CH), 6.72–7.54 (7H, arom), 7.89 (s, H, CH), 11.44 (s, H, NH); 13C-NMR (ppm): 18.6, 65.7, 101.9, 102.1, 111.7, 112.2, 112.8, 113.8, 121.7, 126.8, 127.0, 128.8, 145.3, 149.3, 151.9, 153.8, 155.0, 160.6, 161.9, 163.3, 168.2; MS m/z: 378.20 [M − H]+, (Mr = 379.41).
(E)-N′-((3-(4-Chlorophenyl)-1-(4-fluorophenyl)-1H-pyrazol-4-yl)methylene)-2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (30). Yield 75%; m.p. = 200 °C; Rf = 0.5; 1H-NMR (ppm): 2.40 (s, 3H, CH3), 4.82–5.20 (s, 2H, CH2), 6.22 (s, H, CH), 6.93–7.86 (12H, arom), 8.49 (s, H, CH), 11.82 (s, H, NH); 13C-NMR (ppm): 18.6, 65.5, 102.2, 111.7,113.9, 123.4,123.8, 126.8, 128.6, 129.3, 135.6, 142.9, 146.5, 153.8, 154.9, 160.6, 161.7, 164.3, 168.8; MS m/z: 529.30 [M + H]+, (Mr = 530.94).
(E)-N′-((3-(4-Bromophenyl)-1-(p-tolyl)-1H-pyrazol-4-yl)methylene)-2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (31). Yield 83%; m.p. = 248–251 °C; Rf = 0.47; 1H-NMR (ppm): 2.35 (s, 3H, CH3), 2.49 (s, 3H, CH3), 4.76–5.11 (s, 2H, CH2), 6.22 (s, H, CH), 6.88–7.87 (12H, arom), 8.97 (s, H, CH), 11.47 (s, H, NH); 13C-NMR (ppm): 18.1, 20.4, 65.2, 101,7, 111.4,118.4, 121.8, 126.4, 129.9, 130.4, 131.4, 131.7, 136.5, 153.3, 154.5, 160.1; MS m/z: 570.80 [M − H]+, (Mr = 571.43).
(E)-N′-((3-(4-iodophenyl)-1-(p-tolyl)-1H-pyrazol-4-yl)methylene)-2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (32). Yield 73%; m.p. = 265 °C; Rf = 0.45; 1H-NMR (ppm): 2.26 (s, 3H, CH3), 2.41 (s, 3H, CH3), 5.32 (s, 2H, CH2), 6.21 (s, H, CH), 6.95–7.79 (12H, arom), 8.97 (s, H, CH), 10.91 (s, H, NH); 13C-NMR (ppm): 18.1, 65.2, 95.8, 101.6, 111.2,112.3, 113.3, 126.3, 128.3, 129.5, 130.4, 137.5,147.8, 153.2, 154.6, 160.0, 161.4; MS m/z: 617.20 [M − H]+, (Mr = 618.08)
(E)-N′-((3-(4-methoxyphenyl)-1-(p-tolyl)-1H-pyrazol-4-yl)methylene)-2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (33). Yield 93%; m.p. = 238 °C; Rf = 0.41; 1H-NMR (ppm): 2.36 (s, 3H, CH3), 2.40 (s, 3H, CH3), 3.79-3.83 (s, 3H, OCH3), 4.78–5.18 (s, 2H, CH2), 6.22 (s, H, CH), 7.05-8.12 (12H, arom), 8.94 (s, H, CH), 11.48 (s, H, NH); 13C-NMR (ppm): 18.6, 20.9, 55.7, 101.9, 111.7,112.7, 114.4, 118.9, 126.9, 128.4, 130.2, 130.4, 136.7, 137.3, 147.8, 153.8, 168.3; MS m/z: 521.40 [M − H]+, (Mr = 522.2).
(E)-N′-((3-(4-Chlorophenyl)-1-(p-tolyl)-1H-pyrazol-4-yl)methylene)-2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (34). Yield 65%; m.p. = 255 °C; Rf = 0.47; 1H-NMR (ppm): 2.36 (s, 3H, CH3), 2.40 (s, 3H, CH3), 4.79–5.13 (s, 2H, CH2), 6.23 (s, H, CH), 6.90–8.12 (12H, arom), 8.98 (s, H, CH), 11.50 (s, H, NH); 13C-NMR (ppm): 18.6, 20.9, 101.9, 102.2, 111.7, 112.7, 119.1, 119.3, 126.9, 129.0, 129.2, 130.5, 130.6, 153.8; MS m/z: 525.30 [M − H]+, (Mr = 526.98).
(E)-N′-((1-(4-Chlorophenyl)-3-(4-nitrophenyl)-1H-pyrazol-4-yl)methylene)-2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (35). Yield 69%; m.p. = 281 °C; Rf = 0.42; 1H-NMR (ppm): 2.40 (s, 3H, CH3), 4.81–5.09 (s, 2H, CH2), 6.23 (s, H, CH), 6.86–8.47 (12H, arom), 9.11 (s, H, CH), 11.59 (s, H, NH); 13C-NMR (ppm): 18.6, 101.9, 111.8, 112.6, 121.5, 124.1, 124.2, 126.8, 130.0, 130.2, 168.4; MS m/z: 556.30 [M − H]+, (Mr = 557.94).
(E)-N′-((4-Chloro-2-oxo-2H-chromen-3-yl)methylene)-2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)acetohydrazide (36). Yield 78%; m.p. = 233 °C; Rf = 0.28; 1H-NMR (ppm): 2.40 (s, 3H, CH3), 4.78–5.25 (s, 2H, CH2), 6.21 (s, H, CH), 6.91–8.03 (12H, arom), 8.21 (s, H, CH), 11.98 (s, H, NH); 13C-NMR (ppm): 18.1, 65.2, 101.4, 111.3, 112.1, 113.4, 116.5, 118.4, 125.3, 126.1, 126.4, 133.6, 1370.5, 153.3, 154.5; MS m/z: 439.20 [M + H]+, (Mr = 438.82).
3.5. Determination of DPPH Scavenging Activity
DPPH scavenging activity for previously synthesized compounds [2,5] and new compounds (30–36) was performed according to the procedure described in our previous work [4]. For each sample, antioxidant activity determination was performed in triplicate and expressed as % scavenging activity (% DPPH).
3.6. QSAR Studies
3.6.1. Data Set
The dataset used for building QSAR models consists of 36 molecules whose antioxidant activities were measured and described in the present study. Antioxidant activity, expressed as % DPPH), were converted in the form of the logarithm (log % DPPH) and presented in Table 1 together with structures of molecules (While transformation of the experimental data to both logit % DPPH and log %DPPH afforded a normal distribution, the latter approach afforded a QSAR model, which appears to provide a better relationship between the structures of the molecules and their activities).
3.6.2. Descriptor Calculation and Selection
The 3D structures of 36 molecules were optimized applying the Avogadro 1.2.0. (University of Pittsburgh, Pittsburgh, PA, USA) [46] using the molecular mechanics force field (MM+) [47]. Subsequently, all structures were submitted to geometry optimization using the semiempirical AM1 method [48]. Two sets of descriptors were generated using Parameter Client (Virtual Computational Chemistry Laboratory, an electronic remote version of the Dragon program [49]). In order to reduce the huge number of calculated descriptors (about 1260), firstly, zero value descriptors were excluded from the initial pool. Further exclusion was performed using QSARINS-Chem 2.2.1 (University of Insubria, Varese, Italy) [50]: constant and semi-constant descriptors, i.e., those having chemical compounds with a constant value for more than 80%, and descriptors that are too inter-correlated (>85%) were rejected. Data sets were randomly divided into training (80%, Ntrain = 29) and test (20%, Ntest = 7) set using QSARINS.
3.6.3. Regression Analysis and Validation of Models
The best QSAR models were obtained by Genetic Algorithm (GA) using QSARINS. The number of descriptors (I) in the multiple regression equation was limited to three. The models have been assessed by: fitting criteria; internal cross-validation using the leave-one out (LOO) method and Y-scrambling; and external validation. Fitting criteria included: the coefficient of determination (R2), adjusted (R2adj), cross-validate R2 using the leave-one-out method (Q2LOO), global correlation among descriptors (Kxx), the difference between global correlation between molecular descriptors and y the response variable, and global correlation among descriptors (ΔK), standard deviation of regression (s), and Fisher ratio (F) [32,51,52]. Internal and external validations also included the following parameters: root-mean-square error of the training set (RMSEtr); root-mean-square error of the training set determined through the cross validated LOO method (RMSEcv), root-mean-square error of the external validation set (RMSEex), squared correlation coefficients between the observed and (leave-one-out) predicted values of the compounds with and without intercept (); concordance correlation coefficient of the training set (CCCtr), test set using LOO cross validation (CCCcv), and of the external validation set (CCCex), mean absolute error of the training set (MAEtr), mean absolute error of the internal validation set (MAEcv) and mean absolute error of the external validation set (MAEex) predictive residual sum of squares determined through cross-validated LOO method (PRESScv) in the training set and in the external prediction set (PRESSex) [23,51]. The analysed external validation parameters also include the coefficient of determination (R2ex). Robustness of QSAR models was tested by a Y-randomisation test.
Investigation of the applicability domain of a prediction model was performed by leverage plot (plotting residuals vs. leverage of training compounds). The warning leverage h* is defined as 3p′/n, where n is the number of training compounds and p′ is the number of model adjustable parameters [37]. Tools of regression diagnostics as residual plots and Williams plots were used to check the quality of the best models and define their applicability domain using QSARINS.
3.6.4. Visualization of Electrostatic Potential Surface
Electrostatic potential surface has been generated from optimized structures by ArgusLab 4.0.1 (Mark A. Thompson, Planaria Software LLC, Seattle, WA, USA). In an ESP-mapped density surface, the electron density surface gives the shape of the surface, while the value of the ESP on that surface gives the colors. Maximum mapped surface was set to 0.05, while the minimum to −0.05 a.u.
4. Conclusions
A series of novel coumarinyl Schiff bases in DES have been synthesized and evaluated for antioxidant activity. Compared to previously synthesized coumarin derivatives, novel compounds have not showed an improvement in antioxidant effect. QSAR study has clarified the importance of two hydroxyl groups in ortho position on phenyl ring and hydrophilicity for enhanced antioxidant activity coumarin derivatives. Electrostatic potential surface provides a visualization of possible regions in molecules that allow easy electron transfer to DPPH radicals.
Supplementary Materials
Supplementary materials are available online. File S1: 1H- and 13C-NMR spectra of synthesized compounds; Table S2: Values of the descriptors included in models (1–2), as experimental and calculated log %DPPH.
Acknowledgments
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7 2007-2013) under Grant No. 291823 Marie Curie FP7-PEOPLE-2011-COFUND (the new International Fellowship Mobility Programme for Experienced Researchers in Croatia—NEWFELPRO). This presentation has been prepared as a part of a project “Synthesis and characterization of some chalcone based heterocyclic compounds and their biological screening as potential in vitro antioxidant agent”, which has received funding through the NEWFELPRO project under Grant No. 84.
Author Contributions
Maja Molnar, Mario Komar and Harshad Brahmbhatt conceived, designed the experiments and performed the experiments; Vesna Rastija performed QSAR analysis and wrote the paper together with Maja Molnar, while Jurislav Babić and Stela Jokić helped with experiment design and feasibility.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Bourgaud, F.; Hehn, A.; Larbat, R.; Doerper, S.; Gontier, E.; Kellner, S.; Matern, U. Biosynthesis of coumarins in plants: A major pathway still to be unravelled for cytochrome P450 enzymes. Phytochem. Rev. 2006, 5, 293–308. [Google Scholar] [CrossRef]
- Molnar, M.; Šarkanj, B.; Čačić, M.; Gille, L.; Strelec, I. Antioxidant properties and growth-inhibitory activity of coumarin Schiff bases against common foodborne fungi. Der Pharm. Chem. 2014, 6, 313–320. [Google Scholar]
- Čačić, M.; Pavić, V.; Molnar, M.; Šarkanj, B.; Has-Schön, E. Design and synthesis of some new 1,3,4-thiadiazines with coumarin moieties and their antioxidative and antifungal activity. Molecules 2014, 19, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Šarkanj, B.; Molnar, M.; Čačić, M.; Gille, L. 4-Methyl-7-hydroxycoumarin antifungal and antioxidant activity enhancement by substitution with thiosemicarbazide and thiazolidinone moieties. Food Chem. 2013, 139, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Čačić, M.; Molnar, M.; Šarkanj, B.; Has-Schön, E.; Rajković, V. Synthesis and antioxidant activity of some new coumarinyl-1,3-thiazolidine-4-ones. Molecules 2010, 15, 6795–6809. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I.M.; Ramon, D.J. Deep eutectic solvents: The organic reaction medium of the century. Eur. J. Org. Chem. 2016, 4, 612–632. [Google Scholar] [CrossRef]
- Lobo, H.R.; Singh, B.S.; Shankarling, G.S. Bio-compatible eutectic mixture for multi-component synthesis: A valuable acidic catalyst for synthesis of novel 2.3-dihydroquinazolin-4(1H)-one derivatives. Catal. Commun. 2012, 27, 179–183. [Google Scholar] [CrossRef]
- Khandelwal, S.; Tailor, Y.K.; Kumar, M. Deep eutectic solvents (DESs) as eco-friendly and sustainable solvent/catalyst systems in organic transformations. J. Mol. Liq. 2016, 215, 345–386. [Google Scholar] [CrossRef]
- Liu, P.; Hao, J.; Mo, L.; Zhang, Z. Recent advances in the application of deep eutectic solvents as sustainable media as well as catalysts in organic reactions. RSC Adv. 2015, 5, 48675–48704. [Google Scholar] [CrossRef]
- Abbott, P.A.; Boothby, D.; Capper, G.; Davies, D.L.; Rasheed, R.K. Deep eutectic solvents formed between choline chloride and carboxylic acids: Versatile alternatives to ionic liquids. J. Am. Chem. Soc. 2004, 126, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; van Spronsen, J.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y.H. Natural deep eutectic solvents as new potential media for green technology. Anal. Chim. Acta 2013, 766, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Capua, M.; Perrone, S.; Perna, F.M.; Vitale, P.; Troisi, L.; Salomone, A.; Capriati, V. An expeditious and greener synthesis of 2-aminoimidazoles in deep eutectic solvents. Molecules 2016, 21, 924. [Google Scholar] [CrossRef] [PubMed]
- Massolo, E.; Palmieri, S.; Benaglia, M.; Capriati, V.; Perna, F.M. Stereoselective organocatalysed reactions in deep eutectic solvents: Highly tunable and biorenewable reaction media for sustainable organic synthesis. Green Chem. 2016, 18, 792–797. [Google Scholar] [CrossRef]
- Azizi, N.; Edrisi, M. Deep eutectic solvent catalyzed eco-friendly synthesis of imines and hydrobenzamides. Monatsh. Chem. 2015, 146, 1695–1698. [Google Scholar] [CrossRef]
- Martínez, R.; Berbegal, L.; Guillena, G.; Ramón, D.J. Bio-renewable enantioselective aldol reaction in natural deep eutectic solvents. Green Chem. 2016, 18, 1724–1730. [Google Scholar] [CrossRef]
- Dilauro, G.; Cicco, L.; Perna, F.M.; Vitale, P.; Capriati, V. Solvent-catalyzed umpolung carbonesulfur bond-forming reactions by nucleophilic addition of thiolate and sulfinate ions to in situederived nitrosoalkenes in deep eutectic solvents. C. R. Chim. 2017, 1–7. [Google Scholar] [CrossRef]
- Maka, H.; Spychaj, T.; Adamus, J. Lewis acid type deep eutectic solvents as catalysts for epoxy resin rosslinking. RSC Adv. 2015, 5, 82813–82821. [Google Scholar] [CrossRef]
- García-Argüelles, S.; Ferrer, M.L.; Iglesias, M.; Del Monte, F.; Gutiérrez, M.C. Study of superbase-sased deep eutectic solvents as the catalyst in the chemical fixation of CO2 into cyclic carbonates under mild conditions. Materials 2017, 10, 759. [Google Scholar] [CrossRef] [PubMed]
- Amić, A.; Molnar, M. An improved and efficient N-acetylation of amines using choline chloride based deep eutectic solvents. Org. Prep. Proc. Int. 2017, 49, 249–257. [Google Scholar] [CrossRef]
- Kontogiorgis, A.C.; Pontiki, A.E.; Hadjipavlou-Litina, D. A Review on quantitative structure-activity relationships (QSARs) of natural and synthetic antioxidants compounds. Mini Rev. Med. Chem. 2005, 5, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Worachartcheewan, A.; Suvannang, N.; Prachayasittikul, S.; Prachayasittikul, V.; Nantasenamat, C. Probing the origins of aromatase inhibitory activity of disubstituted coumarins via QSAR and molecular docking. EXCLI J. 2014, 13, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Erzincan, P.; Saҫan, M.T.; Yüce-Durson, Ö.; Daniş, Ö.; Demir, S.; Erdern, S.S.; Ogan, A. QSAR models for antioxidant activity of new coumarine derivatives. SAR QSAR Environ. Res. 2015, 26, 721–737. [Google Scholar] [CrossRef] [PubMed]
- Mitra, I.; Saha, A.; Roy, K. Predictive modeling of antioxidant coumarin derivatives using multiple approaches: Descriptor-based QSAR. 3D-Pharmacophore mapping and HQSAR. Sci. Pharm. 2013, 81, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Razo-Hernández, R.S.; Pineda-Urbina, K.; Velazco-Medel, M.A.; Villanueva-García, M.; Sumaya-Martínez, M.T.; Sumaya-Martínez, M.T.; Martínez-Martínez, F.J.; Gómez-Sandoval, Z. QSAR study of the DPPH• radical scavenging activity of coumarin derivatives and xanthine oxidase inhibition by molecular docking. Cent. Eur. J. Chem. 2014, 12, 1067–1080. [Google Scholar] [CrossRef]
- Martínez-Martínez, F.J.; Razo-Hernández, R.S.; Peraza-Campos, A.L.; Villanueva-García, M.; Sumaya-Martínez, M.T.; Cano, D.J.; Gómez-Sandoval, Z. Synthesis and in vitro antioxidant activity evaluation of 3-carboxycoumarin derivatives and QSAR study of their DPPH• radical scavenging activity. Molecules 2012, 17, 14882–14898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Y.; Wang, L.-F. Theoretical elucidation of structure–activity relationship for coumarins to scavenge peroxyl radical. J. Mol. Struct. Theochem. 2004, 673. [Google Scholar] [CrossRef]
- Jorge, E.G.; Rayar, A.M.; Barigye, S.J.; Rodríguez, M.E.J.; Veitía, M.S.-I. Development of an in silico model of DPPH free radical scavenging capacity: Prediction of antioxidant activity of coumarin type compounds. Int. J. Mol. Sci. 2016, 17, 881. [Google Scholar] [CrossRef] [PubMed]
- Gacche, R.N.; Jadhav, S.G. Antioxidant activities and cytotoxicity of selected coumarin derivatives: Preliminary results of a structure activity relationship study using computational tools. J. Exp. Clin. Med. 2012, 4, 165–169. [Google Scholar] [CrossRef]
- Mladenović, M.; Mihailović, M.; Bogojević, D.; Matić, S.; Nićiforović, N.; Mihailović, V.; Vuković, N.; Sukdolak, S.; Solujić, S. In vitro antioxidant activity of selected 4-hydroxy-chromene-2-one derivatives—SAR, QSAR and DFT studies. Int. J. Mol. Sci. 2011, 12, 2822–2841. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.N.; Shankarling, G.S. Room temperature ionic liquid choline chloride-oxalic acid: A versatile catalyst for acid-catalyzed transformation in organic reactions. J. Mol. Liq. 2014, 191, 137–141. [Google Scholar] [CrossRef]
- Sokeman, B.B.; Aydin, G.; Gumus, A.; Karadeniz, S.; Ugras, H.I. Synthesis, antielastase, antioxidant and radical scavenging activities of 4-(aza substituted) methylene substituted dihydroxy coumarines. J. Enzym. Inhib. Med. Chem. 2013, 28, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V.; Maiocchi, A. The K correlation index: Theory development and its application in chemometrics. Chemom. Intell. Lab. Syst. 1999, 46, 13–29. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. J. Comput. Aided Mol. Des. 2002, 16, 358–369. [Google Scholar] [CrossRef]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.-D.; Lee, K.-H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Masand, V.H.; Mahajan, D.T.; Nazeruddin, G.M.; Hadda, T.B.; Rastija, V.; Alfeedy, A.M. Effect of information leakage and method of splitting (rational and random) on external predictive ability and behavior of different statistical parameters of QSAR model. Med. Chem. Res. 2015, 24, 1241–1264. [Google Scholar] [CrossRef]
- Roy, K.; Mitra, I. On the use of the metric rm(2) as an effective tool for validation of QSAR models in computational drug design and predictive toxicology. Mini Rev. Med. Chem. 2012, 491–504. [Google Scholar] [CrossRef]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.D.; McDowell, R.M.; Gramatica, P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef]
- Tominaga, H.; Kobayashi, Y.; Goto, T.; Kasemura, K.; Nomura, M. DPPH radical-scavenging effect of several phenylpropanoid compounds and their glycoside derivatives. Yakugaku Zasshi 2005, 125, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Devinyak, O.; Havrylyuk, D.; Lesyk, R. 3D-MoRSE descriptors explained. J. Mol. Graph. Model. 2014, 54, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Jukić, M.; Rastija, V.; Opačak-Bernardi, T.; Stolić, I.; Krstulović, L.; Bajić, M.; Glavaš-Pbrovoac, L. Antitumor activity of 3,4-ethylenedioxythiophene derivatives and quantitative structure-activity relationship analysis. J. Mol. Struct. 2017, 1133, 66–73. [Google Scholar] [CrossRef]
- Moran, P.A.P. Notes on continuous stochastic phenomena. Biometrika 1950, 37, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Vighi, M.; Finizio, A.; Gramatica, P. 30-Modelling and Prediction by WHIM Descriptors. Part 8. Toxicity and Physico-chemical Properties of Environmental Priority Chemicals by 2D-TI and 3D-WHIM Descriptors. SAR QSAR Environ. Res. 1997, 7, 173–193. [Google Scholar] [CrossRef] [PubMed]
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef]
- Weiner, P.K.; Langridge, R.; Blaney, J.M.; Schaefer, R.; Kollman, P.A. Electrostatic potential molecular surface. Proc. Natl. Acad. Sci. USA 1982, 79, 3754–3758. [Google Scholar] [CrossRef] [PubMed]
- Kidwai, M.; Poddar, R.; Jain, A.; Kumar, R.; Luthar, P.M. Synthesis and SARs of coumarin fused 1,5-benzothiazepines as novel anticancer and antioxidant agents. Mini-Rev. Org. Chem. 2015, 12, 24–33. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Hocquet, A.; Langgård, M. An evaluation of the MM+ force field. Mol. Model. 1998, 4, 94–112. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. AM1: A new general purpose quantum mechanical model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory-design and description. J. Comput. Aided. Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. Mol. Inf. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inf. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 3–36 are available from the authors. |





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).