Deep Eutectic Solvents as Convenient Media for Synthesis of Novel Coumarinyl Schiff Bases and Their QSAR Studies



 and
and
Abstract
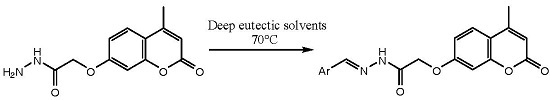
:
1. Introduction
2. Results
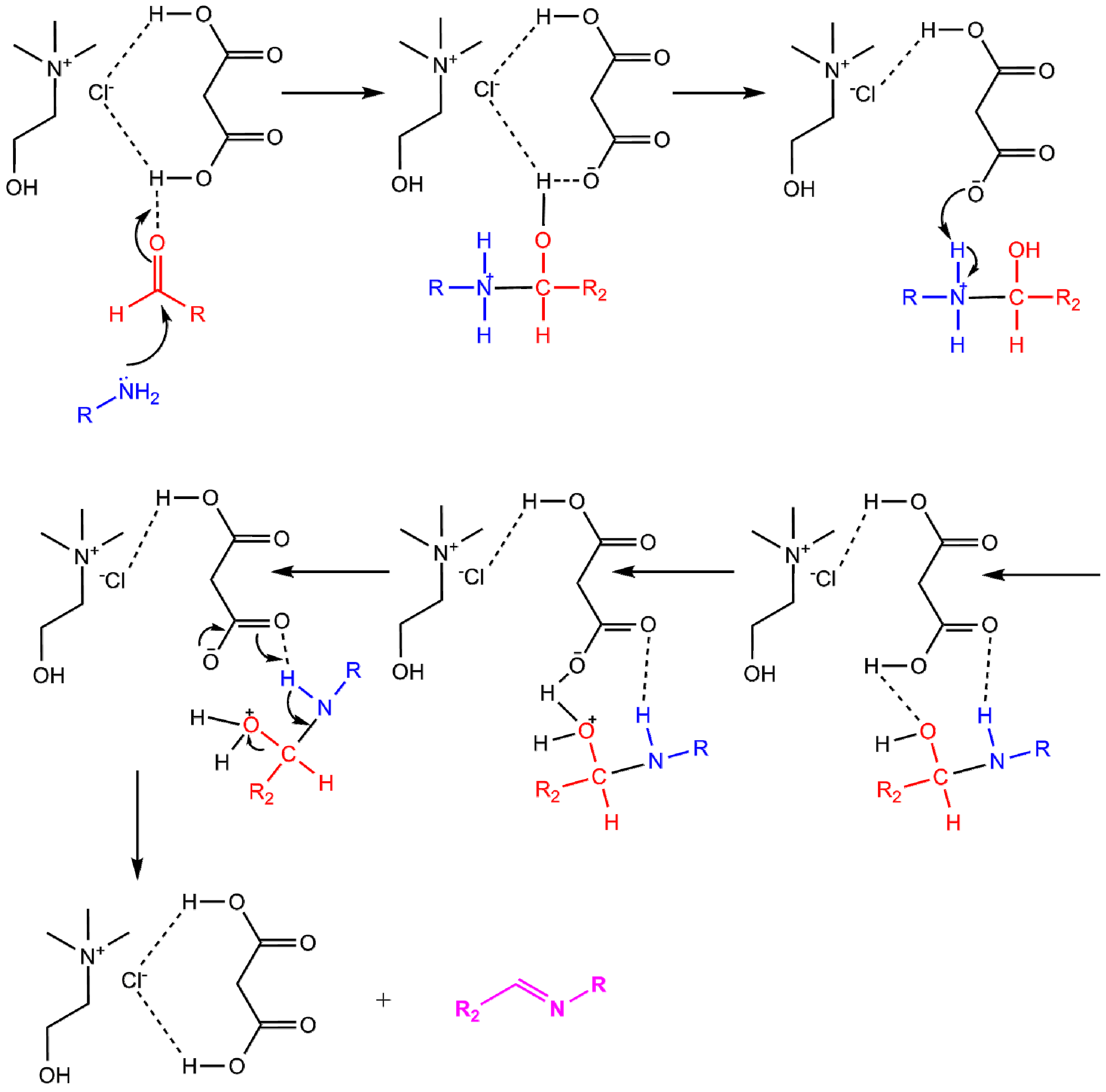
2.1. Synthesis
2.2. Antioxidant Activity
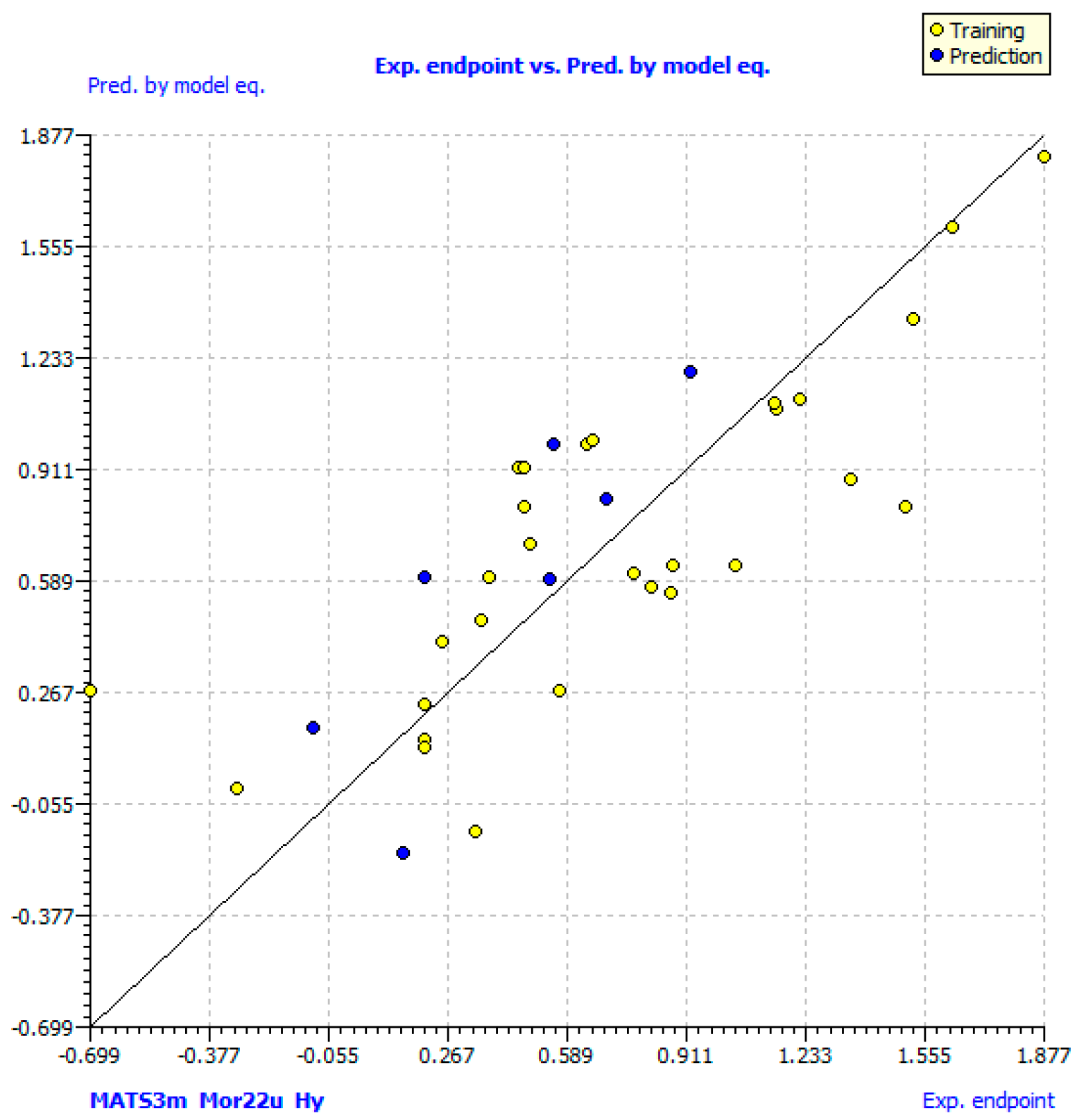
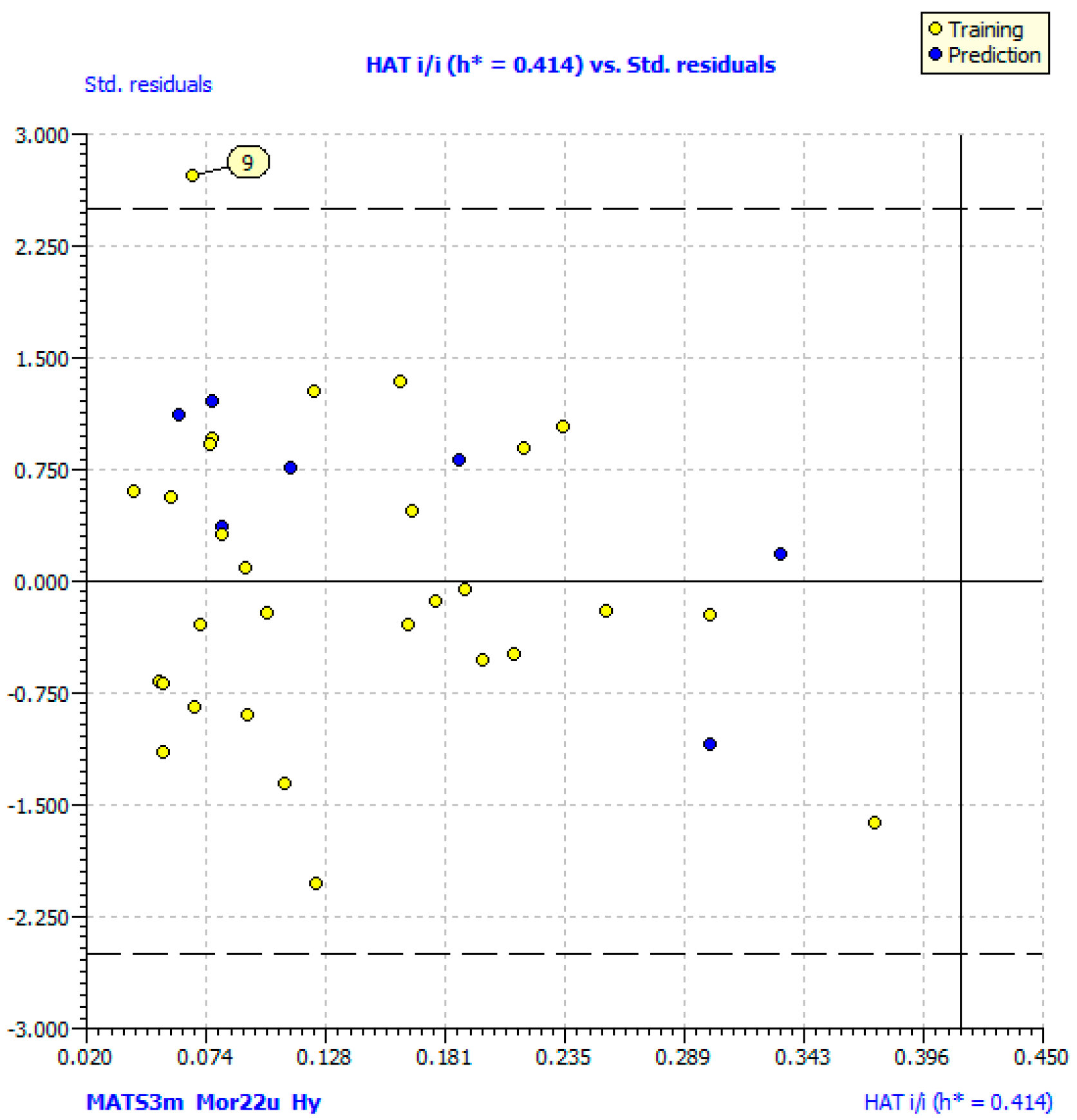
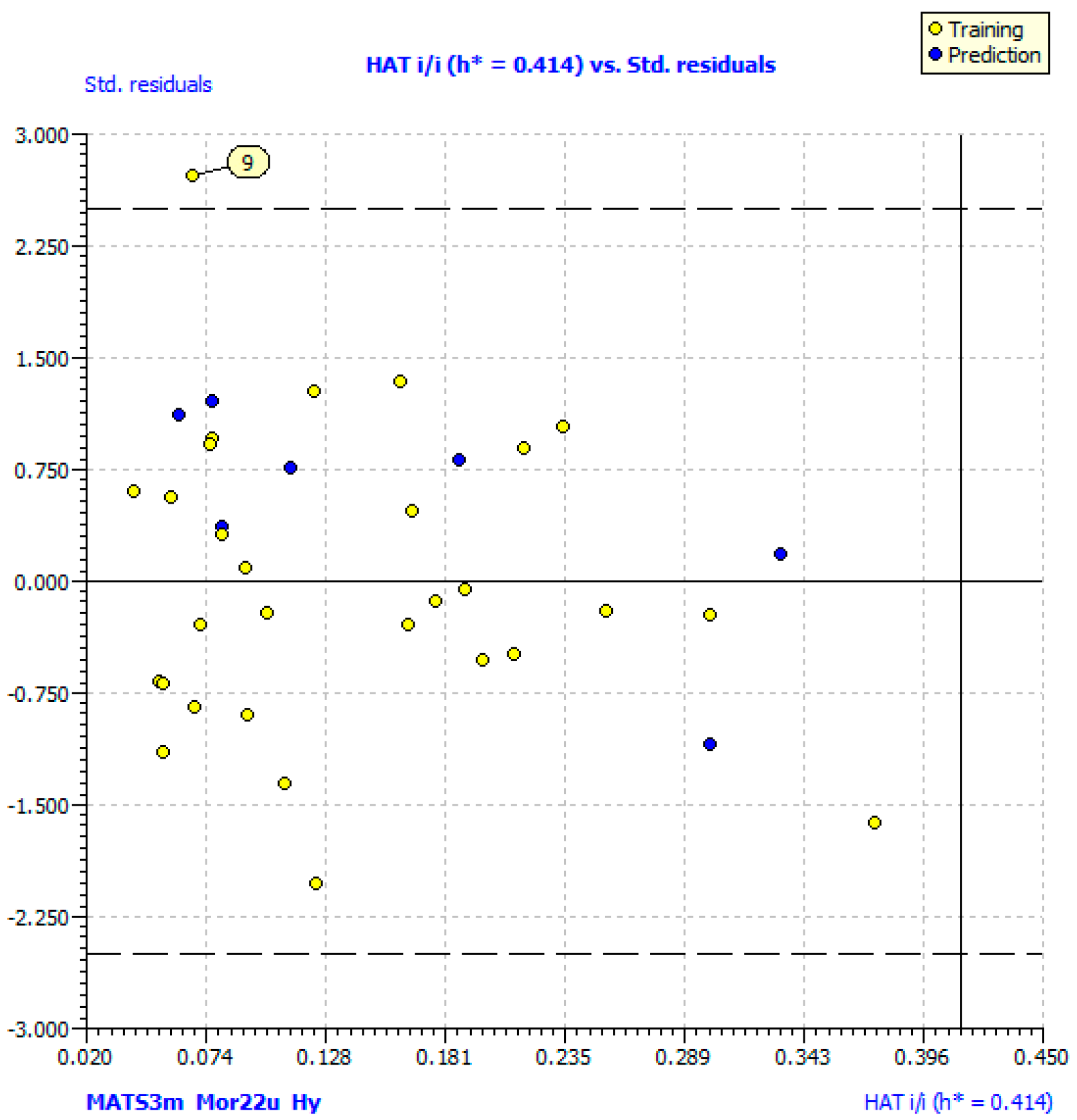
2.3. QSAR Models
N(training) = 29; N(test) = 7 (6,12,19,24,28,33,34)
N(training) = 28; N(test) = 7 (6,12,19,24,28,33,34) (β in brackets)
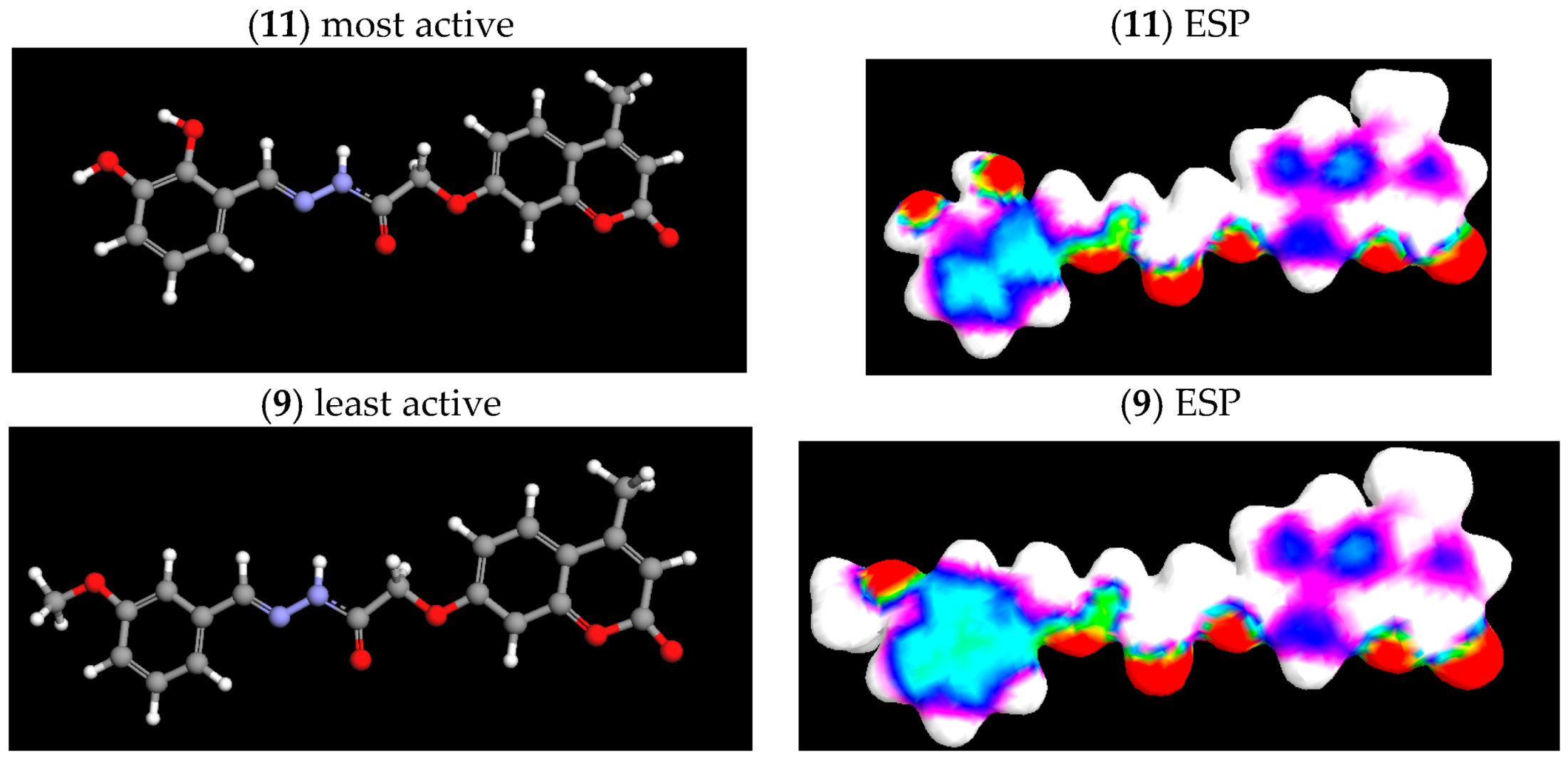
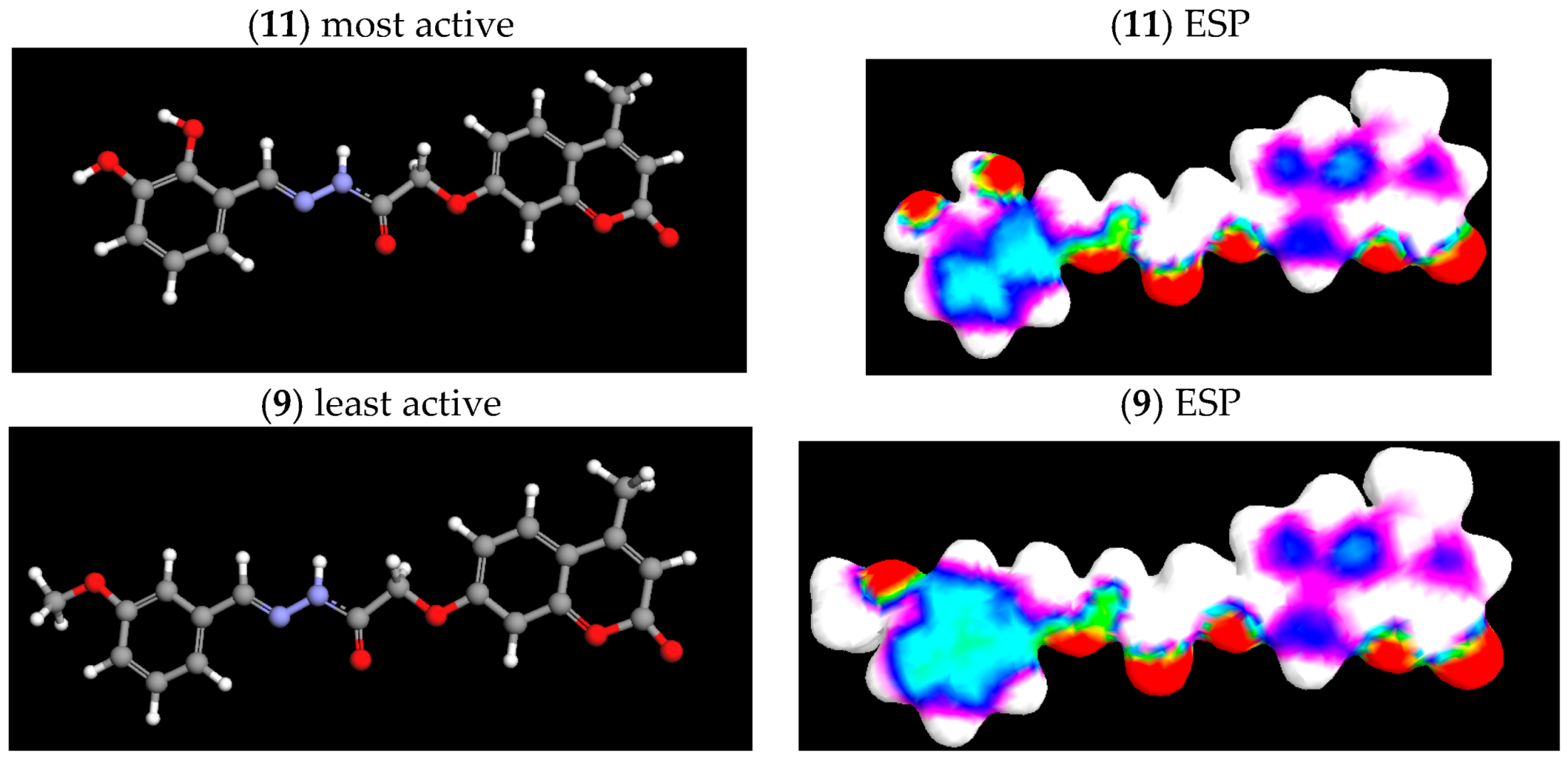
2.4. Electrostatic Potential (ESP) Surface
3. Materials and Methods
3.1. Chemistry
3.2. Preparation of DES
3.3. Synthesis of Schiff Bases in DES Choline Chloride:Malonic Acid (1:1)
3.4. Characterization of Compounds
3.5. Determination of DPPH Scavenging Activity
3.6. QSAR Studies
3.6.1. Data Set
3.6.2. Descriptor Calculation and Selection
3.6.3. Regression Analysis and Validation of Models
3.6.4. Visualization of Electrostatic Potential Surface
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bourgaud, F.; Hehn, A.; Larbat, R.; Doerper, S.; Gontier, E.; Kellner, S.; Matern, U. Biosynthesis of coumarins in plants: A major pathway still to be unravelled for cytochrome P450 enzymes. Phytochem. Rev. 2006, 5, 293–308. [Google Scholar] [CrossRef]
- Molnar, M.; Šarkanj, B.; Čačić, M.; Gille, L.; Strelec, I. Antioxidant properties and growth-inhibitory activity of coumarin Schiff bases against common foodborne fungi. Der Pharm. Chem. 2014, 6, 313–320. [Google Scholar]
- Čačić, M.; Pavić, V.; Molnar, M.; Šarkanj, B.; Has-Schön, E. Design and synthesis of some new 1,3,4-thiadiazines with coumarin moieties and their antioxidative and antifungal activity. Molecules 2014, 19, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Šarkanj, B.; Molnar, M.; Čačić, M.; Gille, L. 4-Methyl-7-hydroxycoumarin antifungal and antioxidant activity enhancement by substitution with thiosemicarbazide and thiazolidinone moieties. Food Chem. 2013, 139, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Čačić, M.; Molnar, M.; Šarkanj, B.; Has-Schön, E.; Rajković, V. Synthesis and antioxidant activity of some new coumarinyl-1,3-thiazolidine-4-ones. Molecules 2010, 15, 6795–6809. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I.M.; Ramon, D.J. Deep eutectic solvents: The organic reaction medium of the century. Eur. J. Org. Chem. 2016, 4, 612–632. [Google Scholar] [CrossRef]
- Lobo, H.R.; Singh, B.S.; Shankarling, G.S. Bio-compatible eutectic mixture for multi-component synthesis: A valuable acidic catalyst for synthesis of novel 2.3-dihydroquinazolin-4(1H)-one derivatives. Catal. Commun. 2012, 27, 179–183. [Google Scholar] [CrossRef]
- Khandelwal, S.; Tailor, Y.K.; Kumar, M. Deep eutectic solvents (DESs) as eco-friendly and sustainable solvent/catalyst systems in organic transformations. J. Mol. Liq. 2016, 215, 345–386. [Google Scholar] [CrossRef]
- Liu, P.; Hao, J.; Mo, L.; Zhang, Z. Recent advances in the application of deep eutectic solvents as sustainable media as well as catalysts in organic reactions. RSC Adv. 2015, 5, 48675–48704. [Google Scholar] [CrossRef]
- Abbott, P.A.; Boothby, D.; Capper, G.; Davies, D.L.; Rasheed, R.K. Deep eutectic solvents formed between choline chloride and carboxylic acids: Versatile alternatives to ionic liquids. J. Am. Chem. Soc. 2004, 126, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; van Spronsen, J.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y.H. Natural deep eutectic solvents as new potential media for green technology. Anal. Chim. Acta 2013, 766, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Capua, M.; Perrone, S.; Perna, F.M.; Vitale, P.; Troisi, L.; Salomone, A.; Capriati, V. An expeditious and greener synthesis of 2-aminoimidazoles in deep eutectic solvents. Molecules 2016, 21, 924. [Google Scholar] [CrossRef] [PubMed]
- Massolo, E.; Palmieri, S.; Benaglia, M.; Capriati, V.; Perna, F.M. Stereoselective organocatalysed reactions in deep eutectic solvents: Highly tunable and biorenewable reaction media for sustainable organic synthesis. Green Chem. 2016, 18, 792–797. [Google Scholar] [CrossRef]
- Azizi, N.; Edrisi, M. Deep eutectic solvent catalyzed eco-friendly synthesis of imines and hydrobenzamides. Monatsh. Chem. 2015, 146, 1695–1698. [Google Scholar] [CrossRef]
- Martínez, R.; Berbegal, L.; Guillena, G.; Ramón, D.J. Bio-renewable enantioselective aldol reaction in natural deep eutectic solvents. Green Chem. 2016, 18, 1724–1730. [Google Scholar] [CrossRef] [Green Version]
- Dilauro, G.; Cicco, L.; Perna, F.M.; Vitale, P.; Capriati, V. Solvent-catalyzed umpolung carbonesulfur bond-forming reactions by nucleophilic addition of thiolate and sulfinate ions to in situederived nitrosoalkenes in deep eutectic solvents. C. R. Chim. 2017, 1–7. [Google Scholar] [CrossRef]
- Maka, H.; Spychaj, T.; Adamus, J. Lewis acid type deep eutectic solvents as catalysts for epoxy resin rosslinking. RSC Adv. 2015, 5, 82813–82821. [Google Scholar] [CrossRef]
- García-Argüelles, S.; Ferrer, M.L.; Iglesias, M.; Del Monte, F.; Gutiérrez, M.C. Study of superbase-sased deep eutectic solvents as the catalyst in the chemical fixation of CO2 into cyclic carbonates under mild conditions. Materials 2017, 10, 759. [Google Scholar] [CrossRef] [PubMed]
- Amić, A.; Molnar, M. An improved and efficient N-acetylation of amines using choline chloride based deep eutectic solvents. Org. Prep. Proc. Int. 2017, 49, 249–257. [Google Scholar] [CrossRef]
- Kontogiorgis, A.C.; Pontiki, A.E.; Hadjipavlou-Litina, D. A Review on quantitative structure-activity relationships (QSARs) of natural and synthetic antioxidants compounds. Mini Rev. Med. Chem. 2005, 5, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Worachartcheewan, A.; Suvannang, N.; Prachayasittikul, S.; Prachayasittikul, V.; Nantasenamat, C. Probing the origins of aromatase inhibitory activity of disubstituted coumarins via QSAR and molecular docking. EXCLI J. 2014, 13, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Erzincan, P.; Saҫan, M.T.; Yüce-Durson, Ö.; Daniş, Ö.; Demir, S.; Erdern, S.S.; Ogan, A. QSAR models for antioxidant activity of new coumarine derivatives. SAR QSAR Environ. Res. 2015, 26, 721–737. [Google Scholar] [CrossRef] [PubMed]
- Mitra, I.; Saha, A.; Roy, K. Predictive modeling of antioxidant coumarin derivatives using multiple approaches: Descriptor-based QSAR. 3D-Pharmacophore mapping and HQSAR. Sci. Pharm. 2013, 81, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Razo-Hernández, R.S.; Pineda-Urbina, K.; Velazco-Medel, M.A.; Villanueva-García, M.; Sumaya-Martínez, M.T.; Sumaya-Martínez, M.T.; Martínez-Martínez, F.J.; Gómez-Sandoval, Z. QSAR study of the DPPH• radical scavenging activity of coumarin derivatives and xanthine oxidase inhibition by molecular docking. Cent. Eur. J. Chem. 2014, 12, 1067–1080. [Google Scholar] [CrossRef]
- Martínez-Martínez, F.J.; Razo-Hernández, R.S.; Peraza-Campos, A.L.; Villanueva-García, M.; Sumaya-Martínez, M.T.; Cano, D.J.; Gómez-Sandoval, Z. Synthesis and in vitro antioxidant activity evaluation of 3-carboxycoumarin derivatives and QSAR study of their DPPH• radical scavenging activity. Molecules 2012, 17, 14882–14898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Y.; Wang, L.-F. Theoretical elucidation of structure–activity relationship for coumarins to scavenge peroxyl radical. J. Mol. Struct. Theochem. 2004, 673. [Google Scholar] [CrossRef]
- Jorge, E.G.; Rayar, A.M.; Barigye, S.J.; Rodríguez, M.E.J.; Veitía, M.S.-I. Development of an in silico model of DPPH free radical scavenging capacity: Prediction of antioxidant activity of coumarin type compounds. Int. J. Mol. Sci. 2016, 17, 881. [Google Scholar] [CrossRef] [PubMed]
- Gacche, R.N.; Jadhav, S.G. Antioxidant activities and cytotoxicity of selected coumarin derivatives: Preliminary results of a structure activity relationship study using computational tools. J. Exp. Clin. Med. 2012, 4, 165–169. [Google Scholar] [CrossRef]
- Mladenović, M.; Mihailović, M.; Bogojević, D.; Matić, S.; Nićiforović, N.; Mihailović, V.; Vuković, N.; Sukdolak, S.; Solujić, S. In vitro antioxidant activity of selected 4-hydroxy-chromene-2-one derivatives—SAR, QSAR and DFT studies. Int. J. Mol. Sci. 2011, 12, 2822–2841. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.N.; Shankarling, G.S. Room temperature ionic liquid choline chloride-oxalic acid: A versatile catalyst for acid-catalyzed transformation in organic reactions. J. Mol. Liq. 2014, 191, 137–141. [Google Scholar] [CrossRef]
- Sokeman, B.B.; Aydin, G.; Gumus, A.; Karadeniz, S.; Ugras, H.I. Synthesis, antielastase, antioxidant and radical scavenging activities of 4-(aza substituted) methylene substituted dihydroxy coumarines. J. Enzym. Inhib. Med. Chem. 2013, 28, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V.; Maiocchi, A. The K correlation index: Theory development and its application in chemometrics. Chemom. Intell. Lab. Syst. 1999, 46, 13–29. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. J. Comput. Aided Mol. Des. 2002, 16, 358–369. [Google Scholar] [CrossRef]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.-D.; Lee, K.-H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Masand, V.H.; Mahajan, D.T.; Nazeruddin, G.M.; Hadda, T.B.; Rastija, V.; Alfeedy, A.M. Effect of information leakage and method of splitting (rational and random) on external predictive ability and behavior of different statistical parameters of QSAR model. Med. Chem. Res. 2015, 24, 1241–1264. [Google Scholar] [CrossRef]
- Roy, K.; Mitra, I. On the use of the metric rm(2) as an effective tool for validation of QSAR models in computational drug design and predictive toxicology. Mini Rev. Med. Chem. 2012, 491–504. [Google Scholar] [CrossRef]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.D.; McDowell, R.M.; Gramatica, P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef]
- Tominaga, H.; Kobayashi, Y.; Goto, T.; Kasemura, K.; Nomura, M. DPPH radical-scavenging effect of several phenylpropanoid compounds and their glycoside derivatives. Yakugaku Zasshi 2005, 125, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Devinyak, O.; Havrylyuk, D.; Lesyk, R. 3D-MoRSE descriptors explained. J. Mol. Graph. Model. 2014, 54, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Jukić, M.; Rastija, V.; Opačak-Bernardi, T.; Stolić, I.; Krstulović, L.; Bajić, M.; Glavaš-Pbrovoac, L. Antitumor activity of 3,4-ethylenedioxythiophene derivatives and quantitative structure-activity relationship analysis. J. Mol. Struct. 2017, 1133, 66–73. [Google Scholar] [CrossRef]
- Moran, P.A.P. Notes on continuous stochastic phenomena. Biometrika 1950, 37, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Vighi, M.; Finizio, A.; Gramatica, P. 30-Modelling and Prediction by WHIM Descriptors. Part 8. Toxicity and Physico-chemical Properties of Environmental Priority Chemicals by 2D-TI and 3D-WHIM Descriptors. SAR QSAR Environ. Res. 1997, 7, 173–193. [Google Scholar] [CrossRef] [PubMed]
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef]
- Weiner, P.K.; Langridge, R.; Blaney, J.M.; Schaefer, R.; Kollman, P.A. Electrostatic potential molecular surface. Proc. Natl. Acad. Sci. USA 1982, 79, 3754–3758. [Google Scholar] [CrossRef] [PubMed]
- Kidwai, M.; Poddar, R.; Jain, A.; Kumar, R.; Luthar, P.M. Synthesis and SARs of coumarin fused 1,5-benzothiazepines as novel anticancer and antioxidant agents. Mini-Rev. Org. Chem. 2015, 12, 24–33. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Hocquet, A.; Langgård, M. An evaluation of the MM+ force field. Mol. Model. 1998, 4, 94–112. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. AM1: A new general purpose quantum mechanical model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory-design and description. J. Comput. Aided. Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. Mol. Inf. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inf. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 3–36 are available from the authors. |
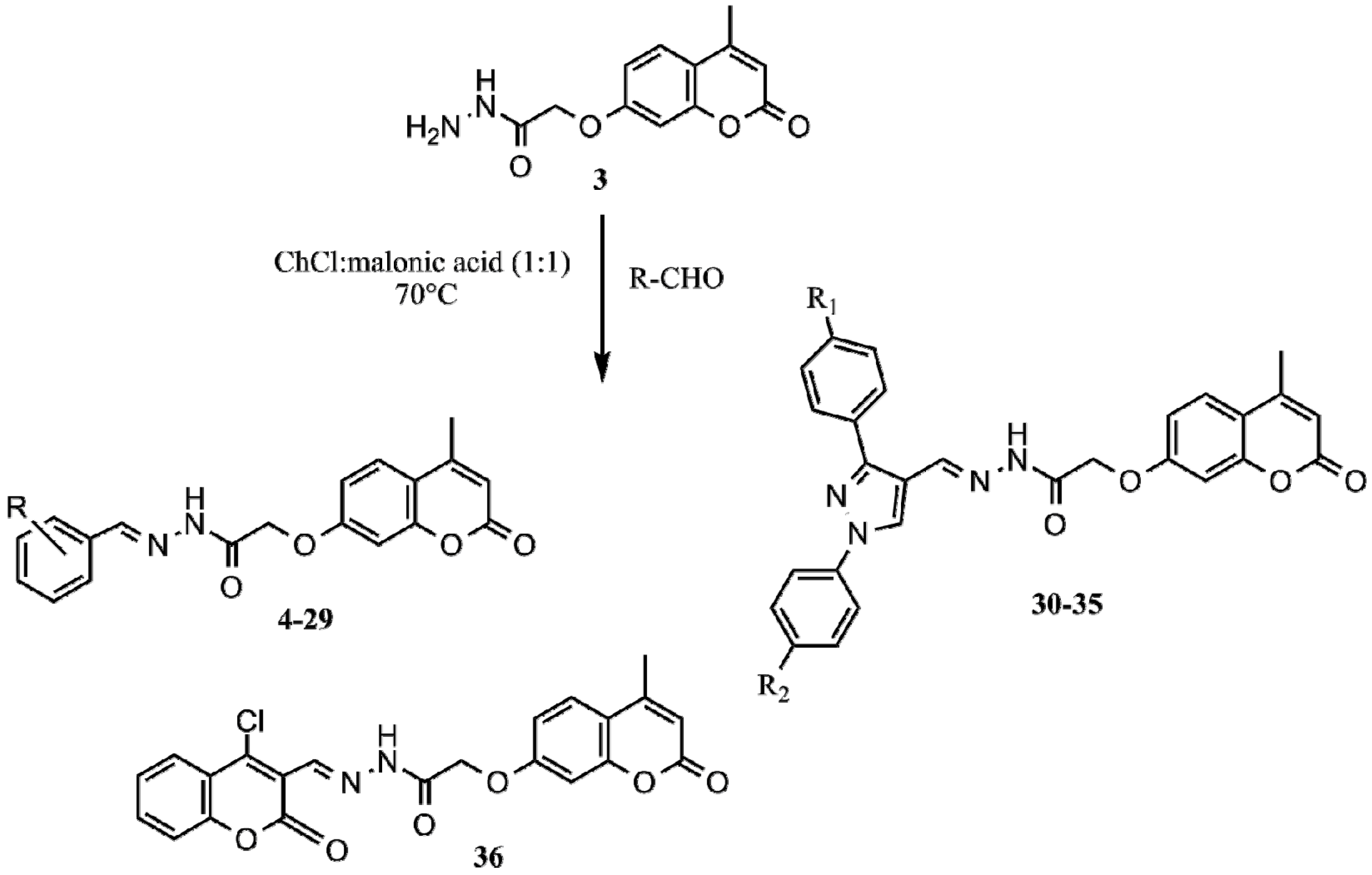
| Compound | R | Compound | R | Compound | R |
|---|---|---|---|---|---|
| 4 | H | 17 | 3-O-Ph | 30 | R1 = Cl; R2 = F |
| 5 | 2-OH | 18 | 2-OH; 5-NO2 | 31 | R1 = Br; R2 = CH3 |
| 6 | 3-OH | 19 | 3,4,5-OCH3 | 32 | R1 = I; R2 = CH3 |
| 7 | 4-OH | 20 | 2-Cl | 33 | R1 = OCH3; R2 = CH3 |
| 8 | 2-OCH3 | 21 | 3-Cl | 34 | R1 = Cl; R2 = CH3 |
| 9 | 3-OCH3 | 22 | 2-Br | 35 | R1 = NO2; R2 = Cl |
| 10 | 4-OCH3 | 23 | 3-Br | ||
| 11 | 2,3-OH | 24 | 4-Br | ||
| 12 | 2,4-OH | 25 | 2-F | ||
| 13 | 2,5-OH | 26 | 3-F | ||
| 14 | 3,4-OH | 27 | 4-F | ||
| 15 | 3,5-OH | 28 | styryl | ||
| 16 | 3-OCH3; 4-OH | 29 | 4-N(CH3)2 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
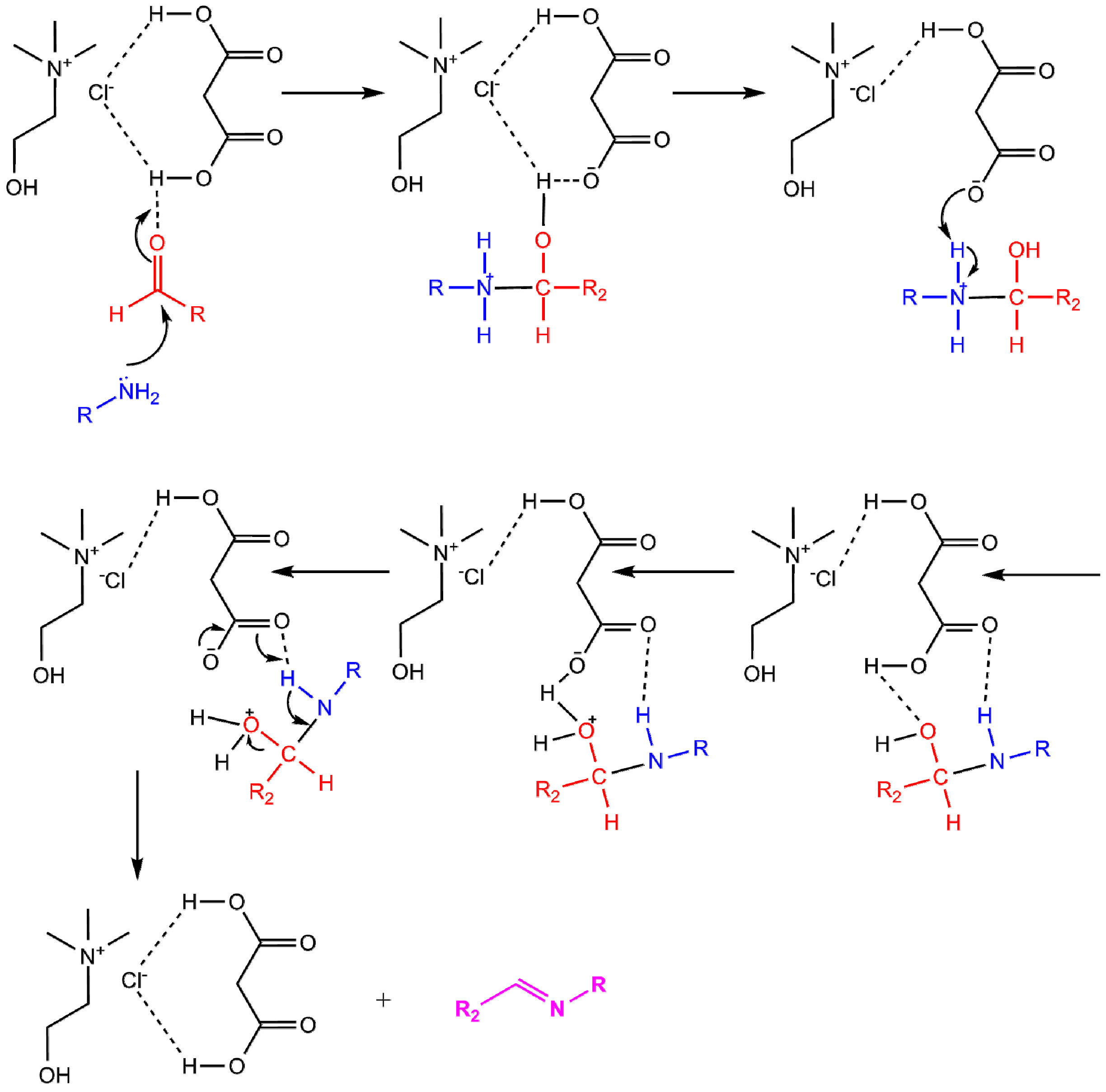{kind=link}
 |  | ||||
| Mol. ID | R1/R2 | % DPPH | Mol. ID | R | % DPPH |
| 1 | H-/H- | 1.8 ± 0.22 | 3 | H2N- | 14.4 ± 0.81 |
| 2 | -CH3/-OH | 2.4 ± 0.12 | 28 |  | 1.6 ± 0.00 |
| 36 |  | 14.1 ± 0.10 | |||
 | |||||
|---|---|---|---|---|---|
| Mol. ID | Supstituents | % DPPH | Mol. ID | Supstituents | % DPPH |
| 4 | H | 22.9 ± 0.50 | 16 | 3-OCH3; 4-OH | 32.0 ± 0.25 |
| 5 | 2-OH | 16.6 ± 0.56 | 17 | 3-O-Ph | 2.2 ± 0.68 |
| 6 | 3-OH | 3.6 ± 0.24 | 18 | 2-OH; 5-NO2 | 2.9 ± 0.99 |
| 7 | 4-OH | 4.4 ± 0.37 | 19 | 3,4,5-OCH3 | 3.5 ± 0.42 |
| 8 | 2-OCH3 | 3.1 ± 0.86 | 20 | 2-Cl | 11.1 ± 0.87 |
| 9 | 3-OCH3 | 0.2 ± 0.08 | 21 | 3-Cl | 7.5 ± 0.37 |
| 10 | 4-OCH3 | 3.7 ± 0.23 | 22 | 2-Br | 6.6 ± 0.62 |
| 11 | 2,3-OH | 75.4 ± 2.62 | 23 | 3-Br | 3.0 ± 0.32 |
| 12 | 2,4-OH | 8.4 ± 0.12 | 24 | 4-Br | 5.0 ± 0.27 |
| 13 | 2,5-OH | 33.4 ± 3.74 | 25 | 2-F | 3.0 ± 0.76 |
| 14 | 3,4-OH | 42.6 ± 4.10 | 26 | 3-F | 2.3 ± 0.68 |
| 15 | 3,5-OH | 4.6 ± 2.17 | 27 | 4-F | 7.4 ± 0.12 |
| 29 | 4-N(CH3)2 | 5.9 ± 0.62 | |||
 | |||||
| Mol. ID | R | % DPPH | Mol. ID | R | % DPPH |
| 30 | R1 = Cl; R2 = F | 1.6 ± 0.23 | 33 | R1 = OCH3; R2 = CH3 | 0.8 ± 0.09 |
| 31 | R1 = Br; R2 = CH3 | 0.5 ± 0.01 | 34 | R1 = Cl; R2 = CH3 | 1.4 ± 0.19 |
| 32 | R1 = I; R2 = CH3 | 1.6 ± 0.18 | 35 | R1 = NO2; R2 = Cl | 1.6 ± 0.31 |
| Model (1) | Model (2) | |
|---|---|---|
| Fittinig criteria | ||
| R2 | 0.636 | 0.673 |
| R2adj | 0.592 | 0.632 |
| s | 0.369 | 0.315 |
| F | 14.559 | 16.467 |
| Kxx | 0.242 | 0.241 |
| ΔK | 0.170 | 0.178 |
| RMSEtr | 0.342 | 0.292 |
| MAEtr | 0.271 | 0.241 |
| CCCtr | 0.778 | 0.805 |
| Internal validation criteria | ||
| Q2LOO | 0.512 | 0.544 |
| RMSEcv | 0.397 | 0.345 |
| MAEcv | 0.314 | 0.285 |
| PRESScv | 4.560 | 3.326 |
| CCCcv | 0.710 | 0.733 |
| R2Y scr | 0.108 | 0.112 |
| Q2Y scr | −0.214 | −0.221 |
| External validation criteria | ||
| RMSEext | 0.299 | 0.311 |
| MAEext | 0.271 | 0.283 |
| PRESSext | 0.627 | 0.677 |
| R2ext | 0.709 | 0.712 |
| Q2F1 | 0.523 | 0.558 |
| Q2F 2 | 0.169 | 0.103 |
| Q2F3 | 0.722 | 0.629 |
| CCCext | 0.732 | 0.701 |
| 0.619 | 0.600 | |
| 0.050 | 0.197 | |
| Applicability domain (h* = 0.4138) | ||
| N compounds outlier | 1 (9) | 0 |
| N compounds out of app.dom. | 0 | 0 |
| MATS3m | Mor22u | Hy | |
|---|---|---|---|
| MATS3m | 1 | ||
| Mor22u | −0.260 | 1 | |
| Hy | 0.194 | 0.309 | 1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molnar, M.; Komar, M.; Brahmbhatt, H.; Babić, J.; Jokić, S.; Rastija, V. Deep Eutectic Solvents as Convenient Media for Synthesis of Novel Coumarinyl Schiff Bases and Their QSAR Studies. Molecules 2017, 22, 1482. https://doi.org/10.3390/molecules22091482
Molnar M, Komar M, Brahmbhatt H, Babić J, Jokić S, Rastija V. Deep Eutectic Solvents as Convenient Media for Synthesis of Novel Coumarinyl Schiff Bases and Their QSAR Studies. Molecules. 2017; 22(9):1482. https://doi.org/10.3390/molecules22091482
Chicago/Turabian StyleMolnar, Maja, Mario Komar, Harshad Brahmbhatt, Jurislav Babić, Stela Jokić, and Vesna Rastija. 2017. "Deep Eutectic Solvents as Convenient Media for Synthesis of Novel Coumarinyl Schiff Bases and Their QSAR Studies" Molecules 22, no. 9: 1482. https://doi.org/10.3390/molecules22091482
APA StyleMolnar, M., Komar, M., Brahmbhatt, H., Babić, J., Jokić, S., & Rastija, V. (2017). Deep Eutectic Solvents as Convenient Media for Synthesis of Novel Coumarinyl Schiff Bases and Their QSAR Studies. Molecules, 22(9), 1482. https://doi.org/10.3390/molecules22091482