Coordination Mechanism and Bio-Evidence: Reactive γ-Ketoenal Intermediated Hepatotoxicity of Psoralen and Isopsoralen Based on Computer Approach and Bioassay


Abstract
:1. Introduction
2. Results
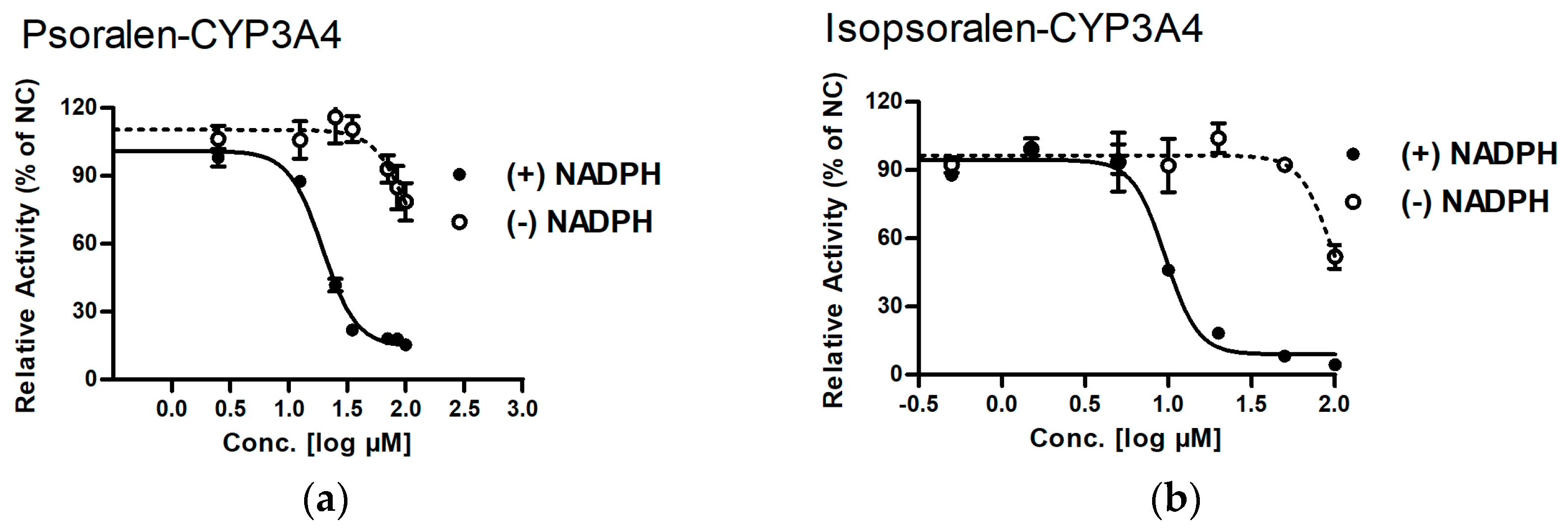
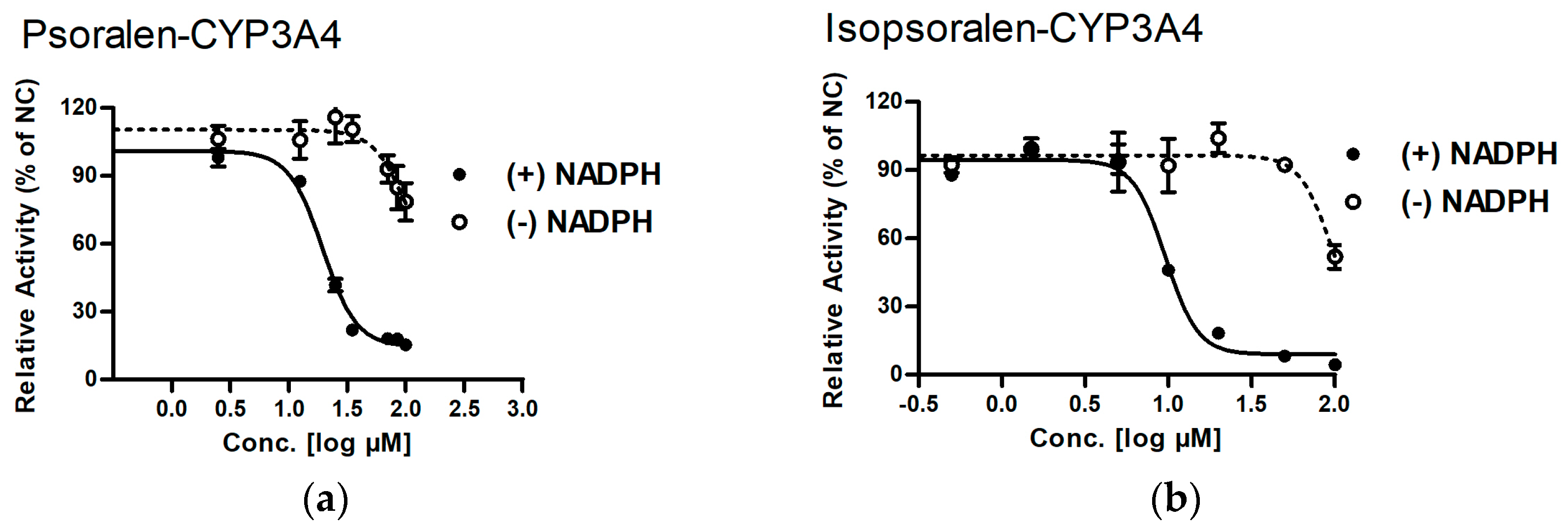
2.1. MBI Evaluation of Psoralen and Isopsoralen
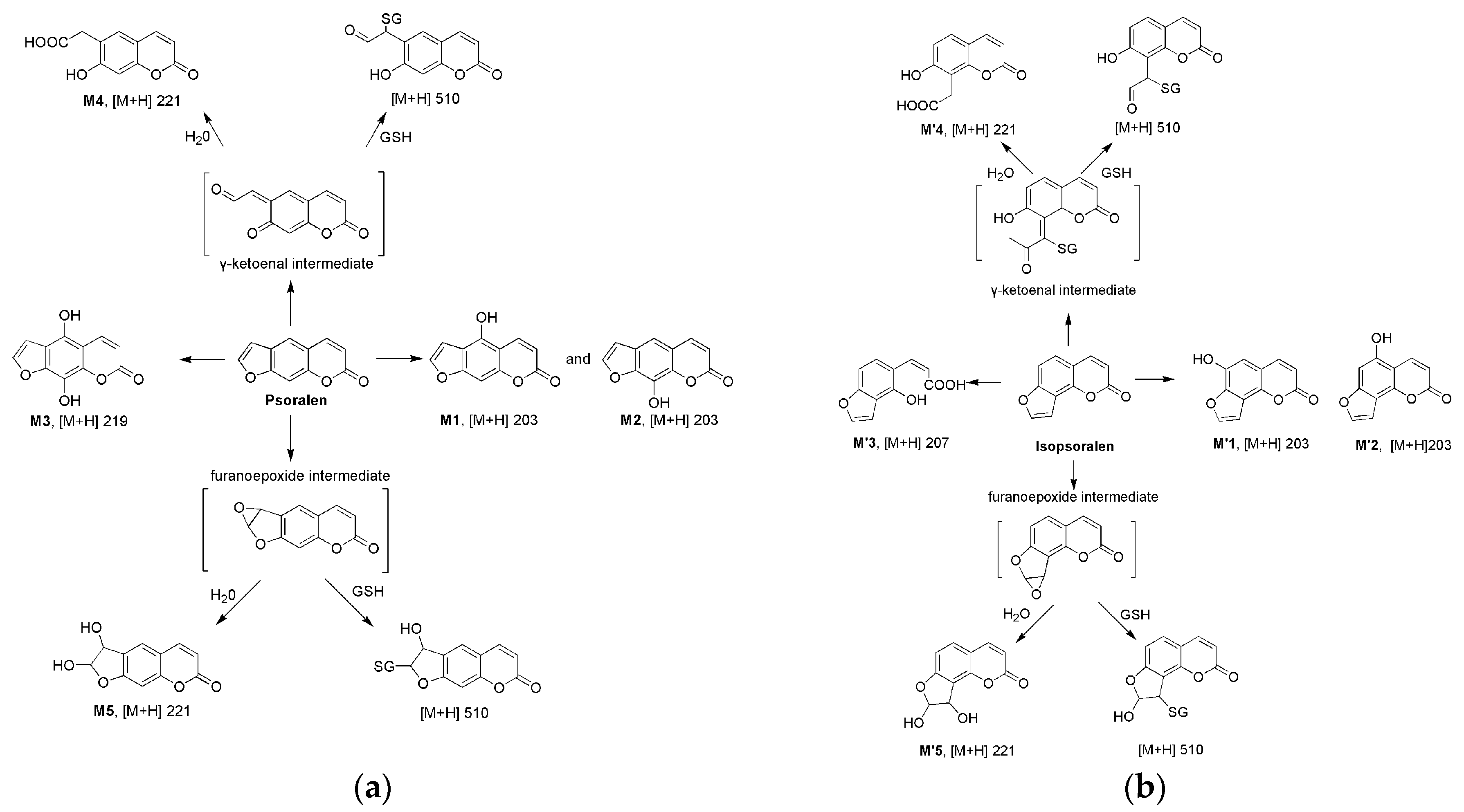
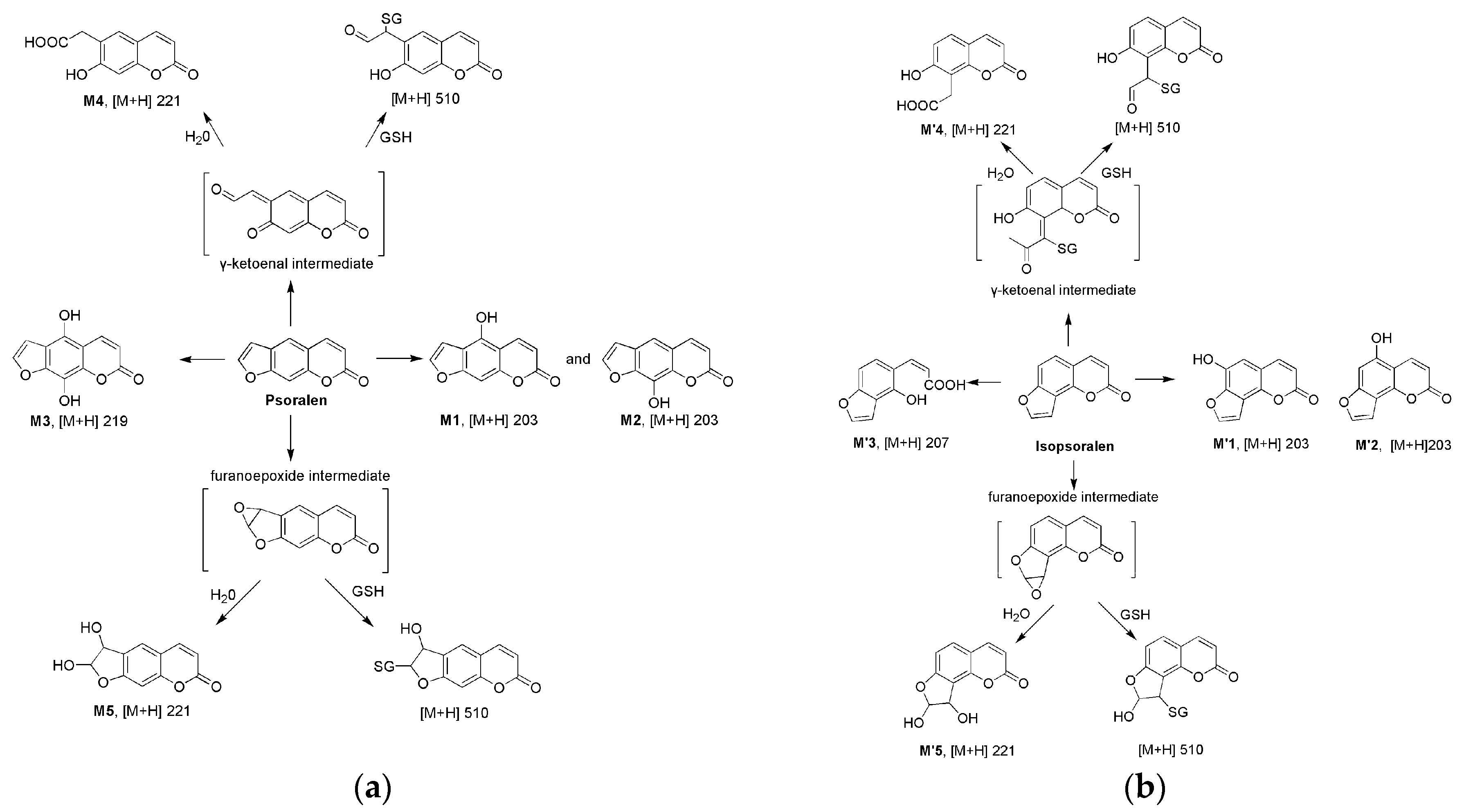
2.2. Metabolism of Psoralen and Isopsoralen in Liver Microsomes
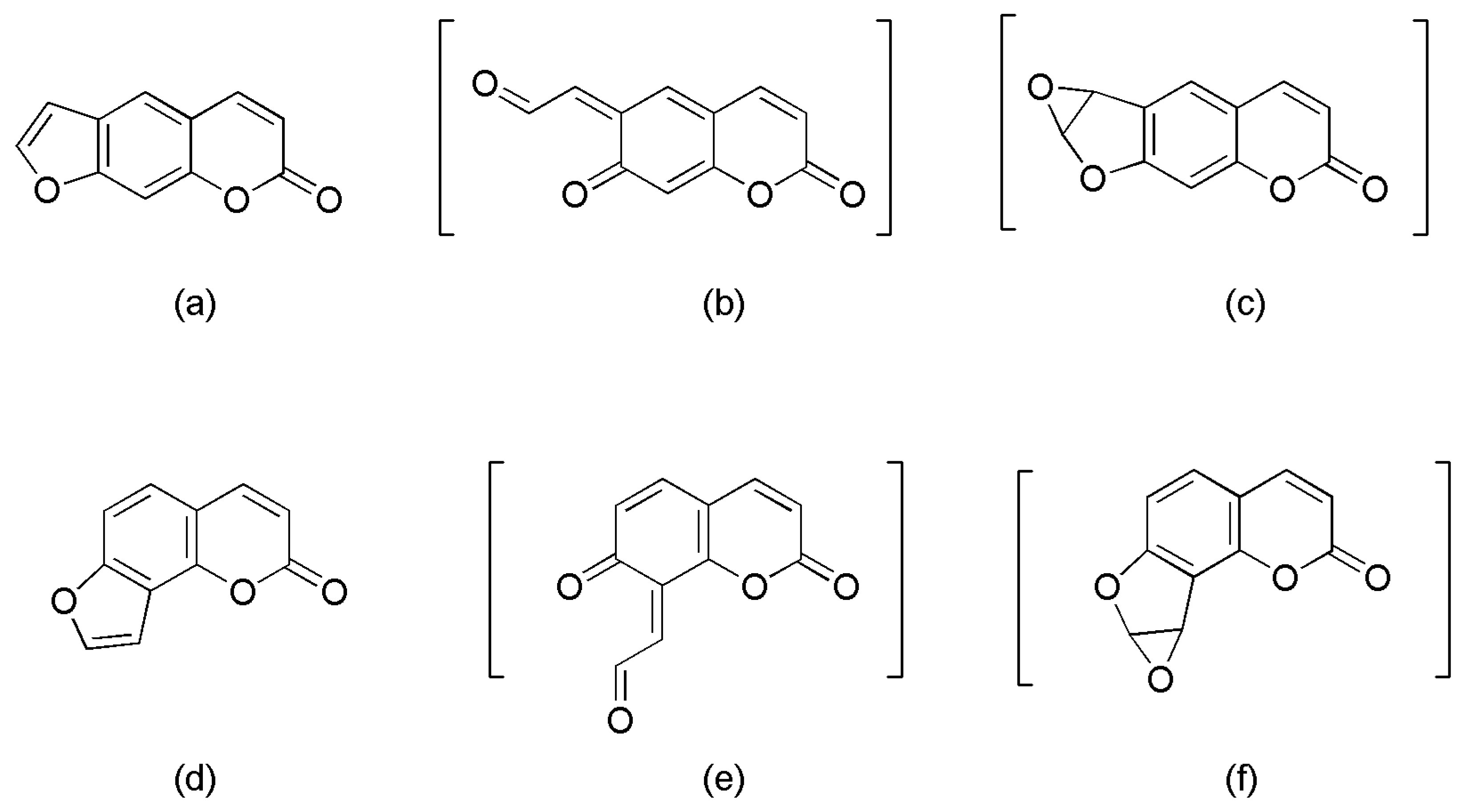
2.3. Identifying Reactive Metabolites Based on Mechanism-Based Inhibition
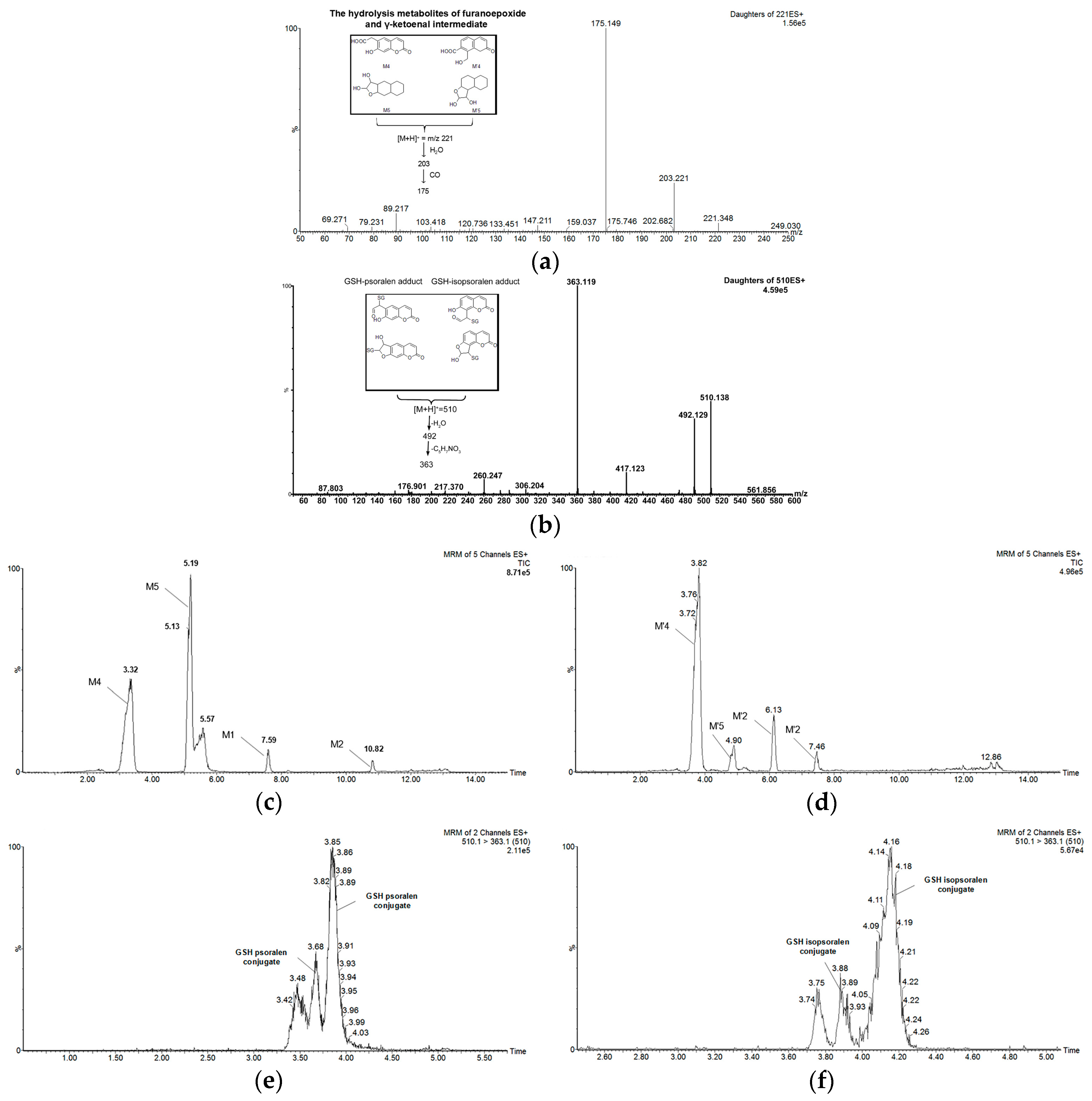
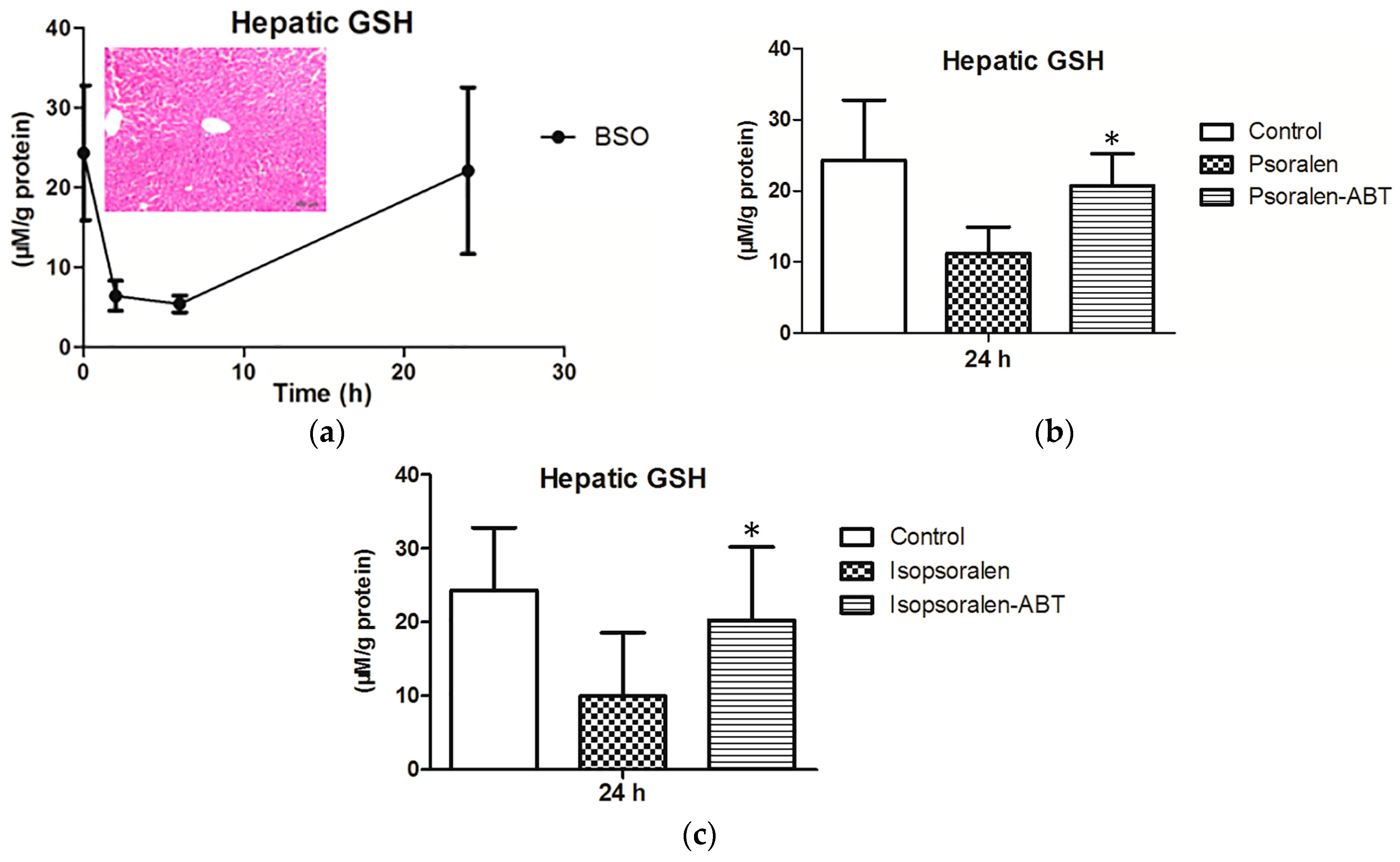
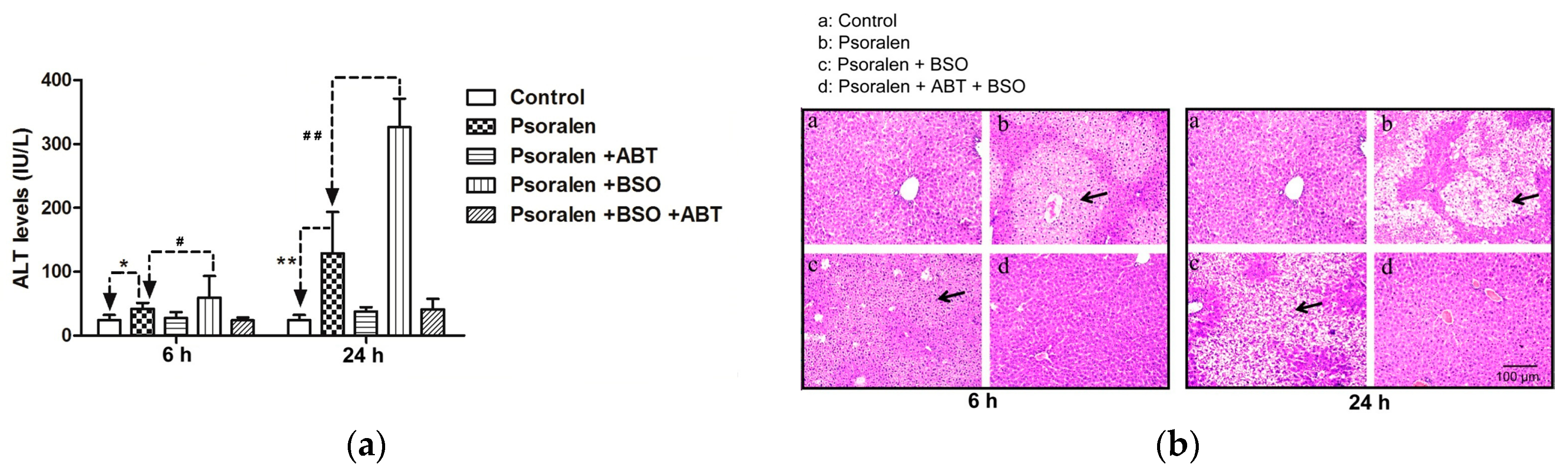
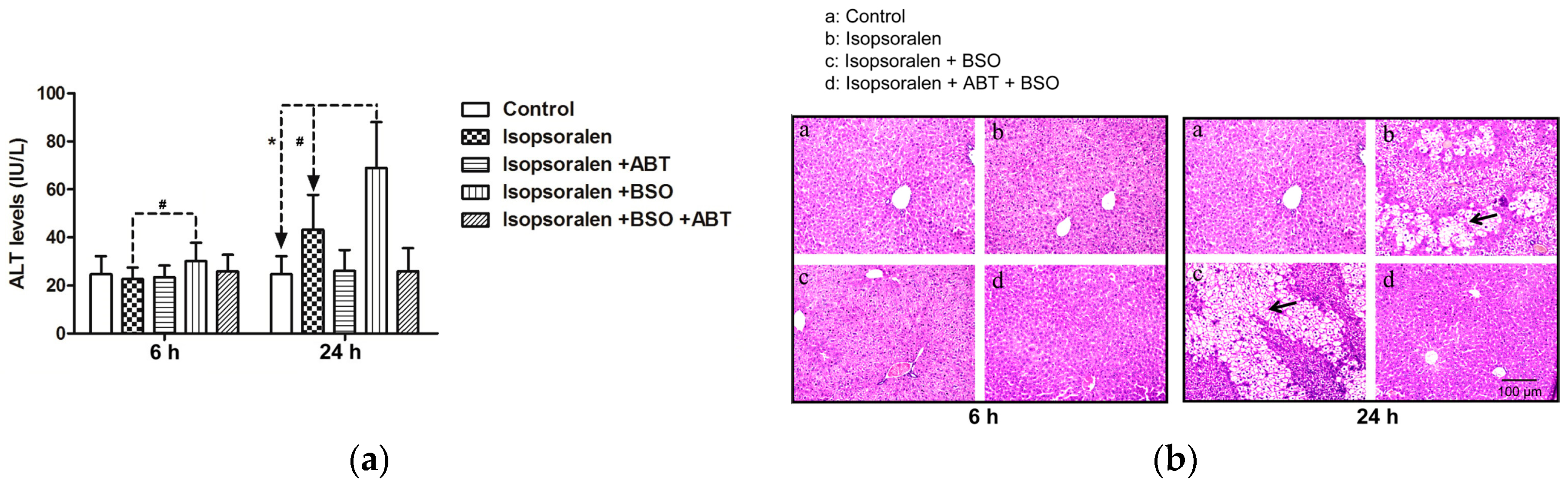
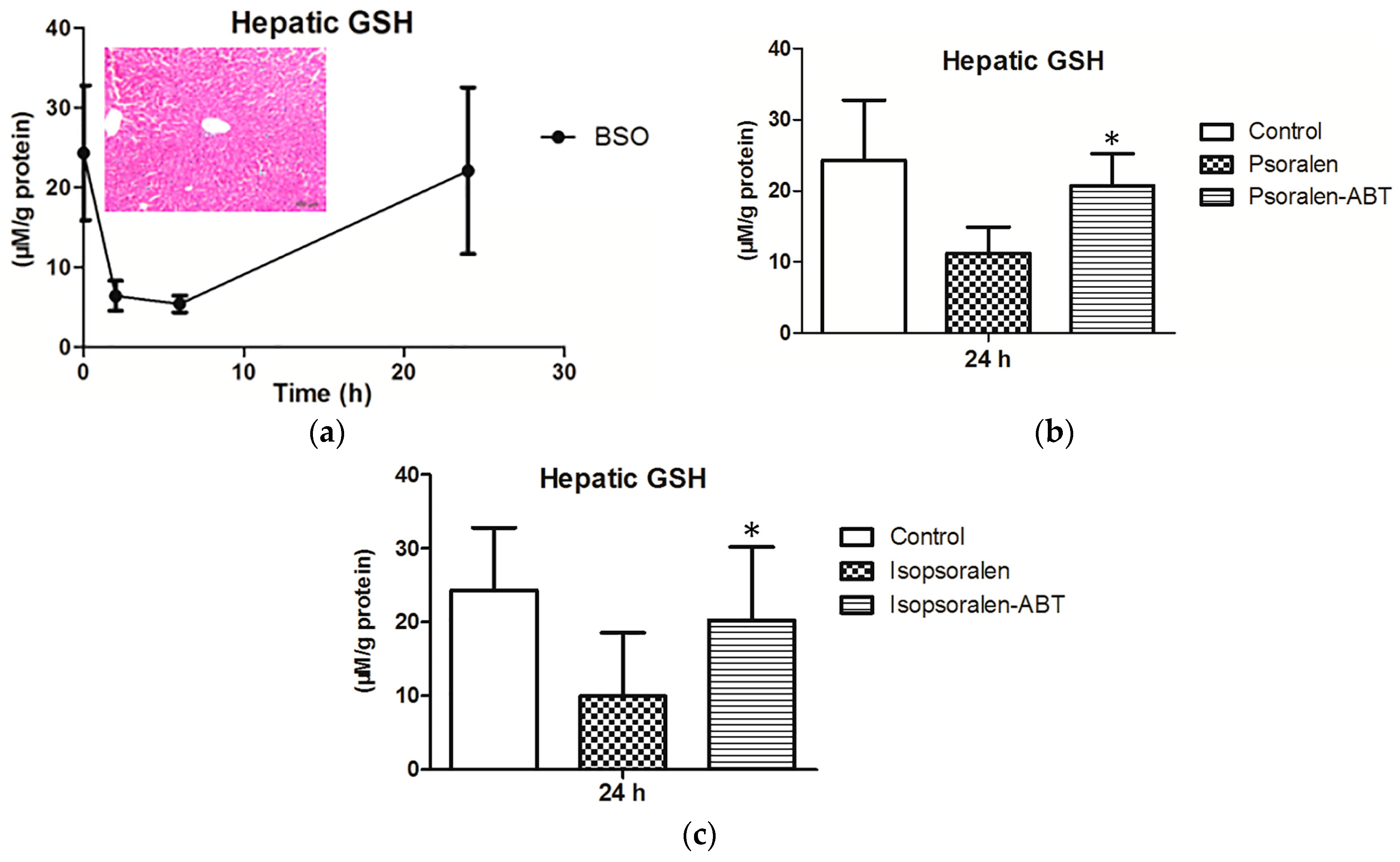
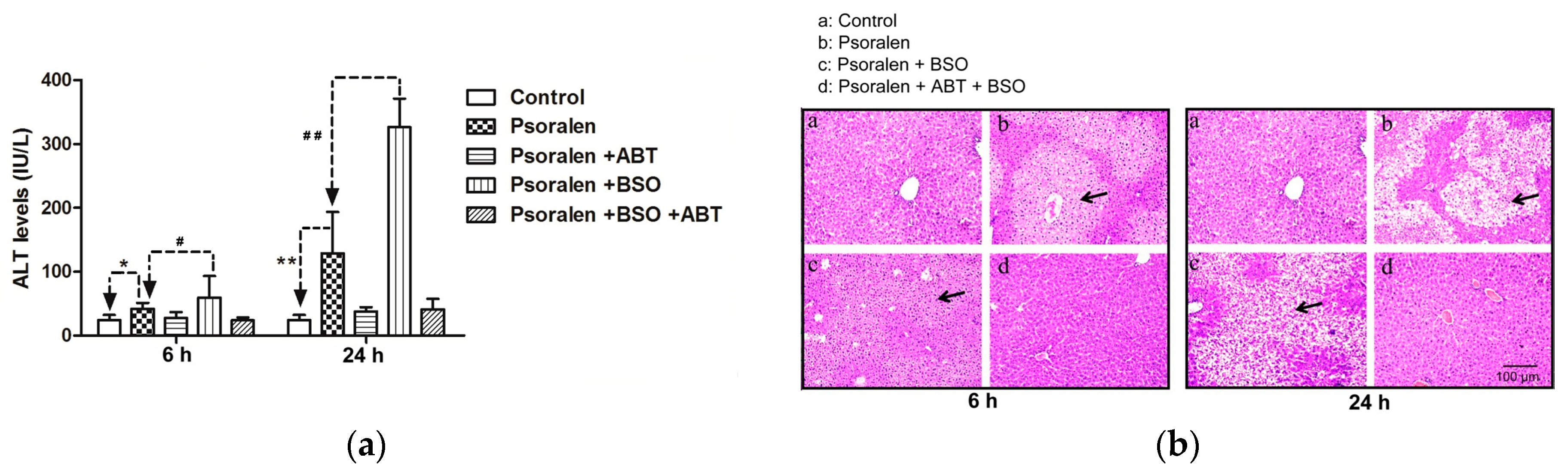
2.4. Evidence for Reactive Metabolites-Induced Hepatotoxicity
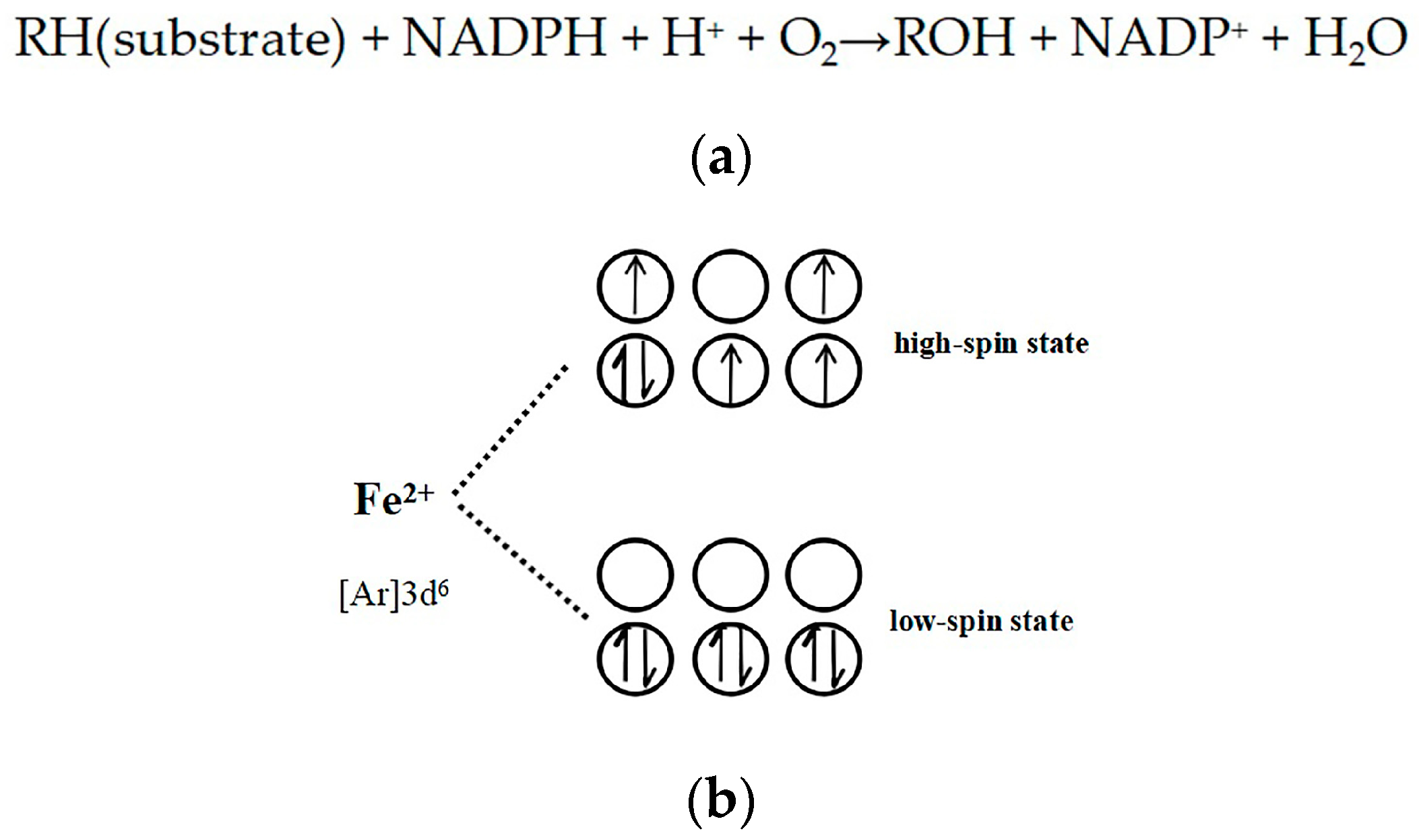
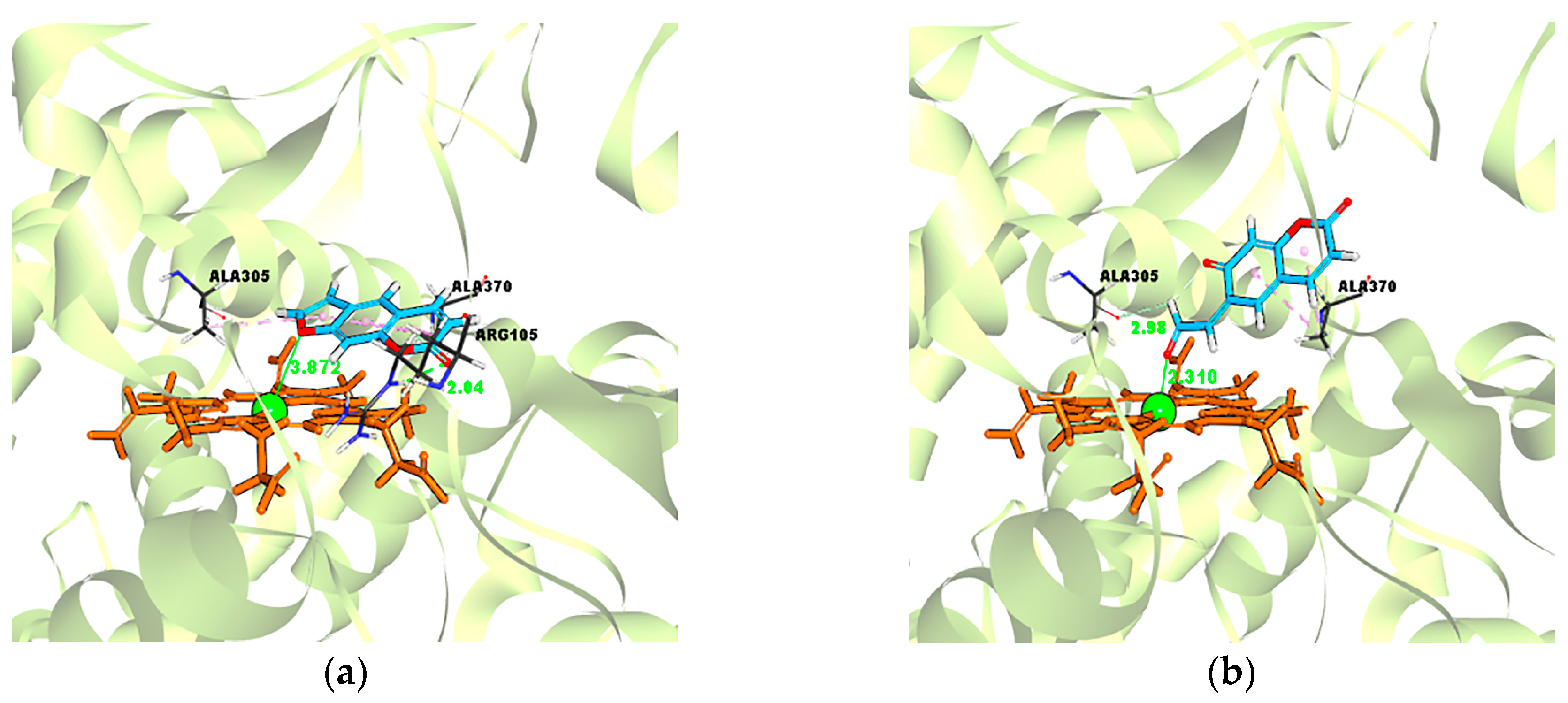
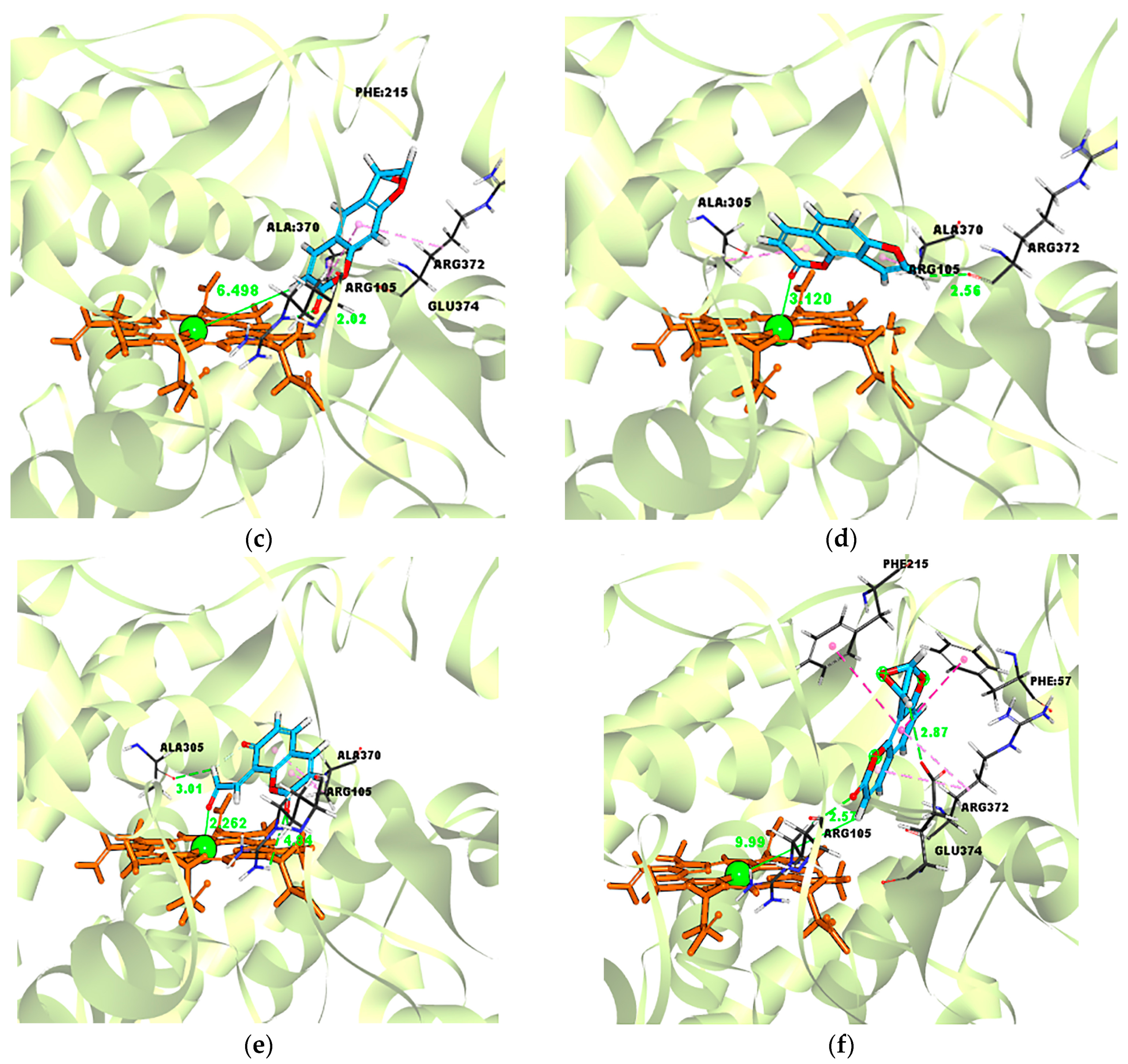
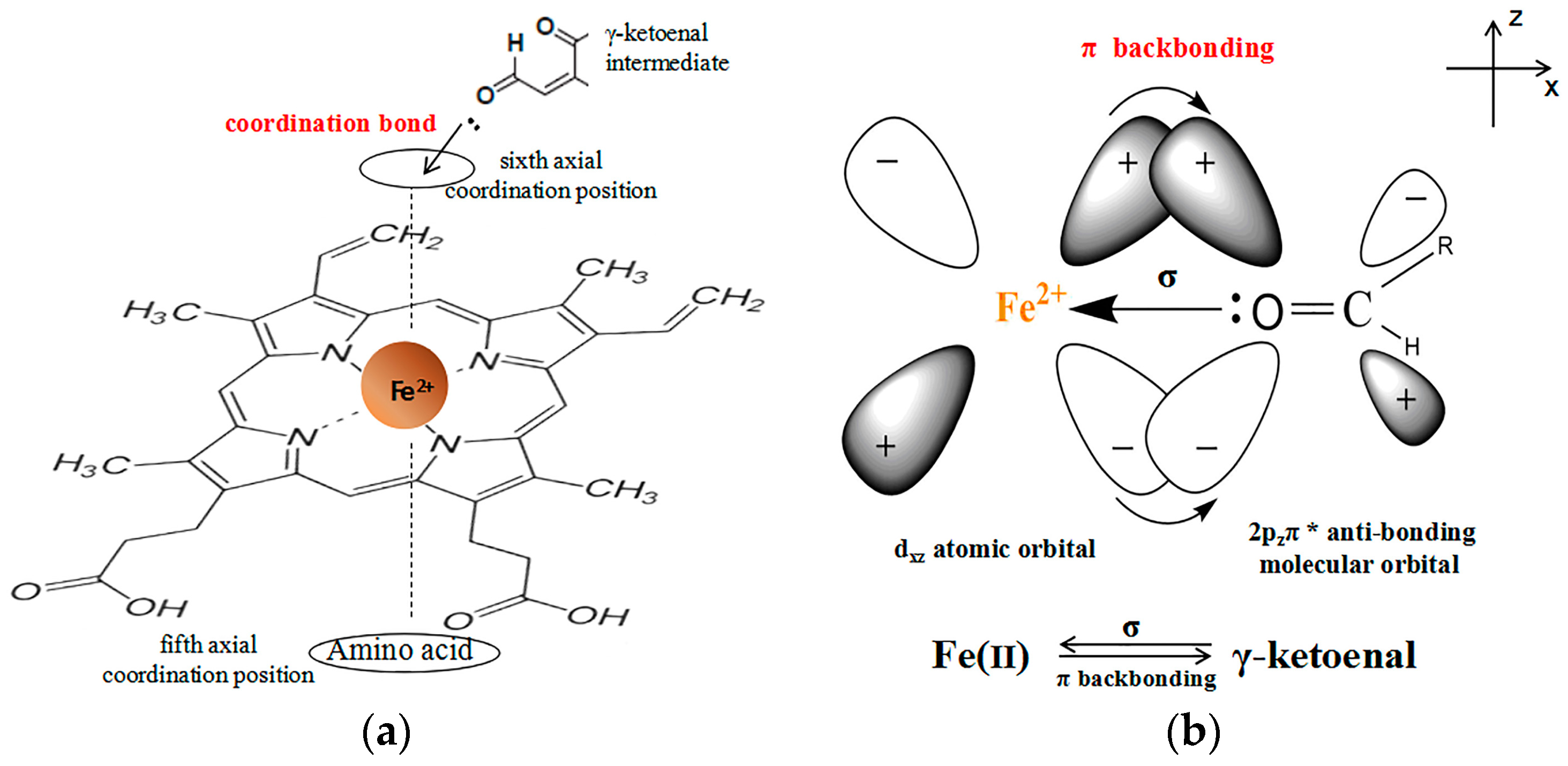
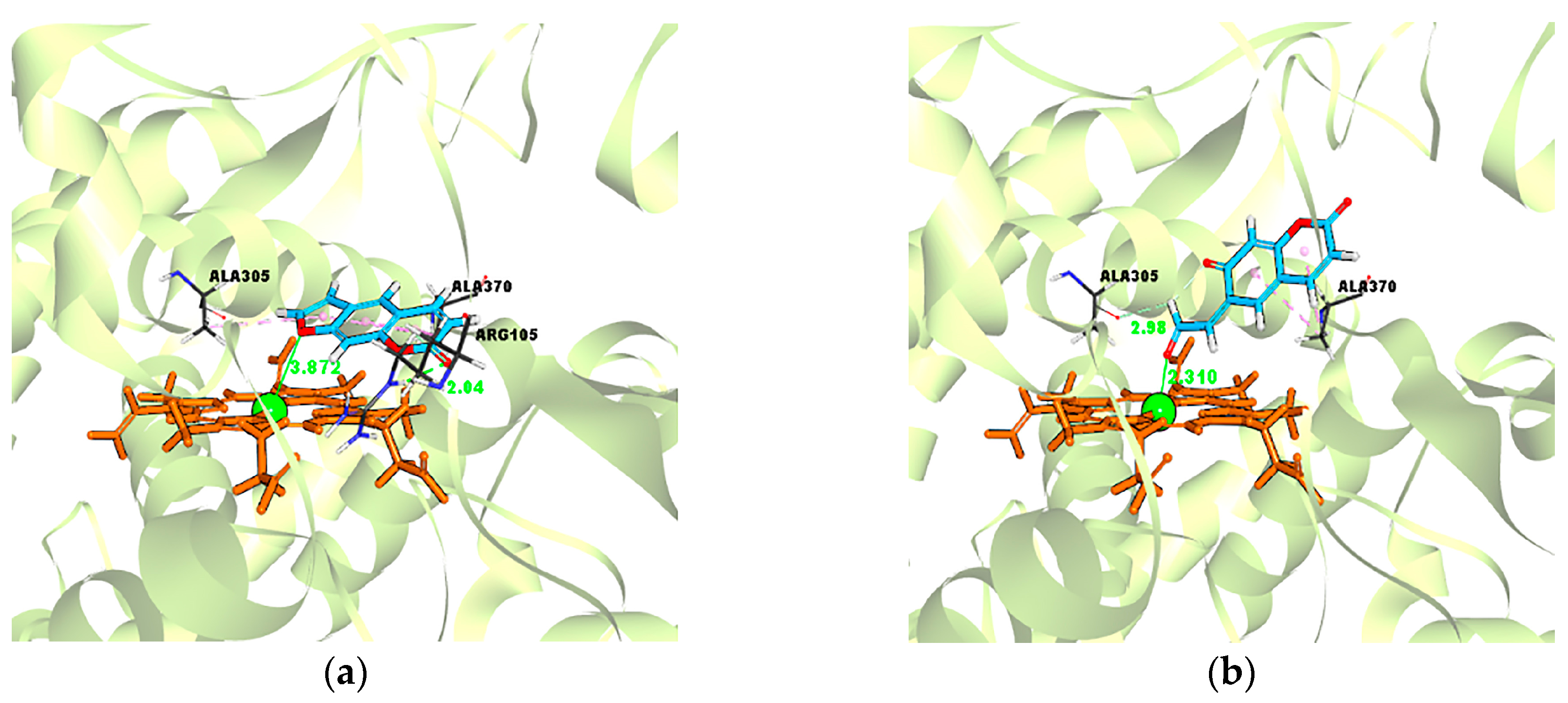
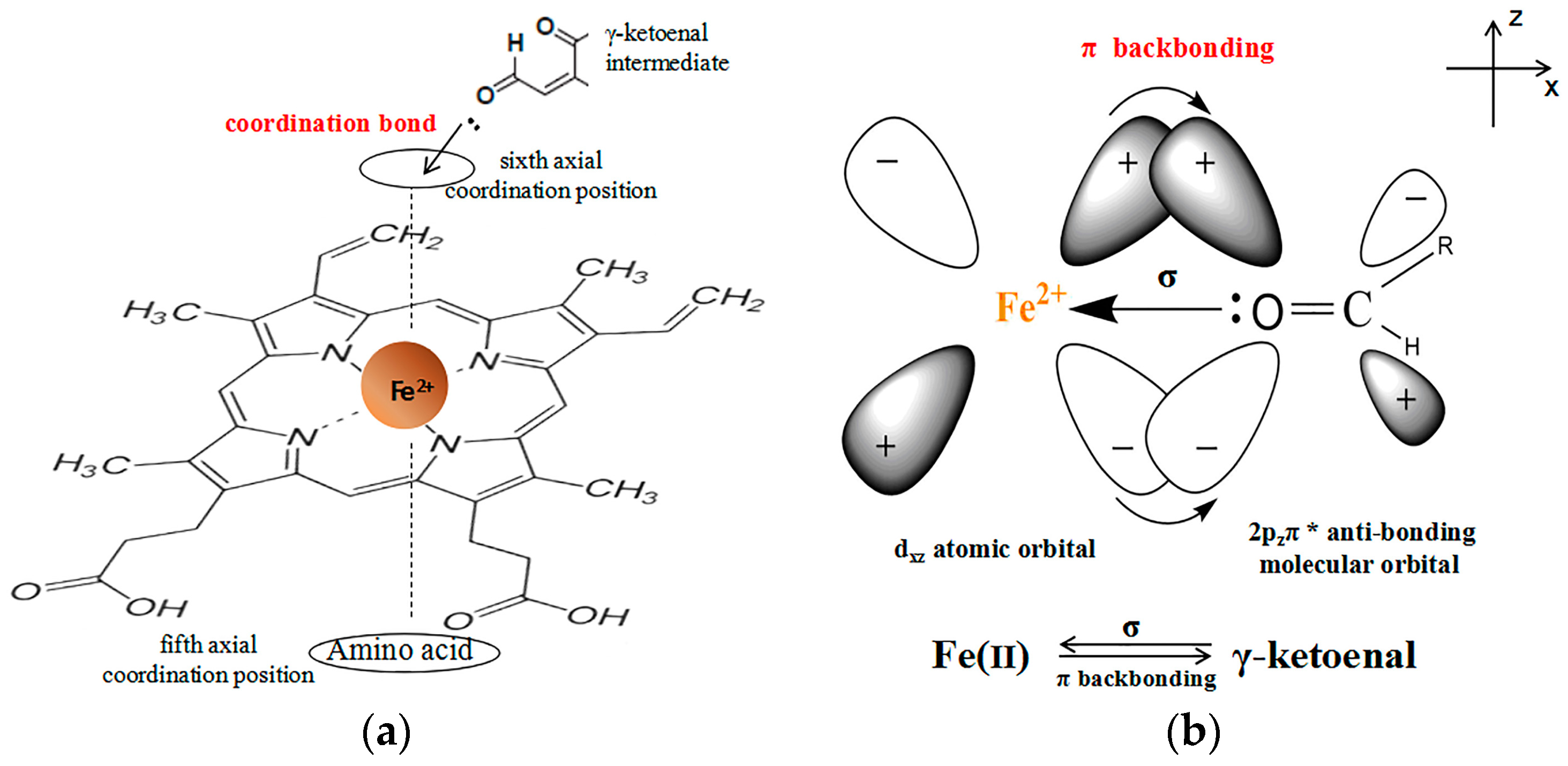
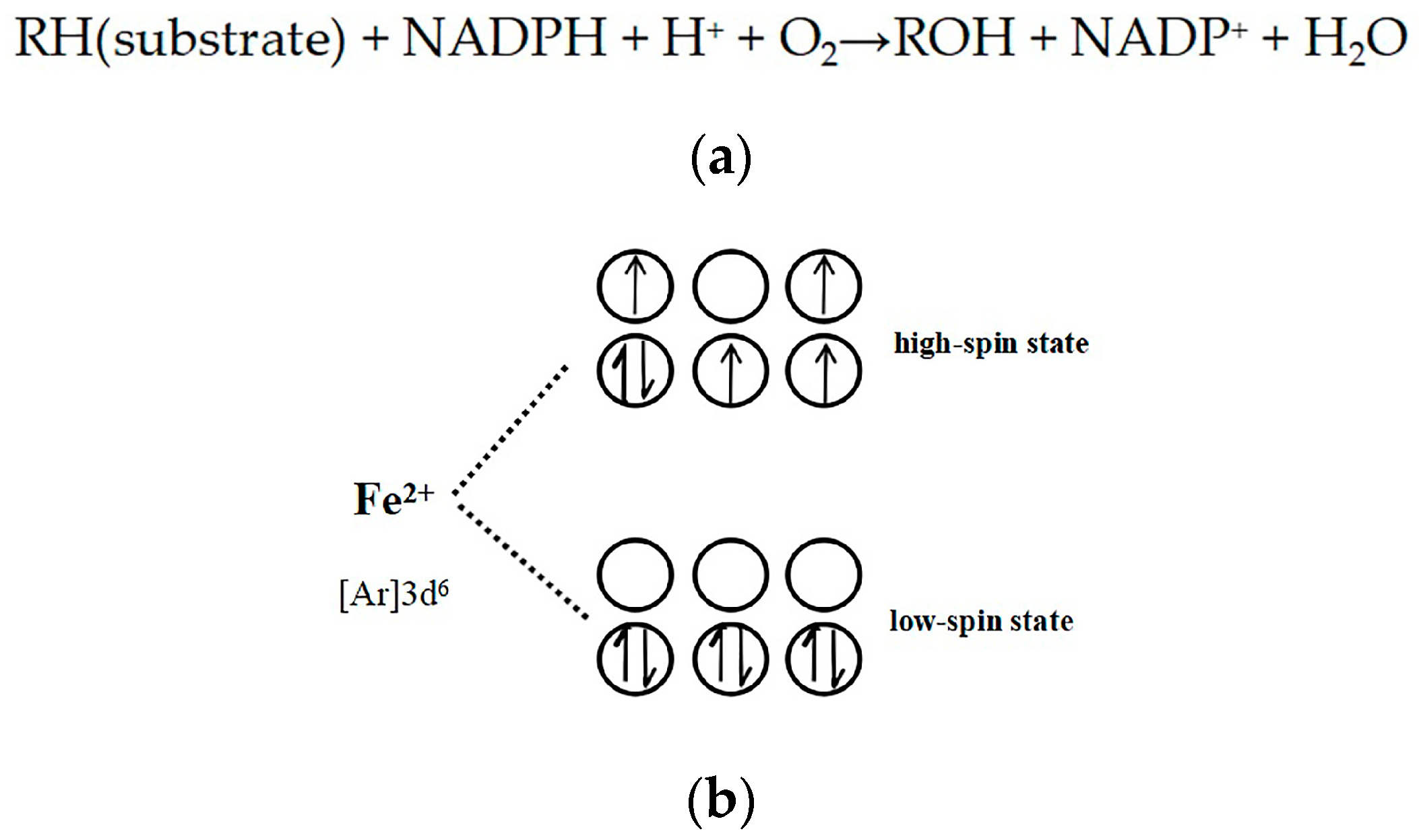
2.5. Evidence for the Reactive γ-Ketoenal Metabolite–CYP3A4 Heme Fe(II) Coordination Complex
3. Discussion
4. Materials and Methods
4.1. Inhibitory Effects of Psoralen and Isopsoralen on CYP3A4
4.2. MBI Evaluation of Psoralen and Isopsoralen
4.3. Effects of Scavenging Agents (GSH) on Inhibition Ability of Psoralen and Isopsralen
4.4. Metabolism of Psoralen and Isopsoralen in Liver Microsome
4.5. Animals, Treatment, and Sampling
4.6. Molecular Docking Simulation
4.7. Data Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Conforti, F.; Marrelli, M.; Menichini, F.; Bonesi, M.; Statti, G.; Provenzano, E.; Menichini, F. Natural and synthetic furanocoumarins as treatment for vitiligo and psoriasis. Curr. Drug Ther. 2009, 4, 38–58. [Google Scholar] [CrossRef]
- Lapolla, W.; Yentzer, B.A.; Bagel, J.; Halvorson, C.R.; Feldman, S.R. A review of phototherapy protocols for psoriasis treatment. J. Am. Acad. Dermatol. 2011, 64, 936–949. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, H.; Inoue, J.; Tamura, Y.; Mizutani, K. Antioxidative components of Psoralea corylifolia (Leguminosae). Phytother. Res. 2010, 16, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Ha, T.Y.; Ahn, J.; Kim, S. Estrogenic activities of Psoralea corylifolia L. seed extracts and main constituents. Phytomedicine 2011, 18, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Sun, Z.; Ye, Y.; Han, X.; Song, X.; Liu, S. Psoralen inhibits bone metastasis of breast cancer in mice. Fitoterapia 2013, 91, 205–210. [Google Scholar] [CrossRef] [PubMed]
- China Food and Drug Administration Website. Available online: http://www.cdr-adr.org.cn/xxtb_255/ypblfyxxtb/200806/t20080626_2823.html (accessed on 26 June 2008).
- China Food and Drug Administration Website. Available online: http://www.cdr-adr.org.cn/xxtb_255/ypblfyxxtb/200812/t20081216_2816.html (accessed on 16 December 2008).
- Danan, G.; Teschke, R. RUCAM in drug and herb induced liver injury: The update. Int. J. Mol. Sci. 2016, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Eickhoff, A. Herbal hepatotoxicity in traditional and modern medicine: Actual key issues and new encouraging steps. Front. Pharmacol. 2015, 6, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Bahre, R. Severe hepatotoxicity by Indian Ayurvedic herbal products: A structured causality assessment. Ann. Hepatol. 2009, 8, 258–266. [Google Scholar] [PubMed]
- Pariser, D.M.; Wyles, R.J. Toxic hepatitis from oral methoxsalen photochemotherapy (PUVA). J. Am. Acad. Dermatol. 1980, 3, 248–250. [Google Scholar] [CrossRef]
- Nyfors, A.; Dahl-Nyfors, B.; Hopwood, D. Liver biopsies from patients with psoriasis related to photochemotherapy (PUVA): Findings before and after 1 year of therapy in twelve patients. A blind study and review of literature on hepatotoxicity of PUVA. J. Am. Acad. Dermatol. 1986, 14, 43–48. [Google Scholar] [CrossRef]
- Diawara, M.M.; Williams, D.E.; Oganesian, A.; Spitsbergen, J. Dietarypsoralens induce hepatotoxicity in C57 mice. J. Nat. Toxins 2000, 9, 179. [Google Scholar] [PubMed]
- Wang, X.; Lou, Y.J.; Wang, M.X.; Shi, Y.W.; Xu, H.X.; Kong, L.D. Furocoumarins affect hepatic cytochrome P450 and renal organic ion transporters in mice. Toxicol. Lett. 2012, 209, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Mays, D.C.; Hilliard, J.B.; Wong, D.D.; Chambers, M.A.; Park, S.S.; Gelboin, H.V.; Gerber, N. Bioactivation of 8-Methoxypsoralen and irreversible inactivation of cytochrome P-450 in mouse liver microsomes: Modification by monoclonal antibodies, inhibition of drug metabolism and distribution of covalent adducts. J. Pharmacol. Exp. Ther. 1990, 254, 720–731. [Google Scholar] [PubMed]
- Koenigs, L.L.; Trager, W.F. Mechanism-based inactivation of P450 2A6 by furanocoumarins. Biochemistry 1998, 37, 10047–10061. [Google Scholar] [CrossRef] [PubMed]
- Koenigs, L.L.; Trager, W.F. Mechanism-Based inactivation of cytochrome P450 2B1 by 8-Methoxypsoralen and several other furanocoumarins. Biochemistry 1998, 37, 13184–13193. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.L.; Kenaan, C.; Hollenberg, P.F. Identification of the residue in human CYP3A4 that is covalently modified by bergamottin and the reactive intermediate that contributes to the grapefruit juice effect. Drug Metab. Dispos. 2012, 40, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.M.; Zhong, Y.H.; Xiao, W.B.; Li, H.; Lu, C. Identification and characterization of psoralen and isopsoralen as potent CYP1A2 reversible and time-dependent inhibitors in human and rat preclinical studies. Drug Metab. Dispos. 2013, 41, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Lu, D.; Cao, J.; Zheng, L.; Peng, Y.; Zheng, J. Psoralen, a mechanism-based inactivator of CYP2B6. Chem.-Biol. Interact. 2015, 240, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Dan, L.; Lin, J.; Zheng, L.; Cao, J.; Ying, P.; Jiang, Z. Mechanism-based inactivation of cytochrome P450 2B6 by isopsoralen. Chem.-Biol. Interact. 2015, 226, 23–29. [Google Scholar]
- Zhou, S.F.; Xue, C.C.; Yu, X.Q.; Wang, G. Metabolic activation of herbal and dietary constituents and its clinical and toxicological implications: An update. Curr. Drug Metab. 2007, 8, 526–553. [Google Scholar] [CrossRef] [PubMed]
- Essigmann, J.M.; Croy, R.G.; Bennett, R.A.; Wogan, G.N. Metabolic activationof aflatoxin B1: Patterns of DNA adduct formation, removal, and excretion in relationto carcinogenesis. Drug Metab. Rev. 2008, 13, 581. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Pang, X.; Wu, P.; Yan, R.; Gao, L.; Li, C.; Lian, W.; Wang, Q.; Liu, A.; Du, G. Molecular modeling on berberine derivatives toward BuChE: An integrated study with quantitative structure-activity relationships models, molecular docking, and molecular dynamics simulations. Chem. Biol. Drug Des. 2016, 87, 649–663. [Google Scholar] [PubMed]
- Feng, R.; Zhou, X.; Or, P.M.Y.; Ma, J.; Tan, X.; Fu, J.; Ma, C.; Shi, J.; Che, C.; Wang, Y.; et al. Enzyme kinetic and molecular docking studies on the metabolic interactions of 1-hydroxy-2,3,5-trimethoxy-xanthone, isolated from Halenia elliptica D. Don, with model probe substrates of human cytochrome P450 enzymes. Phytomedicine 2012, 19, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hai, Y.; An, L.; Chen, J.; Liang, R.; He, X. Rapid screening the potential mechanism-based inhibitors of cyp3a4 from tripterygium wilfordi based on computer approaches combined with in vitro bioassay. Bioorg. Med. Chem. 2017, 25, 2689–2700. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.H.; Andersson, L.A.; Sono, M. Spectroscopic investigations of ferric cytochrome p-450-cam ligand complexes. Identification of the ligand trans to cysteinate in the native enzyme. J. Biol. Chem. 1982, 257, 3606–3617. [Google Scholar] [PubMed]
- Mansuy, D. Cytochrome P-450 and synthetic models. Pure Appl. Chem. 1987, 59, 759–770. [Google Scholar] [CrossRef]
- Pauling, L. The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [Google Scholar] [CrossRef]
- Pauling, L. Nature of the chemical bond, and the structure of molecules and crystals. J. Chem. Phys. 1948, 2, 482. [Google Scholar] [CrossRef]
- Landis, C.R.; Weinhold, F. Valence and extra-valence orbitals in main group and transition metal bonding. J. Comput. Chem. 2007, 28, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.A.; Sono, M.; Dawson, J.H. Circular dichroism studies of low-spin ferric cytochrome P-450 CAM, ligand complexes. Biochim. Biophy. Acta 1983, 748, 341–352. [Google Scholar] [CrossRef]
- Sono, M.; Dawson, J.H. Formation of low spin complexes of ferric cytochrome P-450-CAM with anionic ligands. Spin state and ligand affinity comparison to myoglobin. J. Biol. Chem. 1982, 257, 5496–5502. [Google Scholar] [PubMed]
- Sono, M.; Andersson, L.A.; Dawson, J.H. Sulfur donor ligand binding to ferric cytochrome P-450-CAM and myoglobin. Ultraviolet-visible absorption, magnetic circular dichroism, and electron paramagnetic resonance spectroscopic investigation of the complexes. J. Biol. Chem. 1982, 257, 8308–8320. [Google Scholar] [PubMed]
- Grimm, S.W.; Einolf, H.J.; Hall, S.D.; He, K.; Lim, H.K.; Ling, K.H.; Lu, C.; Nomeir, A.A.; Seibert, E.; Skordos, K.W.; et al. The conduct of in vitro studies to address time-dependent inhibition of drug-metabolizing enzymes: A perspective of the pharmaceutical research and manufacturers of America. Drug Metab. Dispos. 2009, 37, 1355. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Zhong, D.F.; Chen, X.Y. Applications of liquid chromatography-tandem mass spectrometry to detection and characterization of reactive metabolites. J. Chin. Mass Spectrom. Soc. 2011, 32, 1–12. [Google Scholar]
- Masini, A.; Gallesi, D.; Giovannini, F.; Trenti, T.; Ceccarelli, D. Membrane potential of hepatic mitochondria after acute cocaine administration in rats—The role of mitochondrial reduced glutathione. Hepatology 1997, 25, 385–390. [Google Scholar] [PubMed]
- Mizutani, T.; Yoshida, K.; Murakami, M.; Shirai, M.; Kawazoe, S. Evidence for the involvement of N-methylthiourea, a ring cleavage metabolite, in the hepatotoxicity of methimazole in glutathione-depleted mice: Structure-toxicity and metabolic studies. Chem. Res. Toxicol. 2000, 13, 170. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, T.; Mori, K.; Hattori, C.; Kai, K.; Kataoka, H.; Masubuchi, N.; Jindo, T.; Mnabe, M. The crucial protective role of glutathione against tienilic acid hepatotoxicity in rats. Toxicol. Appl. Pharmacol. 2008, 232, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Atsumi, R.; Nakazawa, T.; Izumi, T.; Sudo, K.; Okazaki, O.; Saji, H. Ticlopidine-induced hepatotoxicity in a GSH-depleted rat model. Arch. Toxicol. 2011, 85, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Mays, D.C.; Rogers, S.L.; Guiler, R.C.; Sharp, D.E.; Hecht, S.G.; Staubus, A.E.; Gerber, N. Disposition of 8-Methoxypsoralen in the Rat: Methodology for measurement, dose-dependent pharmacokinetics, tissue distribution and identification of metabolites. J. Pharmacol. Exp. Ther. 1986, 236, 364. [Google Scholar] [PubMed]
- Zhou, S.; Chan, E.; Duan, W.; Huang, M.; Chen, Y.Z. Drug bioactivation, covalent binding to target proteins and toxicity relevance. Drug Metab. Rev. 2005, 37, 41. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; He, X. Mechanism-Based inhibition of CYP450: An indicator of drug-induced hepatotoxicity. Curr. Drug Metab. 2013, 14, 921. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C.; Bjornsson, E.; Niklasson, A.; Chalasani, N. Oral medications with significant hepatic metabolism at higher risk for hepatic adverse events. Hepatology 2010, 51, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.D.; Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the inorganic crystal-structure database. Acta Crystallogr. 2010, 41, 244–247. [Google Scholar] [CrossRef]
- Brese, N.E.; O’Keeffe, M. Bond-valence parameters for solids. Acta Crystallogr. 2010, 47, 192–197. [Google Scholar] [CrossRef]
- Hu, S.Z.; Zhou, Z.H. The correlation between metal oxidation state and bond valence parameters for m—o bonds (m = v, Fe and Cu). A simple method to search for the metal oxidation—State independent parameter pairs. Z. Kristallographie Cryst. Mater. 2004, 219, 614–620. [Google Scholar] [CrossRef]
- Bergerhoff, G.; Brown, I.D. Inorganic crystal structure database. Data Sci. Technol. 1981, 37, 324–326. [Google Scholar]
- Pauling, L. The principles determining the structure of complexionic crystals. J. Am. Chem. Soc. 1929, 51, 1010–1026. [Google Scholar] [CrossRef]
- Miessler, G.L.; Tarr, D.A. Inorganic Chemistry; Prentice-Hall: Upper Saddle River, Japan, 1999; ISBN 9780138418915. [Google Scholar]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A. Advanced Inorganic Chemistry; Wiley: Japan, 1999; ISBN 9780471199571. [Google Scholar]
- Cheung, W.I.; Tse, M.L.; Ngan, T.; Lin, J.; Lee, W.K.; Poon, W.T.; Mak, T.W.; Leung, V.K.; Chau, T.N. Liver injury associated with the use of Fructus Psoraleae(Bol-gol-zhee or Bu-gu-zhi) and its related proprietary medicine. Clin. Toxicol. 2009, 47, 683. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Kim, S.; Hann, S.K.; Park, Y.K. The effect on liver transaminases of 5-methoxypsoralen used in systemic photochemotherapy. Ann. Dermatol. 1995, 7, 51–53. [Google Scholar] [CrossRef]
- Peterson, S.; Lampe, J.W.; Bammler, T.K.; Gross-Steinmeyer, K.; Eaton, D.L. Apiaceous vegetable constituents inhibit human cytochrome P-450 1A2 (hCYP1A2) activity and hCYP1A2-mediated mutagenicity of aflatoxin B1. Food Chem. Toxicol. 2006, 44, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.W.; Baek, J.T.; Lee, D.S.; Kang, S.B.; Ahn, B.M.; Chung, K.W. A case of acute cholestatic hepatitis associated with the seeds of Psoralea corylifolia (Boh-Gol-Zhee). Clin. Toxicol. 2005, 43, 589. [Google Scholar] [CrossRef]
- Takizawa, T.; Imai, T.; Mitsumori, K.; Takagi, H.; Onodera, H.; Yasuhara, K.; Ueda, M.; Tamura, T.; Hirose, M. Gonadal toxicity of an ethanol extract of Psoralea corylifolia in a rat 90-day repeated dose study. J. Toxicol. Sci. 2002, 27, 97. [Google Scholar] [CrossRef] [PubMed]
- Hiroi, T.; Chow, T.; Imaoka, S.; Funae, Y. Catalytic specificity of CYP2D isoforms in rat and human. Drug Metab. Dispos. 2002, 30, 970. [Google Scholar] [CrossRef] [PubMed]
- Venhorst, J.; Ter Laak, A.M.; Commandeur, J.N.; Funae, Y.; Hiroi, T.; Vermeulen, N.P. Homology modeling of rat and human cytochrome P450 2D (CYP2D) isoforms and computational rationalization of experimental ligand-binding specificities. J. Med. Chem. 2003, 46, 74. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Bi, H.C.; Xie, Z.Y.; Zuo, Z.; Li, J.K.; Li, X.; Zhao, L.Z.; Chen, X.; Huang, M. Rapid determination of six metabolites from multiple cytochrome P450 probe substrates in human liver microsome by liquid chromatography/mass spectrometry: Application to high-throughput inhibition screening of terpenoids. Rapid Commun. Mass. Spectrom. 2007, 21, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2010, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.A.; Jalaie, M.; Robertson, D.H.; Lewis, R.A.; Vieth, M. Lessons in Molecular Recognition: The Effects of Ligand and Protein Flexibility on Molecular Docking Accuracy. J. Med. Chem. 2004, 47, 45–55. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) | ||||
|---|---|---|---|---|
| Enzyme | Compound | (−) NADPH | (+) NADPH | Shift (fold) |
| CYP3A4 | Psoralen | >200 | 19.50 ± 1.04 ** | >10.3 |
| Isopsoralen | 96.83 ± 2.21 | 9.59 ± 1.04 ** | 10.1 | |
| Formula | [M + H]+ m/z | Retention Time (min) | Prominent Fragment Ion (m/z) | Peak Area 1 | ||
|---|---|---|---|---|---|---|
| HLM | MLM | |||||
| Psoralen | C11H6O3 | 187 | 7.72 | 159, 143, 131, 115 | ||
| M1 | C11H6O4 | 203 | 5.19 | 175.1, 147.0, 119.2 | 557 | 1019 |
| M2 | C11H6O4 | 203 | 7.58 | 175.2, 159.1, 147.1, 131.2 | 502 | 8655 |
| M3 | C11H6O5 | 219 | 7.23 | 201.1, 174.8, 157.5 | 529 | 164 |
| M4 | C11H8O5 | 221 | 3.34 | 203.1, 175.1, 158.9, 147.0 | 66,831 | 98,864 |
| M5 | C11H8O5 | 221 | 5.18 | 203.2, 175.1, 147.2 | 61,016 | 102,180 |
| Isopsoralen | C11H6O3 | 187 | 8.06 | 158.6, 142.6, 130.6, 114.6 | ||
| M′1 | C11H6O4 | 203 | 6.12 | 175.1, 147.0, 119.2 | 1288 | 14,513 |
| M′2 | C11H6O4 | 203 | 7.45 | 175.3, 159.0, 147.1, 131.2 | 1349 | 4707 |
| M′3 | C11H10O4 | 205 | 5.23 | 177.1,148.8, 133.0, 131.2 | 312 | 418 |
| M′4 | C11H8O5 | 221 | 3.80 | 203.3, 174.9, 159.4, 146.8 | 34,274 | 96,828 |
| M′5 | C11H8O5 | 221 | 5.01 | 203.3, 175.4 | 1627 | 7725 |
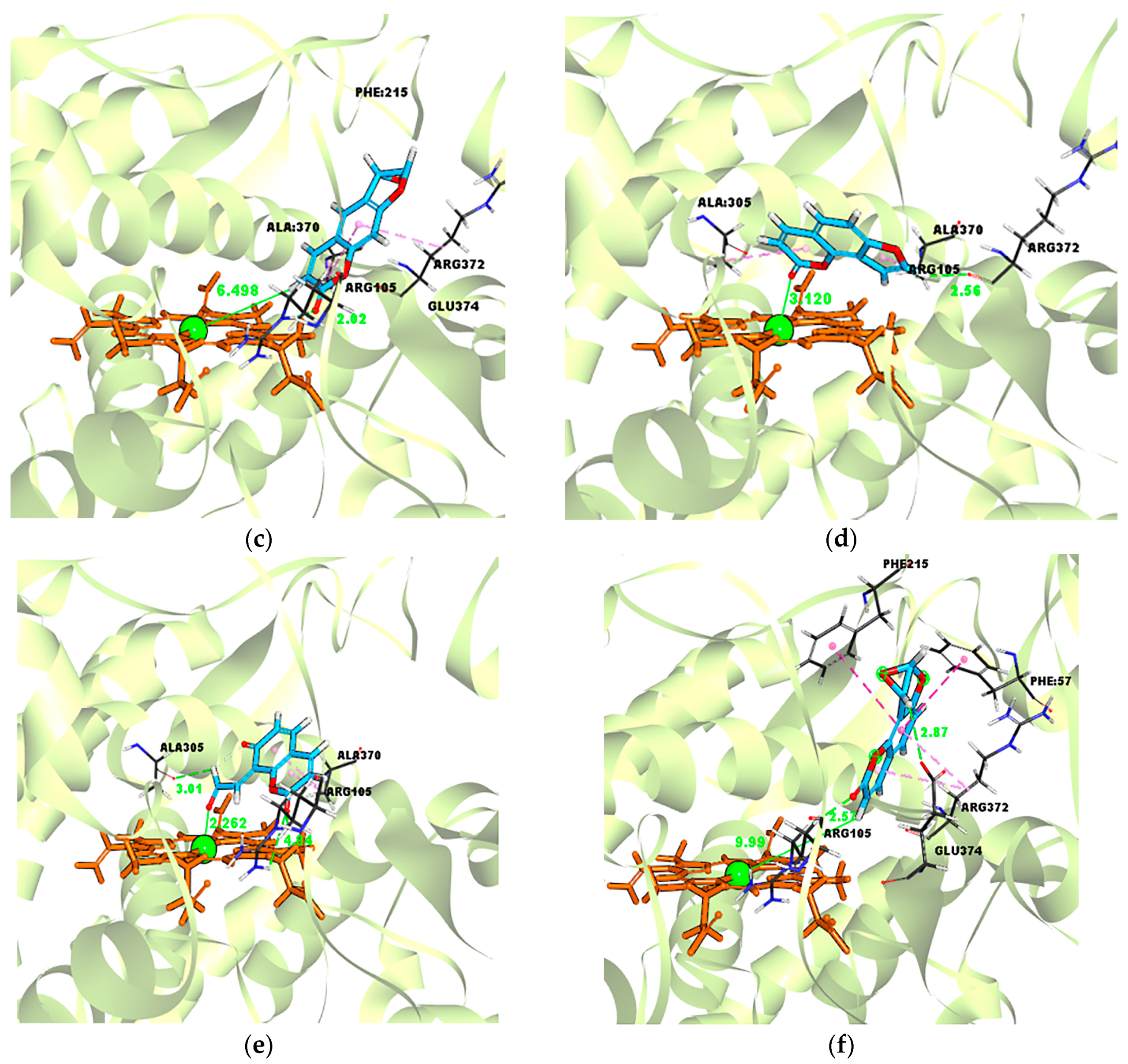
| Ligand–Enzyme Complex | −Cdocker Energy | Ligand–Central Ion Distance |
|---|---|---|
| Psoralen–CYP3A4 | 19.4697 kcal/mol | 3.872 Å |
| γ-ketoenal intermediate of psoralen–CYP3A4 | 21.4426 kcal/mol | 2.310 Å |
| Furanoepoxide intermediate of psoralen–CYP3A4 | −21.3421 kcal/mol | 6.498 Å |
| Isopsoralen–CYP3A4 | 18.5824 kcal/mol | 3.120 Å |
| γ-ketoenal intermediate of isopsoralen–CYP3A4 | 19.635 kcal/mol | 2.226 Å |
| furanoepoxide intermediate of isopsoralen–CYP3A4 | −23.4627 kcal/mol | 9.993 Å |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hai, Y.; Feng, S.; Wang, L.; Ma, Y.; Zhai, Y.; Wu, Z.; Zhang, S.; He, X. Coordination Mechanism and Bio-Evidence: Reactive γ-Ketoenal Intermediated Hepatotoxicity of Psoralen and Isopsoralen Based on Computer Approach and Bioassay. Molecules 2017, 22, 1451. https://doi.org/10.3390/molecules22091451
Hai Y, Feng S, Wang L, Ma Y, Zhai Y, Wu Z, Zhang S, He X. Coordination Mechanism and Bio-Evidence: Reactive γ-Ketoenal Intermediated Hepatotoxicity of Psoralen and Isopsoralen Based on Computer Approach and Bioassay. Molecules. 2017; 22(9):1451. https://doi.org/10.3390/molecules22091451
Chicago/Turabian StyleHai, Yue, Shan Feng, Lili Wang, Yetao Ma, Yiran Zhai, Zijun Wu, Sichao Zhang, and Xin He. 2017. "Coordination Mechanism and Bio-Evidence: Reactive γ-Ketoenal Intermediated Hepatotoxicity of Psoralen and Isopsoralen Based on Computer Approach and Bioassay" Molecules 22, no. 9: 1451. https://doi.org/10.3390/molecules22091451