1. Introduction
Lipases are α/β hydrolases which catalyze the hydrolysis of acylglycerols to release fatty acids and glycerol. These enzymes can be divided into two main groups: (i) carboxylesterases (EC 3.1.1.1) and (ii) “true” lipases or triacylglycerol lipases (EC 3.1.1.3), which differ in several biochemical features [
1,
2]. After determining the three-dimensional structure of this latter type, the presence of a “lid” was verified. This lid is known as a protein surface loop that covers the enzyme active site and is directly related to the “interfacial activation”. This promotes a conformational change in the enzyme structure causing the movement of the lid which is in contact with this interface, exposing the active site and activating the “true” lipases. Its activity should sharply increase as soon as it is in contact with the triglyceride substrate [
2,
3,
4].
Lipases are ubiquitous in Nature and they are produced by plants [
5], microorganisms [
6] and mammals [
7]. Lipases from microorganisms are widely used for biotechnological applications, because they are usually more stable than the ones obtained from animals and plants, which makes their production more convenient, safer, easier, less expensive and they are generally more stable in organic solvents. Moreover, lipases from this source show versatility in their properties with respect to the enzymatic activity and substrate specificity [
2,
8,
9]. Fungi are able to secrete large amounts of enzymes in the extracellular environment. Therefore, microbial lipases from fungi have been widely used to produce lipases in the last decades [
9].
The use of lipases in industry has continuously grown, especially in industries such as food, dairy, pharmaceuticals, agrochemicals, detergents, oleo-chemicals, tea industries, cosmetics, leather, as well as in several bioremediation processes [
8,
9,
10]. The most recent application of lipases is in the biosensor industry [
11,
12]. Manufacturers in many other important industries, including health care, pharmaceuticals [
13] and chemicals [
14], are increasingly taking advantage of Nature’s amazing catalysts. For the last few years, enzymes have widely been used in the production of biofuels such as biodiesel [
15]. One of the best uses of lipases in modern life is in the treatment of wastes in general [
16], especially in the treatment of solid wastes [
17] and in the purification of waste water [
18]. In some cases, industrial applications of enzymes in organic solvents are also developed [
19]. Moreover, microbial enzymes, including lipases, can be produced from renewable raw materials. In addition, the mild operating conditions of enzymatic processes mean that they can be operated in relatively simple and totally controlled equipment. However, all these desirable characteristics of enzymes, and their widespread industrial applications are often hampered by their lack of long-term operational stability and shelf-storage life, low thermal and chemical stability, and by their cumbersome recovery and re-use [
12,
20].
In order to enhance the economy of biocatalytic processes it is possible to perform enzyme immobilization [
21]. Improvements in current immobilization strategies have progressed by using supports that increase the effectiveness and stability of the connection through multipoint attachments. Novel methods of enzyme self-immobilization have been developed (CLEC, CLEA, Spherezyme), as well as carrier materials (Dendrispheres), encapsulation (PEI microspheres), and entrapment. Apart from retention, recovery and stabilization, other advantages of enzyme immobilization have emerged, such as enhanced enzyme activity, modification of substrate selectivity and enantioselectivity, and multi-enzyme reactions. Protein immobilization commonly alters the natural molecular environment of the enzymes, and often affects their catalytic activity due to modification of the substrate’s accessibility to the active center, decreased enzyme molecular mobility and changes in its conformational integrity [
22]. These advances promise to enhance the role of enzyme immobilization in industry, opening the way to novel applications [
23].
Immobilization allows re-using the enzyme for an extended period and enables easier separation of the catalyst from the product. Additionally, immobilization improves many properties of enzymes such as their performance in organic solvents, pH tolerance, and heat stability. The increase of the structural rigidity of the protein and stabilization of multimeric enzymes prevent dissociation-related to inactivation [
21]. Immobilization is attractive for lipases because, besides all the advantages common to other proteins, it can also considerably increase the catalytic activity, probably due to conformational changes that better expose the active site of the enzyme [
24].
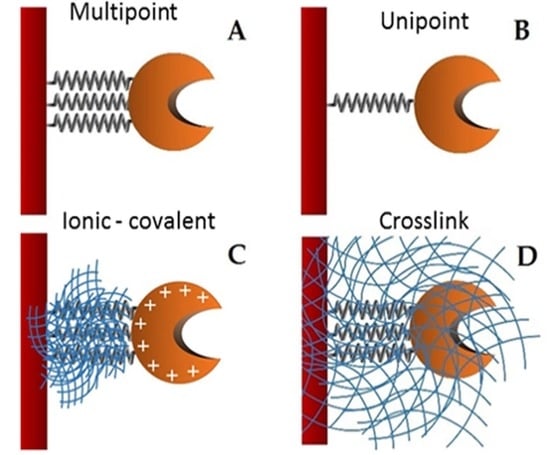
Supports activated with glutaraldehyde or primary amino groups can be used for enzyme immobilization [
25]. Among them, chitin and chitosan are the most important functional materials for immobilization. They offer biocompatibility, biodegradability, nontoxicity, physiological inertness, antibacterial properties, heavy metal ion chelation, gel forming properties and hydrophilicity, and remarkable affinity to proteins [
26]. The cationic nature of their surface allows the rapid ionic attachment of the protein at low-ionic strengths [
27]. Besides, there are two different possibilities: (1) direct covalent-attachment of the enzyme on glutaraldehyde-activated supports, and (2) adsorption of the proteins on amino-activated supports followed by glutaraldehyde cross-linking of both the enzyme and the support [
28]. In this work, these two different attaching chemistries are employed for the stabilization of the studied lipase. The immobilization of the lipase from
Hypocrea pseudokoningii, which has been scarcely studied, is presented, thus contributing with its exploitable catalytic properties. This work also shows an enzymatic biocatalyst highly stable in organic solvents, high temperatures and under denaturing conditions, providing many important contributions for the future application of this enzyme.
3. Material and Methods
3.1. Microorganism, Culture Conditions and Lipase Production
Hypocrea pseudokoningii was maintained at 30 °C in slants of 4% PDA. Conidia from 7-day old culture were inoculated into 50 mL of Adams liquid medium [
46] (final concentration of 10
5 spores/mL). Cultures were incubated in a rotational shaker (110 rpm), for 96 h, at 30 °C [
6]. After that, the mycelia were separated from the extracellular medium using vacuum filtration on no. 1 Whatman filter paper, and the crude filtrate was used as a source of extracellular lipase activity.
3.2. Measurement of Lipase Activity and Protein
3.2.1. Hydrolysis of p-Nitrophenyl Butyrate (pNPB)
This assay was performed by measuring the increase in the absorbance at 348 nm produced by the release of p-nitrophenol in the hydrolysis of 0.4 mM pNPB in 25 mM sodium phosphate buffer at pH 7 and 25 °C, using a spectrophotometer equipped with a thermostatized chamber and continuous magnetic stirring to keep the immobilized enzyme homogenously suspended. The beginning of the reaction occurred with 0.1 mL of lipase solution or a suspension added to 2.5 mL of substrate solution. The presence of solids during the assay only produced a marginal increase in the noise of readings and did not affect the measurement of absorbance.
3.2.2. Hydrolysis of p-Nitrophenyl Palmitate (pNPP)
This assay was performed by measuring the increase in the absorbance at 400 nm produced by the release of
p-nitrophenol in the hydrolysis of 0.4 mM
pNPP in 25 mM acetate sodium buffer at pH 6 and 45 °C. For that, it was used a spectrophotometer equipped with a thermostatized chamber and continuous magnetic stirring to keep the immobilized enzyme homogenously suspended. The beginning of the reaction occurred with 0.1 mL of lipase solution or a suspension added to 2.5 mL of substrate solution. The presence of solids during the assay only produced a marginal increase in the noise of readings and did not affect the measurement of the absorbance. One unit (U) of enzyme activity was defined as that catalyzing the conversion of 1 μmol of substrate (or the formation of 1 μmol of product) in 1 min, in the assay conditions. Proteins were measured according to Bradford method [
47], using bovine serum albumin as standard and expressed by mg protein/mL. Specific activity (SA) was expressed as Units/mg protein.
3.3. Purification of Lipase from Hypocrea Pseudokoningii
Lipase from
H. pseudokoningii was purified using immobilization on Octyl Sepharose support in 5 mM sodium phosphate buffer solution, pH 7, at 4 °C [
29]. After the immobilization, adsorbed lipase derivatives were extensively washed with distilled water and the purity was determined. Afterwards, the immobilized enzyme was desorbed with 2% Triton X-100, resulting in a purified enzyme extract.
3.4. Support Preparation and Enzyme Immobilization
Glyoxyl agarose gel was prepared according to Guisan [
48]. Monoaminoethyl-
N-ethylagarose (MANAE) was synthesized according to Fernandez-Lafuente et al. [
49] MANAE activated with glutaraldehyde and crosslinked were prepared according to Betancor [
28]. The biocatalyst obtained by the immobilization through this support was named GA-crosslink.
A mass of 1 g of covalent support (glyoxyl—agarose and MANAE activated with glutaraldehyde) was added to 10 mL of the purified enzyme solution (with a maximum enzyme activity of 5 U/mL and 0.25 mg/mL) in different buffers according to each immobilization. The immobilization occurred overnight. After this period, the derivative (support containing the enzyme) was separated of the supernatant by filtration. The biocatalyst immobilization obtained from lipase plus glyoxyl-agarose and MANAE activated with glutaraldehyde were called GX and GA derivatives, respectively. After enzyme immobilization, linkages between glyoxyl and amine groups were reduced with 1.0 mg/mL sodium borohydride for 30 min at room temperature. The derivatives were washed with distilled water and maintained at 4 °C.
Samples of the supernatants and suspensions were periodically withdrawn, and the enzyme activity was measured. The immobilization was considered completed when no activity was detected in the supernatant. Sodium phosphate buffer (25 mM) at pH 7.0 and 8.0, at 25 °C as well as 25 mM sodium bicarbonate buffer at pH 10.5, 4 °C, were used for lipase immobilization in MANAE-crosslink, MANAE activated with glutaraldehyde, and glyoxyl agarose, respectively. The yield of immobilization was considered to be the rate between the activities in the supernatant compared to the activity in the blank of soluble enzyme (initial solution). In all cases, the activity of the blank was 100% during the immobilization process. Activity recovery was calculated through the ratio of the activity in the derivative after the immobilization process and the initial activity of the offered enzyme.
3.5. Immobilization of Lipases on Cyanogen Bromide (CNBr-Activated) Support
This derivative was prepared using the Pharmacia
® protocol at pH 7.0. The immobilization should occur via the most reactive group: the amino terminal [
47,
50]. One gram of CNBr-activated support was added to a solution of 10 mL of the pure lipase (0.25 mg/mL) in the present of 25 mM sodium phosphate buffer containing 2% (
v/
v) Triton X-100 at pH 7.0, 4 °C. After 15 min, 100% of the lipase became immobilized. The enzyme immobilization on CNBr was ended by blocking the amine reactive groups of the support in the presence of ethanolamine 1 M at pH 8.0. After 2 h, the immobilized preparation was washed with abundant water.
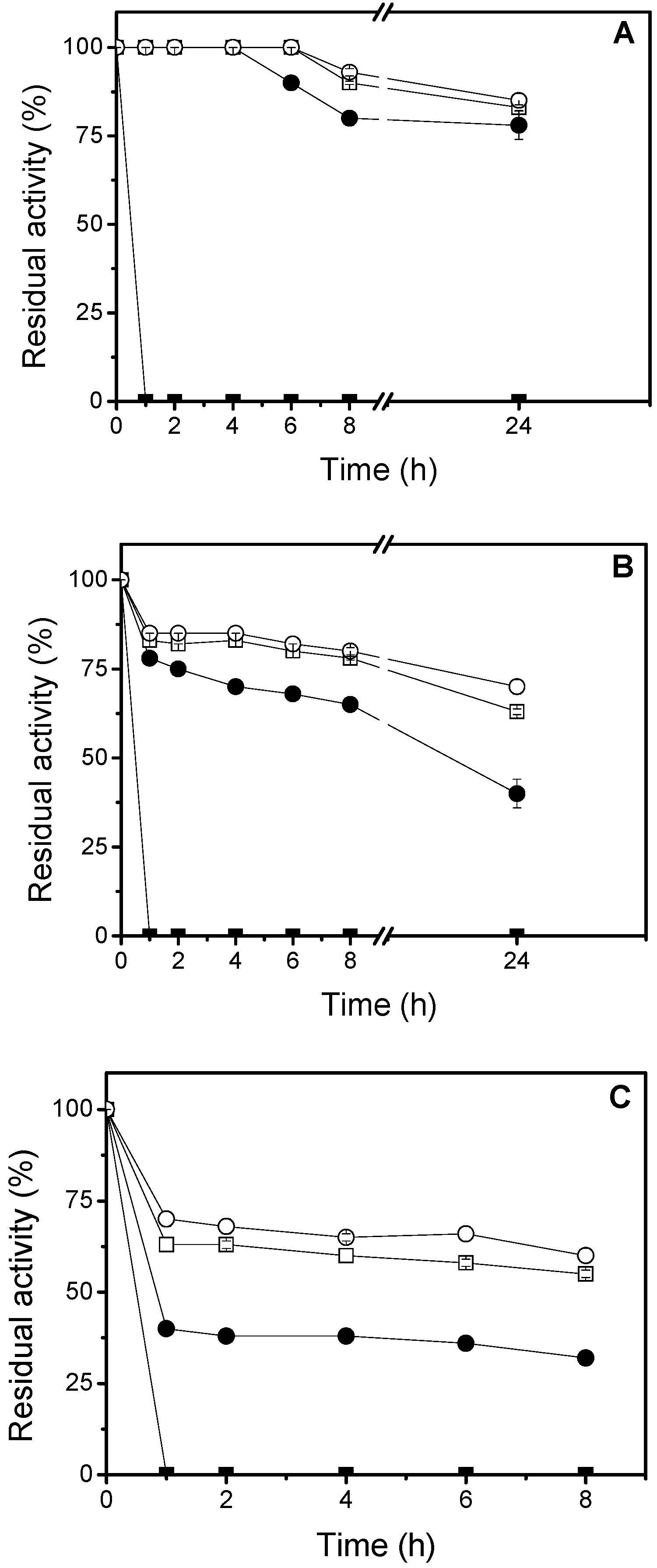
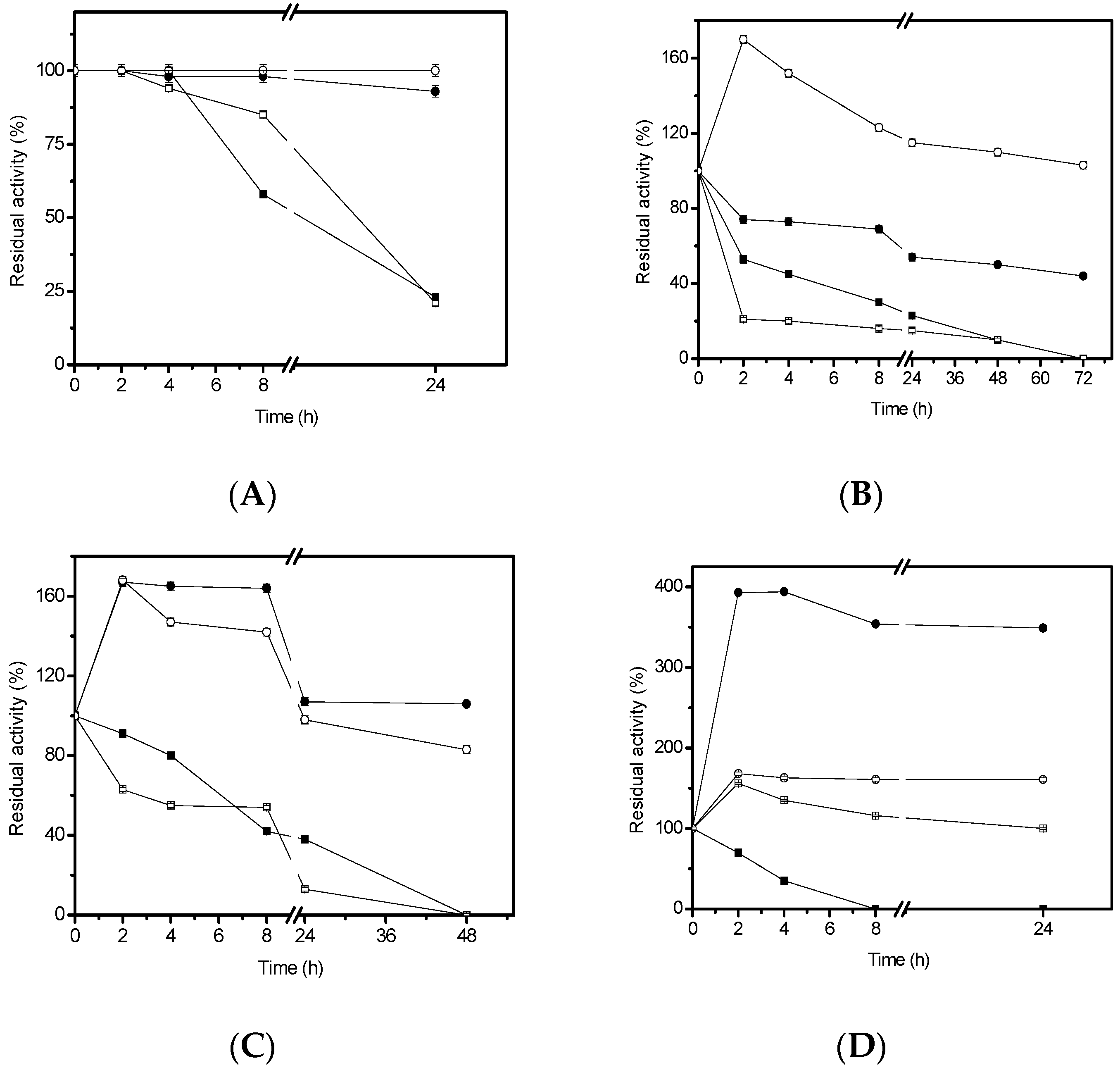
3.6. Thermal Stability Studies
In order to study the thermal stability, 4.68 U/g of the immobilized lipase were used. The inactivation was carried out at 50 °C, 60 °C and 70 °C, in 25 mM sodium phosphate buffer, at pH 7.0. At different times, samples were withdrawn and their activities were tested as previously described. The remaining activity was calculated as the ratio between the activity at a given time and the activity at time “zero” of incubation. Half-lives were calculated from the observed inactivation courses.
3.7. Organic Solvents Stability Studies
In order to study the lipase stability in organic solvents, 4.68 U/g of the immobilized lipase were incubated with 50% (v/v) ethanol, methanol, n-propanol, or cyclohexane. The inactivation was carried out in 25 mM sodium phosphate buffer at pH 7.0 and room temperature. At different times, samples were withdrawn and their activities were tested. The remaining activity was calculated between the activity at a given time and activity at time zero of incubation.
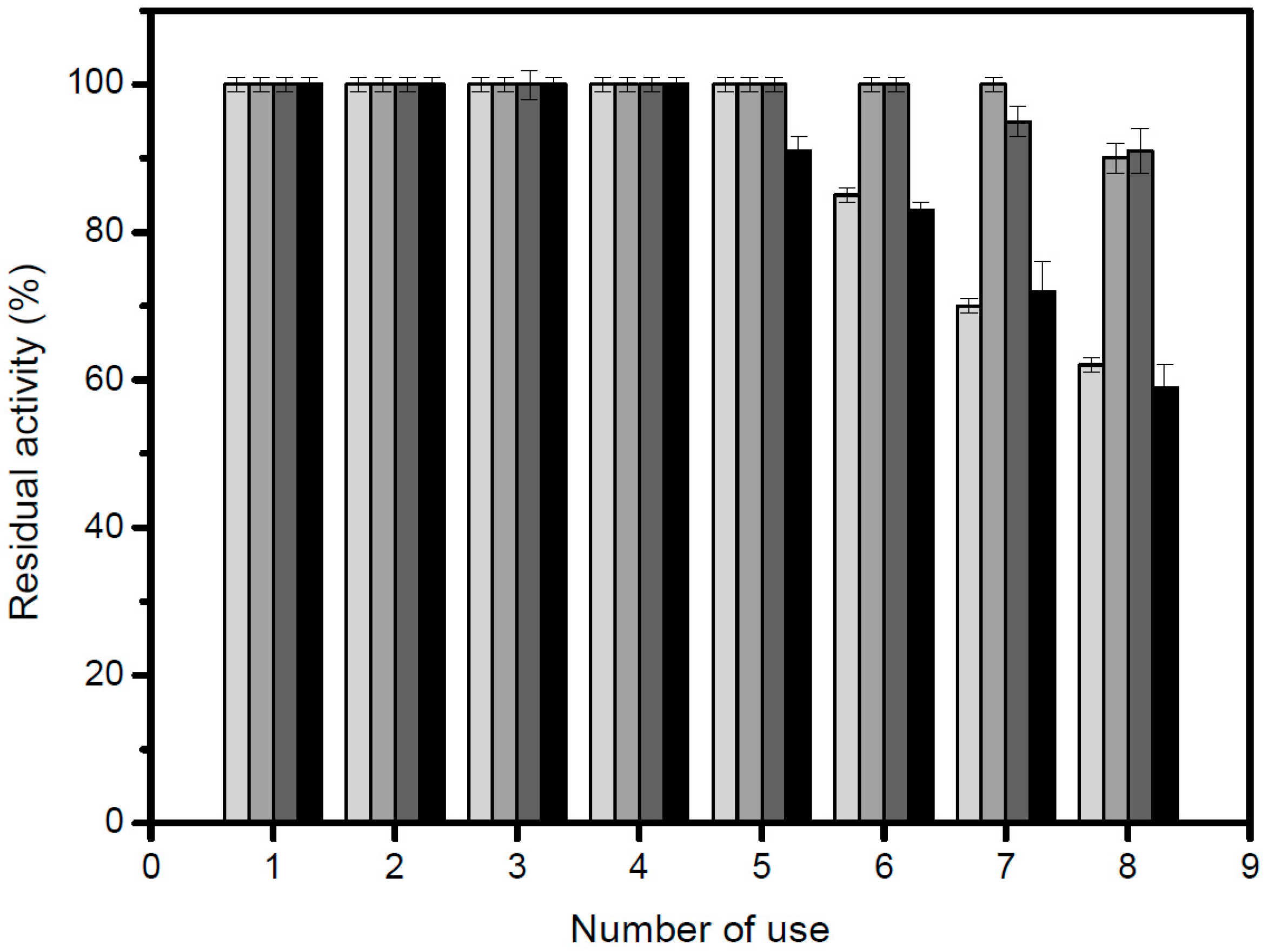
3.8. Hydrolysis of Oils
The hydrolysis of sardine oil was performed in a water-organic solvent two-phase system as described [
51] (4.5 mL of cyclohexane, 5 mL of 0.1 M Tris buffer pH 6.0 and 0.5 mL of sardine oil). The reaction was initiated by adding 0.4 g of lipase derivatives, and the mixture was mechanically stirred at 600 rpm for 24 h, at 25 °C. A volume of 100 μL of the sample present in the organic phase were removed and mixed with 400 μL acetonitrile. Reactants and products were analyzed by HPLC (SP 100 coupled with a SP 8450 UV detector, Spectra Physics, Santa Clara, CA, USA) using a Kromasil C8 column (150 × 4.6 mm). Products were eluted at a flow rate of 1.5 mL/min with acetonitrile/water/acetic acid (70:30:0.1
v:
v:
v) and pH 3.0. The UV detection was performed at 215 nm. The retention times for the unsaturated fatty acids were 9.4 min for EPA and 13.5 min for DHA.
The hydrolysis of açaí (Euterpe oleracea), grape seed and cotton seed oils was performed in 25 mM sodium acetate buffer, pH 5.0, using an automatic titrator. The reaction was performed as follows: 6.0 mL of oil, 8.0 mL of 25 mM sodium acetate buffer, and 0.5 mL of Triton X-100. The reaction was initiated by adding 300 mg of the immobilized lipase, and the mixture was mechanically stirred at 250 rpm, 45 °C, for 16 h. The reaction was interrupted by the addition of a solution constituted by ethanol: acetone (1:1). The total unit (Total U) is defined as the amount of enzyme required to release one μmol of fatty acid in one minute (μmols/min), considering the total volume of the reaction.
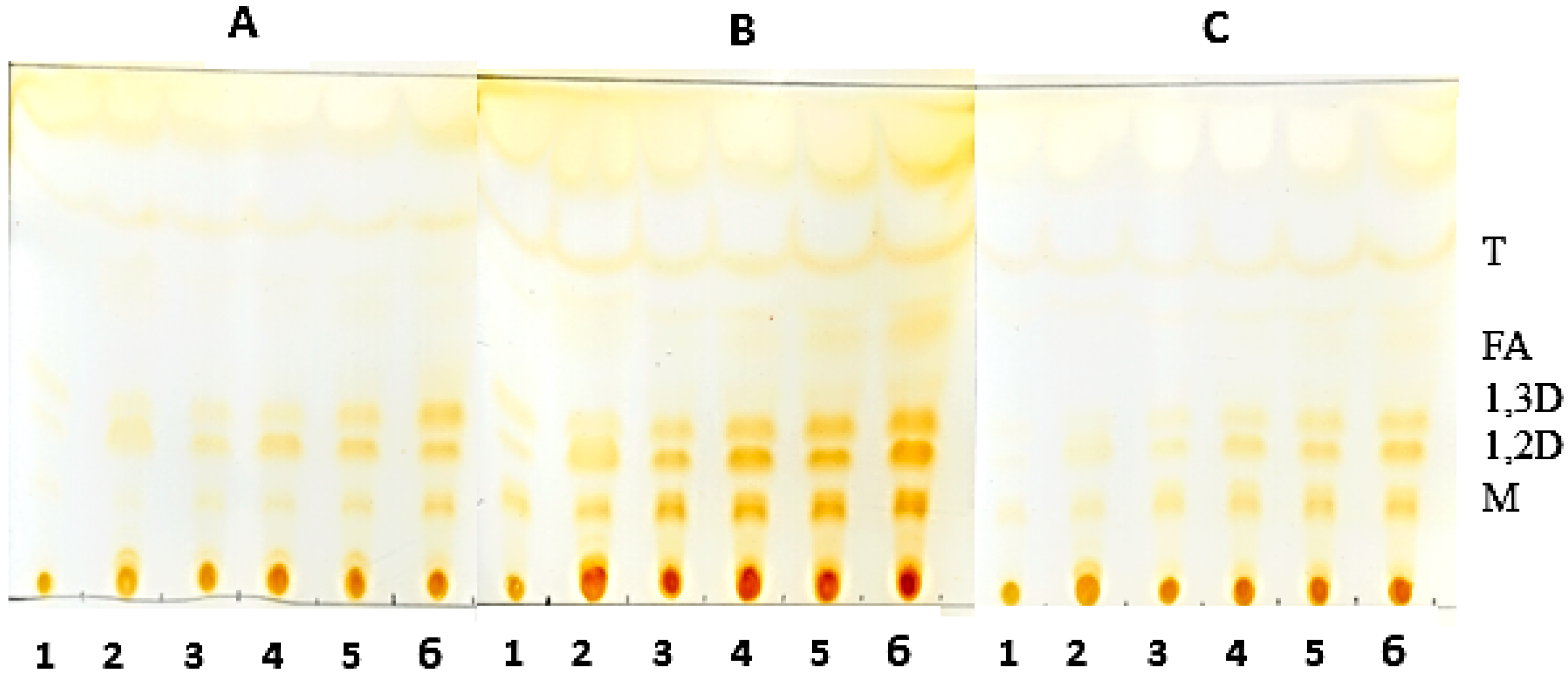
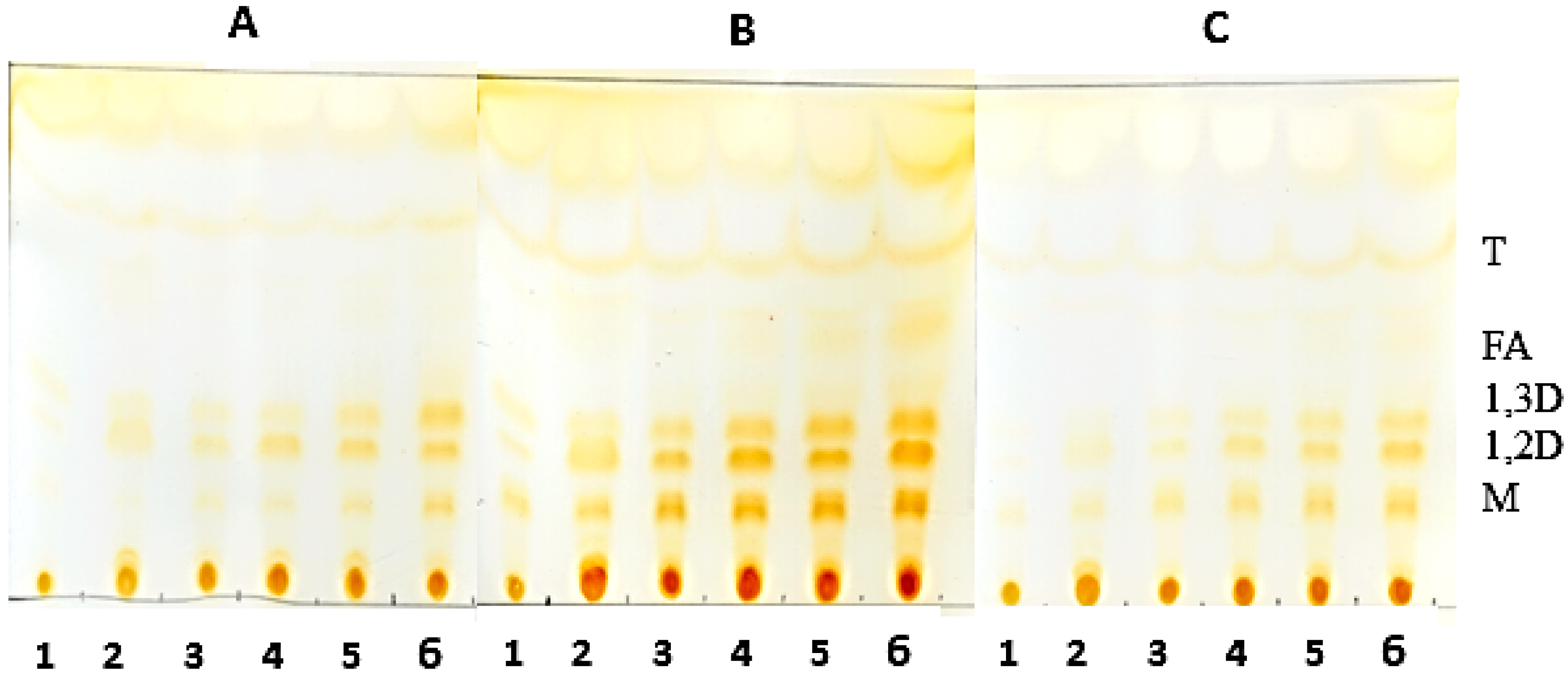
3.9. Thin-Layer Chromatography of the Hydrolysis Product of Oils Catalyzed by Derivatives
Analysis of the hydrolysis products was obtained by thin layer chromatography (TLC) on silica plates (DC-Alufolien Kieselgel 60 without fluorescence indicator, Merck®, Darmstadt, Germany). The reaction mixture consisted of 2.0 mL of oil, 300 mg of derivative, 0.3 mL of Triton X-100 and, 0.7 mL 500 mM MES buffer, pH 6.0. Incubation of the mixture was carried out at 45 °C under constant agitation. Aliquots were removed and 5.0 μL were applied to a silica plate. The run was carried out twice with a mixture of hexane-ethyl acetate-acetic acid (90:10:1). The visualization of the hydrolysis products was performed by exposure to iodine vapor until the appearance of bands corresponding to the products of triglyceride degradation.
3.10. Enzymatic Hydrolysis of Racemic Mixtures
The activities of different preparations of lipase from
H. pseudokoningii on the hydrolysis of racemic mixtures were investigated by adding the enzyme preparations (0.3 g) to 3.0 mL of 5.0 mM (
R,
S)-substrate under mechanical stirring. At different times, the degree of hydrolysis was measured by reverse-phase HPLC (Spectra Physics SP 100 coupled with a Spectra Physics SP 8450UV detector) on a Kromasil C18 (25 cm × 0.4 cm) column supplied by Analysis Vinicos (Tomelloso, Spain) using the corresponding mobile phase and flow rates listed in
Section 3.11 below. The elution was monitored by recording the absorbance at 254 nm. Triplicates (at least) of each assay were made and experimental error was never higher than 5%.
3.11. Mobile Phases
Enzymatic Hydrolysis of Racemic-2-hydroxy-4-phenylbutanoic Acid Ethyl Ester (rac-HPBE)
The mobile phase consisted of acetonitrile (40%) and 10 mM ammonium phosphate buffer (60%) at pH 2.9 and a flow rate of 1.0 mL/min.
Enzymatic Hydrolysis of Racemic Butyryl-2-phenylacetic Acid (rac-BPA)
The mobile phase consisted of acetonitrile (35%) and 10 mM ammonium phosphate buffer (65%) at pH 2.5 and a flow rate of 1.5 mL/min.
Enzymatic Hydrolysis of Racemic Methyl Mandelate (rac-MEMA)
The mobile phase consisted of acetonitrile (40%) and 10 mM ammonium phosphate buffer (60%) at pH 2.9 and a flow rate of 1.0 mL/min.
Enzymatic Hydrolysis of Racemic 1-Phenylethanol Acetate (rac-AFE)
The mobile phase consisted of acetonitrile (40%) and 10 mM ammonium phosphate buffer (60%) at pH 2.9 and a flow rate of 1.0 mL/min.
3.12. Determination of Enantiomeric Excess (ee)
At different degrees of conversion, the ee of the produced acid was analyzed by chiral reverse-phase HPLC. The column was a Chiracel OD-R, the mobile phase was an isocratic mixture of 5% acetonitrile and 95% ammonium phosphate 10 mM, at pH 2.3 and the analyzes were performed at a flow rate of 0.5 mL/min by recording the absorbance at 225 nm. Except by rac-MEMA, the mobile phase was an isocratic mixture of 30% acetonitrile and 70% ammonium phosphate 10 mM, pH 2.3.
3.13. Calculation of E (Enantiomeric Ratio)-Values
The
E-value was calculated from the ratio between the enantiomers of both released acid. Conversion degree was always between 10% and 15%. The formula used was the one reported by Chen et al. [
42]:
where
X: conversion;
ee: enantiomeric excess.
3.14. Reproducibility of Experiments
All experiments were performed at least three times to confirm the results obtained and the standard.



 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}