Novel Methylselenoesters as Antiproliferative Agents


 , and
, and 
Abstract
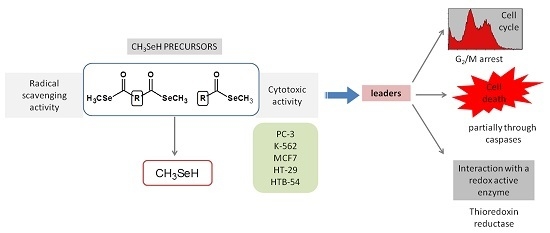
:
1. Introduction
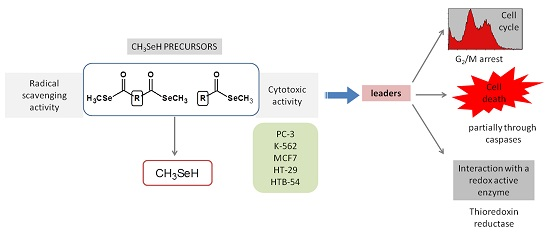
2. Results and Discussion
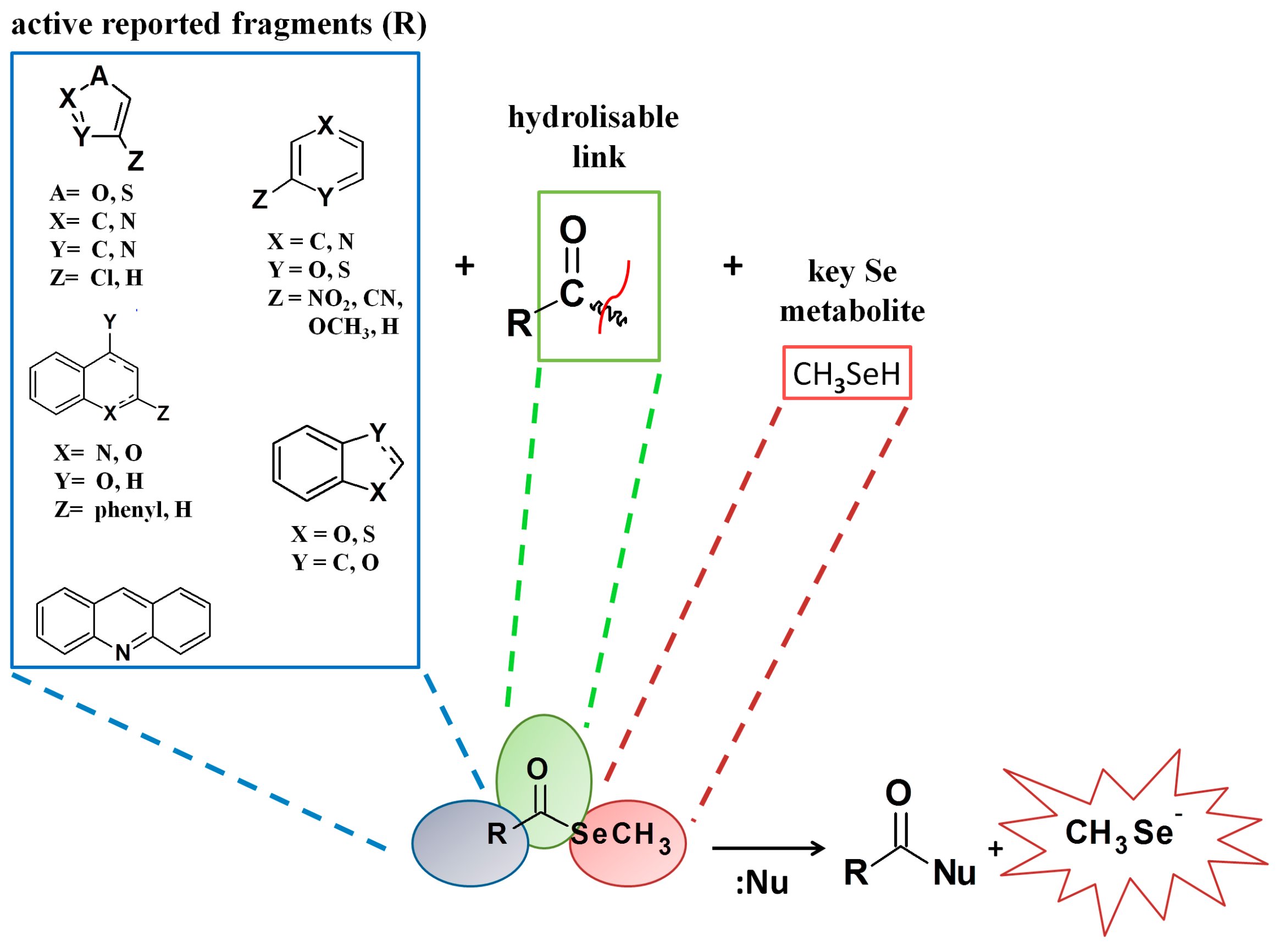
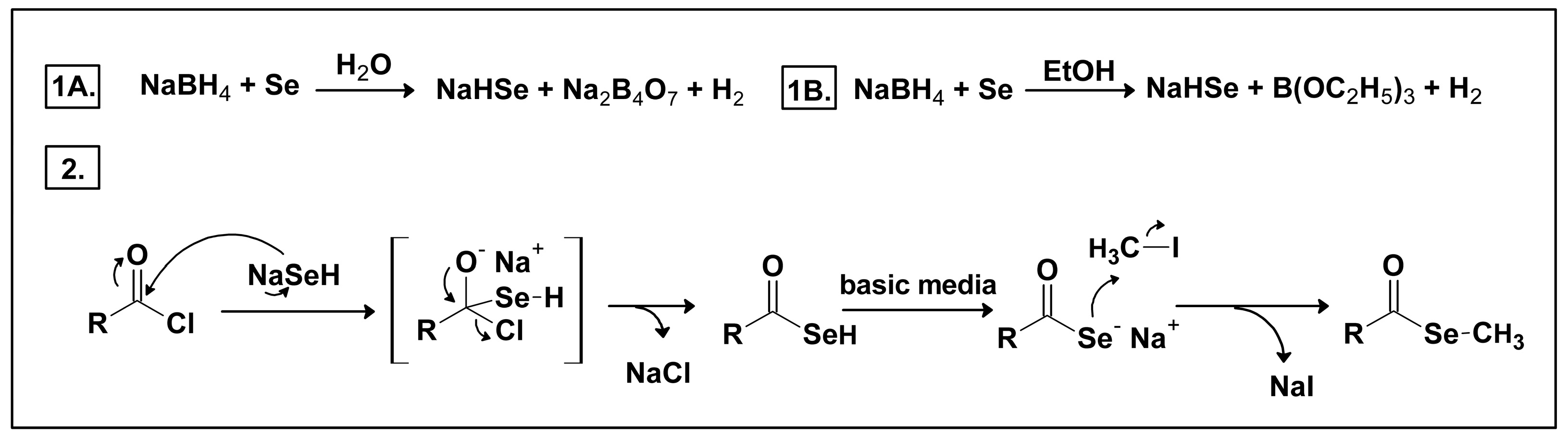
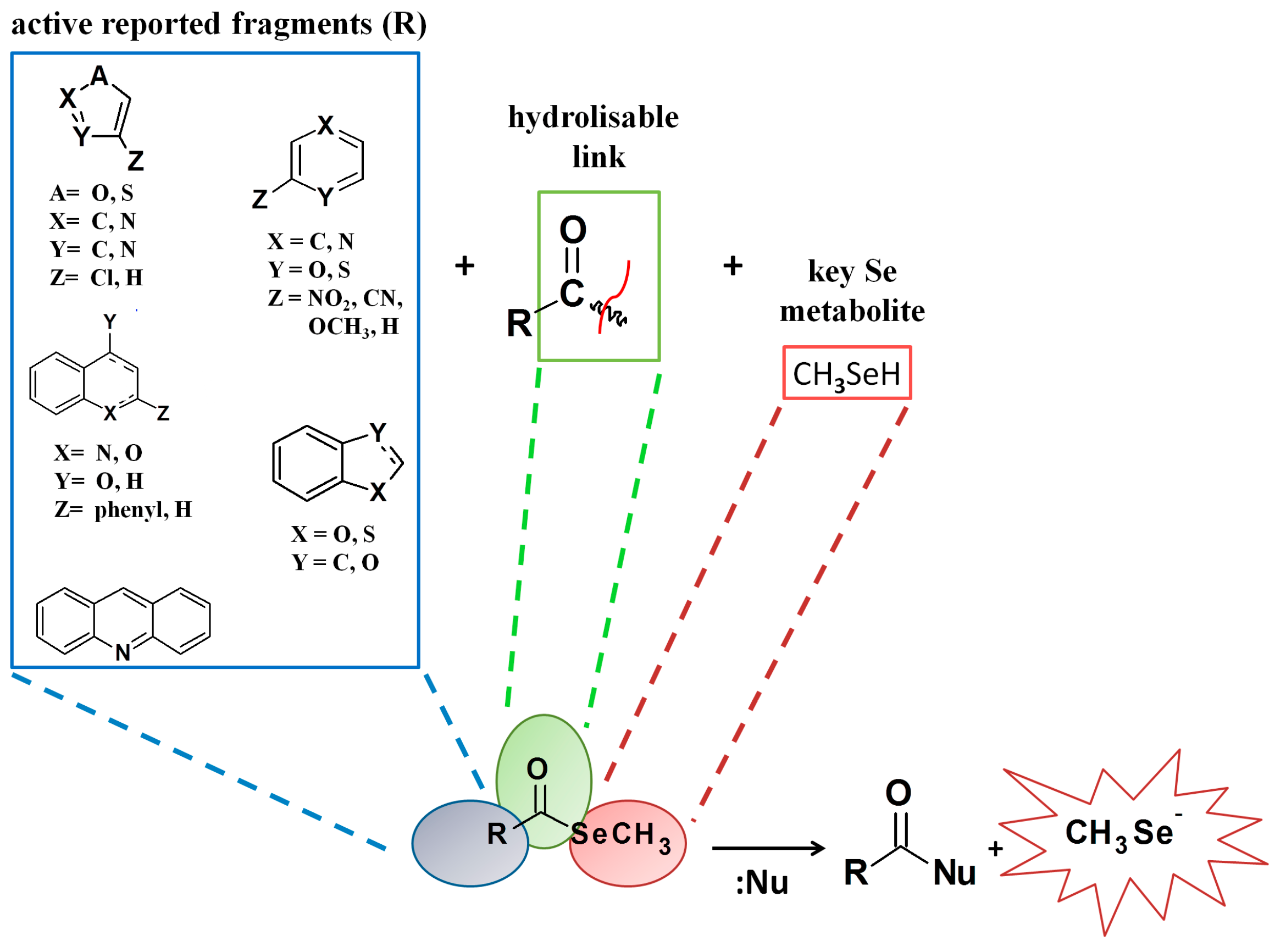
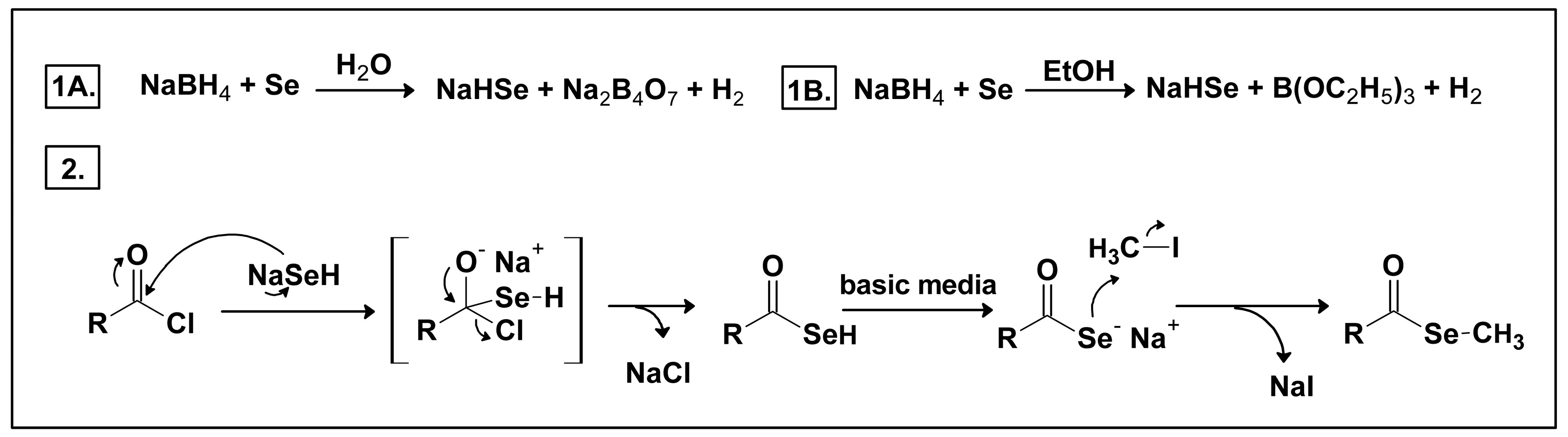
2.1. Chemistry
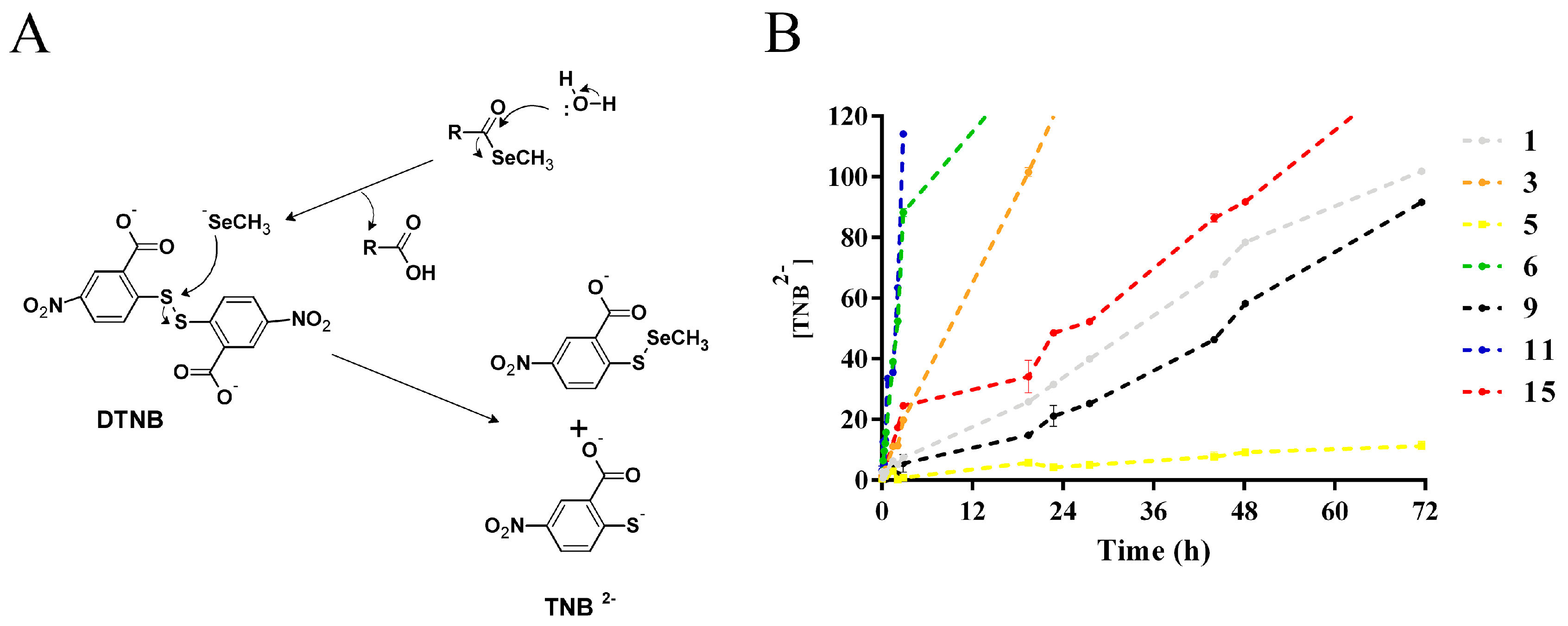
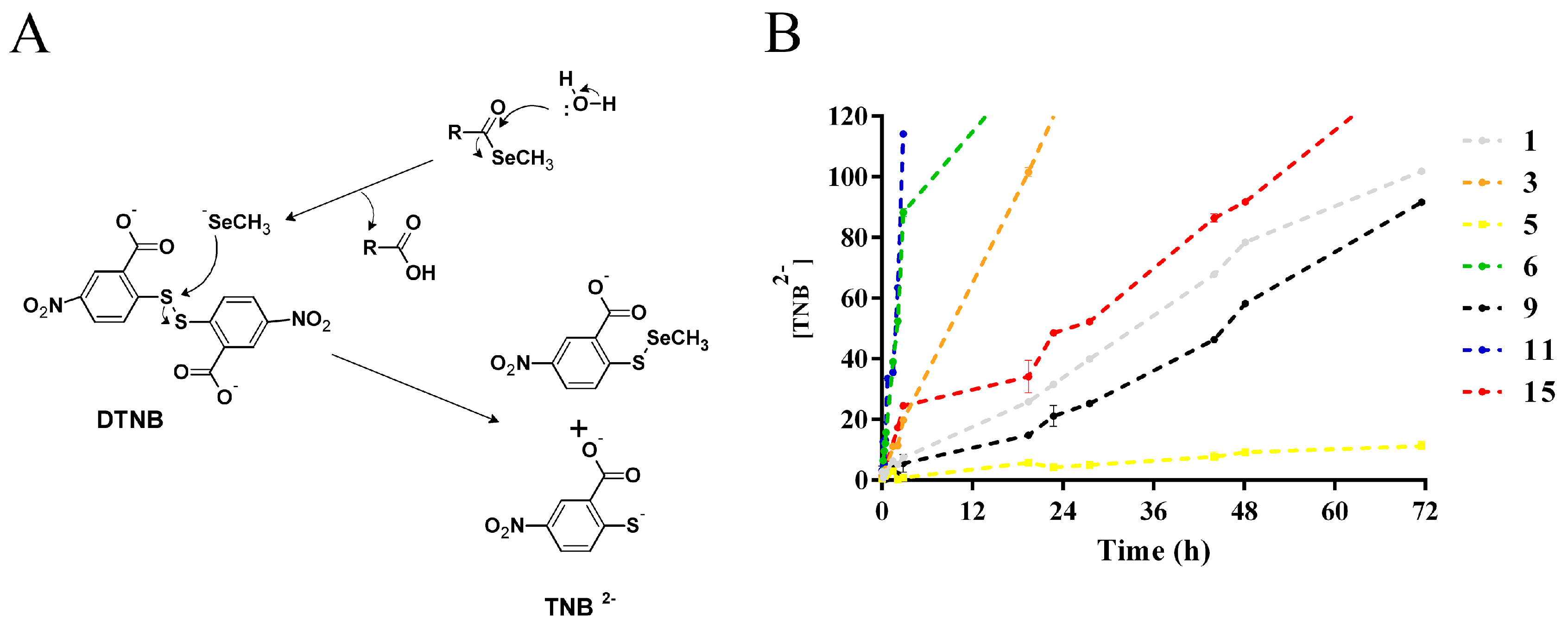
2.2. Methylselenol Release Studies
2.3. Theoretical Calculations of Molecular Properties
2.3.1. Molinspiration Calculations
2.3.2. Osiris DataWarrior Calculations
2.4. Biological Studies
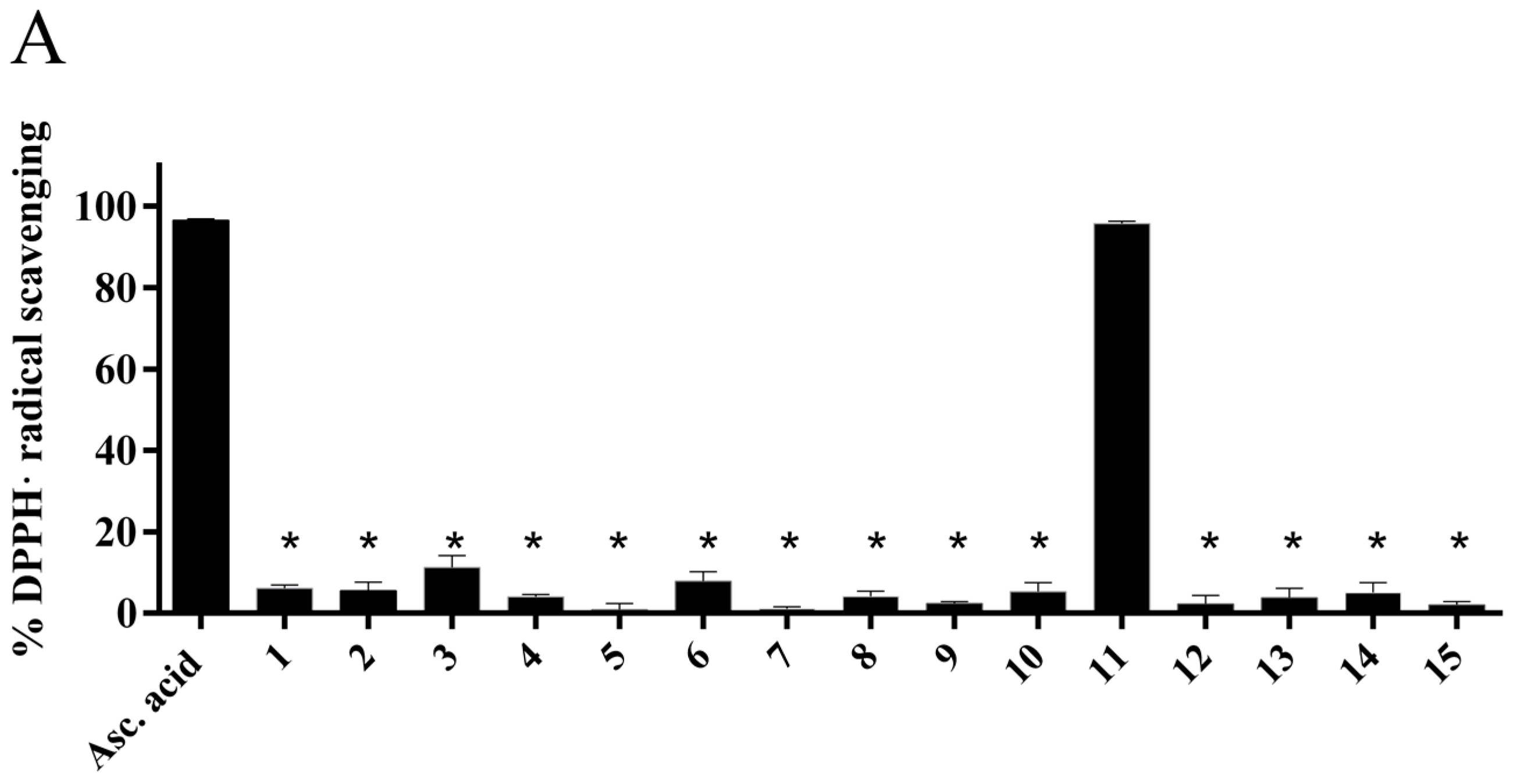
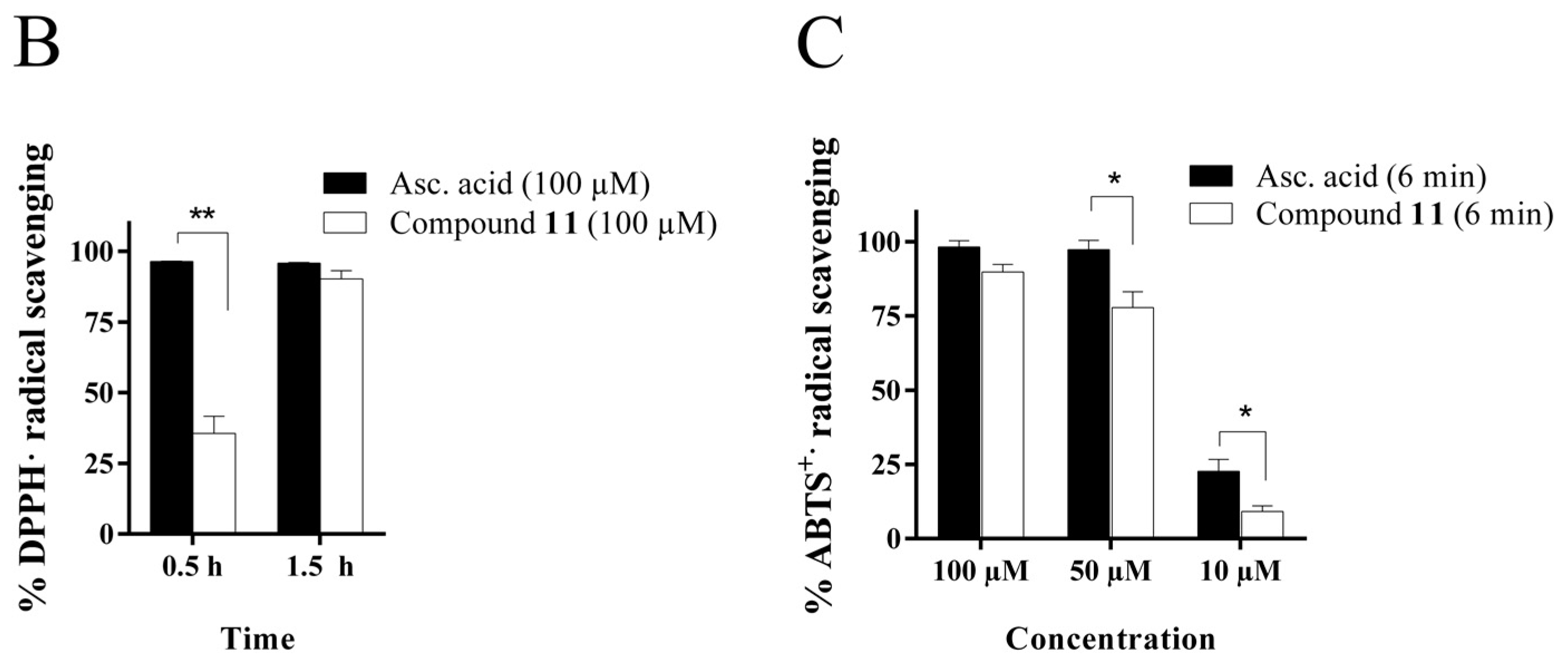
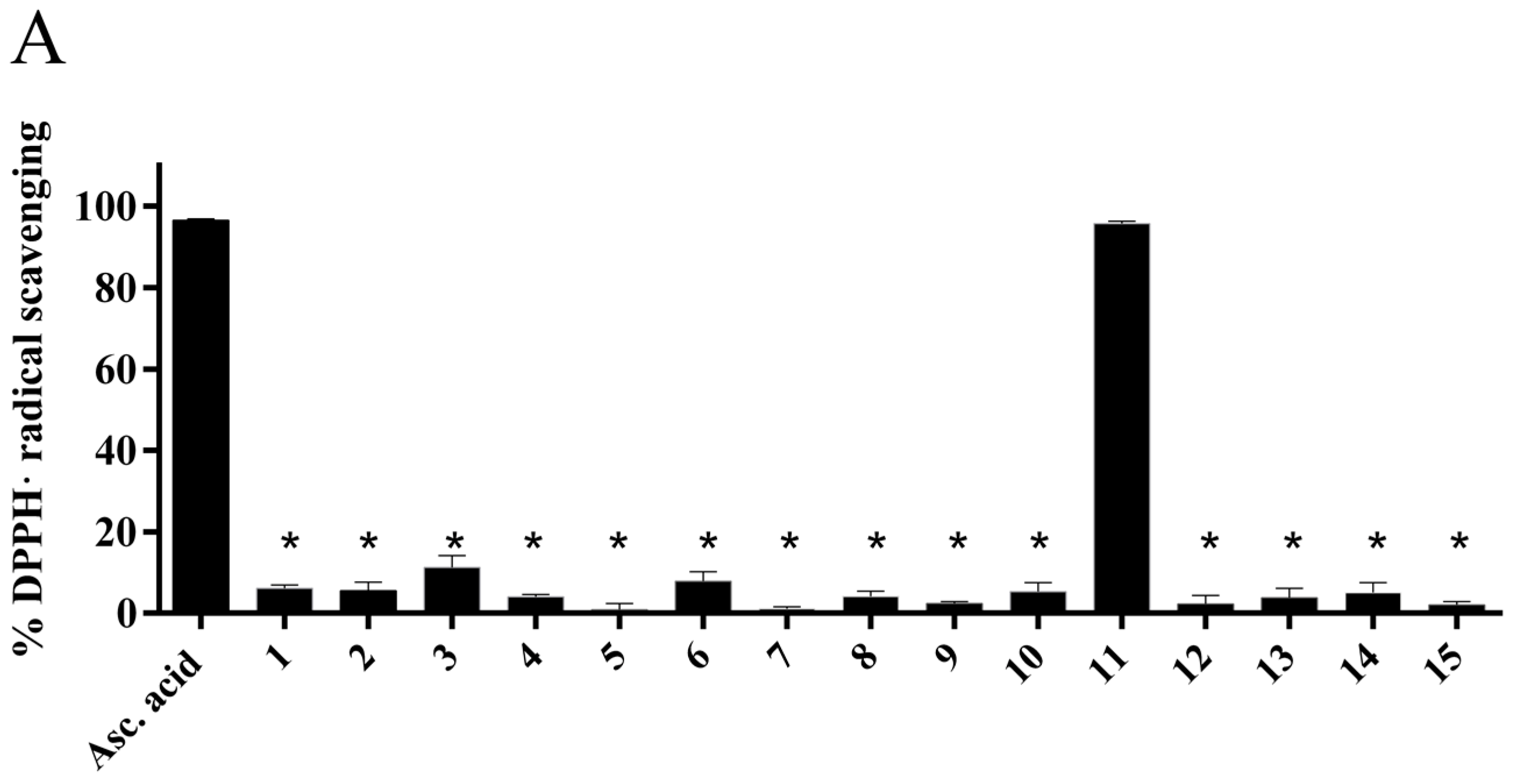
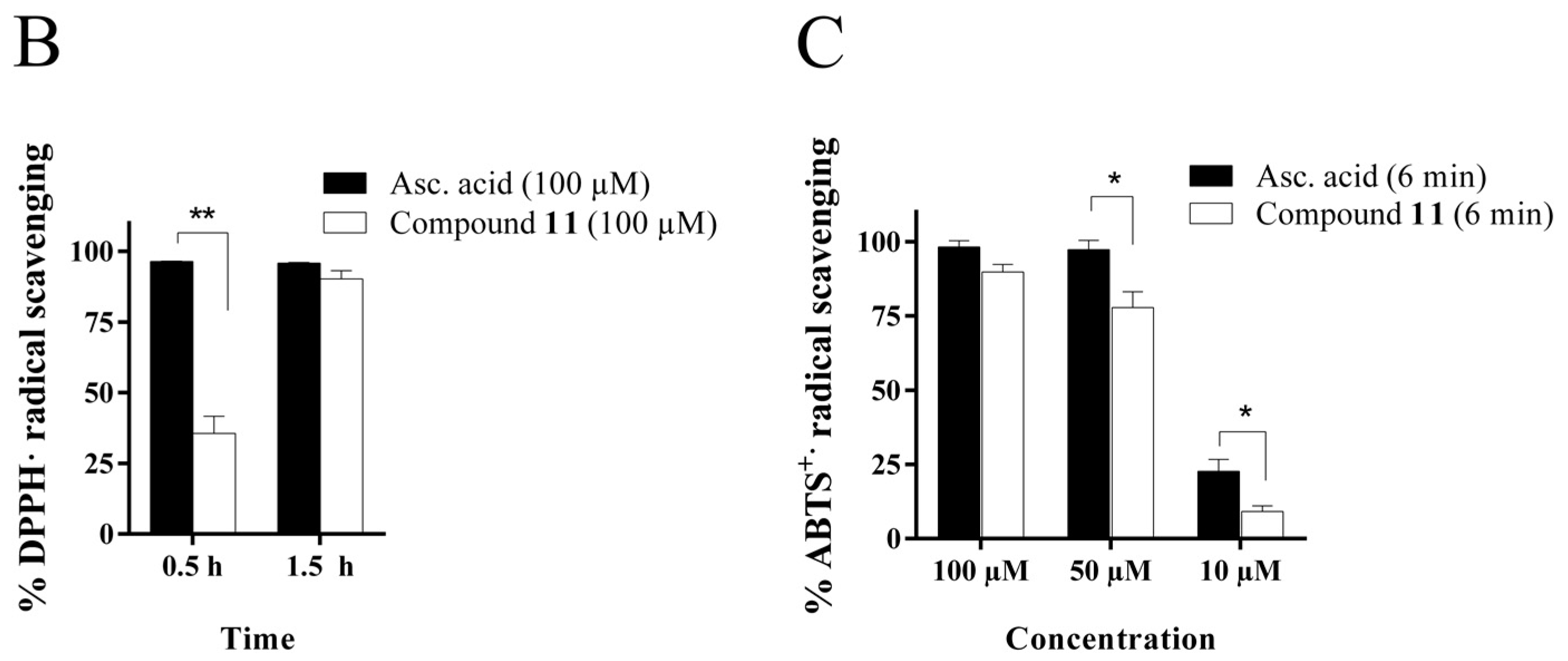
2.4.1. Radical Scavenging Activity of the New Methylselenoesters
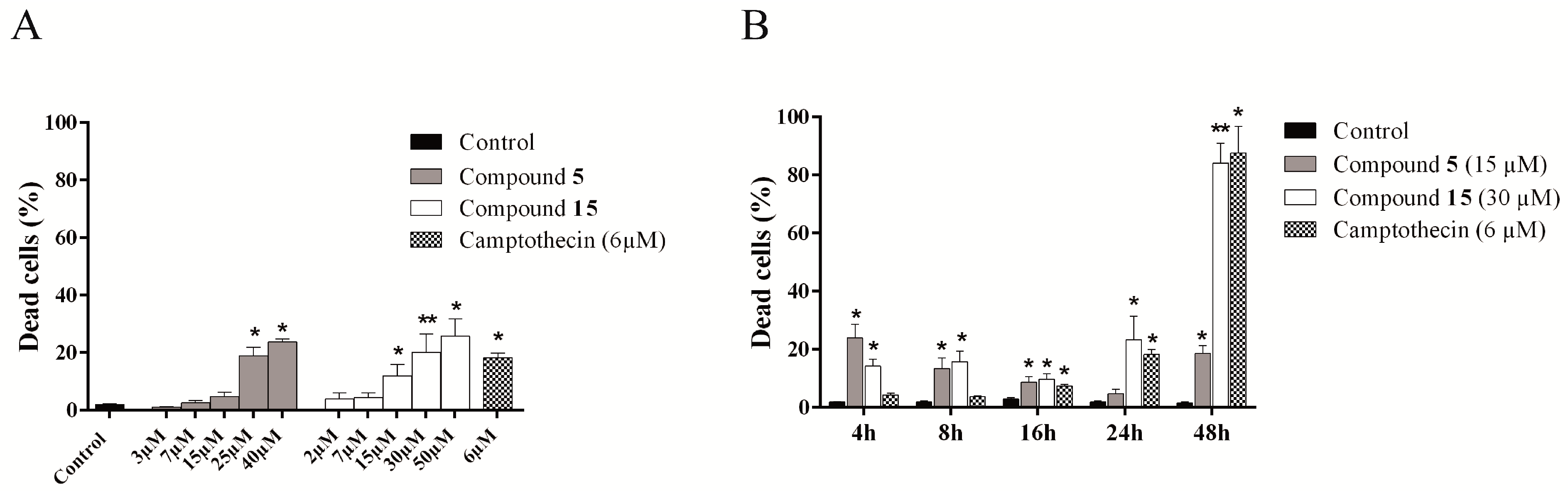
2.4.2. Cytotoxic Activity of the Novel Methylselenoesters
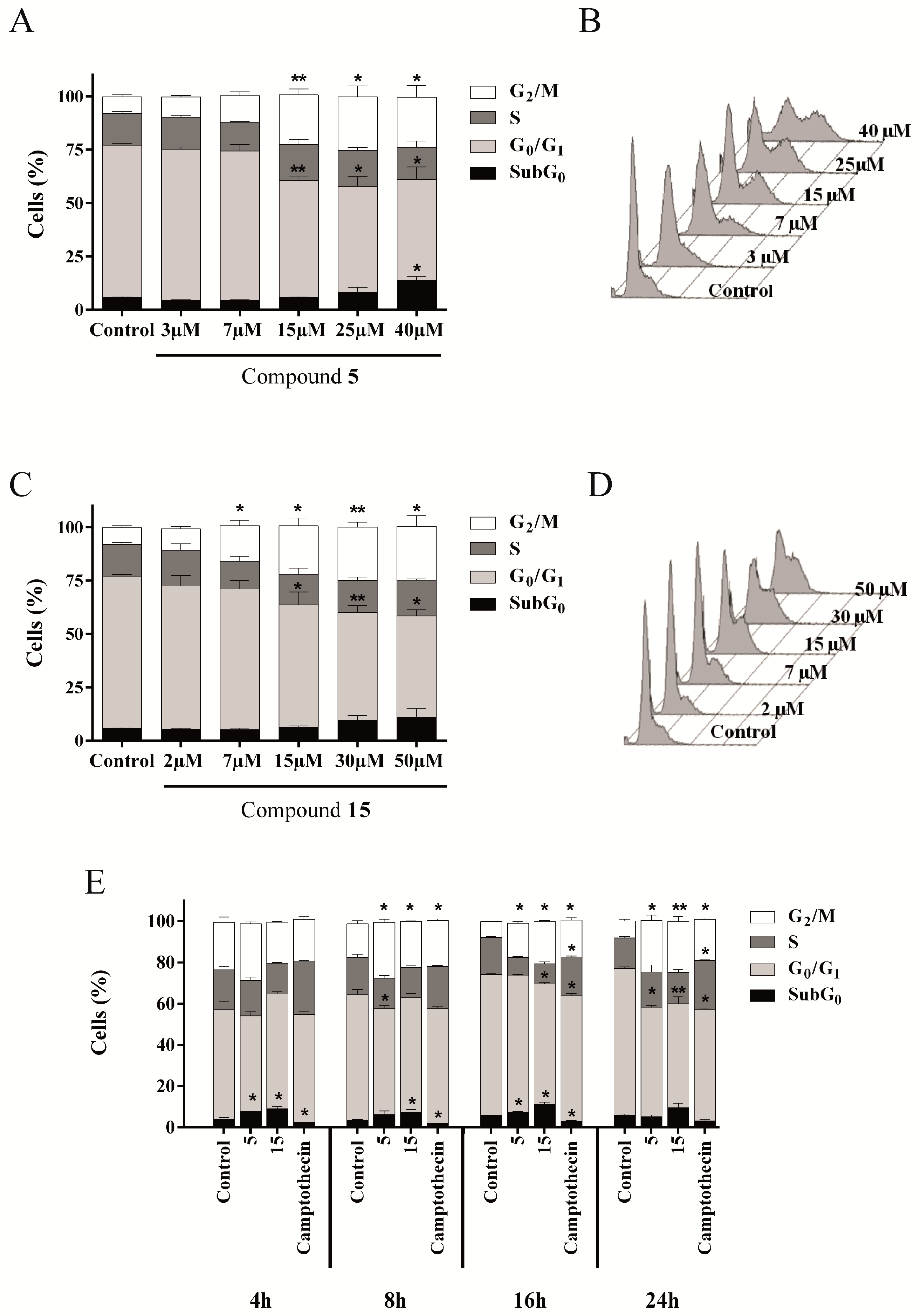
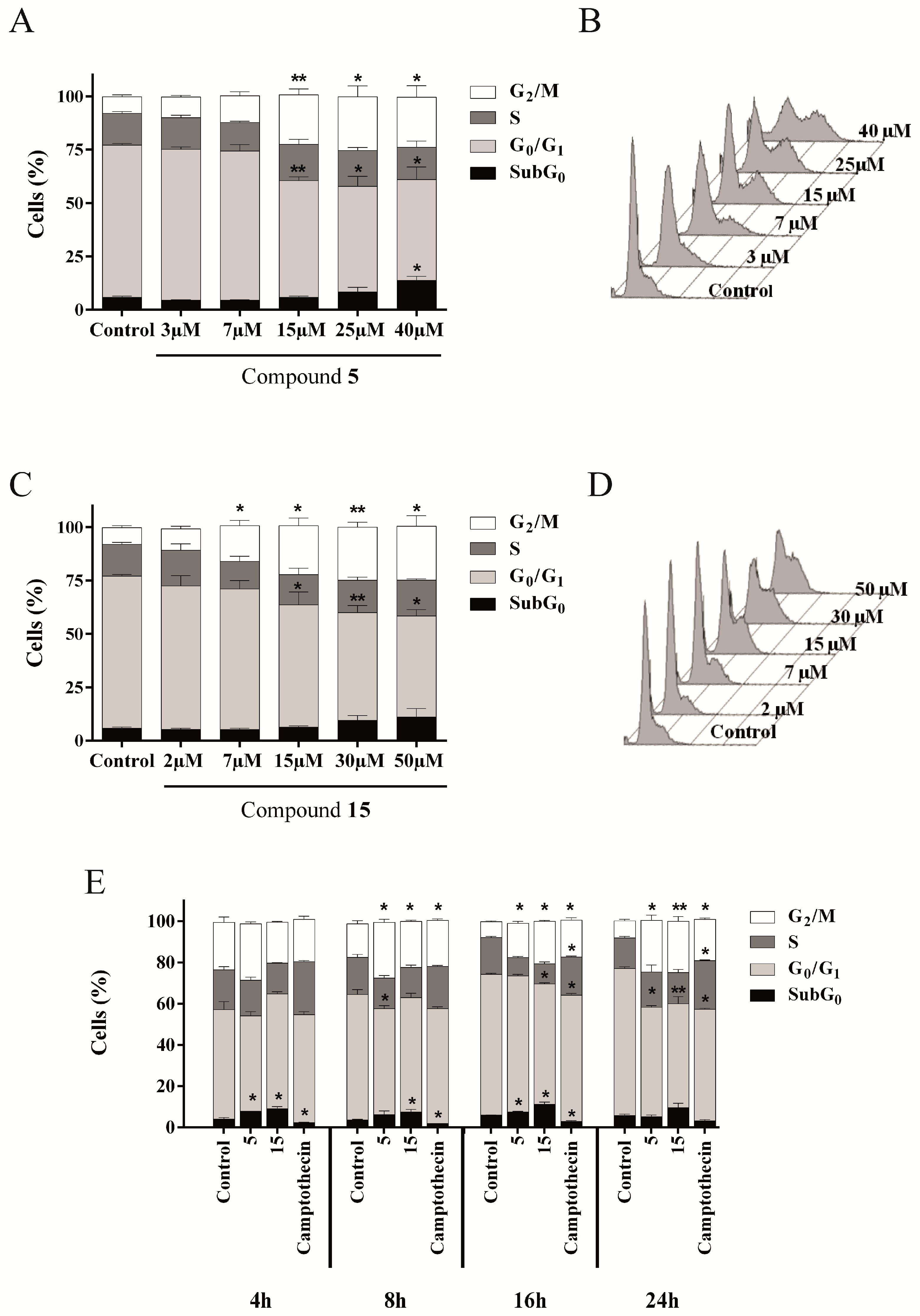
2.4.3. Compounds 5 and 15 Lead to Cell Cycle Arrest in G2/M Phase
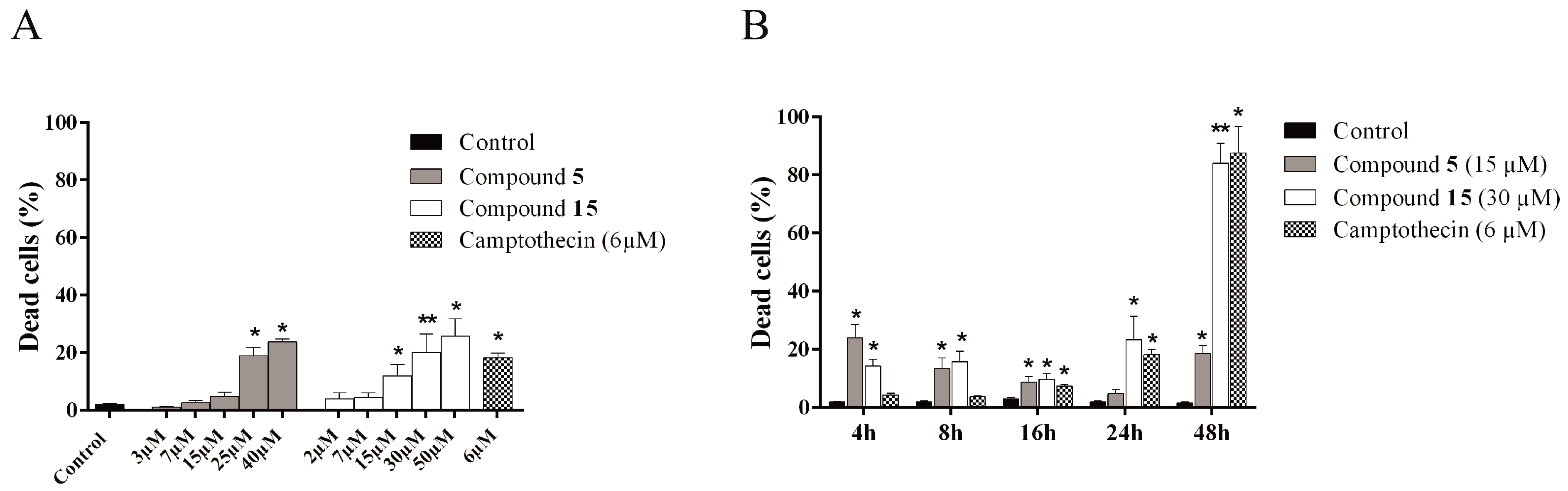
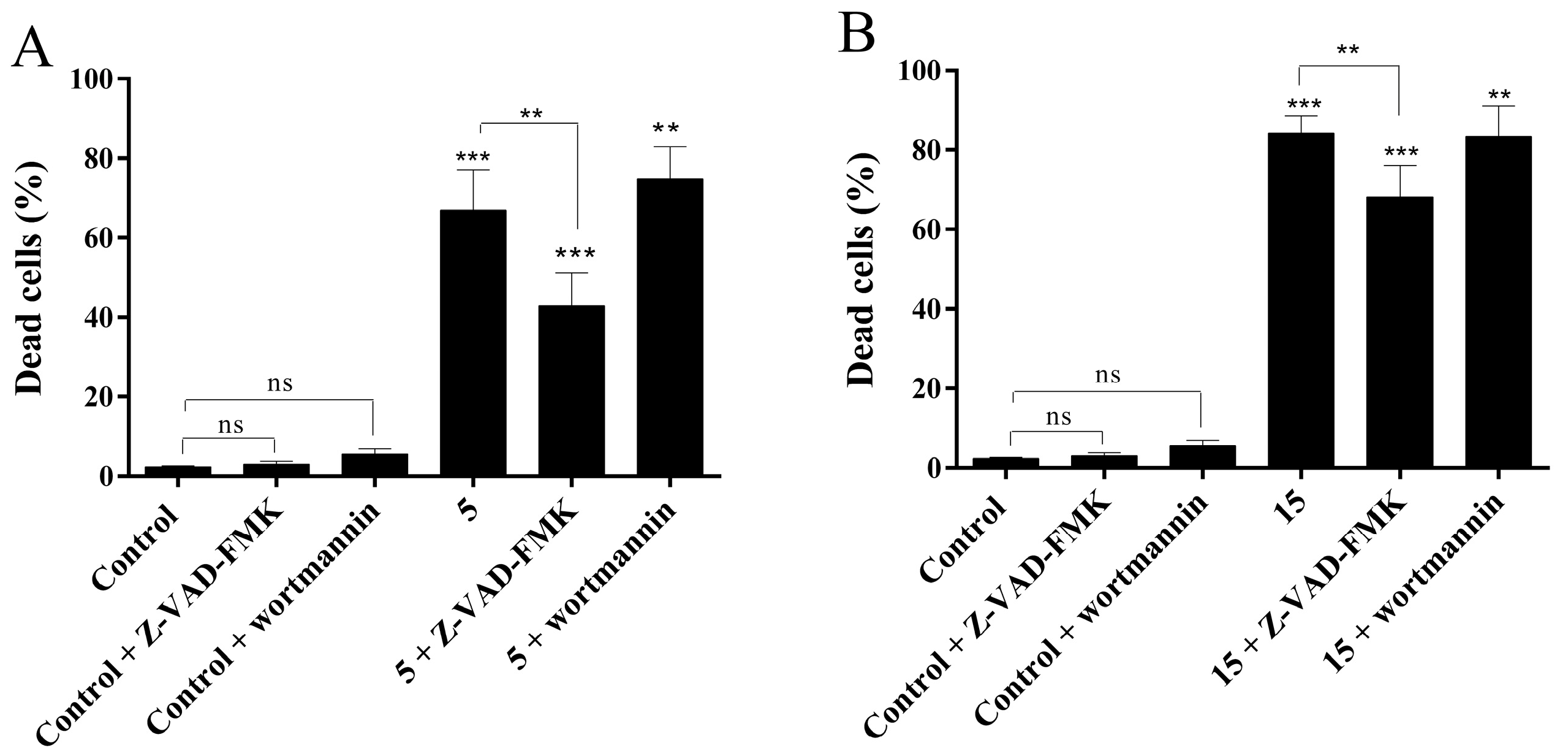
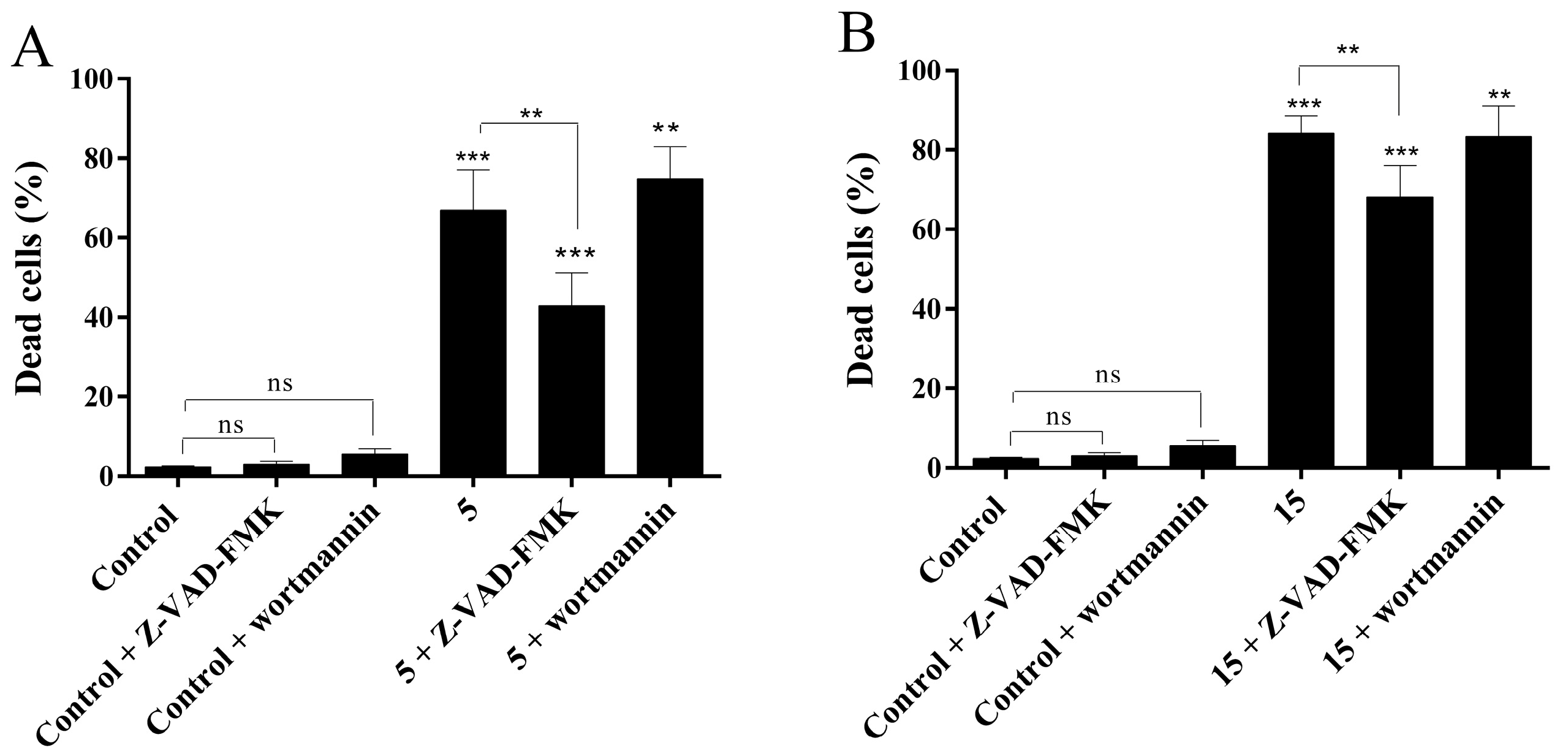
2.4.4. Evaluation of Cell Death Mechanism Induced by Compounds 5 and 15
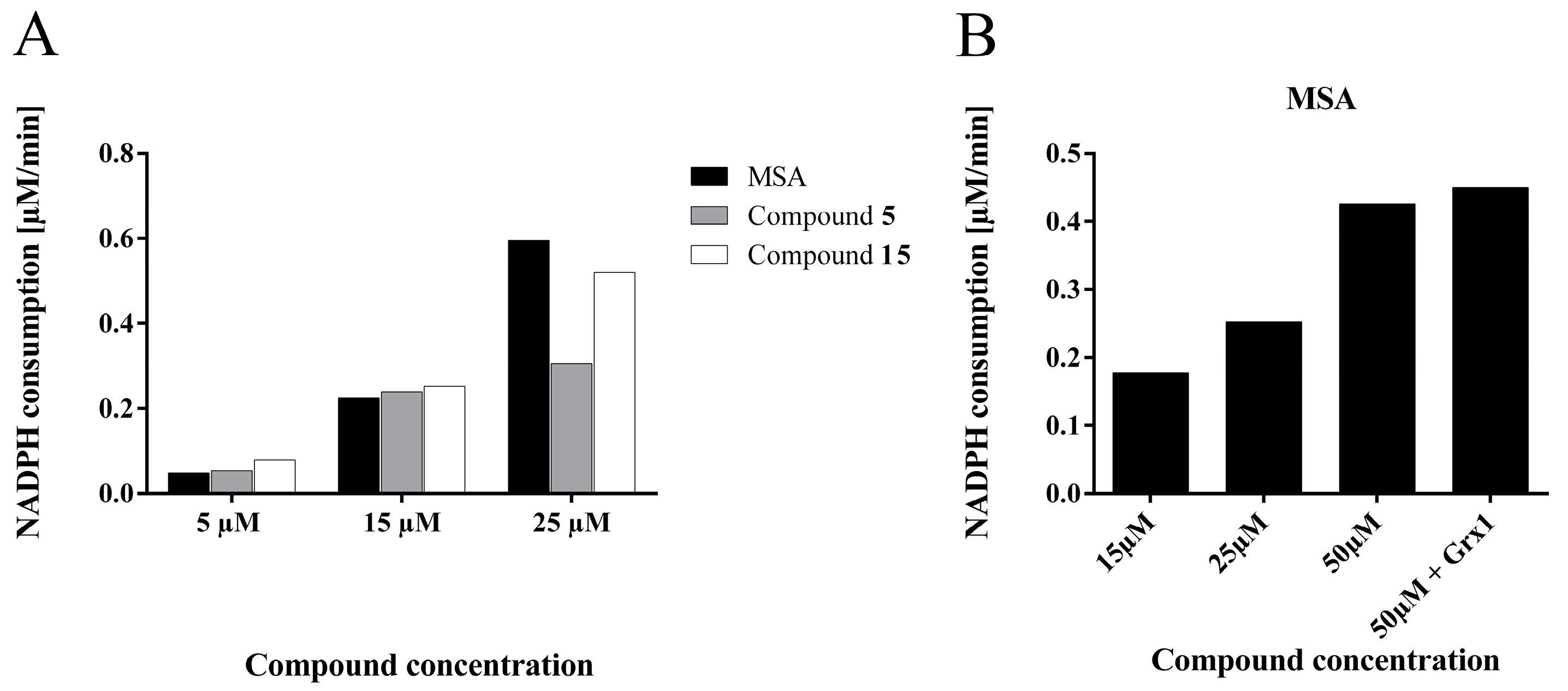
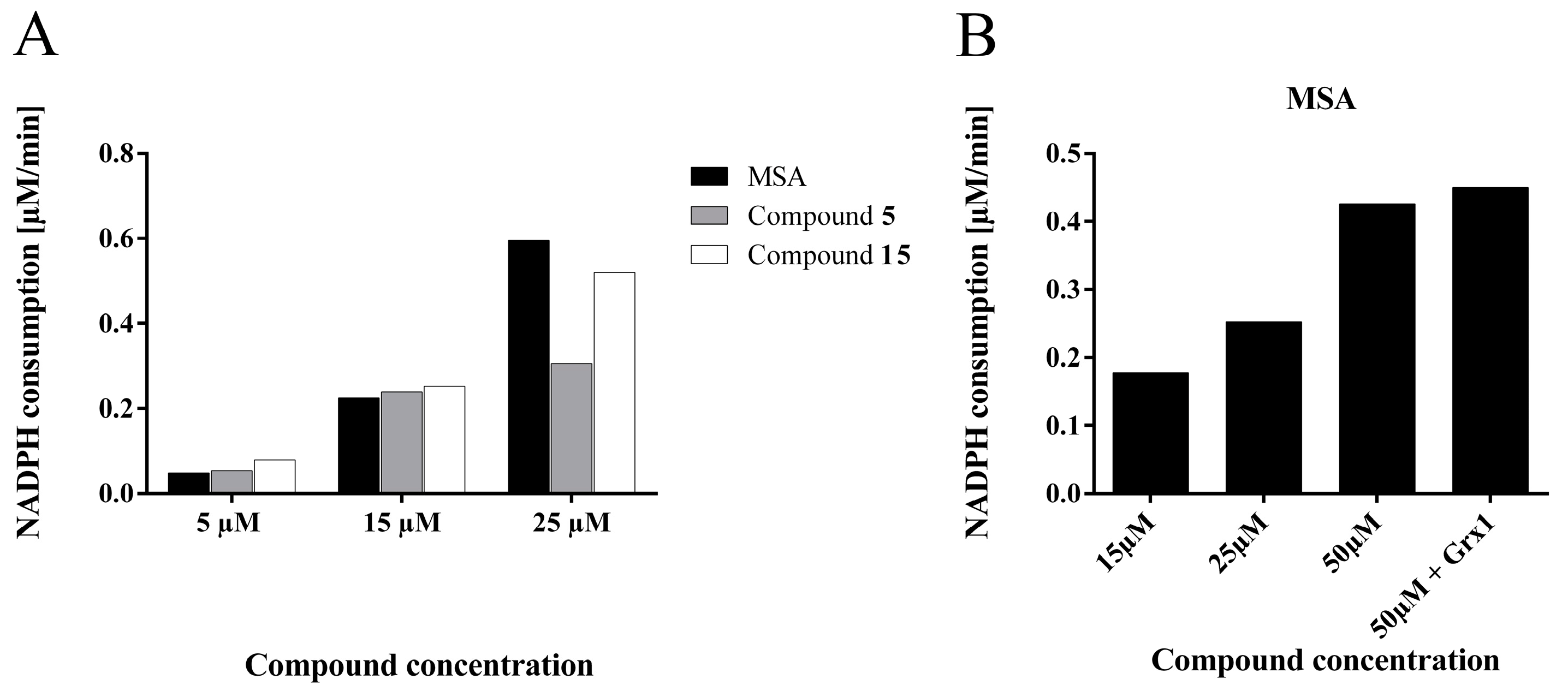
2.4.5. Compounds 5 and 15 are Substrates for Thioredoxin Reductase But not for the Glutathione- Glutaredoxin System
3. Material and Methods
3.1. General Information
3.2. Chemistry
3.2.1. General Procedure
3.2.2. General Procedure for Compounds 4, 6, 8, 9, 11, 14 and 15
3.2.3. General Procedure for Compounds 1–3, 5, 7, 10, 12 and 13
3.3. Methylselenol Release
3.4. Theoretical Calculations of Molecular Properties
3.5. Biology
3.5.1. Radical Scavenging Assays
DPPH assay
ABTS assay
3.5.2. Cell Culture
3.5.3. Viability Assay
3.5.4. Cell Cycle and Cell Death Analysis
3.5.5. Caspase and Autophagy Inhibitors Assay
3.5.6. Enzymatic Assays
Thioredoxin Reductase Activity Assay
Glutaredoxin/Glutathione Assay
3.5.7. Statistics
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA. Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.P.; Gandin, V. Selenium compounds as therapeutic agents in cancer. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 1642–1660. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.; Zhang, J.; Jiang, C.; Deng, Y.; Özten, N.; Bosland, M.C. Cancer chemoprevention research with selenium in the post-SELECT era: Promises and challenges. Nutr. Cancer 2016, 68, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, B. Redox-active Selenium in Health and Disease: A Conceptual Review. Mini Rev. Med. Chem. 2016. [Google Scholar] [CrossRef]
- Wallenberg, M.; Misra, S.; Björnstedt, M. Selenium cytotoxicity in cancer. Basic Clin. Pharmacol. Toxicol. 2014, 114, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Boylan, M.; Selvam, A.; Spallholz, J.E.; Björnstedt, M. Redox-active selenium compounds—From toxicity and cell death to cancer treatment. Nutrients 2015, 7, 3536–3556. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Combs, G.F. Consumption of a high-fat diet abrogates inhibitory effects of methylseleninic acid on spontaneous metastasis of Lewis lung carcinoma in mice. Carcinogenesis 2014, 35, 2308–2313. [Google Scholar] [CrossRef] [PubMed]
- Hagemann-Jensen, M.; Uhlenbrock, F.; Kehlet, S.; Andresen, L.; Gabel-Jensen, C.; Ellgaard, L.; Gammelgaard, B.; Skov, S. The selenium metabolite methylselenol regulates the expression of ligands that trigger immune activation through the lymphocyte receptor NKG2D. J. Biol. Chem. 2014, 289, 31576–31590. [Google Scholar] [CrossRef] [PubMed]
- Marschall, T.A.; Bornhorst, J.; Kuehnelt, D.; Schwerdtle, T. Differing cytotoxicity and bioavailability of selenite, methylselenocysteine, selenomethionine, selenosugar 1 and trimethylselenonium ion and their underlying metabolic transformations in human cells. Mol. Nutr. Food Res. 2016, 2622–2632. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. Selenium in cancer prevention: A review of the evidence and mechanism of action. Proc. Nutr. Soc. 2005, 64, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Roman, M.; Jitaru, P.; Barbante, C. Selenium biochemistry and its role for human health. Metallomics 2014, 6, 25–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liu, X.; Guo, Y.; Liang, Z.; Tian, Y.; Lu, L.; Zhao, X.; Sun, Y.; Zhao, X.; Zhang, H.; Dong, Y. Methylselenocysteine preventing castration-resistant progression of prostate cancer. Prostate 2015, 75, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Durrani, F.A.; Tóth, K.; Rustum, Y.M. Se-methylselenocysteine offers selective protection against toxicity and potentiates the antitumour activity of anticancer drugs in preclinical animal models. Br. J. Cancer 2014, 110, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Weekley, C.M.; Harris, H.H. Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease. Chem. Soc. Rev. 2013, 42, 8870–8894. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Guo, X.; Wang, J.; Jiang, C.; Bosland, M.C.; Lü, J.; Deng, Y. Methylseleninic acid superactivates p53-senescence cancer progression barrier in prostate lesions of pten-knockout mouse. Cancer Prev. Res. 2016, 9, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-M.; Kim, D.-H.; Na, H.-K.; Surh, Y.-J. Methylseleninic acid induces NAD(P)H: Quinone oxidoreductase-1 expression through activation of NF-E2-related factor 2 in Chang liver cells. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Tarrado-Castellarnau, M.; Cortés, R.; Zanuy, M.; Tarragó-Celada, J.; Polat, I.H.; Hill, R.; Fan, T.W. M.; Link, W.; Cascante, M. Methylseleninic acid promotes antitumour effects via nuclear FOXO3a translocation through Akt inhibition. Pharmacol. Res. 2015, 102, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Plano, D.; Sanmartín, C.; Moreno, E.; Prior, C.; Calvo, A.; Palop, J.A. Novel potent organoselenium compounds as cytotoxic agents in prostate cancer cells. Bioorg. Med. Chem. Lett. 2007, 17, 6853–6859. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, E.; Plano, D.; Font, M.; Calvo, A.; Prior, C.; Palop, J.A.; Sanmartín, C. Synthesis and antiproliferative activity of novel symmetrical alkylthio- and alkylseleno-imidocarbamates. Eur. J. Med. Chem. 2011, 46, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, E.; Agliano, A.; Prior, C.; Nguewa, P.; Redrado, M.; González-Zubeldia, I.; Plano, D.; Palop, J.A.; Sanmartín, C.; Calvo, A. The quinoline imidoselenocarbamate EI201 blocks the AKT/mTOR pathway and targets cancer stem cells leading to a strong antitumor activity. Curr. Med. Chem. 2012, 19, 3031–3043. [Google Scholar] [CrossRef] [PubMed]
- Lamberto, I.; Plano, D.; Moreno, E.; Font, M.; Palop, J.A.; Sanmartin, C.; Encio, I. Bisacylimidoselenocarbamates cause G2/M arrest associated with the modulation of CDK1 and Chk2 in human breast cancer MCF-7 cells. Curr. Med. Chem. 2013, 20, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Romano, B.; Font, M.; Encío, I.; Palop, J.A.; Sanmartín, C. Synthesis and antiproliferative activity of novel methylselenocarbamates. Eur. J. Med. Chem. 2014, 83, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Zuazo, A.; Plano, D.; Ansó, E.; Lizarraga, E.; Font, M.; Martínez Irujo, J.J. Cytotoxic and proapototic activities of imidoselenocarbamate derivatives are dependent on the release of methylselenol. Chem. Res. Toxicol. 2012, 25, 2479–2489. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Álvarez, E.; Plano, D.; Font, M.; Calvo, A.; Prior, C.; Jacob, C.; Palop, J.A.; Sanmartín, C. Synthesis and antiproliferative activity of novel selenoester derivatives. Eur. J. Med. Chem. 2014, 73, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, N.K.; Kim, H.S.; Chae, Y.J.; Lee, Y.N.; Kwon, G.C.; Choi, E.H.; Kim, I.T. Synthesis and anticancer activity of di(3-thienyl)methanol and di(3-thienyl)methane. Molecules 2012, 17, 11456–11468. [Google Scholar] [CrossRef] [PubMed]
- Racané, L.; Sedić, M.; Ilić, N.; Aleksić, M.; Pavelić, S.K.; Karminski-Zamola, G. Novel 2-Thienyl- and 2-Benzothienyl-Substituted 6-(2-Imidazolinyl)Benzothiazoles: Synthesis; in vitro Evaluation of Antitumor Effects and Assessment of Mitochondrial Toxicity. Anti-Cancer Agents Med. Chem. 2017, 17, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Im, D.; Jung, K.; Yang, S.; Aman, W.; Hah, J.M. Discovery of 4-arylamido 3-methyl isoxazole derivatives as novel FMS kinase inhibitors. Eur. J. Med. Chem. 2015, 102, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Ananda, H.; Kumar, K.S. S.; Hegde, M.; Rangappa, K.S. Induction of apoptosis and downregulation of ERα in DMBA-induced mammary gland tumors in Sprague–Dawley rats by synthetic 3,5-disubstituted isoxazole derivatives. Mol. Cell. Biochem. 2016, 420, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Alnabulsi, S.; Santina, E.; Russo, I.; Hussein, B.; Kadirvel, M.; Chadwick, A.; Bichenkova, E.V.; Bryce, R.A.; Nolan, K.; Demonacos, C.; et al. Non-symmetrical furan-amidines as novel leads for the treatment of cancer and malaria. Eur. J. Med. Chem. 2016, 111, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Do, A.; Pires, R.A.; Lecerf-Schmidt, F.; Guragossian, N.; Pazinato, J.; Gozzi, G.J.; Winter, E.; Valdameri, G.; Veale, A.; Ene Boumendjel, A.; et al. New, highly potent and non-toxic, chromone inhibitors of the human breast cancer resistance protein ABCG2. Eur. J. Med. Chem. 2016, 122, 291–301. [Google Scholar] [CrossRef]
- Valdameri, G.; Genoux-Bastide, E.; Peres, B.; Gauthier, C.; Guitton, J.; Terreux, R.; Winnischofer, S.M.B.; Rocha, M.E.M.; Boumendjel, A.; Di Pietro, A. Substituted chromones as highly potent nontoxic inhibitors, specific for the breast cancer resistance protein. J. Med. Chem. 2012, 55, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Dai, G.; Weng, J.; Zhang, Z.; Wang, Q.; Zhou, F.; Jiao, L.; Cui, Y.; Ren, Y.; Fan, S.; et al. Discovery of (S)-1-(1-(Imidazo[1,2-a]pyridin-6-yl)ethyl)-6-(1-methyl-1H-pyrazol-4-yl)-1H-[1,2,3]triazolo[4,5-b]pyrazine (volitinib) as a highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitor in clinical development for tre. J. Med. Chem. 2014, 57, 7577–7589. [Google Scholar] [CrossRef] [PubMed]
- Clausen, D.J.; Smith, W.B.; Haines, B.E.; Wiest, O.; Bradner, J.E.; Williams, R.M. Modular synthesis and biological activity of pyridyl-based analogs of the potent Class i Histone Deacetylase Inhibitor Largazole. Bioorg. Med. Chem. 2015, 23, 5061–5074. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zhong, Q.; Xi, Y.; Mottamal, M.; Zhang, Q.; Schroeder, R.L.; Sridhar, J.; He, L.; McFerrin, H.; Wang, G. Modification and biological evaluation of thiazole derivatives as novel inhibitors of metastatic cancer cell migration and invasion. J. Med. Chem. 2014, 57, 6653–6667. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Lopez-Cara, C.; Preti, D.; Aghazadeh Tabrizi, M.; Balzarini, J.; Bassetto, M.; Brancale, A.; Fu, X.-H.; Gao, Y.; et al. Concise synthesis and biological evaluation of 2-Aroyl-5-amino benzo[b]thiophene derivatives as a novel class of potent antimitotic agents. J. Med. Chem. 2013, 56, 9296–9309. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.; Huang, H.; Huangfu, W.; Hsu, K.; Liu, Y.; Liou, P.; Teng, C.; Yang, C. An oral quinoline derivative, MPT0B392, causes leukemic cells mitotic arrest and overcomes drug resistant cancer cells. Oncotarget 2017, 1–14. [Google Scholar]
- Hussaini, S.M. A. Therapeutic significance of quinolines: A patent review (2013–2015). Expert Opin. Ther. Pat. 2016, 26, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.J.; Lena, R.; Bannister, T.; Blake, N.; Pierceall, W.E.; Carlson, N.E.; Keller, C.E.; Koenig, M.; He, Y.; Minond, D.; et al. Hydroxyquinoline-derived compounds and analoguing of selective Mcl-1 inhibitors using a functional biomarker. Bioorg. Med. Chem. 2013, 21, 6642–6649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, B.; Zhang, W.; Yang, T.; Wang, N.; Gao, C.; Tan, C.; Liu, H.; Jiang, Y. Synthesis and antiproliferative activity of 9-benzylamino-6-chloro-2-methoxy-acridine derivatives as potent DNA-binding ligands and topoisomerase II inhibitors. Eur. J. Med. Chem. 2016, 116, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Tam, A.B.; Alagappan, M.; Hay, M.P.; Gupta, A.; Kozak, M.M.; Solow-Cordero, D.E.; Lum, P.Y.; Denko, N.C.; Giaccia, A.J.; et al. Acridine Derivatives as Inhibitors of the IRE1 -XBP1 Pathway Are Cytotoxic to Human Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Romano, B.; Plano, D.; Encío, I.; Palop, J.A.; Sanmartín, C. In vitro radical scavenging and cytotoxic activities of novel hybrid selenocarbamates. Bioorg. Med. Chem. 2015, 23, 1716–1727. [Google Scholar] [CrossRef] [PubMed]
- Klayman, D.L.; Griffin, T.S. Reaction of Selenium with Sodium Borohydride in Protic Solvents. A Facile Method for the Introduction of Selenium into Organic Molecules. J. Am. Chem. Soc. 1973, 2, 197–199. [Google Scholar] [CrossRef]
- Athayde-Filho, P.F. De Synthesis and characterization of three new organo-selenium compounds. A convenient synthesis of aroylselenoglycolic acids. Arkivoc 2004, 2004, 22–26. [Google Scholar] [CrossRef]
- Ellman, G.L. A colorimetric method for determining low concentrations of mercaptans. Arch. Biochem. Biophys. 1958, 74, 443–450. [Google Scholar] [CrossRef]
- Ip, C.; Thompson, H.J.; Zhu, Z.; Ganther, H.E. In vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000, 60, 2882–2886. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y.; Pei, Z.; Chen, S.; Yang, X.; Chen, Y.; Lin, D.; Ma, R.Z. Methylseleninic acid restricts tumor growth in nude mice model of metastatic breast cancer probably via inhibiting angiopoietin-2. BMC Cancer 2012, 12, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Wu, M. The Inhibitory Efficacy of Methylseleninic Acid Against Colon Cancer Xenografts in C57BL/6 Mice. Nutr. Cancer 2015, 67, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Wu, M.; Botnen, J.H. Methylselenol, a selenium metabolite, induces cell cycle arrest in G1 phase and apoptosis via the extracellular-regulated kinase 1/2 pathway and other cancer signaling genes. J. Nutr. 2009, 139, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.I.; Combs, G.F. Selenium and anticarcinogenesis: Underlying mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Wallenberg, M.; Olm, E.; Hebert, C.; Björnstedt, M.; Fernandes, A.P. Selenium compounds are substrates for glutaredoxins: A novel pathway for selenium metabolism and a potential mechanism for selenium-mediated cytotoxicity. Biochem. J. 2010, 429, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, H.; Li, Y.; Wu, Z.; Zhu, Y.; Wang, T.; Gao, A.C.; Chen, J.; Zhou, Q. Intracellular glutathione content influences the sensitivity of lung cancer cell lines to methylseleninic acid. Mol. Carcinog. 2012, 51, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Gromer, S.; Gross, J.H. Methylseleninate is a substrate rather than an inhibitor of mammalian thioredoxin reductase. Implications for the antitumor effects of selenium. J. Biol. Chem. 2002, 277, 9701–9706. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Narula, R.; Joshi, K.; Rana, S.; Singh, M. In silico modeling of ligand molecule for non structural 3 (NS3) protein target of flaviviruses. Bioinformation 2012, 8, 123–127. [Google Scholar]
- Sander, T.; Freyss, J.; von Korff, M.; Reich, J.R.; Rufener, C. OSIRIS, an entirely inhouse developed drug discovery informatics system. J. Chem. Inf. Model. 2009, 49, 232–246. [Google Scholar] [CrossRef] [PubMed]
- García-Herreros, C.; García-Iñiguez, M.; Astiasarán, I.; Ansorena, D. Antioxidant activity and phenolic content of water extracts of borago officinalis I.: Influence of plant part and cooking procedure. Ital. J. Food Sci. 2010, 22, 156–164. [Google Scholar]
- Arnér, E.S.J.; Holmgren, A. Measurement of thioredoxin and thioredoxin reductase. In Current Protocols in Toxicology/Editorial Board, Mahin D. Maines; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Holmgren, A.; Aslund, F. Glutaredoxin. Methods Enzym. 1995, 252, 283–292. [Google Scholar]
Sample Availability: Samples of the compounds 1–15 reported in this paper are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
General Structure | |||||
| Ref. a | R | Solvent b | Ref. | R | Solvent |
| 1 |  | EtOH | 8 |  | H2O |
| 2 |  | EtOH | 9 |  | H2O |
| 3 |  | EtOH | 10 |  | EtOH |
| 4 |  | H2O | |||
| 5 |  | EtOH | 11 |  | H2O |
| 6 |  | H2O | 12 |  | EtOH |
| 7 |  | EtOH | 13 |  | EtOH |
| General structure |  | ||||
| Ref. | R | Solvent | Ref. | R | Solvent |
| 14 |  | H2O | 15 |  | H2O |
| Molinspiration Calculations | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | milogP a | PSA b | MW c | nON d | nOHNH e | nViolations f | Nrotb g | Volume |
| 1 | 2.39 | 62.90 | 244.11 | 4 | 0 | 0 | 3 | 166.41 |
| 2 | 2.48 | 26.30 | 229.14 | 2 | 0 | 0 | 3 | 168.62 |
| 3 | 0.96 | 42.85 | 201.09 | 3 | 0 | 0 | 2 | 134.77 |
| 4 | 2.33 | 17.07 | 205.14 | 1 | 0 | 0 | 2 | 133.79 |
| 5 | 2.93 | 17.07 | 239.59 | 1 | 0 | 0 | 2 | 147.33 |
| 6 | 1.15 | 43.10 | 190.09 | 3 | 0 | 0 | 2 | 120.49 |
| 7 | 3.63 | 17.07 | 255.20 | 1 | 0 | 0 | 2 | 177.78 |
| 8 | 2.32 | 35.54 | 243.12 | 3 | 0 | 0 | 2 | 167.01 |
| 9 | 2.41 | 29.96 | 250.16 | 2 | 0 | 0 | 2 | 182.91 |
| 10 | 4.55 | 29.96 | 326.26 | 2 | 0 | 0 | 3 | 254.32 |
| 11 | 3.33 | 47.28 | 346.04 | 3 | 0 | 0 | 2 | 205.51 |
| 12 | 2.23 | 42.85 | 283.21 | 3 | 0 | 0 | 3 | 196.89 |
| 13 | 4.19 | 29.96 | 300.22 | 2 | 0 | 0 | 2 | 226.91 |
| 14 | 2.60 | 57.93 | 345.12 | 3 | 0 | 0 | 4 | 218.97 |
| 15 | 2.41 | 47.28 | 310.07 | 3 | 0 | 0 | 4 | 183.68 |
| MSA | 0.60 | 37.30 | 127.00 | 2 | 1 | 0 | 0 | 68.78 |
| Compound | Toxicity Level | Osiris Calculations | |||
|---|---|---|---|---|---|
| Mutagenic | Tumorigenic | Irritant | RE a | DL b | |
| 1 | −10.80 | ||||
| 2 | −5.34 | ||||
| 3 | −5.25 | ||||
| 4 | −1.96 | ||||
| 5 | −2.77 | ||||
| 6 | −5.52 | ||||
| 7 | −2.67 | ||||
| 8 | −2.91 | ||||
| 9 | −5.25 | ||||
| 10 | −5.25 | ||||
| 11 | −4.86 | ||||
| 12 | −0.85 | ||||
| 13 | −5.25 | ||||
| 14 | −9.53 | ||||
| 15 | −2.76 | ||||
| MSA | −5.69 | ||||
 High risk;
High risk;  Mild risk;
Mild risk;  No risk.
No risk.| Cell Line | ||||||||||||
| No. | PC-3 a | MCF7 b | 184B5 c | |||||||||
| GI50 | TGI | LC50 | GI50 | TGI | LC50 | GI50 | TGI | LC50 | ||||
| 1 | 5.8 ± 0.7 | 10.4 ± 2.3 | 75.7 ± 3.3 | 4.1 ± 0.5 | 7.9 ± 0.9 | 45.0 ± 4.6 | 3.4 ± 0.4 | 6.4 ± 1.1 | 9.3 ± 0.6 | |||
| 2 | 7.3 ± 1.7 | 45.1 ± 4.5 | >100 | 4.1 ± 0.8 | 9.3 ± 4.1 | 74.8 ± 4.0 | 3.7 ± 0.8 | 6.4 ± 1.5 | 9.0 ± 2.1 | |||
| 3 | 5.4 ± 1.1 | 15.3 ± 1.7 | 60.5 ± 4.3 | 3.5 ± 0.8 | 7.2 ± 1.0 | 40.1 ± 4.4 | 3.1 ± 0.5 | 6.6 ± 0.9 | 17.6 ± 4.4 | |||
| 4 | 5.2 ± 0.9 | 19.4 ± 3.6 | 62.0 ± 4.9 | 5.3 ± 1.4 | 22.8 ± 3.8 | 89.6 ± 4.1 | 3.6 ± 0.8 | 7.2 ± 1.4 | 27.9 ± 4.6 | |||
| 5 | 8.6 ± 1.7 | 47.7 ± 2.9 | 91.6 ± 3.9 | 3.1 ± 0.9 | 6.9 ± 1.1 | 35.9 ± 4.4 | 3.5 ± 0.4 | 5.9 ± 0.3 | 8.3 ± 0.2 | |||
| 6 | 4.5 ± 1.3 | 7.7 ± 0.1 | 61.5 ± 14.0 | 3.5 ± 1.0 | 8.8 ± 0.7 | 71.9 ± 3.2 | 2.7 ± 0.2 | 6.0 ± 0.4 | 9.3 ± 2.0 | |||
| 7 | 18.4 ± 4.5 | 47.8 ± 3.9 | 77.3 ± 4.5 | 6.9 ± 1.4 | 61.6 ± 4.1 | >100 | 3.9 ± 1.2 | 6.9 ± 1.7 | 9.9 ± 0.8 | |||
| 8 | 5.7 ± 1.6 | 11.0 ± 3.1 | 96.0 ± 4.7 | 4.6 ± 0.5 | 9.2 ± 1.3 | 68.7 ± 2.5 | 3.7 ± 0.3 | 6.4 ± 0.2 | 9.0 ± 0.4 | |||
| 9 | 5.9 ± 0.5 | 10.0 ± 1.9 | 84.1 ± 8.9 | 4.4 ± 0.8 | 8.1 ± 1.2 | 48.0 ± 7.5 | 3.7 ± 0.5 | 6.2 ± 0.4 | 8.6 ± 0.4 | |||
| 10 | 4.4 ± 1.3 | 9.9 ± 4.7 | 70.9 ± 5.2 | 5.6 ± 1.3 | 51.3 ± 3.7 | >100 | 3.9 ± 0.7 | 6.3 ± 0.5 | 8.7 ± 0.4 | |||
| 11 | 5.3 ± 1.1 | 17.1 ± 5.0 | 66.2 ± 2.4 | 4.2 ± 0.7 | 8.2 ± 1.1 | >100 | 3.3 ± 0.1 | 6.7 ± 0.9 | 14.5 ± 4.5 | |||
| 12 | 16.9 ± 4.1 | 50.9 ± 4.9 | 85.0 ± 4.7 | 4.4 ± 1.0 | 8.1 ± 1.7 | 56.2 ± 9.6 | 3.4 ± 0.3 | 6.5 ± 0.8 | 9.6 ± 2.8 | |||
| 13 | 39.7 ± 5.1 | >100 | >100 | 4.2 ± 1.8 | 35.4 ± 4.7 | 83.4 ± 4.1 | 6.6 ± 1.2 | 26.8 ± 1.0 | 65.0 ± 4.5 | |||
| 14 | 7.2 ± 3.5 | 32.3 ± 3.7 | 68.7 ± 3.0 | 4.9 ± 0.8 | 8.4 ± 1.1 | 54.4 ± 2.7 | 3.8 ± 0.7 | 6.5 ± 0.6 | 9.3 ± 0.7 | |||
| 15 | 4.8 ± 0.8 | 8.4 ± 0.6 | 47.8 ± 4.4 | 1.8 ± 0.9 | 7.2 ± 1.9 | 57.0 ± 4.0 | 3.5 ± 1.1 | 6.4 ± 0.5 | 9.2 ± 0.9 | |||
| MSA | 4.4 ± 1.1 | 8.5 ± 2.4 | 47.6 ± 4.6 | 1.5 ± 0.5 | 5.4 ± 0.5 | 9.4 ± 1.0 | 5.3 ± 1.2 | 9.2 ± 1.3 | 13.1 ± 1.3 | |||
| Cell Line | ||||||||||||
| No. | K-562 d | HT-29 e | HTB-54 f | BEAS-2B g | ||||||||
| GI50 | TGI | LC50 | GI50 | TGI | LC50 | GI50 | TGI | LC50 | GI50 | TGI | LC50 | |
| 1 | 50.1 ± 2.3 | 85.3 ± 4.9 | >100 | 5.0 ± 0.7 | 9.3 ± 1.4 | 87.2 ± 5.0 | 22.5 ± 4.5 | 50.5 ± 5.1 | 78.6 ± 2.9 | 3.8 ± 0.3 | 6.4 ± 0.4 | 9.0 ± 0.7 |
| 2 | 40.8 ± 4.8 | 70.6 ± 5.9 | >100 | 4.1 ± 0.1 | 7.5 ± 1.1 | 50.0 ± 2.4 | 6.8 ± 1.7 | 29.7 ± 4.3 | 70.0 ± 2.9 | 3.2 ± 0.5 | 5.8 ± 0.6 | 8.4 ± 1.0 |
| 3 | 30.2 ± 2.2 | 59.1 ± 2.1 | 87.9 ± 2.0 | 4.7 ± 0.6 | 9.1 ± 0.8 | >100 | 6.2 ± 1.5 | 21.1 ±4.1 | 71.1 ± 3.8 | 3.6 ± 0.6 | 6.1 ± 0.5 | 8.6 ± 0.5 |
| 4 | 40.6 ± 4.5 | 74.6 ± 4.9 | >100 | 3.4 ± 0.2 | 7.2 ± 1.2 | 47.5 ± 3.2 | 8.5 ± 3.1 | 39.0 ± 3.1 | 73.9 ± 4.1 | 2.9 ± 0.3 | 5.8 ± 0.7 | 8.7 ± 2.1 |
| 5 | 38.7 ± 2.1 | 67.0 ± 1.9 | 95.2 ± 2.2 | 3.6 ± 0.3 | 6.3 ± 0.2 | 8.9 ± 0.2 | 4.9 ± 0.2 | 9.3 ± 2.7 | 61.5 ± 3.2 | 2.7 ± 0.5 | 5.3 ± 0.7 | 8.0 ± 1.0 |
| 6 | 18.5 ± 7.1 | 58.3 ± 3.5 | 98.1 ± 0.7 | 5.0 ± 0.8 | 15.1 ± 6.7 | 90.8 ± 15.0 | 9.0 ± 2.8 | 43.9 ± 3.9 | 82.0 ± 1.3 | 3.0 ± 0.9 | 5.6 ± 0.7 | 8.2 ± 0.5 |
| 7 | 29.2 ± 4.4 | 58.8 ± 1.5 | 88.3 ± 3.3 | 3.7 ± 0.2 | 7.4 ± 0.9 | 41.8 ± 4.5 | 17.5 ± 4.2 | 55.4 ± 4.4 | 93.4 ± 4.8 | 3.9 ± 0.6 | 6.8 ± 0.8 | 9.8 ± 1.1 |
| 8 | 29.7 ± 4.7 | 62.6 ± 3.9 | 95.4 ± 4.9 | 5.9 ± 0.8 | 27.7 ± 4.5 | 81.1 ± 4.0 | 6.0 ± 0.9 | 23.2 ± 2.6 | 77.1 ± 4.9 | 3.1 ± 0.3 | 5.6 ± 0.6 | 8.0 ± 1.1 |
| 9 | 39.5 ± 3.1 | 72.6 ± 4.1 | >100 | 3.4 ± 0.2 | 7.0 ± 0.2 | 37.2 ± 5.0 | 7.8 ± 1.2 | 33.0 ± 4.5 | 69.2 ± 2.7 | 4.1 ± 1.0 | 7.1 ± 4.5 | 15.3 ± 9.1 |
| 10 | 9.7 ± 4.9 | 59.1 ± 3.5 | >100 | 4.1 ± 0.3 | 7.5 ± 0.7 | 35.0 ± 2.4 | 5.9 ± 1.1 | 16.7 ± 2.3 | 64.4 ± 4.2 | 3.1 ± 0.5 | 5.5 ± 0.4 | 7.8 ± 0.5 |
| 11 | 81.9 ± 4.0 | >100 | >100 | 4.9 ± 0.7 | 9.6 ± 3 | 74.4 ± 4.2 | 9.4 ± 3.5 | 52.3 ± 2.9 | >100 | 2.4 ± 0.9 | 5.0 ± 0.8 | 7.6 ± 0.7 |
| 12 | 82.0 ± 4.4 | >100 | >100 | 5.1 ± 0.1 | 14.2 ± 6.6 | >100 | 9.8 ± 5.1 | 44.9 ± 3.7 | 80.5 ± 3.5 | 3.7 ± 1.3 | 6.4 ± 1.3 | 9.2 ± 1.0 |
| 13 | n.a. h | n.a. | n.a. | 6.0 ± 0.5 | 16.5 ± 1.9 | 66.6 ± 0.1 | n.a. | n.a. | n.a. | 3.8 ± 0.1 | 6.4 ± 0.2 | 8.9 ± 0.4 |
| 14 | 29.9 ± 0.6 | 54.7 ± 0.7 | 79.5 ± 0.7 | 3.8 ± 0.1 | 7.9 ± 0.7 | >100 | 3.8 ± 0.6 | 7.7 ± 1.0 | 66.5 ± 4.5 | 3.2 ± 0.7 | 5.5 ± 0.5 | 7.8 ± 0.3 |
| 15 | 42.0 ± 3.8 | 70.6 ± 3.4 | 99.2 ± 5.3 | 3.0 ± 0.5 | 6.6 ± 0.4 | 35.8 ± 5.0 | 15.6 ± 4.3 | 46.0 ± 3.0 | 76.3 ± 1.1 | 2.6 ± 0.8 | 5.6 ± 0.9 | 8.6 ± 1.2 |
| MSA | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 3.5 ± 0.1 | 6.9 ± 0.2 | 19.9 ± 1.2 | 3.7 ± 0.7 | 6.2 ± 1.2 | 8.7 ± 1.8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Argelich, N.; Encío, I.; Plano, D.; Fernandes, A.P.; Palop, J.A.; Sanmartín, C. Novel Methylselenoesters as Antiproliferative Agents. Molecules 2017, 22, 1288. https://doi.org/10.3390/molecules22081288
Díaz-Argelich N, Encío I, Plano D, Fernandes AP, Palop JA, Sanmartín C. Novel Methylselenoesters as Antiproliferative Agents. Molecules. 2017; 22(8):1288. https://doi.org/10.3390/molecules22081288
Chicago/Turabian StyleDíaz-Argelich, Nuria, Ignacio Encío, Daniel Plano, Aristi P. Fernandes, Juan Antonio Palop, and Carmen Sanmartín. 2017. "Novel Methylselenoesters as Antiproliferative Agents" Molecules 22, no. 8: 1288. https://doi.org/10.3390/molecules22081288