Water-soluble Manganese and Iron Mesotetrakis(carboxyl)porphyrin: DNA Binding, Oxidative Cleavage, and Cytotoxic Activities


Abstract
:1. Introduction
2. Results and Discussion
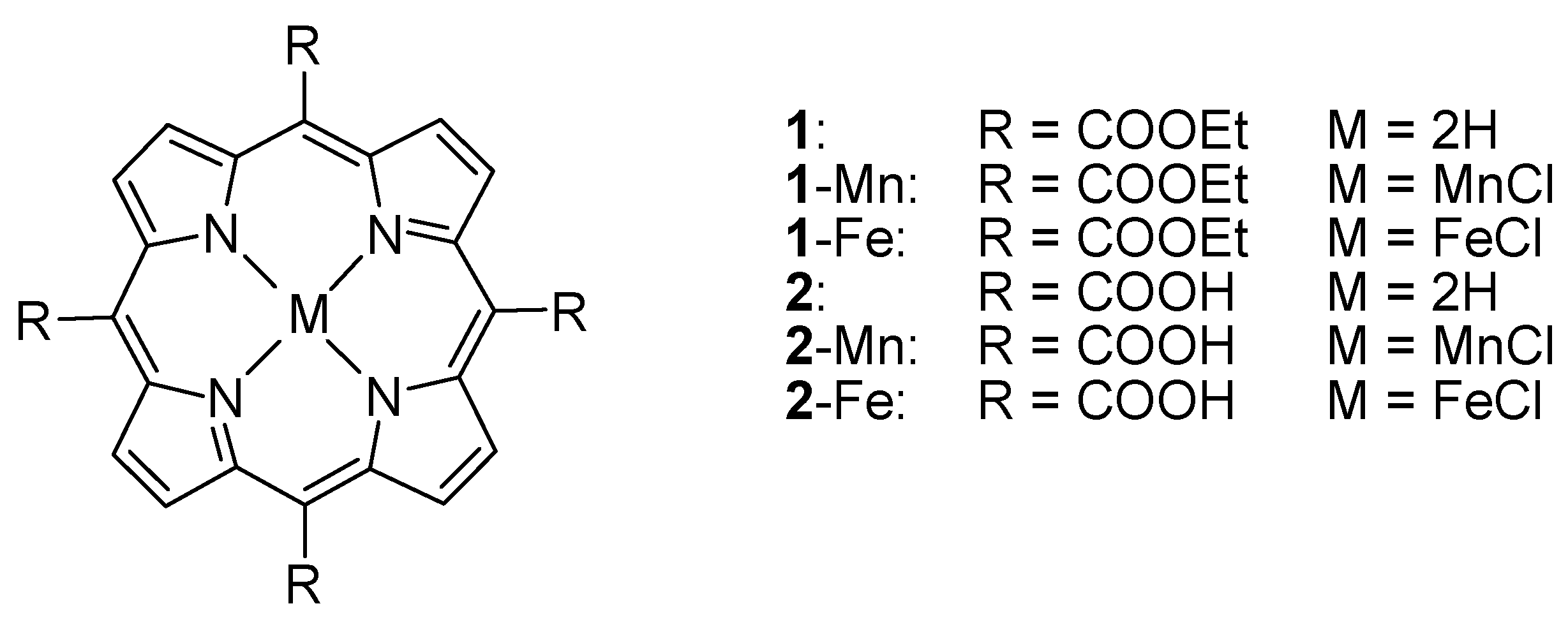
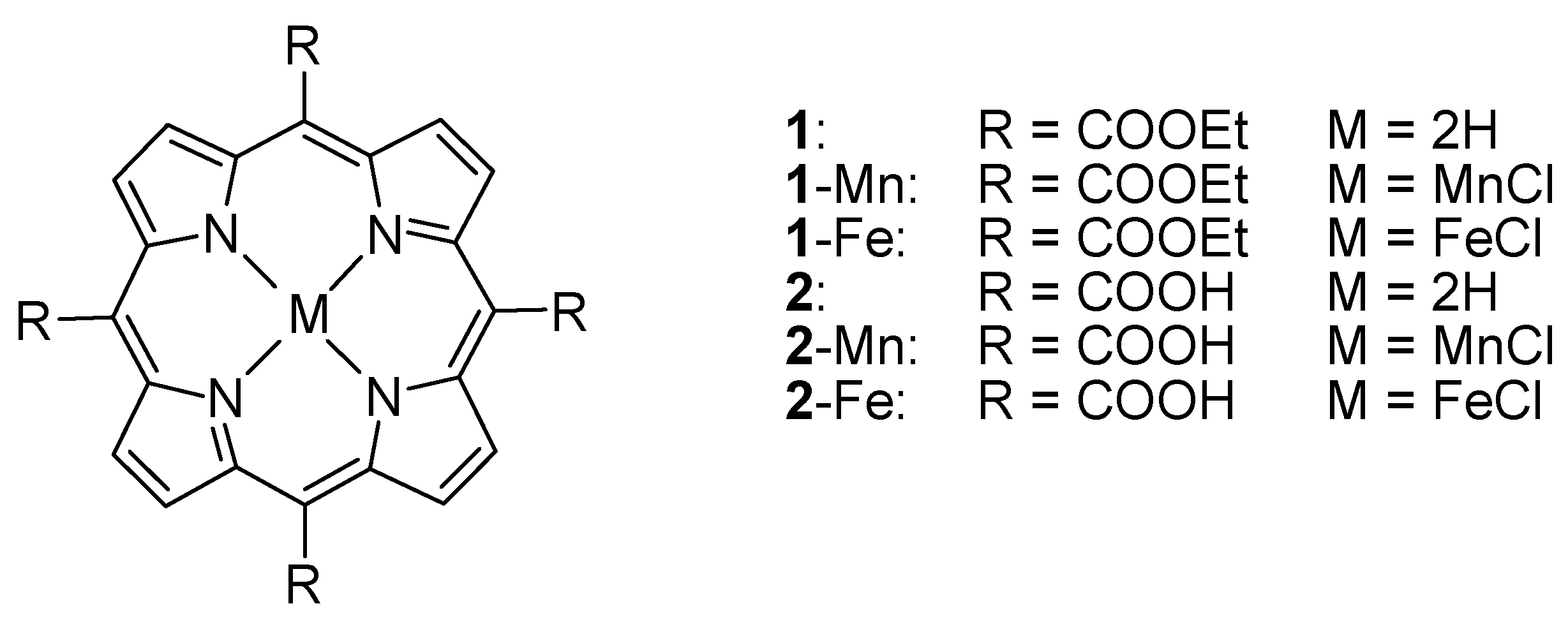
2.1. Synthesis and Characterization
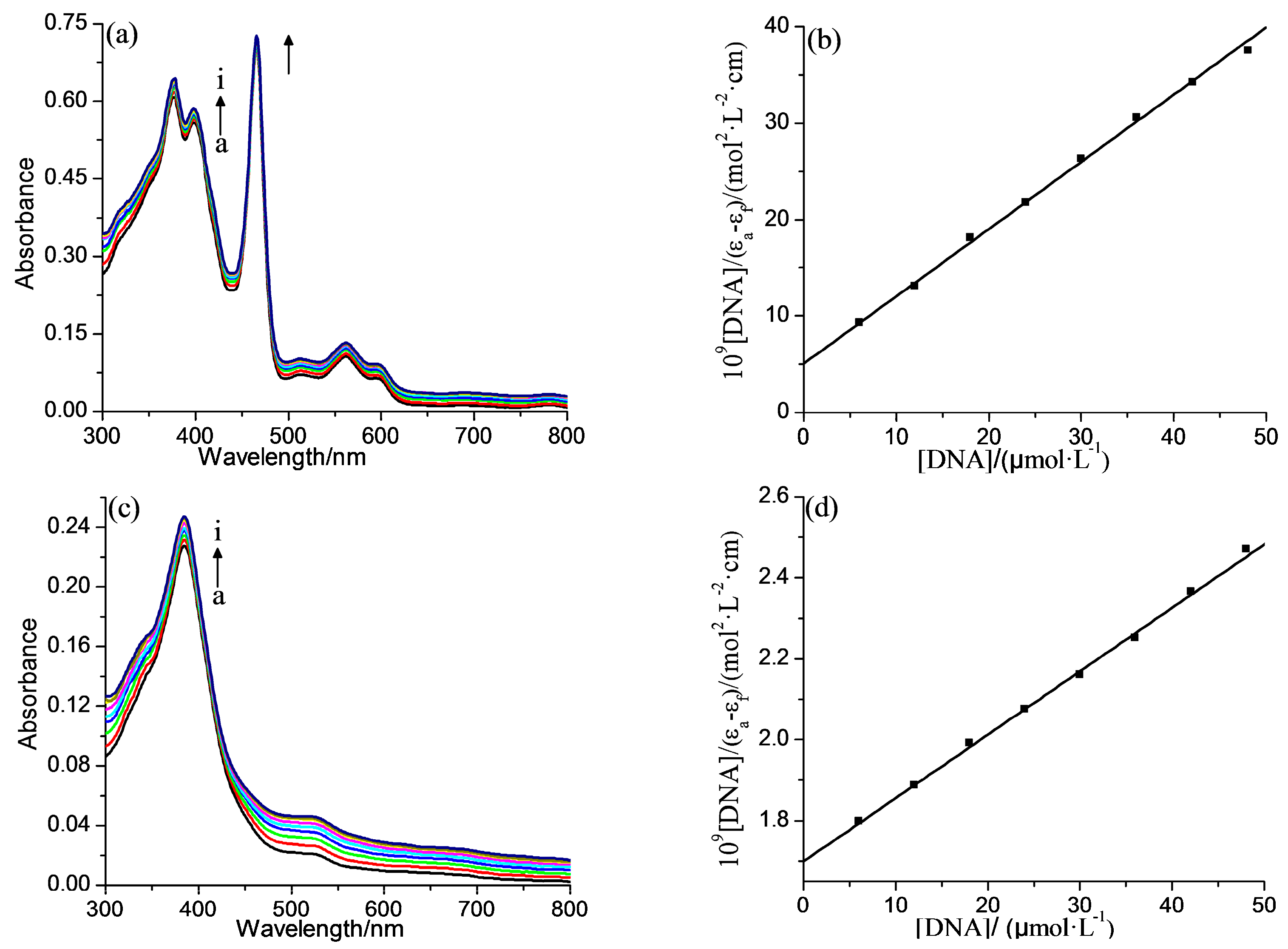
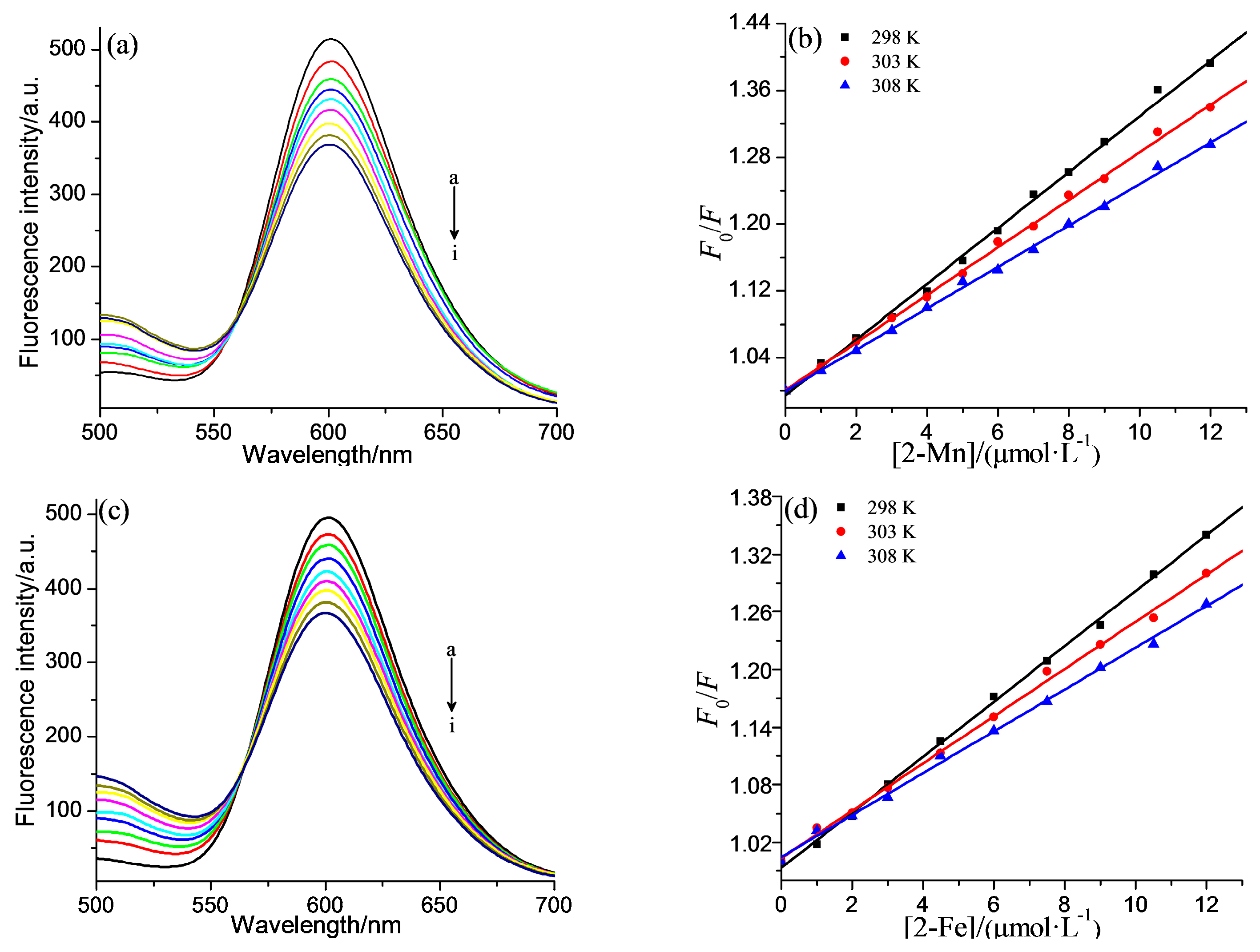
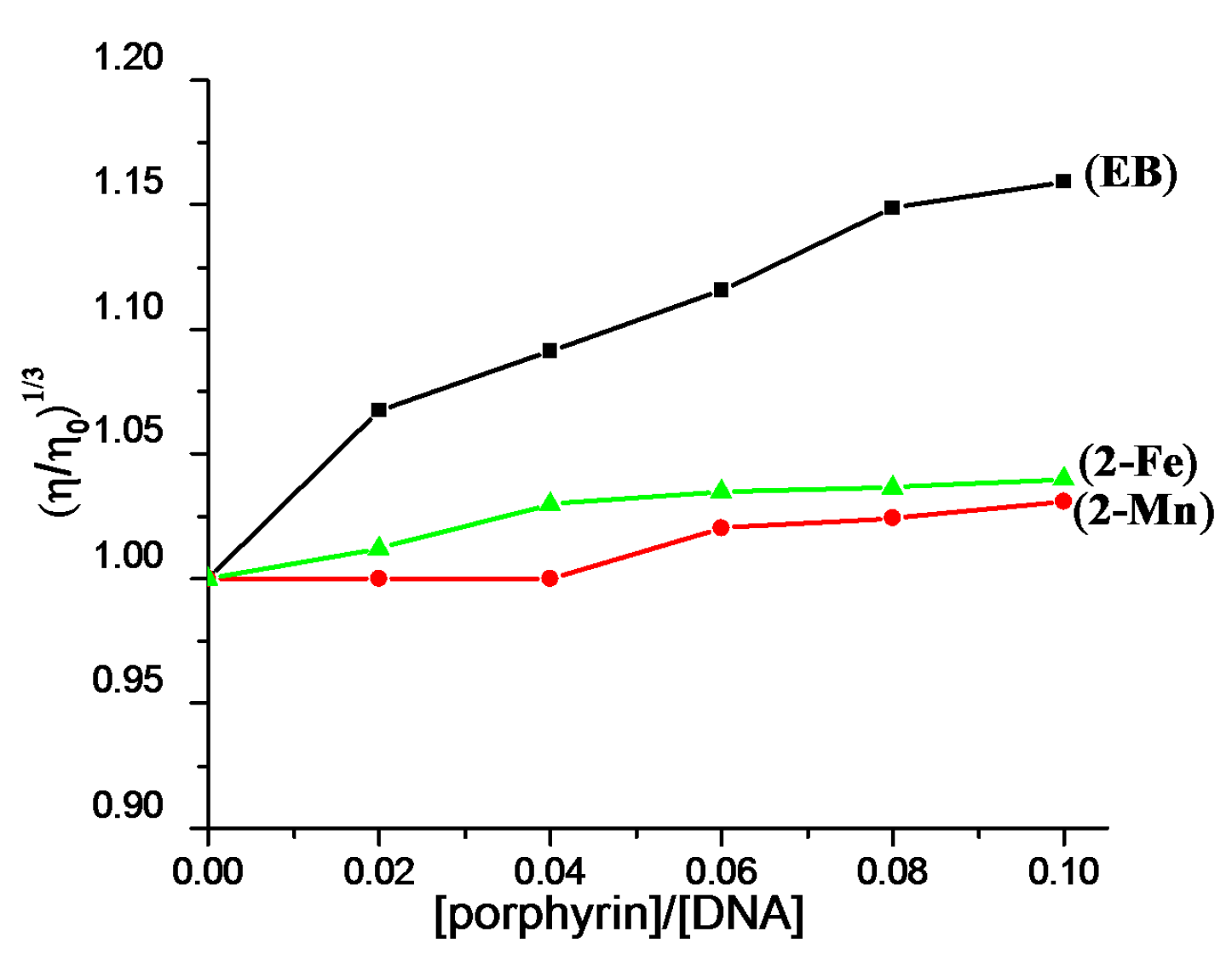
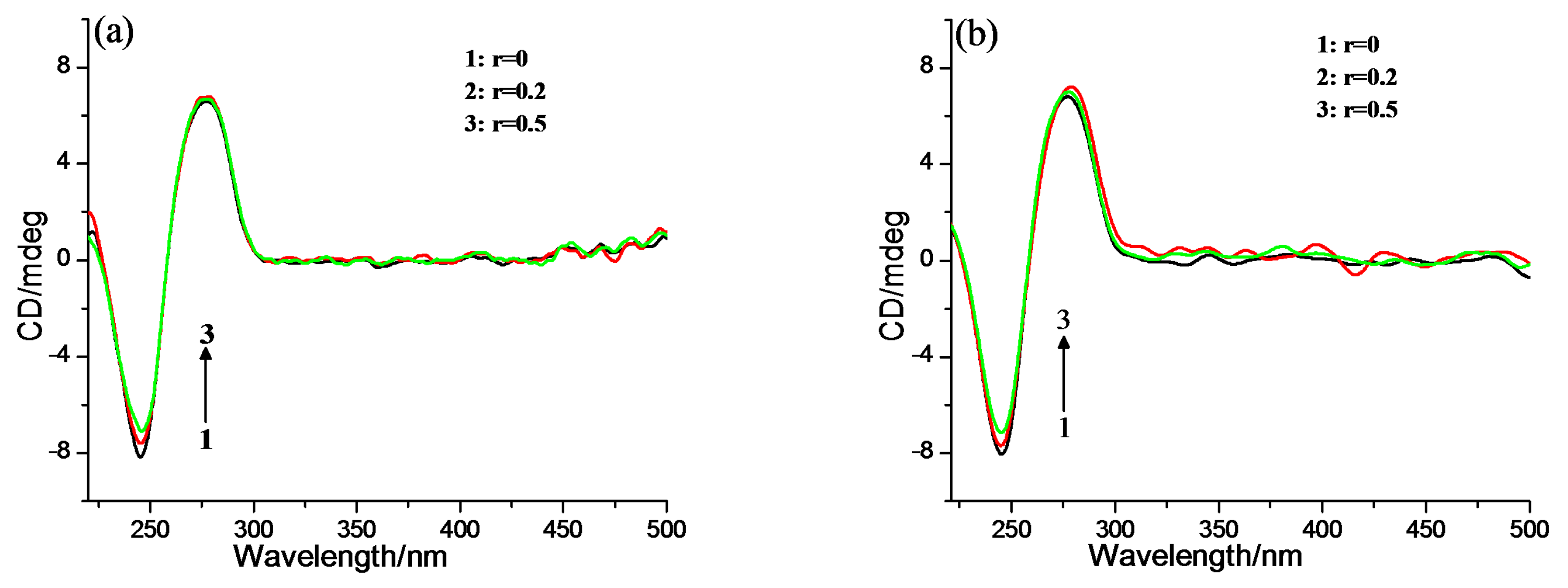
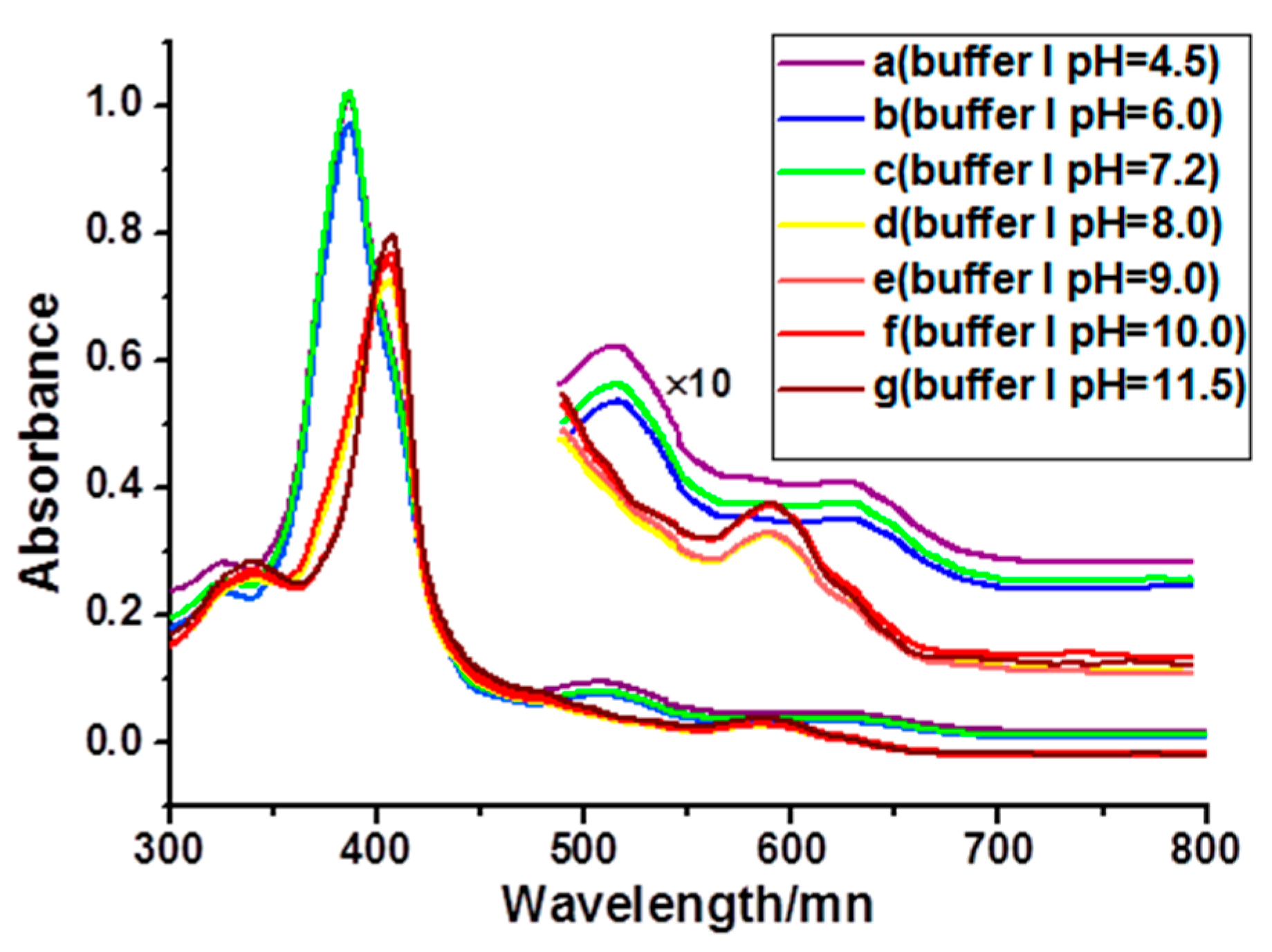
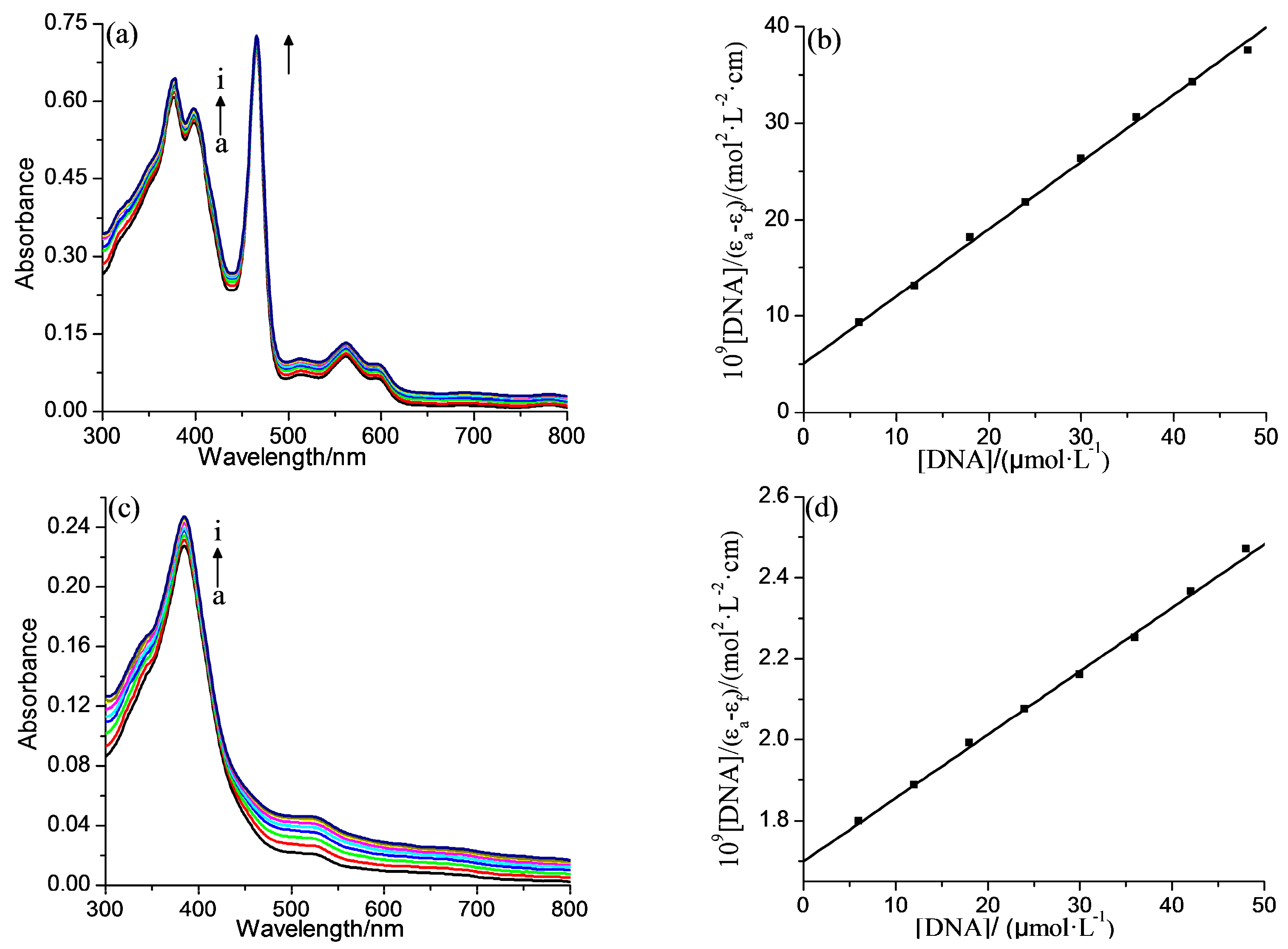
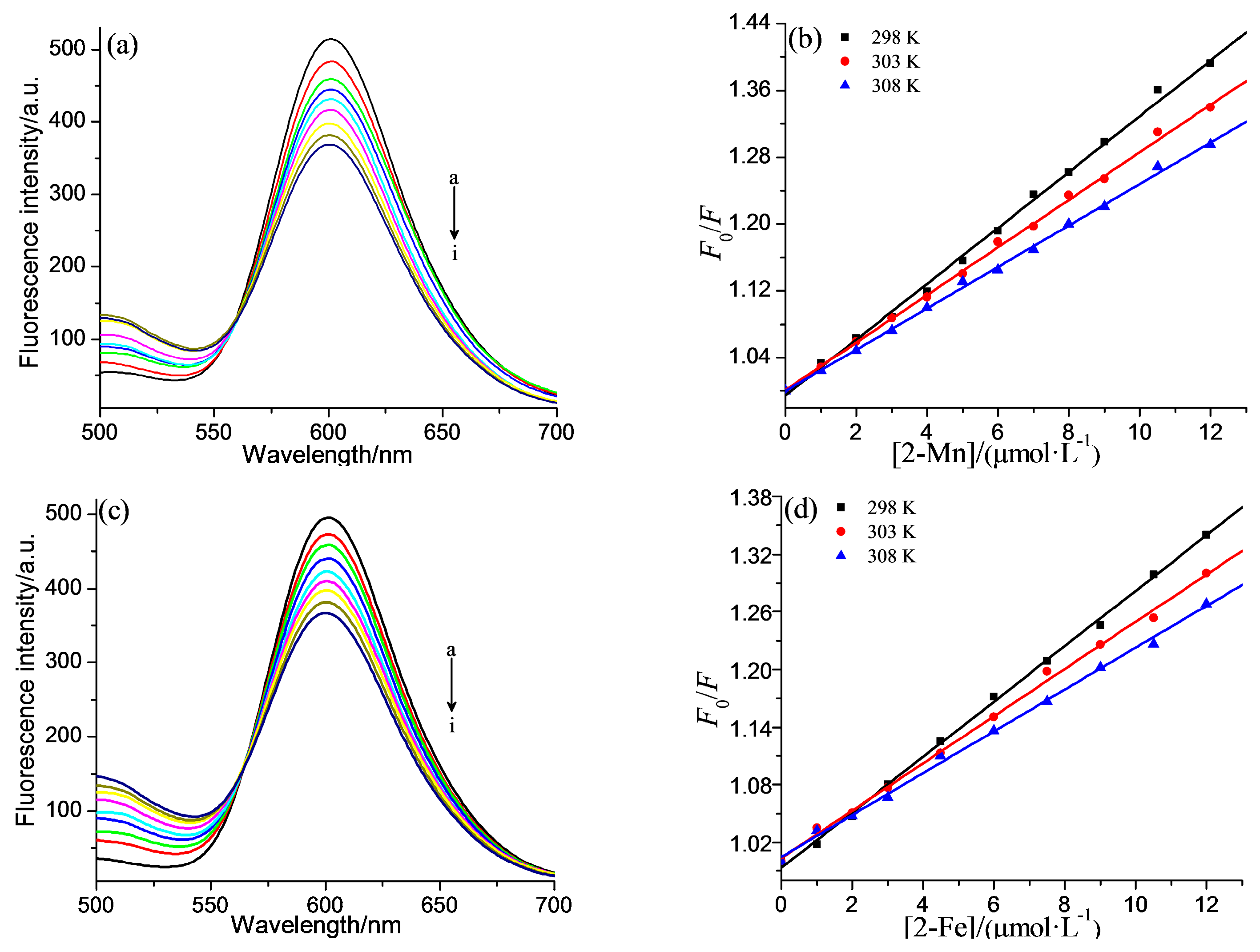
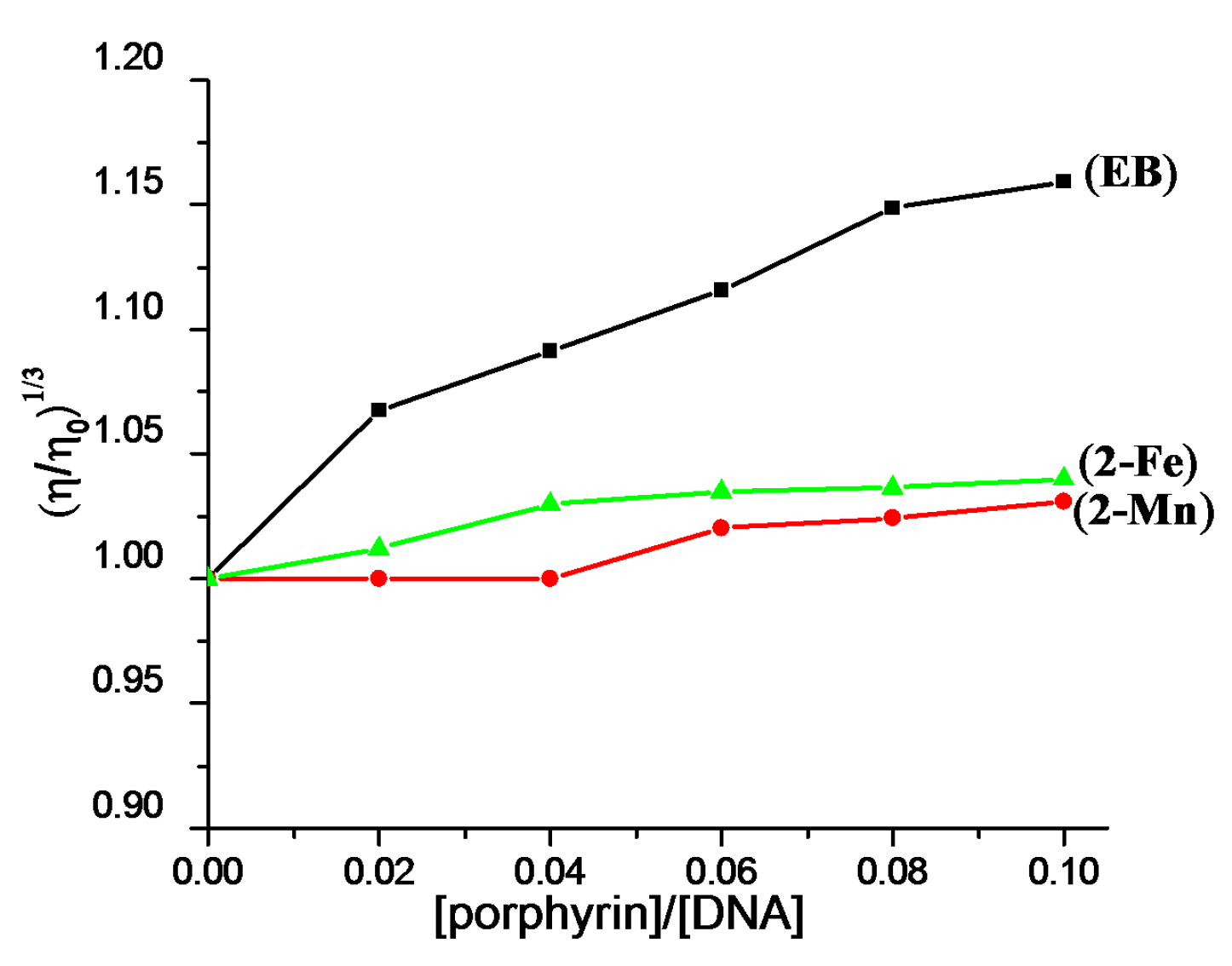
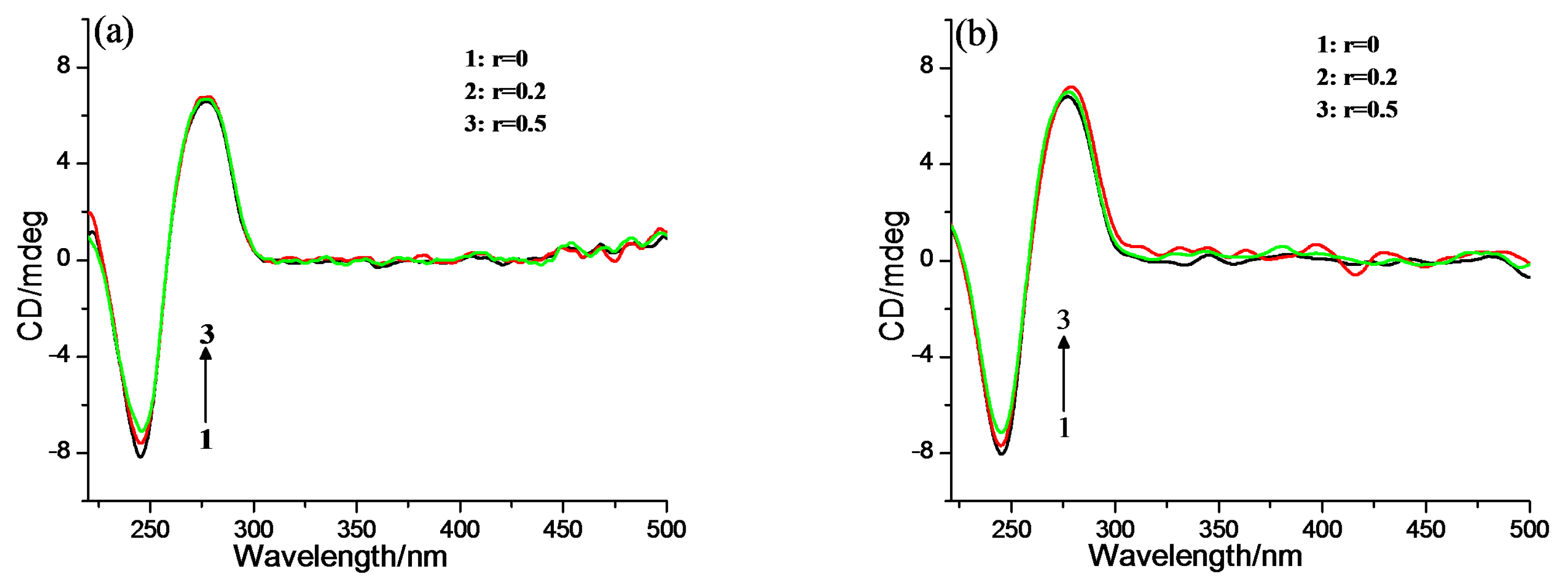
2.2. DNA Binding Properties
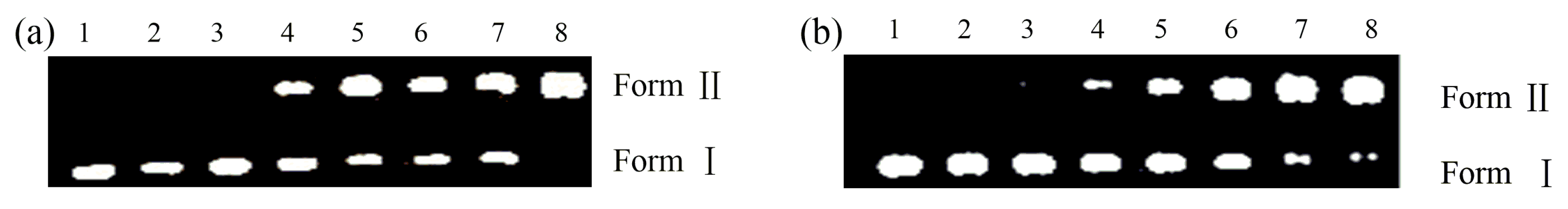
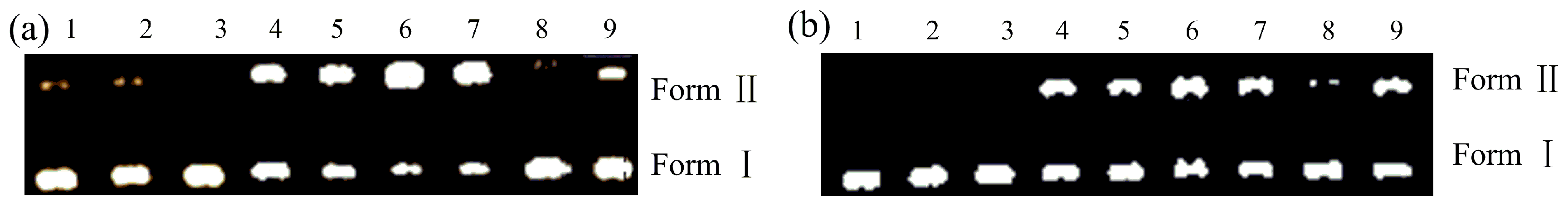
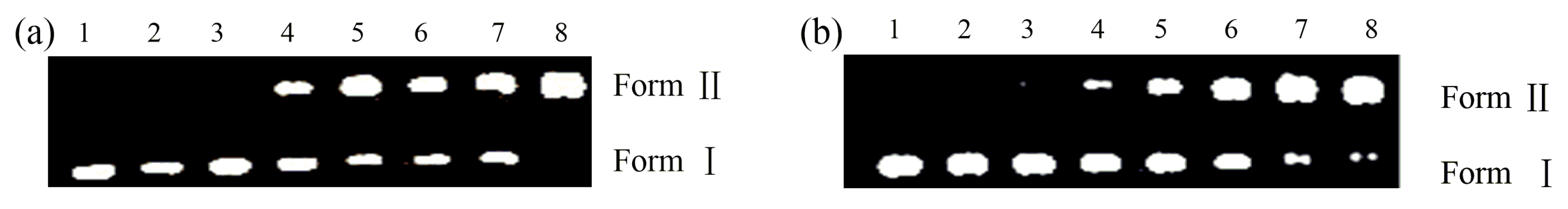
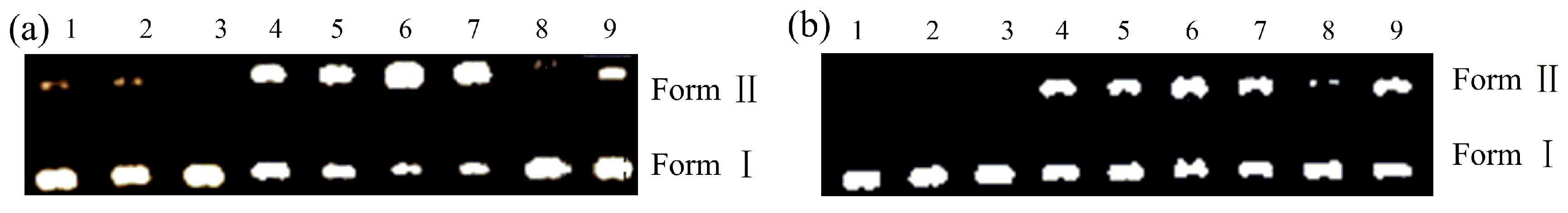
2.3. Nuclease Activities
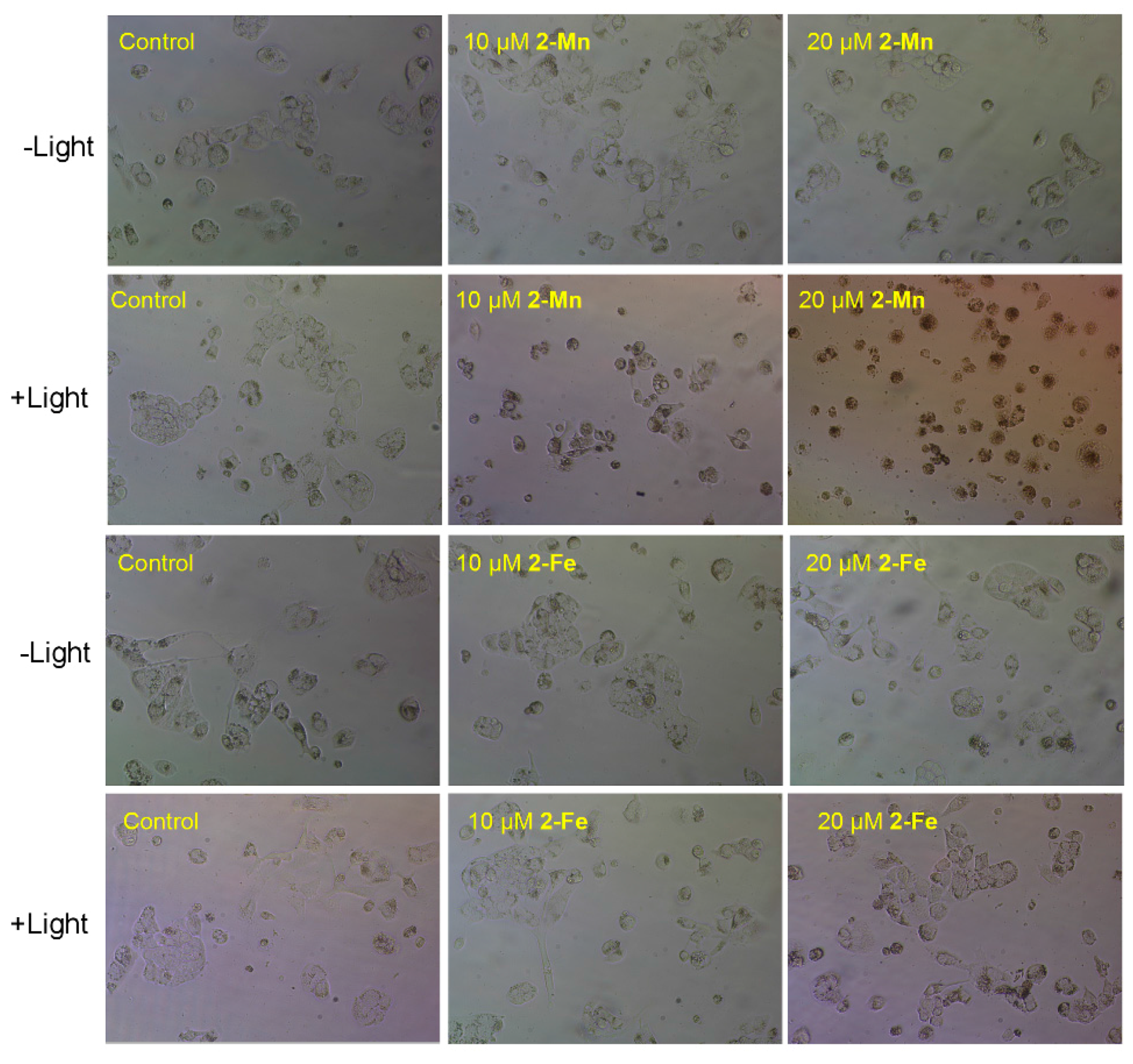
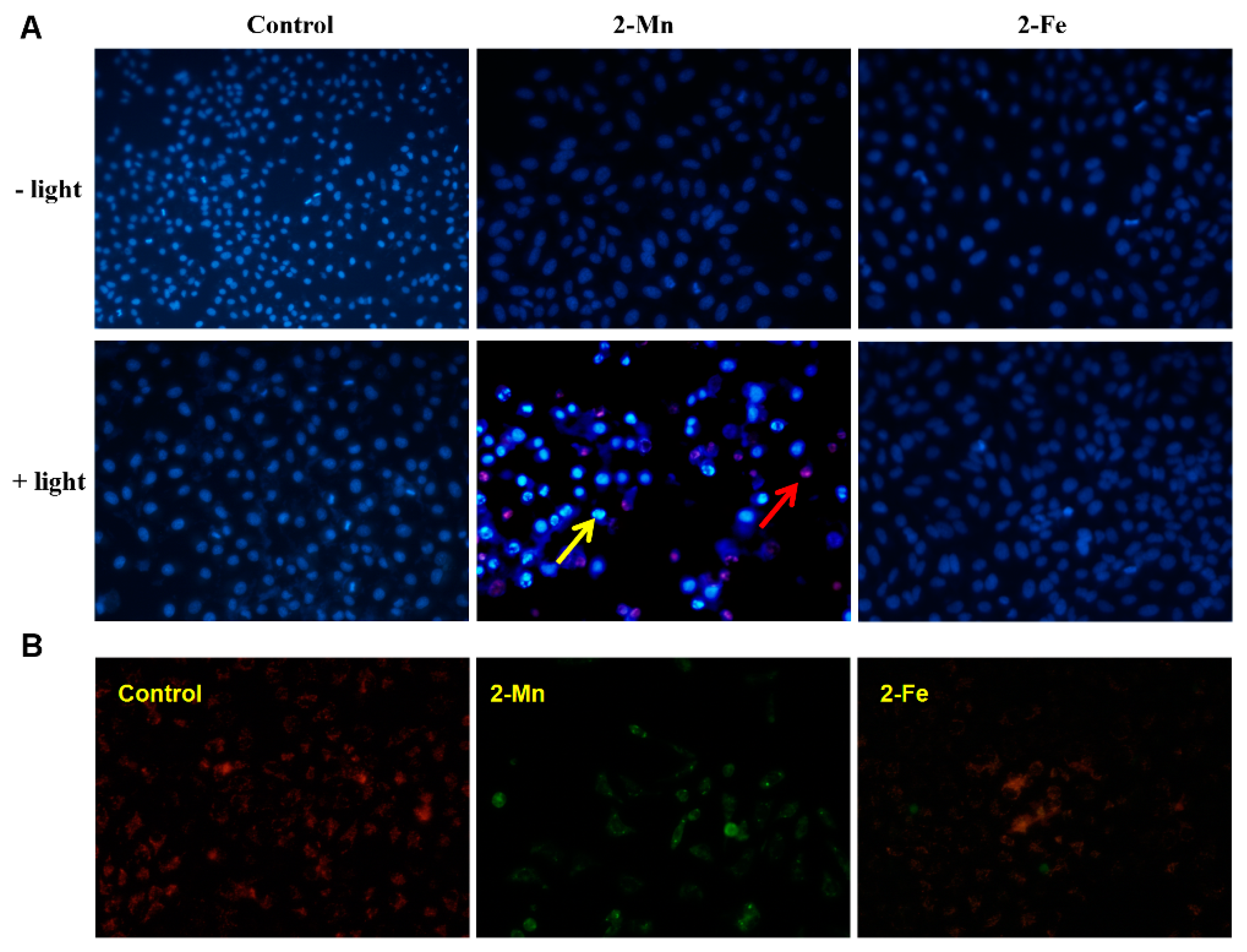
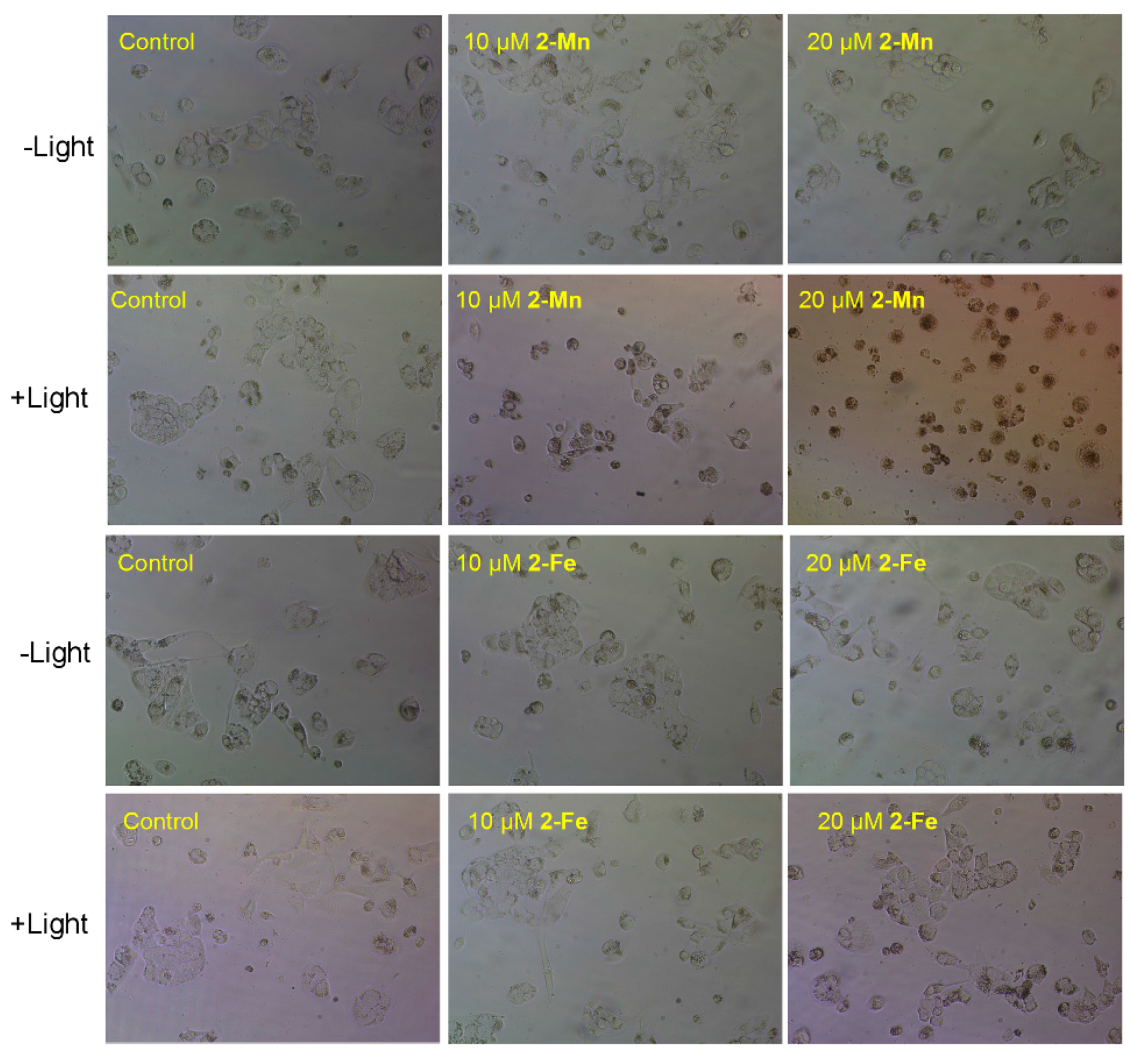
2.4. Cytotoxicity
3. Experimental Section
3.1. General Information
3.2. Synthesis of 2-Mn and 2-Fe
3.3. DNA Binding Experiments
3.3.1. UV Spectroscopy
3.3.2. Fluorescence Spectroscopy
3.3.3. CD Spectroscopy
3.3.4. Viscosity Experiments
3.4. DNA Cleavage Experiments
3.5. Cytotoxic Activity Assay
3.6. Nuclear Staining
3.7. Change of MMP
3.8. ROS Levels Assay
3.9. The EPR Spectra
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Topkaya, D.; Lafont, D.; Poyer, F.; Garcia, G.; Albrieux, F.; Maillard, P.; Bretonnière, Y.; Dumoulin, F. Design of an amphiphilic porphyrin exhibiting high in vitro photocytotoxicity. New J. Chem. 2015, 40, 2044–2050. [Google Scholar] [CrossRef]
- Tovmasyan, A.; Babayan, N.; Poghosyan, D.; Margaryan, K.; Harutyunyan, B.; Grigoryan, R.; Sarkisyan, N.; Spasojevic, I.; Mamyan, S.; Sahakyan, L. Novel amphiphilic cationic porphyrin and its Ag(II) complex as potential anticancer agents. J. Inorg. Biochem. 2014, 140, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Buytaert, E.; Dewaele, M.; Agostinis, P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim. Biophys. Acta 2007, 1776, 86–107. [Google Scholar] [CrossRef] [PubMed]
- Allison, R.R.; Moghissi, K. Photodynamic Therapy (PDT): PDT Mechanisms. Clin. Endosc. 2013, 46, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Gianferrara, T.; Bergamo, A.; Bratsos, I.; Milani, B.; Spagnul, C.; Sava, G.; Alessio, E. Ruthenium-porphyrin conjugates with cytotoxic and phototoxic antitumor activity. J. Med. Chem. 2010, 53, 4678–4690. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.P.; Conceição, D.S.; Calhelha, R.C.; Sousa, T.; Socoteanu, R.; Ferreira, I.C.; Vieira Ferreira, L.F. Porphyrin dye into biopolymeric chitosan films for localized photodynamic therapy of cancer. Carbohyd. Polym. 2016, 151, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Vandresen, C.C.; Gonçalves, A.G.; Ducatti, D.R.; Murakami, F.S.; Noseda, M.D.; Duarte, M.E.; Barreira, S.M. In vitro photodynamic inactivation of conidia of the phytopathogenic fungus colletotrichum graminicola with cationic porphyrins. Photochem. Photobiol. Sci. 2016, 15, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Xing, R.; Zou, Q.; Ma, G.; Möhwald, H.; Yan, X. Simple Peptide-tuned self-assembly of photosensitizers towards anticancer photodynamic therapy. Angew. Chem. Int. Ed. 2016, 128, 3036–3039. [Google Scholar] [CrossRef] [PubMed]
- Kavali, R.R.; Lee, B.C.; Moon, B.S.; Yang, S.D.; Chun, K.S.; Chang, W.C.; Lee, C.H.; Chi, D.Y. Efficient methods for the synthesis of 5-(4-[18F]fluorophenyl)-10,15,20-tris(3-methoxyphenyl)porphyrin as a potential imaging agent for tumor. J. Label. Compd. Radiopharm. 2005, 48, 749–758. [Google Scholar] [CrossRef]
- Dezhampanah, H.; Darvishzad, T.; Aghazadeh, M. Thermodynamic and spectroscopic study on the binding of interaction anionic phthalocyanine with calf thymus DNA. Spectroscopy 2011, 26, 357–365. [Google Scholar] [CrossRef]
- Sweigert, P.; Xu, Z.; Hong, Y.; Swavey, S. Nickel, copper, and zinc centered ruthenium-substituted porphyrins: Effect of transition metals on photoinduced DNA cleavage and photoinduced melanoma cell toxicity. Dalton Trans. 2012, 41, 5201–5208. [Google Scholar] [CrossRef] [PubMed]
- Ranibeeram, S.; Meyer, K.; Mccrate, A.; Hong, Y.; Nielsen, M.; Swavey, S. A fluorinated ruthenium porphyrin as a potential photodynamic therapy agent: Synthesis, characterization, DNA binding, and melanoma cell studies. Inorg. Chem. 2008, 47, 11278–11283. [Google Scholar] [CrossRef] [PubMed]
- Bordbar, A.K.; Mohammadi, K.; Keshavarz, M.; Dezhampanah, H. Interaction of 5, 10, 15, 20-tetrakis (N-benzylpyridilium-4-yl)porphyrin (H2TBzPyP) and its metal derivatives with calf thymus DNA. Acta Chim. Slov. 2007, 336–340. [Google Scholar]
- Meng, S.; Xu, Z.; Hong, G.; Zhao, L.; Zhao, Z.; Guo, J.; Ji, H.; Liu, T. Synthesis, characterization and in vitro photodynamic antimicrobial activity of basic amino acid-porphyrin conjugates. Eur. J. Med. Chem. 2015, 92, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, S.; Al-Asbahy, W.M.; Afzal, M.; Arjmand, F.; Bagchi, V. Molecular drug design, synthesis and structure elucidation of a new specific target peptide based metallo drug for cancer chemotherapy as topoisomerase I inhibitor. Dalton Trans. 2012, 41, 4955–4964. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.M.; Lu, M.F.; Lin, K.C. The interaction of water-soluble Manganese porphyrins with DNA films and their electrocatalytic properties with hydrazine. Electroanalysis 2005, 17, 847–856. [Google Scholar] [CrossRef]
- Meunier, B. Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- An, J.M.; Yang, S.J.; Yi, S.Y.; Jhon, G.J.; Nam, W.W. Oxidative cleavage of DNA by water-soluble iron porphyrin complex and potassium monopersulfate. Bull. Korean Chem. Soc. 1997, 117–119. [Google Scholar]
- Zhao, P.; Liu, M.C.; Madanayake, T.W.; Reena, C.; Zheng, M.; Cheng, Z.F.; Huang, Y.M.; Wang, X.H. Cationic porphyrin@SPION nanospheres as multifunctional anticancer therapeutics: Magnetic targeting, photodynamic potential and bio-safety research. RSC Adv. 2016, 6, 103137–103148. [Google Scholar] [CrossRef]
- Lum, C.T.; Huo, L.; Sun, R.W.Y.; Li, M.; Kung, H.F.; Che, C.M.; Lin, M.C.M. Gold(III) porphyrin 1a prolongs the survival of melanoma-bearing mice and inhibits angiogenesis. Acta Oncol. 2011, 50, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, N.C.; Mendoza, O.; Nicoludis, J.M.; Ruan, T.; Mergny, J.L.; Yatsunyk, L.A. Investigation of the interactions between Pt(II) and Pd(II) derivatives of 5,10,15,20-tetrakis (N-methyl-4-pyridyl) porphyrin and G-quadruplex DNA. J. Biol. Inorg. Chem. 2016, 21, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Lum, C.T.; Liu, X.; Sun, R.W.Y.; Li, X.P.; Peng, Y.; He, M.L.; Kung, H.F.; Che, C.M.; Lin, M.C.M. Gold(III) porphyrin 1a inhibited nasopharyngeal carcinoma metastasisin vivo and inhibited cell migration and invasion in vitro. Cancer Lett. 2010, 294, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Nyarko, E.; Hanada, N.; Habib, A.; Tabata, M. Fluorescence and phosphorescence spectra of Au(III), Pt(II) and Pd(II) porphyrins with DNA at room temperature. Inorg. Chim. Acta 2004, 357, 739–745. [Google Scholar] [CrossRef]
- Sun, R.W.Y.; Li, C.K.L.; Ma, D.L.; Yan, J.J.; Lok, C.N.; Leung, C.H.; Zhu, N.; Che, C.M. Stable anticancer Gold(III)–porphyrin complexes: Effects of porphyrin structure. Chem. Eur. J. 2010, 16, 3097–3113. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, H.; Zhang, Z.; Yang, Q.; Tong, H.; Xu, A.; Wang, C. Synthesis and cancer cell cytotoxicity of water-soluble gold(III) substituted tetraarylporphyrin. J. Inorg. Biochem. 2012, 108, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Liu, Y.; Lai, Y.T.; Tong, K.C.; Fung, Y.M.; Lok, C.N.; Che, C.M. Anticancer Gold(III) porphyrins target mitochondrial chaperone Hsp60. Angew. Chem. Int. Ed. 2015, 128, 1387–1391. [Google Scholar]
- Dixon, D.W.; Gill, A.F.; Giribabu, L.; Vzorov, A.N.; Alam, A.B.; Compans, R.W. Sulfonated naphthyl porphyrins as agents against HIV-1. J. Inorg. Biochem. 2005, 99, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Riou, C.; Calliste, C.A.; Da Silva, A.; Guillaumot, D.; Rezazgui, O.; Sol, V.; Leroy-Lhez, S. Anionic porphyrin as a new powerful cell death inducer of Tobacco Bright Yellow-2 cells. Photochem. Photobiol. Sci. 2014, 13, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Mandal, G.; Ganguly, T. Detailed spectroscopic investigations to reveal the nature of interaction of anionic porphyrin with calf thymus DNA. J. Photochem. Photobiol. B 2010, 101, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Robert, A.; Duarte, V.; Keppler, B.K.; Meunier, B. Synthesis of water-soluble ruthenium porphyrins as DNA cleavers and potential cytotoxic agents. J. Biol. Inorg. Chem. 1997, 2, 427–432. [Google Scholar] [CrossRef]
- Chatterjee, S.R.; Srivastava, T.S.; Kamat, J.P.; Devasagayam, T.P.A. Photocleavage of plasmid pBR322 DNA by some anionic porphyrins. J. Porphyr. Phthalocya. 1998, 2, 337–343. [Google Scholar] [CrossRef]
- Lauceri, R.; Purrello, R.; Shetty, S.J.; Vicente, M.G.H. Interactions of anionic carboranylated porphyrins with DNA. J. Am. Chem. Soc. 2001, 123, 5835–5836. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.K.; D'Urso, A.; Balaz, M. Chiroptical properties of anionic and cationic porphyrins and metalloporphyrins in complex with left-handed Z-DNA and right-handed B-DNA. J. Inorg. Biochem. 2013, 127, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, P.C.; Du, G.; Fukuda, Y.; Sun, D.; Sampath, J.; Mercer, K.E.; Wang, J.; Sosa-Pineda, B.; Murti, K.G.; Schuetz, J.D. Identification of a mammalian mitochondrial porphyrin transporter. Nature 2006, 443, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.; Prydz, K.; Moan, J. Photochemical treatment with the lysosomally localized dye tetra(4-sulfonatophenyl)prophine results in lysosomal release of the dye but not of β-N-acetyl-d-glucosaminidase activity. BBA-Gen. Subj. 1993, 1158, 300–306. [Google Scholar] [CrossRef]
- Kessel, D.; Woodburn, K. Biodistribution of photosensitizing agents. Int. J. Biochem. 1993, 25, 1377–1383. [Google Scholar] [CrossRef]
- Wang, H.H.; Yuan, H.Q.; Mahmood, M.H.R.; Jiang, Y.Y.; Cheng, F.; Shi, L.; Liu, H.Y. Crystal structure, magnetic and catalytic oxidation properties of manganese(III) tetrakis-(ethoxycarbonyl)porphyrin. RSC Adv. 2015, 5, 97391–97399. [Google Scholar] [CrossRef]
- Trova, M.P.; Gauuan, P.J.F.; Pechulis, A.D.; Bubb, S.M.; Bocckino, S.B.; Crapo, J.D.; Day, B.J. Superoxide dismutase mimetics. Part 2: Synthesis and structure–activity relationship of glyoxylate- and glyoxamide-derived metalloporphyrins. Bioorgan. Med. Chem. 2003, 11, 2695–2707. [Google Scholar] [CrossRef]
- Miskelly, G.M.; Webley, W.S.; Clark, C.R.; Buckingham, D.A. Acidity and dimerization of three water-soluble iron(III) porphyrin cations: (meso-α,α,α,α-Tetrakis(o-(N-methylnicotinamido)phenyl)-porphyrinato)iron(III), and (meso-tetrakis(1-methylpyridinium-4-yl)porphyrinato)iron(III). Inorg. Chem. 1988, 27, 3773–3781. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Gibbs, E.J.; Villafranca, J.J. Interactions of porphyrins with nucleic acids. Biochemistry-US 1983, 22, 5409–5417. [Google Scholar] [CrossRef]
- Tovmasyan, A.; Weitner, T.; Sheng, H.; Lu, M.; Rajic, Z.; Warner, D.S.; Spasojevic, I.; Reboucas, J.S.; Benov, L.; Batinic-Haberle, I. Differential coordination demands in Fe versus Mn water-soluble cationic metalloporphyrins translate into remarkably different aqueous redox chemistry and biology. Inorg. Chem. 2013, 52, 5677–5691. [Google Scholar] [CrossRef] [PubMed]
- Monajjemi, M.; Aghaie, H.; Naderi, F. Thermodynamic study of interaction of TSPP, CoTsPc, and FeTsPc with calf thymus DNA. Biochemistry (Moscow) 2007, 72, 652–657. [Google Scholar] [CrossRef]
- Olmsted, J.; Kearns, D.R. Mechanism of ethidium bromide fluorescence enhancement on binding to nucleic acids. Biochemistry 1977, 16, 3647–3654. [Google Scholar] [CrossRef] [PubMed]
- Baguley, B.C.; Le Bret, M. Quenching of DNA-ethidium fluorescence by amsacrine and other antitumor agents: A possible electron-transfer effect. Biochemistry 1984, 23, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Liu, G.; Li, W.; Cui, F. Molecular interaction of ctDNA and HSA with sulfadiazine sodium by multispectroscopic methods and molecular modeling. Luminescence 2013, 28, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Wu, H.; Lu, X.; Wang, Y.; Zhou, L. Study on the interaction of new water-soluble porphyrin with DNA. Spectrochim. Acta A 2005, 61, 2041–2047. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.G.; Zhang, Q.L.; Shi, X.F.; Ji, L.N. Interaction of [Ru(dmp)2(dppz)]2+ and [Ru(dmb)2(dppz)]2+ with DNA: Effects of the ancillary ligands on the DNA-binding behaviors. Inorg. Chem. 2001, 40, 5045–5050. [Google Scholar] [CrossRef] [PubMed]
- Uma Maheswari, P.; Palaniandavar, M. DNA binding and cleavage properties of certain tetrammine ruthenium(II) complexes of modified 1,10-phenanthrolines–effect of hydrogen-bonding on DNA-binding affinity. J. Inorg. Biochem. 2004, 98, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Lee, C.Y.; Kim, S.K. Formation of poly[d(A-T)2] specific Z-DNA by a cationic porphyrin. Sci. Rep. 2015, 5, 9943. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.N.; Zou, X.H.; Liu, J.G. Shape and enantioselective interaction of Ru(II)/Co(III) polypyridyl complexes with DNA. Coord. Chem. Rev. 2001, 216, 513–536. [Google Scholar] [CrossRef]
- Sun, Y.; Hou, Y.J.; Zhou, Q.X.; Lei, W.H.; Chen, J.R.; Wang, X.S.; Zhang, B.W. Dinuclear Cu(II) hypocrellin B complexes with enhanced photonuclease activity. Inorg. Chem. 2010, 49, 10108–10116. [Google Scholar] [CrossRef] [PubMed]
- Maheswari, P.U.; Palaniandavar, M. DNA binding and cleavage activity of [Ru(NH3)4(diimine)]Cl2 complexes. Inorg. Chim. Acta 2004, 357, 901–912. [Google Scholar] [CrossRef]
- Arjmand, F.; Parveen, S.; Mohapatra, D.K. Synthesis, characterization of Cu(II) and Zn(II) complexes of proline-glycine and proline-leucine tetrapeptides: In vitro DNA binding and cleavage studies. Inorg. Chim. Acta 2012, 388, 1–10. [Google Scholar] [CrossRef]
- Haeubl, M.; Reith, L.M.; Gruber, B.; Karner, U.; Müller, N.; Knör, G.; Schoefberger, W. DNA interactions and photocatalytic strand cleavage by artificial nucleases based on water-soluble gold(III) porphyrins. J. Biol. Inorg. Chem. 2009, 14, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Bigey, P.; Sönnichsen, S.H.; Meunier, B.; Nielsen, P.E. DNA binding and cleavage by a cationic Manganese porphyrin-peptide nucleic acid conjugate. Bioconj. Chem. 1997, 8, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Pratviel, G.; Pitieè, M.; Bernadou, J.; Meunier, B. Mechanism of DNA cleavage by cationic manganese porphyrins: Hydroxylations at the 1′-carbon and 5′-carbon atoms of deoxyriboses as initial damages. Nucleic Acids Res. 1991, 19, 6283–6288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wen, J.Y.; Lv, B.B.; Li, X.; Ying, X.; Wang, Y.J.; Zhang, H.T.; Wang, H.; Liu, H.Y.; Chang, C.K. Photocytotoxicity and G-quadruplex DNA interaction of water-soluble gallium(III) tris(N-methyl-4-pyridyl)corrole complex. Appl. Organomet. Chem. 2016, 30, 132–139. [Google Scholar] [CrossRef]
- Kośmider, A.; Czepielewska, E.; Kuraś, M.; Gulewicz, K.; Pietrzak, W.; Nowak, R.; Nowicka, G. Uncaria tomentosa leaves decoction modulates differently ROS production in cancer and normal cells, and effects cisplatin cytotoxicity. Molecules 2017, 22, 620. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; He, H.; Zhang, Z.Y.; Liao, Z.P.; Yin, D.; Liu, D.; Yi, B.; He, M. Long-term sodium ferulate supplementation scavenges oxygen radicals and reverses liver damage induced by Iron overloading. Molecules 2016, 21, 1219. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, M.E.; Rice, S.A.; Thomas, C.A.; Doty, P. A Further Examination of the Molecular Weight and Size of Desoxypentose Nucleic Acid. J. Am. Chem. Soc. 1954, 76, 3047–3053. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | T/K | Ksv/L·mol−1 | Kq/L·mol−1·s−1 | R |
|---|---|---|---|---|
| 2-Mn | 298 | 3.33 × 104 | 3.35 × 1012 | 0.9977 |
| 303 | 2.84 × 104 | 2.85 × 1012 | 0.9985 | |
| 308 | 2.48× 104 | 2.48 × 1012 | 0.9986 | |
| 2-Fe | 298 | 2.89 × 104 | 2.89 × 1012 | 0.9990 |
| 303 | 2.46× 104 | 2.46 × 1012 | 0.9979 | |
| 308 | 2.18 × 104 | 2.18 × 1012 | 0.9980 |
| Compound | IC50 Value /(μM/L) (Dark) | IC50 Value/(μM/L) (Light) | ||||||
|---|---|---|---|---|---|---|---|---|
| MCF-7 | Hep G2 | Hela | BEAS-2B | MCF-7 | Hep G2 | Hela | BEAS-2B | |
| 2 | 174.4 ± 4.9 | 306.7 ± 36.4 | >320 | >320 | >320 | 49.1 ± 13.8 | 74.9 ± 19.6 | 89.4 ± 15.2 |
| 2-Mn | >320 | 100.5 ± 4.7 | 61.7 ± 3.9 | 226.2 ± 15.7 | >320 | 19.0 ± 2.5 | 54.2 ± 4.2 | 245.3 ± 13.9 |
| 2-Fe | >320 | 175.5 ± 7.4 | >320 | >320 | 139.9 ± 7.7 | 137.5 ± 8.9 | >320 | >320 |
| mTHPC | 177.1 ± 19.5 | 129.3 ± 16.2 | 168.3 ± 24.9 | 180.5 ± 17.4 | 1.1 ± 0.3 | 1.3 ± 0.5 | 1.6 ± 0.8 | 1.9 ± 0.8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, L.; Jiang, Y.-Y.; Jiang, T.; Yin, W.; Yang, J.-P.; Cao, M.-L.; Fang, Y.-Q.; Liu, H.-Y. Water-soluble Manganese and Iron Mesotetrakis(carboxyl)porphyrin: DNA Binding, Oxidative Cleavage, and Cytotoxic Activities. Molecules 2017, 22, 1084. https://doi.org/10.3390/molecules22071084
Shi L, Jiang Y-Y, Jiang T, Yin W, Yang J-P, Cao M-L, Fang Y-Q, Liu H-Y. Water-soluble Manganese and Iron Mesotetrakis(carboxyl)porphyrin: DNA Binding, Oxidative Cleavage, and Cytotoxic Activities. Molecules. 2017; 22(7):1084. https://doi.org/10.3390/molecules22071084
Chicago/Turabian StyleShi, Lei, Yi-Yu Jiang, Tao Jiang, Wei Yin, Jian-Ping Yang, Man-Li Cao, Yu-Qi Fang, and Hai-Yang Liu. 2017. "Water-soluble Manganese and Iron Mesotetrakis(carboxyl)porphyrin: DNA Binding, Oxidative Cleavage, and Cytotoxic Activities" Molecules 22, no. 7: 1084. https://doi.org/10.3390/molecules22071084