
Chemical Methods to Knock Down the Amyloid Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
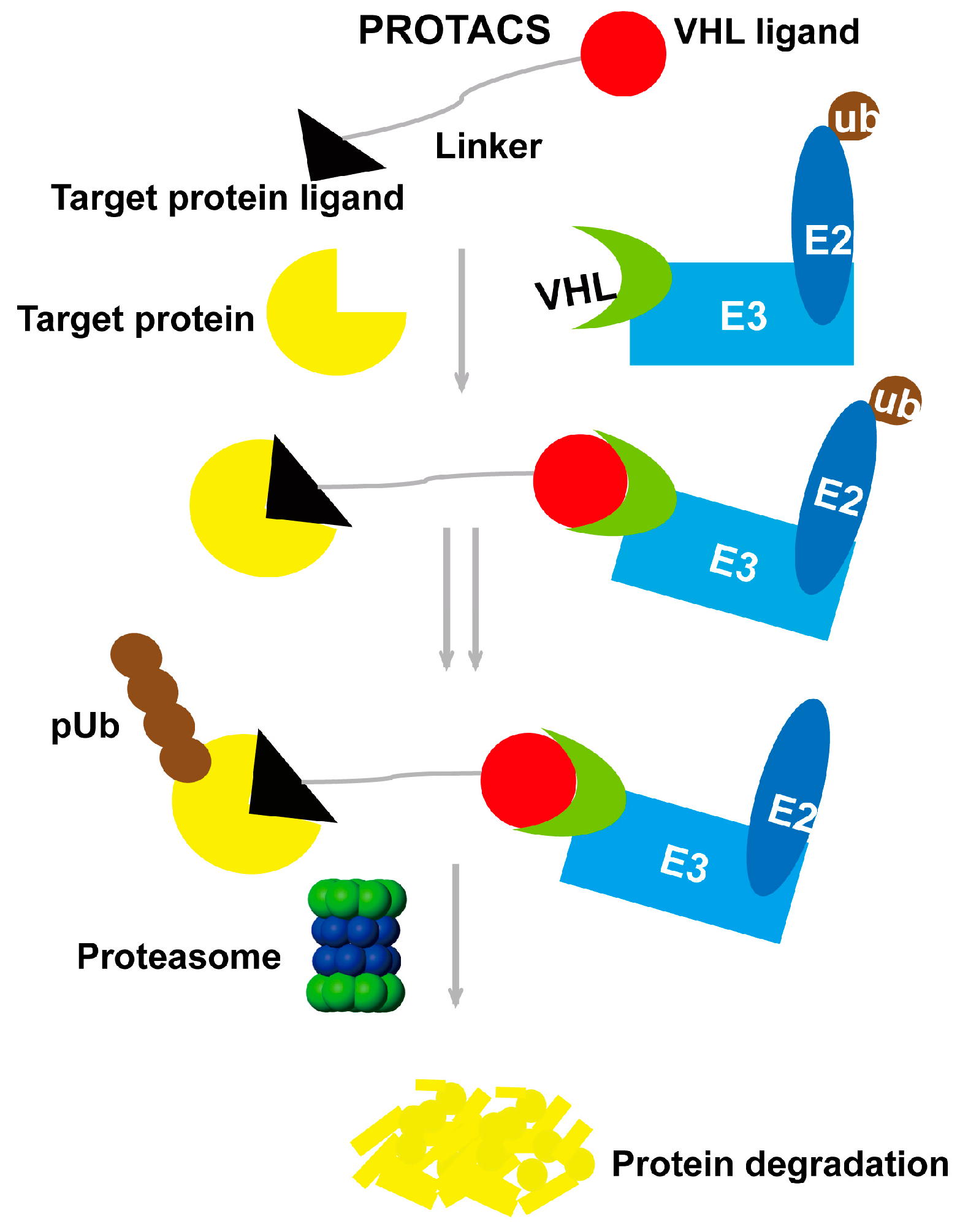
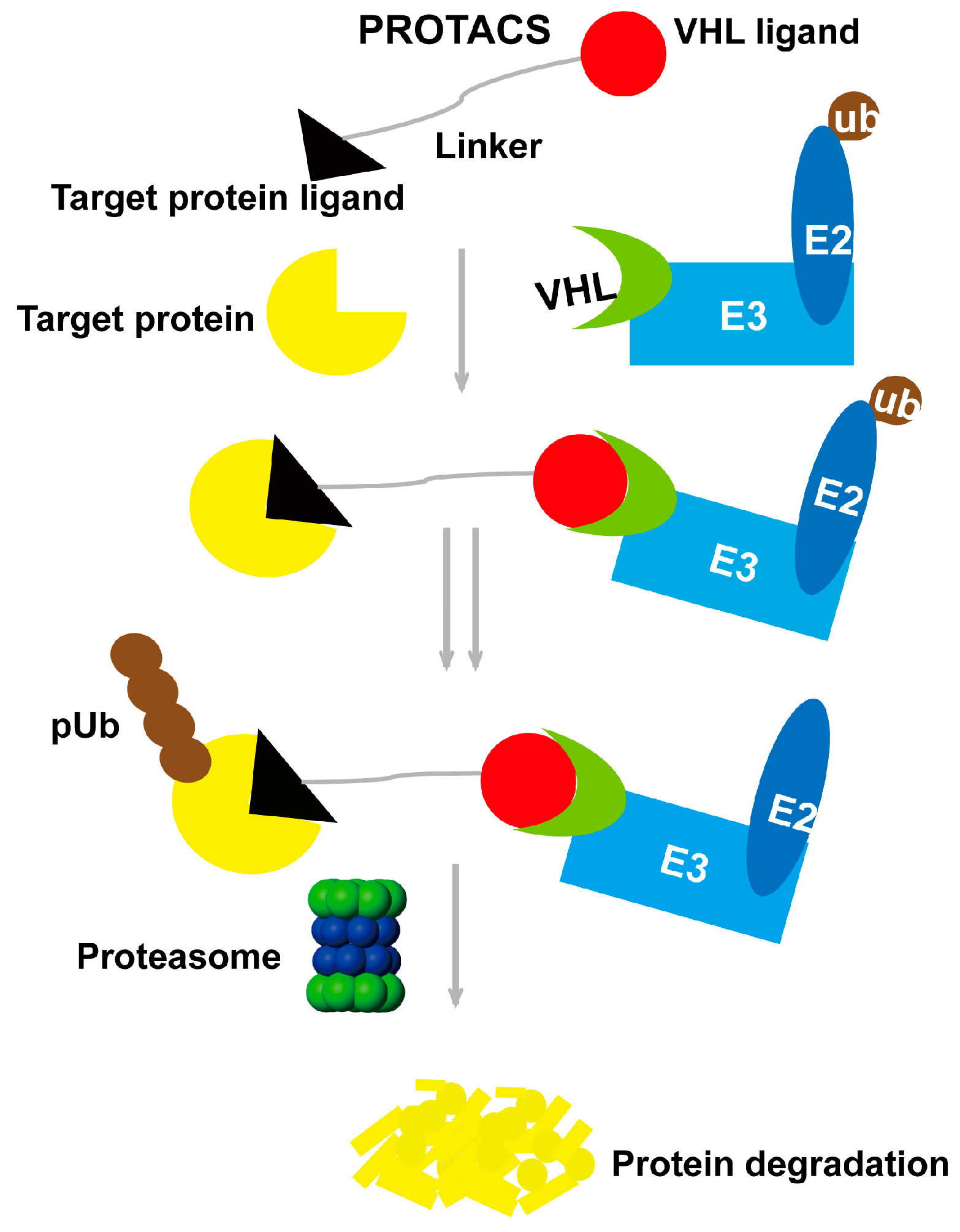
2. Proteolysis-Targeting Chimera Strategy
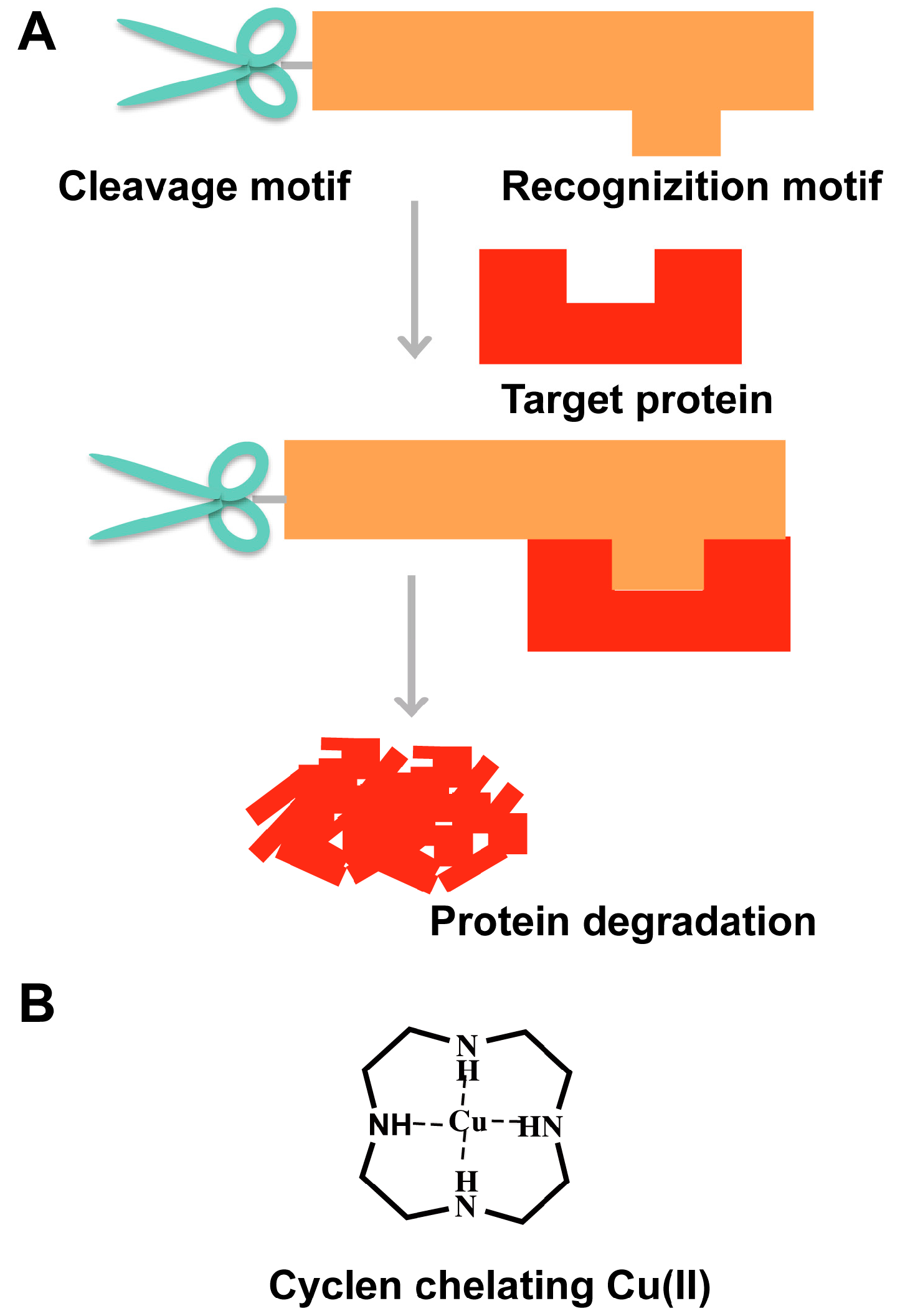
3. “Recognition-Cleavage” Strategy
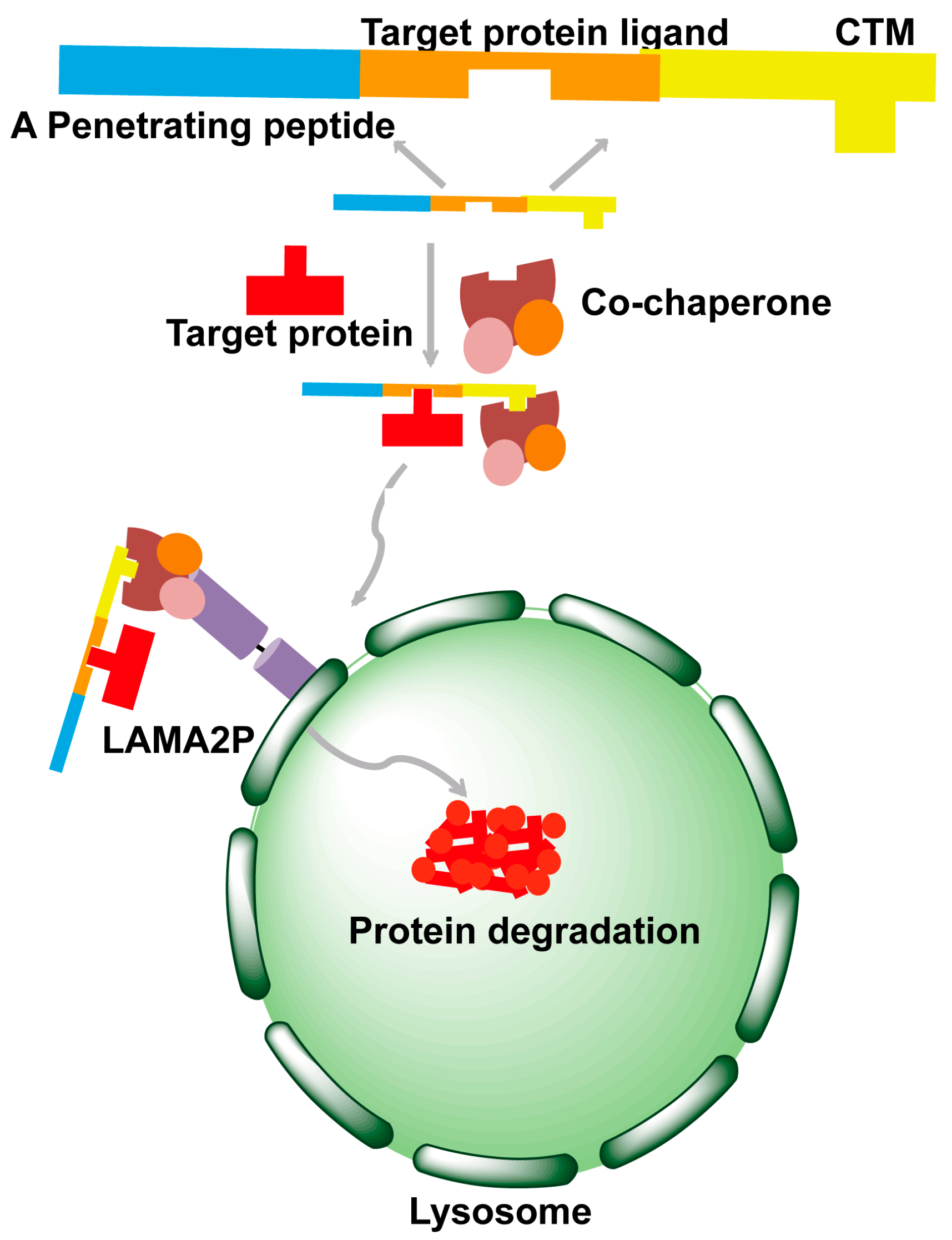
4. Chaperone-Mediated Autophagy Strategy
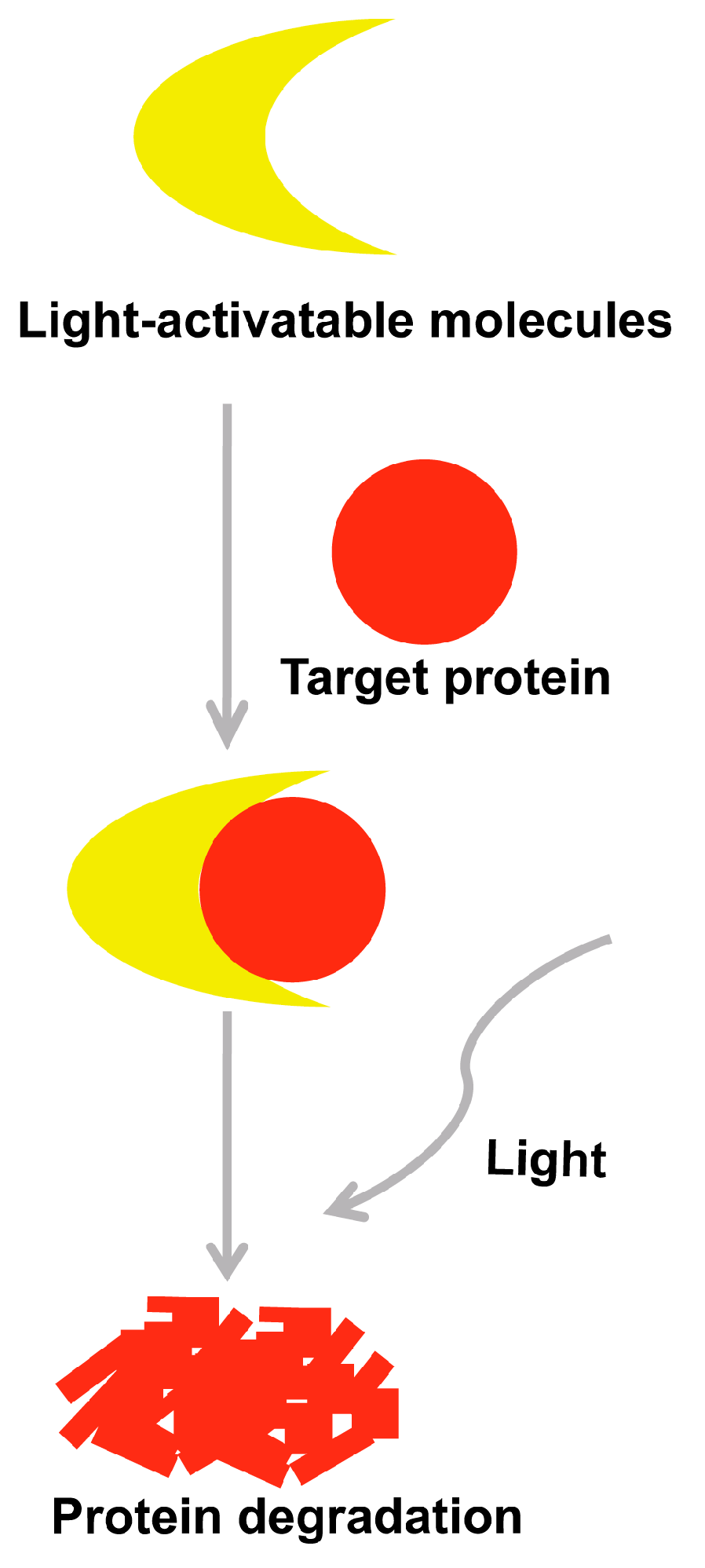
5. Selectively Light-Activatable Organic and Inorganic Molecules Strategy
6. Other Chemical Strategies
7. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Kisilevsky, R. Biology of disease amyloidosis: A familiar problem in light of current pathogenic developments. Lab Investig. 1983, 49, 381–390. [Google Scholar]
- Rochet, J.C.; Lansbury, P.T. Amyloid fibrillogenesis: Themes and variations. Curr. Opin. Struct. Biol. 2000, 10, 60–68. [Google Scholar] [CrossRef]
- Sipe, J.D. Serum amyloid A: From fibril to function. Current status. Amyloid 2000, 7, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Zhao, T.X.; An, B.L.; Liu, X.Y.; Zhong, C. Self-assembly and morphological characterization of two-component functional amyloid proteins. Chin. Chem. Lett. 2016, 12, 008. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genotypes, phenotype and treatments. Science 1997, 275, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Hurle, M.R.; Helms, L.R.; Li, L.; Chan, W.; Wetzel, R. A role for destabilizing amino acid replacements in light chain amyloidosis. Proc. Natl. Acad. Sci. USA 1994, 91, 5446–5450. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Vigushin, D.; Lavender, J.; Pepys, M. Scintigraphic quantification and serial monitoring of human visceral amyloid deposits provide evidence for turnover and regression. Quart. J. Med. 1993, 86, 365–374. [Google Scholar]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W. RNAi therapeutics: A potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Tokatlian, T.; Segura, T. siRNA applications in nanomedicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010, 2, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.S.; Chen, M.S.; Wu, W.H.; Guo, Y.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Investigation on Small Molecules Targeting Cu(I) Preventing Copper-Mediated Neurotoxicity. Acta Chim. Sin. 2015, 73, 799–807. [Google Scholar]
- Sakamoto, K.M. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, J.S. Chemical genetic control of protein levels: Selective in vivo targeted degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Tolnay, M.; Probst, A. REVIEW: Tau protein pathology in Alzheimer’s disease and related disorders. Neuropathol. Appl. Neurobiol. 1999, 25, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Du, J.T.; Yu, C.H.; Zhou, L.X.; Wu, W.H.; Lei, P.; Zhao, Y.F.; Nakanishi, H.; Li, Y.M. Phosphorylation modulates the local conformation and self-aggregation ability of a peptide from the fourth tau microtubule-binding repeat. FEBS J. 2007, 274, 5012–5020. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid ß-induced deficits in an Alzheimer's disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.T.; Gao, N.; Li, Q.Q.; Chen, P.G.; Yang, X.F.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chem. Biol. 2016, 23, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Hon, W.C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.; Schofield, C.J. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002, 417, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Min, J.H.; Yang, H.; Ivan, M. Structure of an HIF-1α-pVHL complex: Hydroxyproline recognition in signaling. Science 2002, 296, 1886–1889. [Google Scholar] [CrossRef] [PubMed]
- Rivas, C.I.; Vera, J.C.; Maccioni, R.B. Anti-idiotypic antibodies that react with microtubule-associated proteins are present in the sera of rabbits immunized with synthetic peptides from tubulin's regulatory domain. Proc. Natl. Acad. Sci. USA 1988, 85, 6092–6096. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Hirose, H.; Nakase, I. Arginine-rich peptides: Methods of translocation through biological membranes. Curr. Pharm. Des. 2013, 19, 2863–2868. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S. Intracellular Delivery Using Arginine Peptides. J. Pharm. Sci. Technol. 2004, 64, 164–167. [Google Scholar]
- Sakamoto, K.; Kim, K.-B.; Verma, R.; Ransick, A.; Stein, B.; Crews, C.M.; Deshaies, R.J. Development of protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell. Proteom. 2003, 2, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C. Targeting the von Hippel–Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef] [PubMed]
- Van Molle, I.; Thomann, A.; Buckley, D.L. Dissecting fragment-based lead discovery at the von Hippel-Lindau protein: Hypoxia inducible factor 1α protein-protein interface. Chem. Biol. 2012, 19, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L.; Gustafson, J.L.; Van Molle, I. Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1α. Angew. Chem. Int. Ed. 2012, 51, 11463–11467. [Google Scholar] [CrossRef] [PubMed]
- Galdeano, C.; Gadd, M.S.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-guided design and optimization of small molecules targeting the protein–protein interaction between the von Hippel–Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 2014, 57, 8657–8663. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.; Galdeano, C.; Soares, P. Potent and selective chemical probe of hypoxic signalling downstream of HIF-α hydroxylation via VHL inhibition. Nat. Commun. 2016, 7, 13312. [Google Scholar] [CrossRef] [PubMed]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.H.; Lei, P.; Liu, Q.; Hu, J.; Gunn, A.P.; Chen, M.S.; Rui, Y.F.; Su, X.Y.; Xie, Z.P.; Zhao, Y.F.; et al. Sequestration of Copper from beta-Amyloid Promotes Selective Lysis by Cyclen-Hybrid Cleavage Agents. J. Biol. Chem. 2008, 283, 31657–31664. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yu, Y.P.; Cui, W.; Fang, C.L.; Wu, W.H.; Zhao, Y.F.; Li, Y.M. Cyclen-hybrid compound captures copper to protect INS-1 cells from islet amyloid polypeptide cytotoxicity by inhibiting and lysing effects. Chem. Commun. 2010, 46, 8023–8025. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.T.; Li, Q.Q.; Qiu, T.; Sun, Z.Y.; Hu, Z.W.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Clearance of the intracellular high level of the tau protein directed by an artificial synthetic hydrolase. Mol. Biosyst. 2014, 10, 3081–3085. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Yoo, S.H.; Kim, M.G. Cleavage agents for soluble oligomers of amyloid β peptides. Angew. Chem. Int. Ed. 2007, 119, 7194–7197. [Google Scholar] [CrossRef]
- Suh, J. Synthetic Artificial Peptidases and Nucleases Using Macromolecular Catalytic Systems. Acc. Chem. Res. 2003, 36, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, L.O.; Naslund, J.; Lindqvist, F.; Johansson, J.; Karlstrom, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambe gaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Yang, P. Molecular dynamics studies of the inhibitory mechanism of copper (II) on aggregation of amyloid β-peptide. Chin. Chem. Lett. 2007, 18, 357–360. [Google Scholar] [CrossRef]
- Von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sawaya, M.R.; Cheng, P.N.; Zheng, J.; Nowick, J.S.; Eisenberg, D. Characteristics of Amyloid-Related Oligomers Revealed by Crystal Structures of Macrocyclic beta-Sheet Mimics. J. Am. Chem. Soc. 2011, 133, 6736–6744. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E. A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990, 15, 305–309. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Martinez-Vicente, M.; Macian, F.; Verkhusha, V.V.; Cuervo, A.M. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2011, 2, 386. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Jin, W.Y.; Lu, J.; Wang, J.; Wang, Y.T. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat. Neurosci. 2014, 17, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Hasegawa, M.; Ishii, M.; Matsumura, S.; Toshima, K. Anthraquinone derivatives as a new family of protein photocleavers. Bioorg. Med. Chem. Lett. 2005, 15, 4624–4627. [Google Scholar] [CrossRef] [PubMed]
- Toshima, K.; Takano, R.; Maeda, Y.; Suzuki, M.; Asai, A.; Matsumura, S. 2-Phenylquinoline–Carbohydrate Hybrids: Molecular Design, Chemical Synthesis, and Evaluation of a New Family of Light-Activatable DNA-Cleaving Agents. Angew. Chem. Int. Ed. 1999, 38, 3733–3735. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic Therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, R.; Prat, F.; Foote, C.S. On the mechanism of DNA cleavage by fullerenes investigated in model systems: Electron transfer from guanosine and 8-oxo-guanosine derivatives to C60. J. Am. Chem. Soc. 1999, 121, 464–465. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, M. Fullerene inhibits beta-amyloid peptide aggregation. Biochem. Biophys. Res. Commun. 2003, 303, 576–579. [Google Scholar] [CrossRef]
- Maggini, M.; Scorrano, G.; Prato, M. Addition of azomethine ylides to C60: Synthesis, characterization, and functionalization of fullerene pyrrolidines. J. Am. Chem. Soc. 1993, 115, 9798–9799. [Google Scholar] [CrossRef]
- Ishida, Y.; Tanimoto, S.; Takahashi, D.; Toshima, K. Photo-degradation of amyloid β by a designed fullerene–sugar hybrid. Med. Chem. Commun. 2010, 1, 212–215. [Google Scholar] [CrossRef]
- Ishida, Y.; Fujii, T.; Oka, K. Inhibition of amyloid β aggregation and cytotoxicity by photodegradation using a designed fullerene derivative. Chem. Asian J. 2011, 6, 2312–2315. [Google Scholar]
- Orvig, C.; Abrams, M. Medicinal Inorganic Chemistry: Introduction. Chem. Rev. 1999, 99, 2201–2204. [Google Scholar] [CrossRef] [PubMed]
- Hasenknopf, B. Polyoxometalates: Introduction to a class of inorganic compounds and their biomedical applications. Front. Biosci. 2005, 10, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Yue, B.; Zhou, Y.; Xu, J.; Wu, Z.; Zhang, X.; Zou, Y.; Jin, S. Photocatalytic Degradation of Aqueous 4-Chlorophenol by Silica-Immobilized Polyoxometalates. Environ. Sci. Technol. 2002, 36, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, G.; Wedd, A.G.; Zhao, C.; Bond, A.M. Photochemical oxidation of water and reduction of polyoxometalate anions at interfaces of water with ionic liquids or diethylether. Proc. Natl. Acad. Sci. USA 2012, 109, 11552–11557. [Google Scholar] [CrossRef] [PubMed]
- Matt, B.; Xiang, X.; Kaledin, A.L.; Han, N.; Moussa, J.; Amouri, H.; Alves, S.; Hill, C.L.; Lian, T.; Musaev, D.G.; et al. Long lived charge separation in iridium (iii)-photosensitized polyoxometalates: Synthesis, photophysical and computational studies of organometallic–redox tunable oxide assemblies. Chem. Sci. 2013, 4, 1737–1745. [Google Scholar] [CrossRef]
- Geng, J.; Li, M.; Ren, J.; Wang, E.; Qu, X. Polyoxometalates as inhibitors of the aggregation of amyloid β peptides associated with Alzheimer’s disease. Angew. Chem. Int. Ed. 2011, 123, 4270–4274. [Google Scholar] [CrossRef]
- Gao, N.; Sun, H.; Dong, K.; Ren, J.; Duan, T.; Xu, C.; Qu, X. Transition-metal-substituted polyoxometalate derivatives as functional anti-amyloid agents for Alzheimer’s disease. Nat. Commun. 2014, 5, 3422. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.I.; Yang, M.; Brender, J.R.; Subramanian, V.; Sun, K.; Joo, N.E.; Kotov, N.A. Inhibition of amyloid peptide fibrillation by inorganic nanoparticles: Functional similarities with proteins. Angew. Chem. Int. Ed. 2011, 50, 5110–5115. [Google Scholar] [CrossRef] [PubMed]
- Kruger, U.; Wang, Y.P.; Kumar, S.; Mandelkow, E.M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol. Aging 2012, 33, 2291–2305. [Google Scholar] [CrossRef] [PubMed]
- Opattova, A.; Filipcik, P.; Cente, M.; Novak, M. Intracellular Degradation of Misfolded Tau Protein Induced by Geldanamycin is Associated with Activation of the Proteasome. J. Alzheimers Dis. 2013, 33, 339–348. [Google Scholar] [PubMed]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Fachinetti, D.; Han, J.S.; Cleveland, D.W. Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc. Natl. Acad. Sci. USA 2012, 109, 3350–3357. [Google Scholar] [CrossRef] [PubMed]
- Bonger, K.M.; Chen, L.C.; Liu, C.W.; Wandless, T.J. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat. Chem. Biol. 2011, 7, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Fukagawa, T.; Takisawa, H.; Kakimoto, T.; Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Method 2009, 6, 917–978. [Google Scholar] [CrossRef] [PubMed]
- Long, M.J.C.; Gollapalli, D.R.; Hedstrom, L. Inhibitor mediated protein degradation. Chem. Biol. 2012, 19, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Long, M.J.C.; Rosenberg, M.M. Boc3Arg-linked ligands induce degradation by localizing target proteins to the 20S proteasome. ACS Chem. Biol. 2016, 11, 3328–3337. [Google Scholar] [CrossRef] [PubMed]
- Neklesa, T.K.; Tae, H.S.; Schneekloth, A.R.; Stulberg, M.J.; Corson, T.W.; Sundberg, T.B.; Raina, K.; Holley, S.A.; Crews, C.M. Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 2011, 7, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Lim, S.M.; Westover, K.D.; Dodge, M.E.; Ercan, D.; Ficarro, S.B.; Udayakumar, D.; Gurbani, D.; Tae, H.S.; Riddle, S.M.; et al. Pharmacological targeting of the pseudokinase Her3. Nat. Chem. Biol. 2014, 10, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Raina, K.; Noblin, D.J.; Serebrenik, Y.V.; Adams, A.; Zhao, C.; Crews, C.M. Targeted protein destabilization reveals an estrogen-mediated ER stress response. Nat. Chem. Biol. 2014, 10, 957–962. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, N.; Chen, Y.-X.; Zhao, Y.-F.; Li, Y.-M. Chemical Methods to Knock Down the Amyloid Proteins. Molecules 2017, 22, 916. https://doi.org/10.3390/molecules22060916
Gao N, Chen Y-X, Zhao Y-F, Li Y-M. Chemical Methods to Knock Down the Amyloid Proteins. Molecules. 2017; 22(6):916. https://doi.org/10.3390/molecules22060916
Chicago/Turabian StyleGao, Na, Yong-Xiang Chen, Yu-Fen Zhao, and Yan-Mei Li. 2017. "Chemical Methods to Knock Down the Amyloid Proteins" Molecules 22, no. 6: 916. https://doi.org/10.3390/molecules22060916
APA StyleGao, N., Chen, Y.-X., Zhao, Y.-F., & Li, Y.-M. (2017). Chemical Methods to Knock Down the Amyloid Proteins. Molecules, 22(6), 916. https://doi.org/10.3390/molecules22060916