3. Experimental Section
3.1. General Information
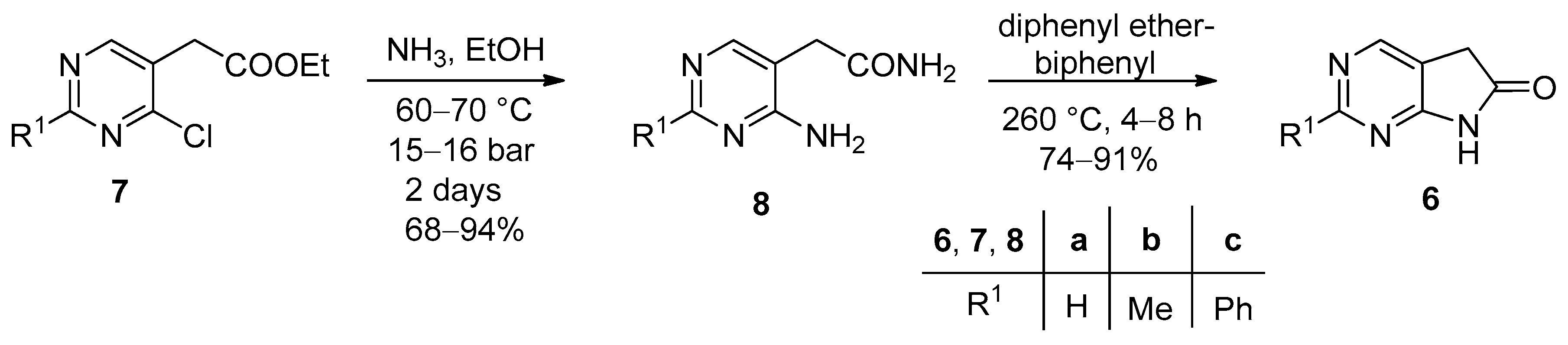
Compounds 24a–c, 25a–c, 26a–f, 27a–f, 28, 29a–c, 30a,b, and 31a,b are new and characterized below. All melting points were determined on an OptiMelt Automated Melting Point System by Stanford Research Systems (Sunnyvale, CA, USA). IR spectra were obtained on a Bruker ALPHA FT-IR spectrometer (Bruker, Billerica, MA, USA). NMR spectra were recorded either on a Bruker AVANCE III 400 (1H and 13C frequencies were 400 and 100 MHz, respectively) or a Bruker AVANCE III HD 600 (1H and 13C frequencies were 600 and 150 MHz, respectively) spectrometer. CDCl3, DMSO-d6 or TFA-d was used as the solvent and tetramethylsilane (TMS) as the internal standard. Chemical shifts (δ) and coupling constants (J) are given in ppm and in Hz, respectively. Some representatives of compound families 26–31 were also investigated using special NMR measurements (1H-1H COSY, 1H-13C HSQC, 1H-13C HMBC, selective NOESY). High-resolution mass spectra (HRMS spectra) were recorded either on a Micromass GCT (Waters, Milford, MA, USA) with EI (direct inlet) or on a Bruker Q-TOF MAXIS Impact mass spectrometer coupled to a Dionex Ultimate 3000 RS HPLC system (Thermo Fisher Scientific, Waltham, MA, USA) with a diode array detector. The reactions were followed by analytical thin layer chromatography on silica gel 60 F254 (Merck, Darmstadt, Germany) and by HPLC-MS on a Shimadzu LC-20 HPLC equipment (Kyoto, Japan) with an SPD-M20A diode-array detector coupled with a LCMS-2020 mass spectrometer. All unspecified reagents were purchased from commercial sources.
3.2. General Procedure I for the Synthesis of Compounds 25a–c
A mixture of the corresponding 1,3-diazaoxindole (6a, 6b or 6c), pyrrolidine (0.60 eq.) and acetone (2.0 eq.) in toluene was stirred at reflux temperature for 2–4 h. The water was removed with a Dean-Stark apparatus. After the reaction was complete, the volatile components were removed in vacuo at 60 °C and the residue was triturated thoroughly with hexane. The precipitate was filtered off, washed with hexane four times and dried.
3.3. General Procedure II for the Synthesis of Compounds 25a–c
A mixture of the corresponding 1,3-diazaoxindole (6a, 6b or 6c), acetic acid (24 eq.) and acetone (46 eq.) was stirred at reflux temperature under argon atmosphere (Ar) for 12–48 h. After the reaction was complete, the volatile components were removed in vacuo at 40 °C and the residue was triturated thoroughly with diethyl ether (DEE) or water. The precipitate was filtered off and dried.
5-(1-Methylethylidene)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (25a). Method A: This compound was prepared according to General Procedure I using 6a (500 mg, 3.7 mmol), pyrrolidine (0.18 mL, 0.16 g, 2.2 mmol), acetone (0.54 mL, 0.43 g, 7.4 mmol) in toluene (10 mL). The crude product was recrystallized from N,N-dimethylformamide (DMF) to give 25a (399 mg, 62%) as pale brown crystals, m.p. 200 °C (decomp., DMF). IR (KBr) ν 1718 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 11.52 (br s, 1H), 8.66 (s, 1H), 8.63 (s, 1H), 2.53 (s, 3H), 2.36 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 167.6, 160.40, 160.36, 156.1, 147.3, 119.1, 116.5, 25.7, 22.7. HRMS calcd. for C9H9N3O [M˙]+ 175.0746; found 175.0762. Method B: This compound was also prepared according to General Procedure II using 6a (3.0 g, 22.2 mmol), acetic acid (30 mL, 524 mmol), acetone (75 mL, 59.25 g, 1022 mmol). The crude product was recrystallized from DMF to give 25a (3.34 g, 86%) as pale brown crystals. Spectral data were identical with those of the product obtained using General Procedure I.
2-Methyl-5-(1-methylethylidene)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (25b). Method A: This compound was prepared according to General Procedure I using 6b (15.00 g, 100 mmol), pyrrolidine (4.96 mL, 4.27 g, 60 mmol), acetone (14.68 mL, 11.60 g, 200 mmol) in toluene (150 mL). The crude product was recrystallized from DMF to give 25b (16.08 g, 85%) as yellow crystals, m.p. 303 °C (decomp., DMF). IR (KBr) ν 1709, 1434 cm−1. 1H-NMR (600 MHz, TFA-d) δ 11.71 (br s, 1H), 8.74 (s, 1H), 2.99 (s, 3H), 2.85 (s, 3H), 2.62 (s, 3H). 13C-NMR (150 MHz, TFA-d) δ 179.8, 171.1, 166.1, 165.6, 138.2, 118.4, 118.1, 27.9, 25.9, 23.1. HRMS calcd. for C10H11N3O [M˙]+ 189.0902; found 189.0891. Method B: This compound was also prepared according to General Procedure II using 6b (180 mg, 1.21 mmol), acetic acid (1.66 mL, 1.74 g, 29.04 mmol), acetone (4.09 mL, 3.23 g, 55.66 mmol). The product 25b was obtained as yellow crystals (98 mg, 42%). Spectral data were identical with those of the product obtained using General Procedure I.
2-Phenyl-5-(1-methylethylidene)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (25c). Method A: This compound was prepared according to General Procedure I using 6c (2.00 g, 9.50 mmol), pyrrolidine (0.47 mL, 0.41 g, 5.7 mmol), acetone (1.39 mL, 1.10 g, 19 mmol) in toluene (20 mL). The crude product was recrystallized from toluene–DMF to give 25c (1.74 g, 73%) as yellow crystals, m.p. 211 °C (decomp., toluene–DMF). IR (KBr) ν 1706, 1592 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 11.60 (br s, 1H), 8.78 (s, 1H), 8.37–8.34 (m, 2H), 7.52−7.50 (m, 3H), 2.54 (s, 3H), 2.38 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 168.0, 161.1, 160.9, 159.4, 147.7, 137.5, 130.8, 128.8, 127.7, 119.3, 114.8, 25.7, 22.6. HRMS calcd. for C15H13N3O [M˙]+ 251.1059; found 251.1056. Method B: This compound was also prepared according to General Procedure II using 6c (120 mg, 0.57 mmol), acetic acid (0.78 mL, 0.82 g, 13.70 mmol), acetone (1.90 mL, 1.50 g, 26.20 mmol). The product 25c was obtained as orange crystals (89 mg, 62%). Spectral data were identical with those of the product obtained using General Procedure I.
3.4. General Procedure III for the Synthesis of Compounds 24a–c
Diazaoxindole derivatives 25a–c were hydrogenated at 60 °C under atmospheric pressure in DMF using activated palladium on charcoal (Pd/C, 10% Pd) catalyst for 12 h. After the reaction was completed, the catalyst was filtered off and washed with DMF three times. The filtrate and the washings were combined and the solvent was removed at 60 °C under reduced pressure. The residue was triturated thoroughly with water, the precipitate was filtered off and dried.
5-(1-Methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a). This compound was prepared according to General Procedure III using 25a (500 mg, 2.85 mmol) and Pd/C catalyst (140 mg) in DMF (50 mL). The crude product was recrystallized from toluene–DMF to give 25a (389 mg, 77%) as colorless crystals, m.p. 127–128 °C (toluene–DMF). IR (KBr) ν 1739, 1596 cm−1. 1H-NMR (600 MHz, CDCl3) δ 10.98 (br s, 1H), 8.87 (d, J = 0.8 Hz, 1H), 8.46 (d, J = 0.8 Hz, 1H), 3.57 (d, J = 4.0 Hz, 1H), 2.78 (m, 1H), 1.22 (d, J = 6.8 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 177.6, 164.3, 157.0, 149.3, 120.3, 50.3, 30.1, 20.2, 17.6. HRMS calcd. for C9H11N3O [M˙]+ 177.0902; found 177.0910.
2-Methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b). This compound was prepared according to General Procedure III using 25b (18.2 g, 96 mmol) and Pd/C catalyst (3.0 g) in DMF (300 mL). The crude product was recrystallized from acetonitrile to give 24b (13.57 g, 74%) as colorless crystals, m.p. 180–181 °C (acetonitrile). IR (KBr) ν 1738, 1618 cm−1. 1H-NMR (400 MHz, CDCl3) δ 10.84 (br s, 1H), 8.31 (s, 1H), 3.49 (d, J = 4.0 Hz, 1H), 2.75 (s, 3H), 2.54 (m, 1H), 1.20 (d, J = 6.8 Hz, 3H), 0.91 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 177.6, 167.0, 164.5, 149.7, 117.0, 50.2, 30.1, 25.5, 20.4, 17.7. HRMS calcd. for C10H13N3O [M˙]+ 191.1059; found 191.1070.
2-Phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24c). This compound was prepared according to General Procedure III using 25c (17.3 g, 68.80 mmol) and Pd/C catalyst (3.0 g) in DMF (250 mL). The crude product was recrystallized from toluene to give 24c (13.77 g, 79%) as colorless crystals, m.p. 178–180 °C (toluene). IR (KBr) ν 1738, 1620 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 11.56 (br s, 1H), 8.54 (s, 1H), 8.35–8.33 (m, 2H), 7.53–7.51 (m, 3H), 3.66 (d, J = 3.7 Hz, 1H), 2.40 (m, 1H), 1.10 (d, J = 6.8 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 178.0, 165.4, 162.3, 149.7, 137.5, 130.8, 128.8, 127.7, 118.6, 49.7, 29.6, 20.0, 18.0. HRMS calcd. for C15H15N3O [M˙]+ 253.1215; found 253.1220.
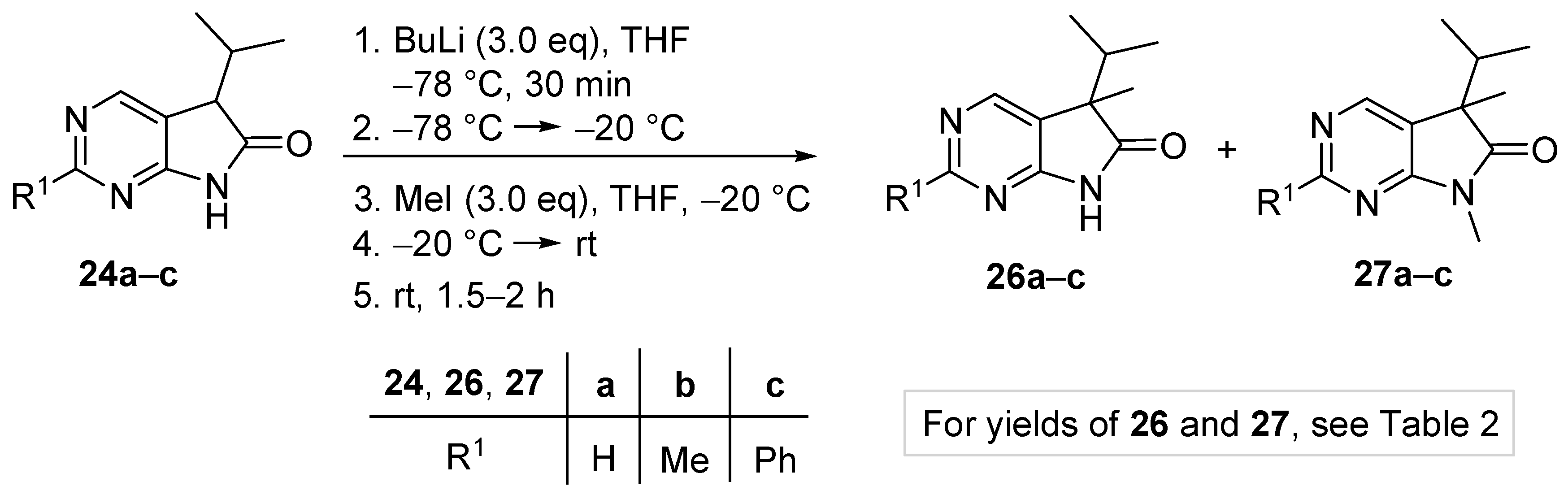
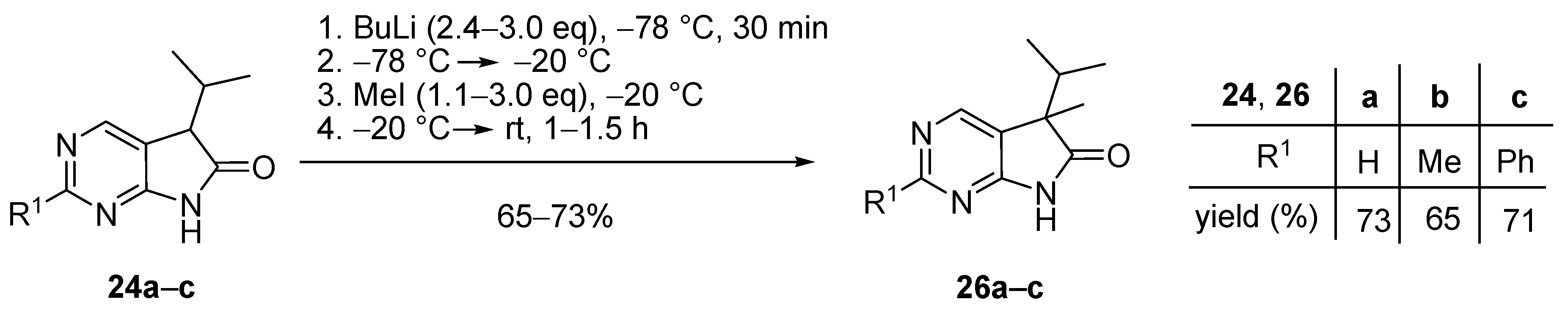
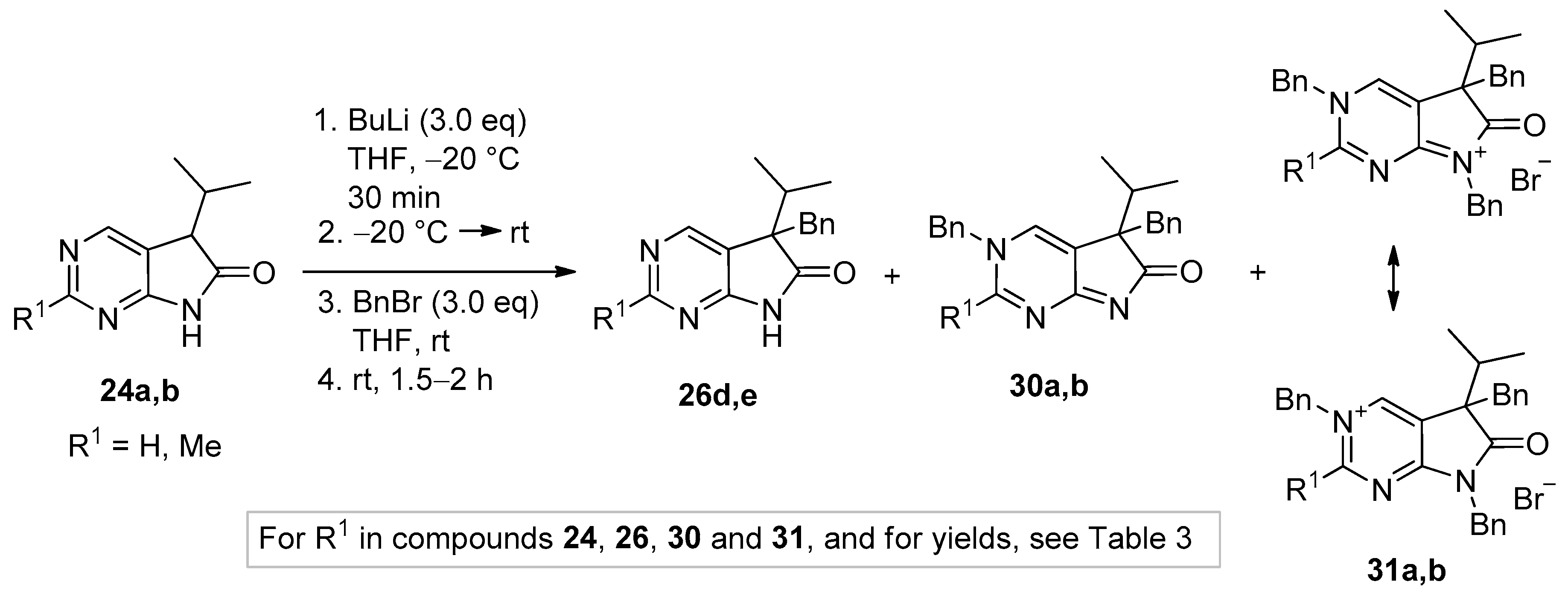
5-Methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26a). To a mixture of BuLi in hexane (5.63 mL, 9.00 mmol, 3.0 eq., 1.6 M) and THF (8 mL), the solution of 5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 532 mg, 3.00 mmol) in THF (10 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to –20 °C and MeI (0.56 mL, 1.28 g, 9.00 mmol, 3.00 eq.) in THF (1 mL) was added dropwise. The acetone-dry ice bath was removed and the stirring was continued for further 1 h. The mixture was quenched with saturated ammonium chloride (NH4Cl) solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with ethyl acetate (EtOAc, 3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane (5 mL), then it was filtered off, washed with diisopropyl ether (DIPE, 2 × 2 mL) and dried. The product 26a was obtained as yellow crystals (420 mg, 73%), m.p. 149–151 °C (hexane–EtOAc, colorless crystals). IR (KBr) ν 1732, 1603 cm−1. 1H-NMR (600 MHz, CDCl3) δ 9.85 (br s, 1H), 8.83 (s, 1H), 8.36 (s, 1H), 2.21 (sp, J = 6.9 Hz, 1H), 1.47 (s, 3H), 1.11 (d, J = 6.9 Hz, 3H), 0.81 (d, J = 6.9 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 181.0, 163.0, 157.0, 149.1, 125.1, 51.6, 34.9, 20.9, 17.7, 17.2. HSQC: 8.83–157.0; 8.36–149.1; 2.21–34.9; 1.47–20.9; 1.11–17.2; 0.81–17.7. HMBC (Jlong-range = 8 Hz; characteristic cross-peaks): 8.83–163.0, 149.1; 8.36–163.0, 157.0, 125.1; 2.21–125.1, 51.6, 17.7, 17.2; 1.47–181.0, 125.1, 51.6, 34.9; 1.11–51.6, 34.9, 17.7; 0.81–51.6, 34.9, 17.2. Selective NOESY: 1.47–8.36, 2.21, 1.11, 0.81; 1.11–8.36, 2.21, 1.47, 0.81; 0.81–8.36, 2.21, 1.47, 1.11. HRMS calcd. for C10H13N3O [M˙]+ 191.1059; found 191.1075.
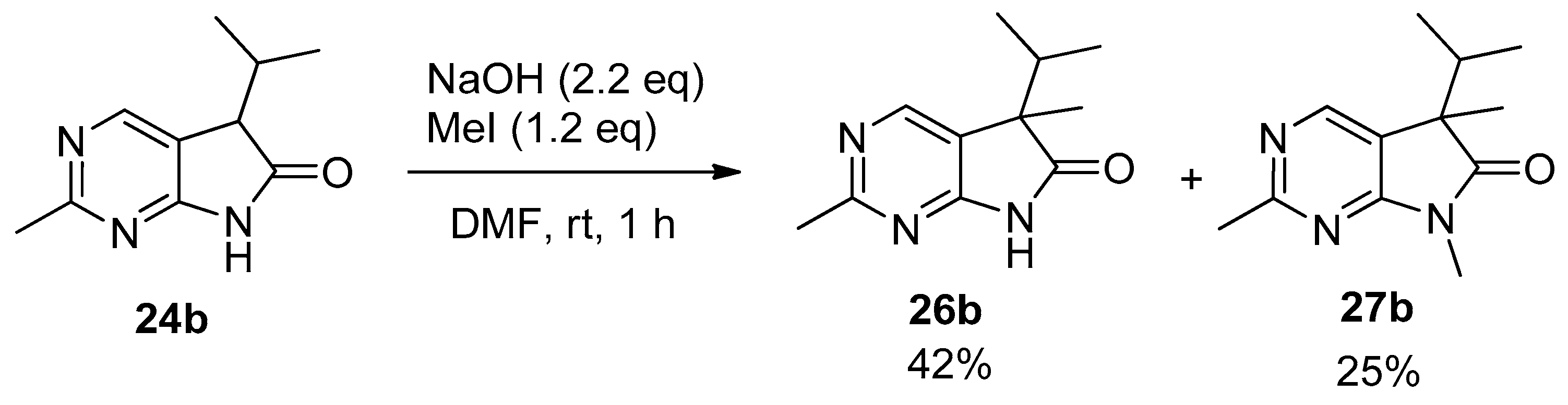
2,5-Dimethyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26b). To a mixture of BuLi in hexane (4.50 mL, 7.20 mmol, 2.4 eq., 1.6 M) and THF (5 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b, 574 mg, 3.00 mmol) in THF (15 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to –20 °C and MeI (0.26 mL, 0.60 g, 4.20 mmol, 1.4 eq.) in THF (1 mL) was added dropwise. The acetone-dry ice bath was removed and the reaction mixture was allowed to warm to room temperature. The stirring was continued for further 1.5 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane (5 mL), then it was filtered off, washed with DIPE (2 × 2 mL) and dried. The product 26b was obtained as yellow crystals (399 mg, 65%), m.p. 190–191 °C (hexane–EtOAc, colorless crystals). IR (KBr) ν 1744, 1621, 1429, 1190 cm−1. 1H-NMR (600 MHz, CDCl3) δ 11.36 (br s, 1H), 8.30 (s, 1H), 2.49 (s, 3H), 2.01 (sp, J = 6.8 Hz, 1H), 1.30 (s, 3H), 0.98 (d, J = 6.8 Hz, 3H), 0.66 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 181.5, 166.1, 163.9, 149.0, 121.4, 50.6, 34.3, 25.8, 20.7, 17.6, 17.2. HRMS calcd. for C11H15N3O [M˙]+ 205.1215; found 205.1219.
5-Methyl-2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26c). To a mixture of BuLi in hexane (2.96 mL, 4.73 mmol, 2.4 eq., 1.6 M) and THF (3 mL), the solution of 2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24c, 500 mg, 1.97 mmol) in THF (8 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to −20 °C, and MeI (0.13 mL, 0.31 g, 2.17 mmol, 1.1 eq.) in THF (1 mL) was added dropwise. The acetone-dry ice bath was removed and the reaction mixture was stirred for further 1 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane (5 mL), then it was filtered off, washed with DIPE (2 × 2 mL) and dried. The product 26c was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product 26c was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give colorless crystals (372 mg, 71%), m.p. 155–156 °C. IR (KBr) ν 1728, 1599, 1405, 1201 cm−1. 1H-NMR (600 MHz, CDCl3) δ 8.58 (br s, 1H), 8.43 (s, 1H), 8.37–8.35 (m, 2H), 7.49–7.48 (m, 3H), 2.23 (sp, J = 6.8 Hz, 1H), 1.49 (s, 3H), 1.13 (d, J = 6.8 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 181.1, 163.7, 163.0, 149.5, 137.1, 130.9, 128.6, 128.1, 122.3, 51.7, 35.0, 21.0, 17.8, 17.3. HRMS calcd. for C16H17N3O [M˙]+ 267.1372; found 267.1370.
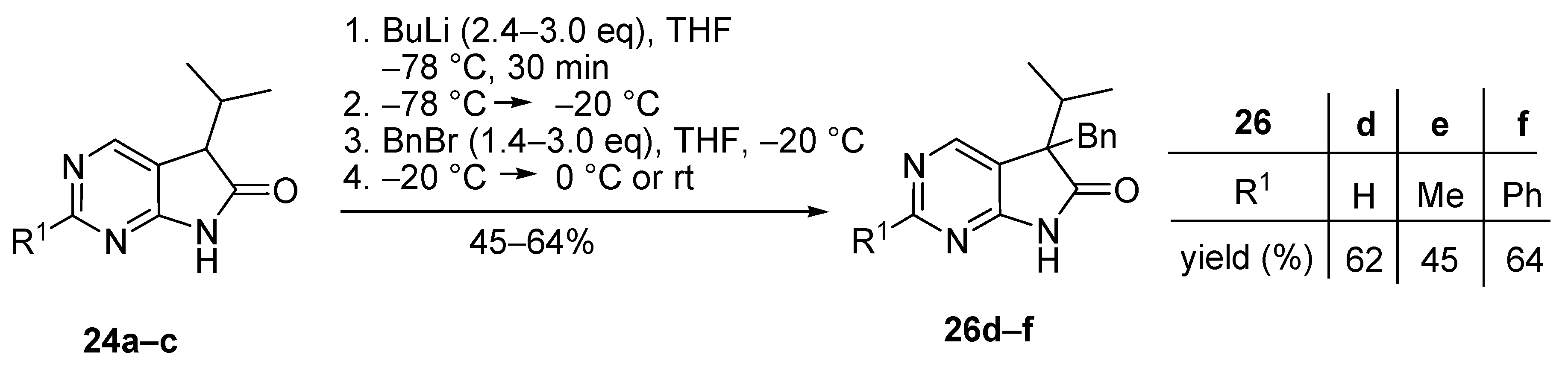
5-Benzyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26d). To a mixture of BuLi in hexane (2.81 mL, 4.50 mmol, 3.0 eq., 1.6 M) and THF (3 mL), the solution of 5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 266 mg, 1.50 mmol) in THF (5 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to −20 °C and BnBr (0.53 mL, 0.77 g, 4.50 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The acetone-dry ice bath was removed and the reaction mixture was stirred for further 1 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane (5 mL), then it was filtered off, washed with DIPE (2 × 2 mL) and dried. The product 26d was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product 26d was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 26d (250 mg, 62%) as colorless crystals, m.p. 195–197 °C. IR (KBr) ν 1731, 1601 cm−1. 1H-NMR (600 MHz, CDCl3) δ 8.70 (br, 1H), 8.65 (s, 1H), 8.43 (s, 1H), 7.09–7.04 (m, 3H), 6.87 (m, 2H), 3.26 (d, J = 13.2 Hz, 1H), 3.21 (d, J = 13.2 Hz, 1H), 2.38 (sp, J = 6.8 Hz, 1H), 1.19 (d, J = 6.8 Hz, 3H), 0.86 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 179.1, 163.0, 157.1, 149.8, 134.9, 129.7, 128.1, 127.0, 122.5, 58.5, 40.9, 35.0, 17.9, 17.6. HSQC: 8.65–157.1; 8.43–149.8, (7.09–7.04)–127.0, 128.1; 6.87–129.7; 3.26–40.9; 3.21–40.9; 2.38–35.0; 1.19–17.6; 0.86–17.9. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 8.65–163.0, 149.8; 8.43–163.0, 157.1, 122.5; (7.09–7.04)–134.9; 6.87–40.9; 3.26–135.0, 129.7, 58.5; 3.21–179.1, 135.0, 129.7, 58.5; 2.38–179.1, 122.5, 58.5, 17.9, 17.6; 1.19–58.5, 35.0, 17.9; 0.86–58.5, 35.0, 17.6. HRMS calcd. for C16H17N3O [M˙]+ 267.1372; found 267.1371.
5-Benzyl-2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26e). To a mixture of BuLi in hexane (2.36 mL, 3.66 mmol, 2.4 eq., 1.6 M) and THF (3 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 300 mg, 1.57 mmol) in THF (7 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to 0 °C and BnBr (0.26 mL, 0.38 g, 2.20 mmol, 1.4 eq.) in THF (1 mL) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for further 3 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane (5 mL), it was filtered off, washed with hexane (2 × 2 mL) and DIPE (2 mL), then dried. The crude product 26e was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. This product 26e was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 26e (199 mg, 45%) as colorless crystals, m.p. 185–187 °C. IR (KBr) ν 1740, 1620, 1426 cm−1. 1H-NMR (400 MHz, CDCl3) δ 9.79 (br s, 1H), 8.29 (s, 1H), 7.07–7.05 (m, 3H), 6.90 (m, 2H), 3.23 (d, J = 13.2 Hz, 1H), 3.19 (d, J = 13.2 Hz, 1H), 2.60 (s, 3H), 2.34 (sp, J = 6.8 Hz, 1H), 1.17 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 179.7, 166.8, 163.6, 149.8, 135.3, 129.8, 128.0, 126.8, 119.2, 58.1, 40.8, 34.9, 25.5, 17.9, 17.6. HRMS calcd. for C17H19N3O [M˙]+ 281.1528; found 281.1524.
5-Benzyl-2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26f). To a mixture of BuLi in hexane (1.89 mL, 4.73 mmol, 2.4 eq., 2.5 M) and THF (3 mL), the solution of 2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24c, 500 mg, 1.97 mmol) in THF (6 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to –20 °C and BnBr (0.70 mL, 1.01 g, 5.91 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The acetone-dry ice bath was removed and the reaction mixture was allowed to warm to 0 °C. The stirring was continued for further 5 h at 0 °C. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane/DIPE = 3/1 (5 mL), then it was filtered off, washed with hexane/DIPE = 3/1 (2 × 2 mL) and dried. The product 26f was obtained as yellow crystals (435 mg, 64%), m.p. 192–193 °C (hexane–EtOAc, colorless crystals). IR (KBr) ν 1728, 1625, 1410 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.52 (s, 1H), 8.31–8.28 (m, 2H), 7.96 (br s, 1H), 7.46–7.44 (m, 3H), 7.07–7.05 (m, 3H), 6.95–6.93 (m, 2H), 3.27 (s, 2H), 2.40 (sp, J = 6.9 Hz, 1H), 1.23 (d, J = 6.9 Hz, 3H), 0.88 (d, J = 6.9 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 179.3, 163.5, 163.2, 150.1, 137.0, 135.2, 130.9, 129.8, 128.6, 128.1, 128.0, 126.9, 119.8, 58.5, 41.0, 35.2, 18.0, 17.7. HRMS calcd. for C22H21N3O [M˙]+ 343.1685; found 343.1660.
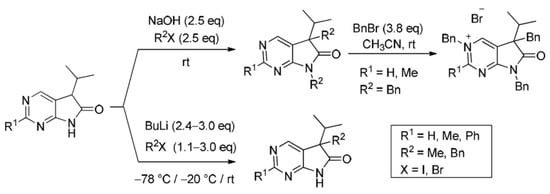
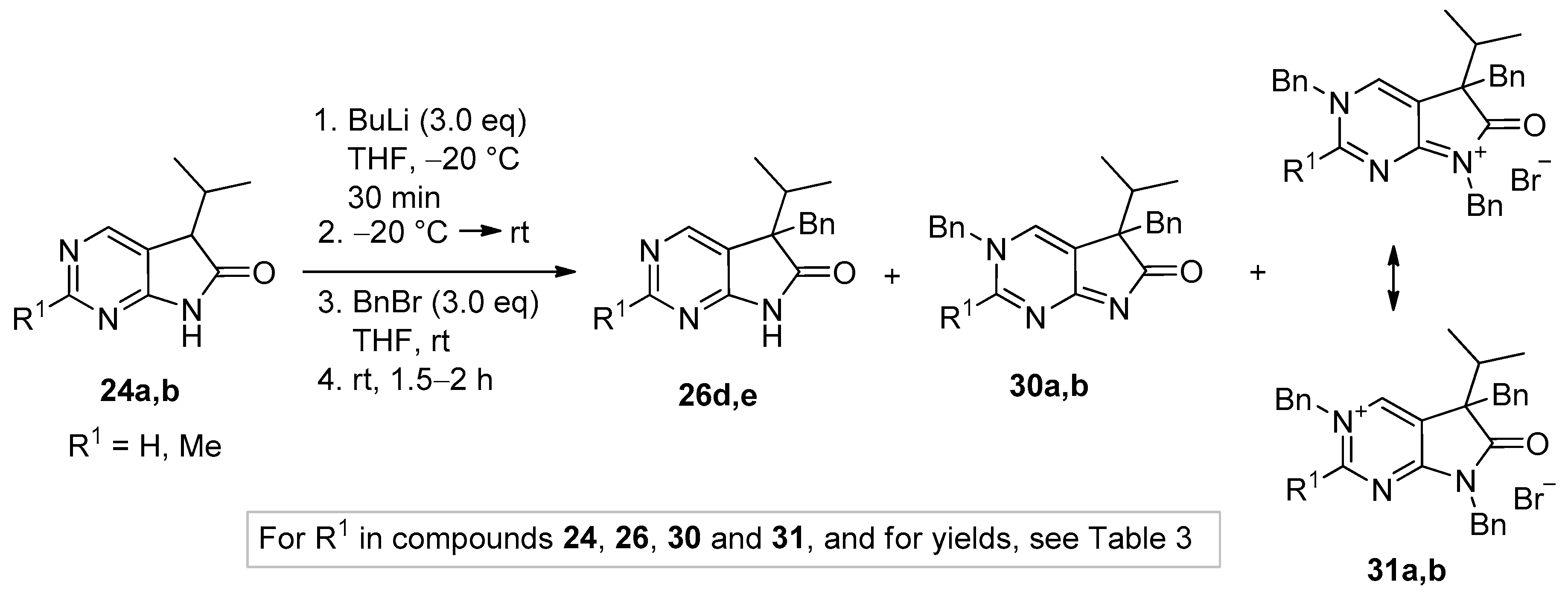
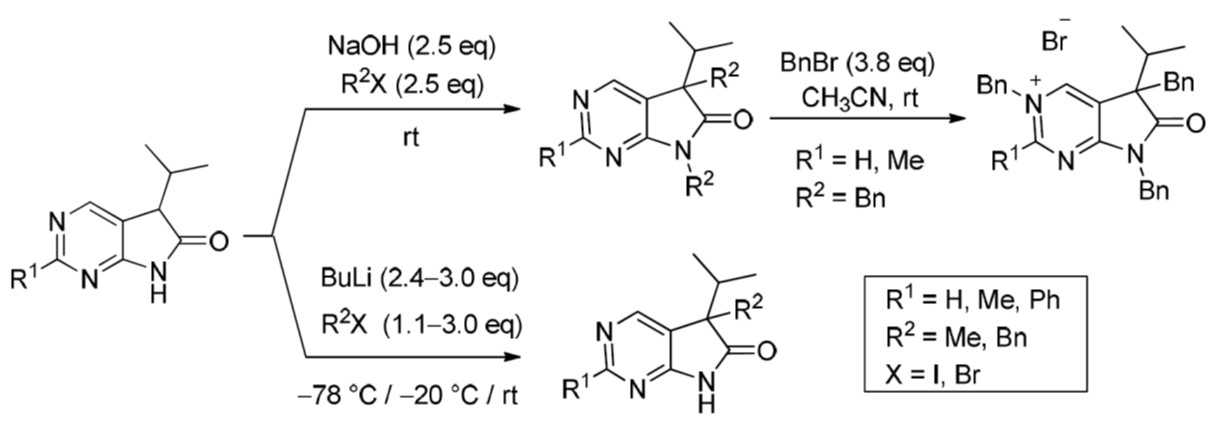
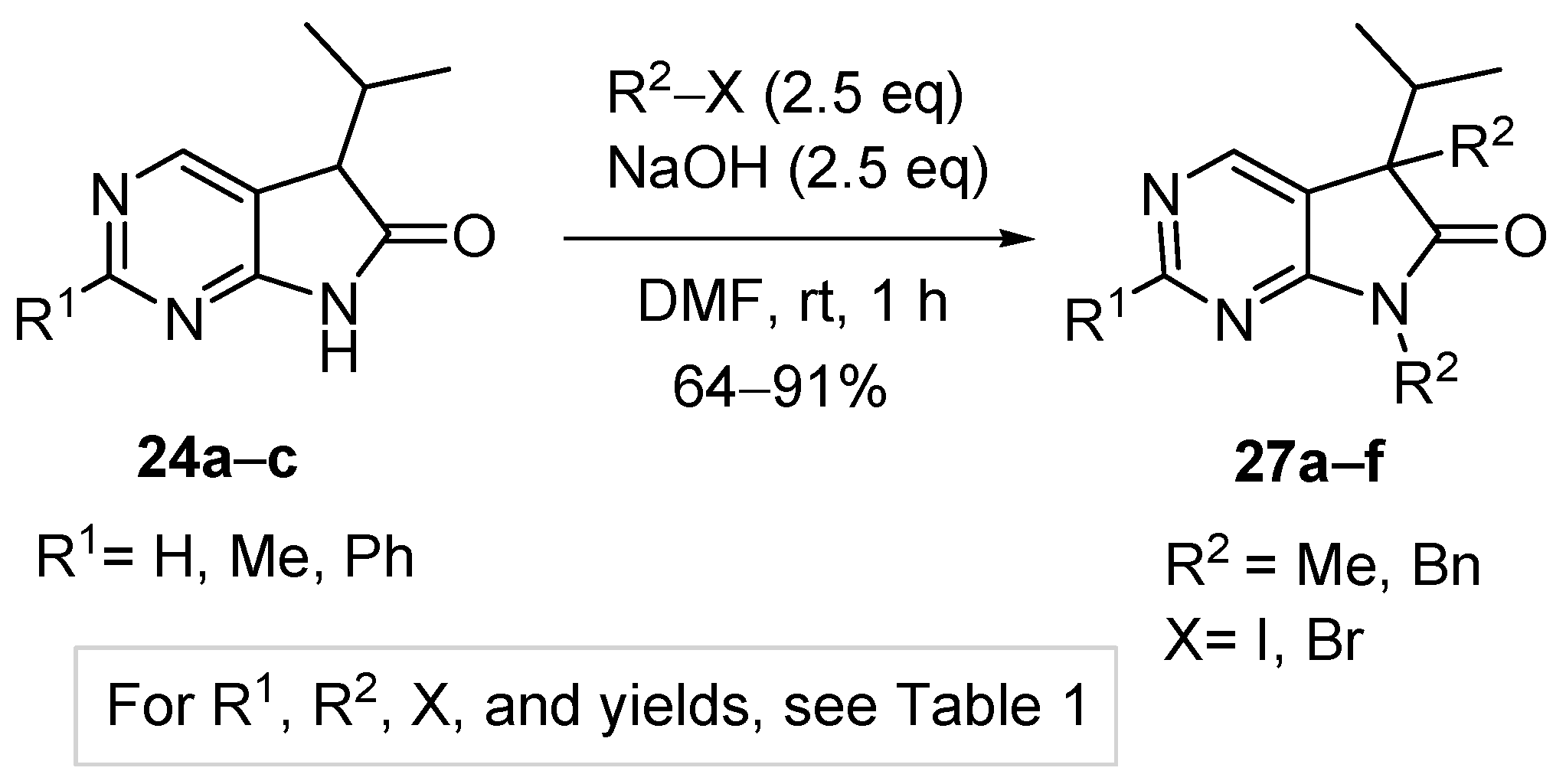
3.5. General Procedure IV for the Synthesis of Compounds 27a–f
To a mixture of NaOH (2.5 eq.) in DMF, the solution of 5-isopropyl-1,3-diazaoxindoles 24a–c in DMF was added dropwise at room temperature, under Ar atmosphere and it was stirred for 30 min. Then the solution of the appropriate alkyl halide (2.5 eq.) in DMF was added dropwise at 15–20 °C. The stirring was continued for further 1 h. The reaction mixture was poured onto ice–water. The precipitate was filtered off or the aqueous layer was extracted with dichloromethane (DCM, 3 × 15 mL). The combined organic layer was washed with brine (5 × 50 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. Purification of the crude products was performed as described below.
5,7-Dimethyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27a). This compound was prepared according to General Procedure IV using 24a (275 mg, 1.55 mmol) in DMF (4 mL), NaOH (155 mg, 3.88 mmol) in DMF (2 mL) and MeI (0.24 mL, 0.55 g, 3.88 mmol) in DMF (1 mL). The residual oil was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 27a (203 mg, 64%) as colorless crystals, m.p. 64–66 °C. IR (KBr) ν 1731, 1586, 1492 cm−1. 1H-NMR (600 MHz, CDCl3) δ 8.81 (s, 1H), 8.29 (s, 1H), 3.28 (s, 3H), 2.20 (sp, J = 7.0 Hz, 1H), 1.43 (s, 3H), 1.07 (d, J = 7.0 Hz, 3H), 0.71 (d, J = 7.0 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 180.2, 163.7, 157.5, 148.1, 124.5, 50.9, 35.0, 25.1, 20.8, 17.6, 17.3. HSQC: 8.81–157.5; 8.29–148.1; 3.28–25.1; 2.20–35.0; 1.43–20.8; 1.07–17.3; 0.71–17.6. HMBC (Jlong-range = 8 Hz; characteristic cross-peaks): 8.81–163.7, 148.1; 8.29–163.7, 157.5, 124.5; 3.28–180.2, 163.7; 1.43–180.2, 124.5, 50.9, 35.0; 1.07–50.9, 35.0, 17.6; 0.71–50.9, 35.0, 17.3. Selective NOESY: 1.43–8.29, 3.28, 2.20, 1.07, 0.71. HRMS calcd. for C11H15N3O [M˙]+ 205.1215; found 205.1223.
2,5,7-Trimethyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27b). This compound was prepared according to General Procedure IV using 24b (574 mg, 3.00 mmol) in DMF (7 mL), NaOH (300 mg, 7.5 mmol) in DMF (2 mL) and MeI (0.47 mL, 1.06 g, 7.5 mmol) in DMF (1 mL). The residual oil was purified by gradient elution column chromatography using hexane and EtOAc as the eluents to give 27b (527 mg, 80%) as a colorless oil. IR (film) ν 1732, 1600, 1488 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 3.26 (s, 3H), 2.67 (s, 3H), 2.17 (sp, J = 6.9 Hz, 1H), 1.40 (s, 3H), 1.10 (d, J = 6.9 Hz, 3H), 0.69 (d, J = 6.9 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 180.6, 167.1, 164.0, 148.0, 121.1, 50.7, 34.9, 26.1, 25.1, 20.9, 17.6, 17.3. HRMS calcd. for C12H17N3O [M˙]+ 219.1372; found 219.1373.
5,7-Dimethyl-2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27c). This compound was prepared according to General Procedure IV using 24c (507 mg, 2 mmol) in DMF (7 mL), NaOH (200 mg, 5.00 mmol) in DMF (2 mL) and MeI (0.31 mL, 0.71 g) in DMF (1 mL). The precipitate was filtered off, washed with water (3 × 5 mL) and dried. The product 27c was obtained as colorless crystals (512 mg, 91%). An analytical sample was obtained by recrystallization from hexane, m.p. 106–107 °C (hexane). IR (KBr) ν 1716, 1678 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.48–8.45 (m, 2H), 8.36 (s, 1H), 7.50–7.49 (m, 3H), 3.36 (s, 3H), 2.21 (sp, J = 6.8 Hz, 1H), 1.45 (s, 3H), 1.10 (d, J = 6.8 Hz, 3H), 0.73 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 180.7, 164.2, 163.6, 148.5, 137.4, 130.8, 128.5, 128.1, 122.1, 51.0, 35.1, 25.2, 20.9, 17.7, 17.3. HRMS calcd. for C17H19N3O [M˙]+ 281.1528; found 281.1522.
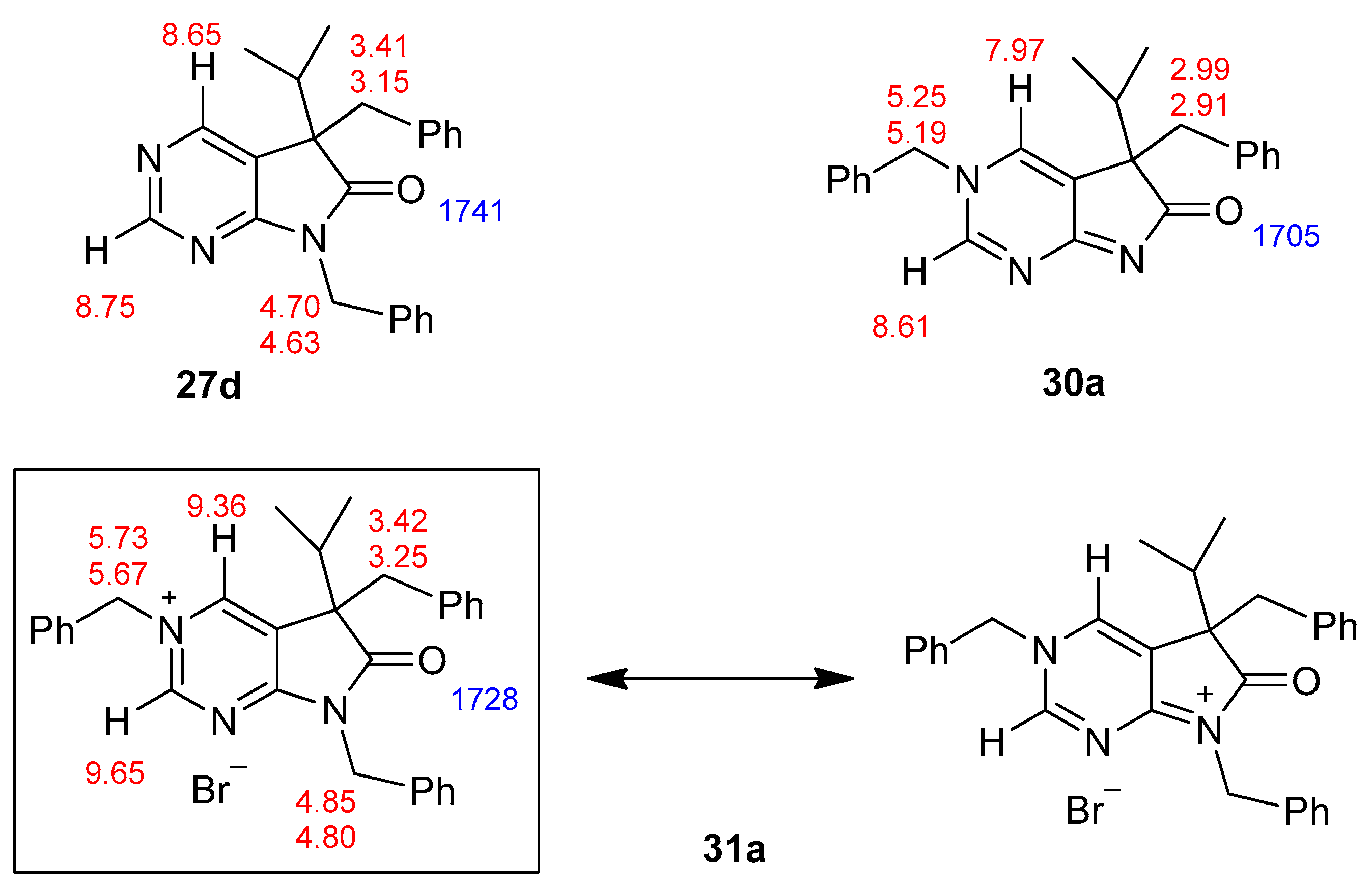
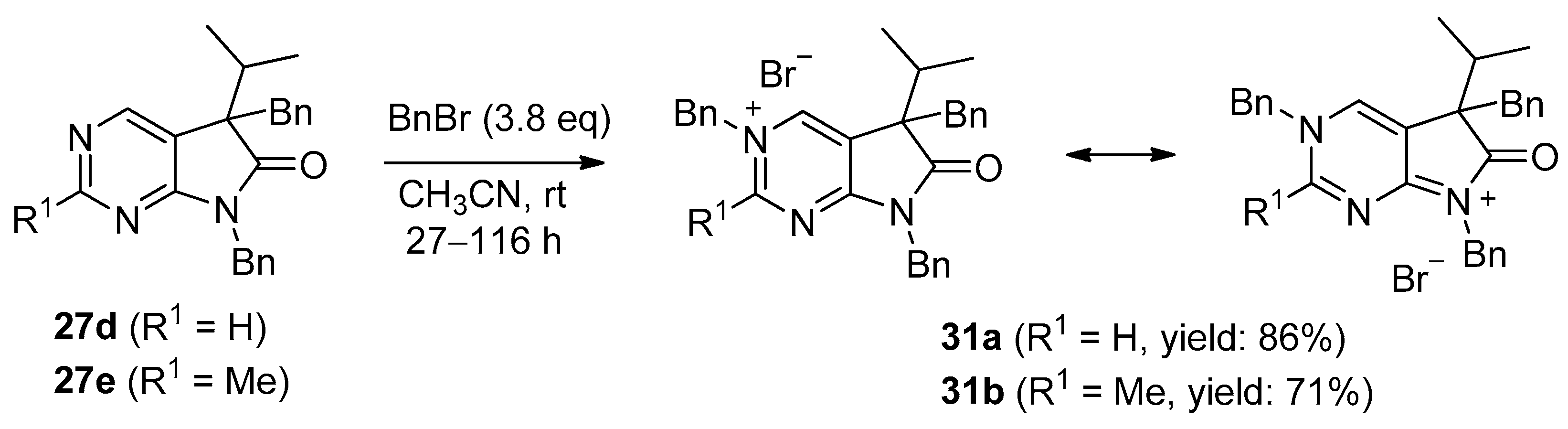
5,7-Dibenzyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27d). This compound was prepared according to General Procedure IV using 24a (250 mg, 1.41 mmol) in DMF (2 mL), NaOH (141 mg, 3.53 mmol) in DMF (2 mL) and BnBr (0.42 mL, 0.60 g, 3.53 mmol) in DMF (1 mL). The residual oil was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 27d (353 mg, 70%) as colorless crystals, m.p. 65–66 °C. IR (KBr) ν 1741, 1496 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 8.75 (s, 1H), 8.65 (s, 1H), 7.20–7.16 (m, 3H), 7.07 (m, 1H), 6.99 (t, J = 7.6 Hz, 2H), 6.83 (m, 2H), 6.81 (m, 2H), 4.70 (d, J = 15.4 Hz, 1H), 4.63 (d, J = 15.4 Hz, 1H), 3.41 (d, J = 13.0 Hz, 1H), 3.15 (d, J = 13.0 Hz, 1H), 2.30 (sp, J = 6.8 Hz, 1H), 1.08 (d, J = 6.8 Hz, 3H), 0.68 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 178.2, 163.1, 157.4, 150.3, 135.8, 135.7, 129.8, 128.5, 128.0, 127.4, 127.2, 126.8, 121.6, 57.3, 41.8, 39.7, 35.2, 17.7, 17.6. COSY: (7.20–7.16)–6.83; 6.99–6.81; 3.41–3.15; 2.30–1.08, 0.68. HSQC: 8.75–150.3; 8.65–157.4; (7.20–7.16)–128.5, 127.4; 7.07–126.8; 6.99–128.0; 6.83–127.2; 6.81–129.8; 4.70–41.8; 4.63–41.8; 3.41–39.7; 3.15–39.7; 2.30–35.2; 1.08–17.7; 0.68–17.6. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 8.75–163.1, 157.4, 121.6; 6.83–41.8; 6.81–39.7; (7.20–7.16)–135.7; 6.99–135.8; 4.70–178.2, 163.1, 135.7, 127.2; 4.63–178.2, 163.1, 135.7, 127.2; 3.41–178.2, 135.8, 129.8, 57.3; 3.15–135.8, 129.8, 121.6, 57.3; 2.30–121.6, 57.3, 17.7, 17.6; 1.08–57.3, 35.2; 0.68–57.3, 35.2. COSY: (7.20–7.16)–6.83; 6.99–6.81; 3.41–3.15; 2.30–1.08, 0.68. HRMS calcd. for C23H23N3O [M˙]+ 357.1841; found 357.1829.
5,7-Dibenzyl-2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27e). This compound was prepared according to General Procedure IV using 24b (200 mg, 1.05 mmol) in DMF (2 mL), NaOH (105 mg, 2.63 mmol) in DMF (2 mL) and BnBr (0.31 mL, 0.45 g, 2.63 mmol) in DMF (1 mL). The precipitate was filtered off, washed with water (3 × 5 mL) and dried. The product 27e was obtained as orange crystals (320 mg, 82%). An analytical sample was obtained by recrystallization from a mixture of hexane and EtOAc to give 27e as colorless crystals, m.p. 84–86 °C (hexane–EtOAc). IR (KBr) ν 1736, 1478 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 8.61 (br s, 1H), 7.21–7.17 (m, 3H), 7.07 (t, J = 7.4 Hz, 1H), 7.00 (t, J = 7.4 Hz, 2H), 6.86–6.80 (m, 4H), 4.67 (d, J = 15.4 Hz, 1H), 4.61 (d, J = 15.4 Hz, 1H), 3.37 (d, J = 13.0 Hz, 1H), 3.12 (d, J = 13.0 Hz, 1H), 2.44 (s, 3H), 2.26 (sp, J = 6.8 Hz, 1H), 1.07 (d, J = 6.8 Hz, 3H), 0.64 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 178.5, 166.3, 163.4, 150.2, 135.90, 135.85, 129.8, 128.5, 128.0, 127.3, 127.1, 126.7, 118.1, 57.0, 41.7, 39.6, 35.2, 25.9, 17.604, 17.596. HRMS calcd. for C24H25N3O [M˙]+ 371.1998; found 371.1989.
5,7-Dibenzyl-5-(1-methylethyl)-2-phenyl-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27f). This compound was prepared according to General Procedure IV using 24c (760 mg, 3.00 mmol) in DMF (10 mL), NaOH (300 mg, 7.50 mmol) in DMF (2 mL) and BnBr (0.89 mL, 1.28 g, 7.50 mmol) in DMF (2 mL). The residual oil was purified by gradient elution column chromatography using hexane and EtOAc as the eluents to give 27f (1062 mg, 82%) as a colorless oil. IR (film) ν 1732, 1596, 1475, 1398 cm−1. 1H-NMR (600 MHz, CDCl3) δ 8.49 (s, 1H), 8.38 (m, 2H), 7.47–7.44 (m, 3H), 7.21–7.17 (m, 3H), 7.13 (m, 2H), 6.98 (m, 1H), 6.90 (m, 2H), 6.82 (m, 2H), 4.80 (s, 2H), 3.27 (d, J = 13.2 Hz, 1H), 3.24 (d, J = 13.2 Hz, 1H), 2.40 (sp, J = 6.8 Hz, 1H), 1.19 (d, J = 6.8 Hz, 3H), 0.72 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 178.7, 164.1, 163.3, 149.3, 137.2, 135.7, 135.1, 130.8, 129.7, 128.5, 128.4, 128.3, 128.0 (signal overlapping), 127.4, 126.7, 119.5, 57.6, 42.5, 40.8, 35.4, 17.8, 17.7. HRMS calcd. for C29H27N3O [M˙]+ 433.2154; found 433.2102.
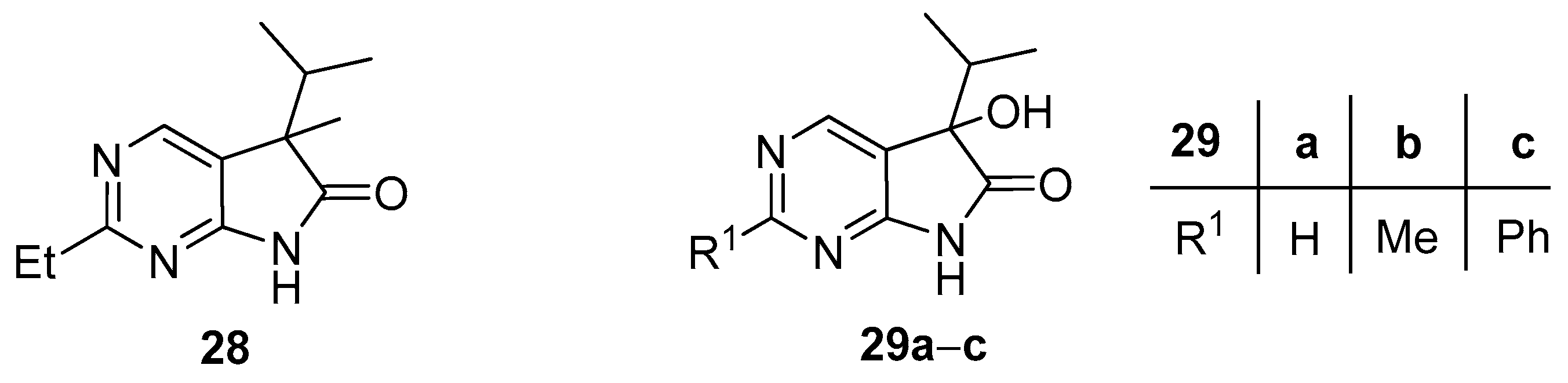
2-Ethyl-5-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (28). To a mixture of BuLi in hexane (2.51 mL, 6.27 mmol, 3.0 eq., 2.5 M) and THF (3 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b, 400 mg, 2.09 mmol) in THF (12 mL) was added dropwise at −20 °C under Ar atmosphere. The temperature was allowed to warm to room temperature and the mixture was stirred for 30 min, then MeI (0.39 mL, 0.89 g, 6.27 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 2 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining yellow crystals were purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 28 (50 mg, 11%) as colorless crystals, m.p. 149–151 °C. IR (KBr) ν 1743, 1620, 1460, 1193 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 11.37 (br s, 1H), 8.34 (s, 1H), 2.77 (q, J = 7.6 Hz, 2H), 2.01 (sp, J = 6.8 Hz, 1H), 1.30 (s, 3H), 1.24 (t, J = 7.6 Hz, 3H), 0.98 (d, J = 6.8 Hz, 3H), 0.66 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 181.5, 170.1, 164.0, 149.1, 121.6, 50.6, 34.4, 32.1, 20.7, 17.6, 17.2, 12.6. HSQC: 8.34–149.1; 2.77–32.1; 2.01–34.4; 1.30–20.7; 1.24–12.6; 0.98–17.3; 0.66–17.6. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 11.37–121.6; 8.34–170.1, 164.0, 121.6; 2.77–170.1; 1.30–181.5, 121.6; 1.24–170.1. HRMS calcd. for C12H17N3O [M˙]+ 219.1372; found 219.1387.
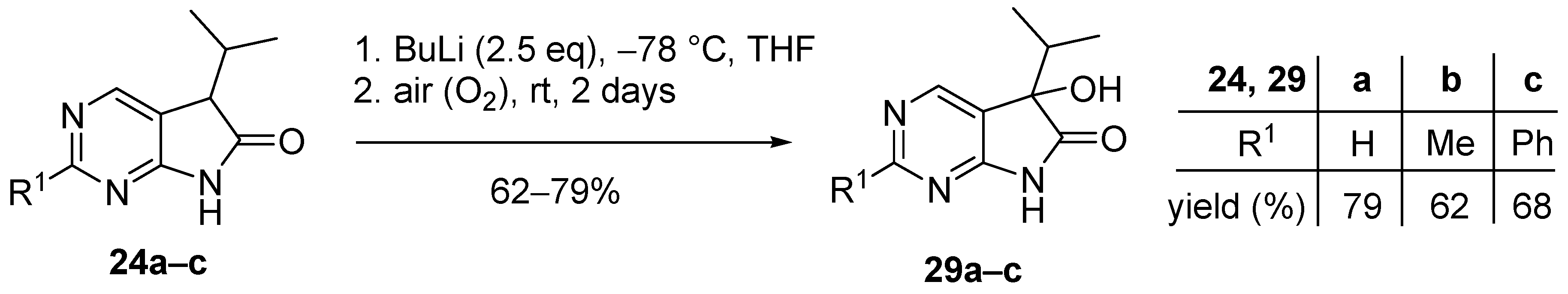
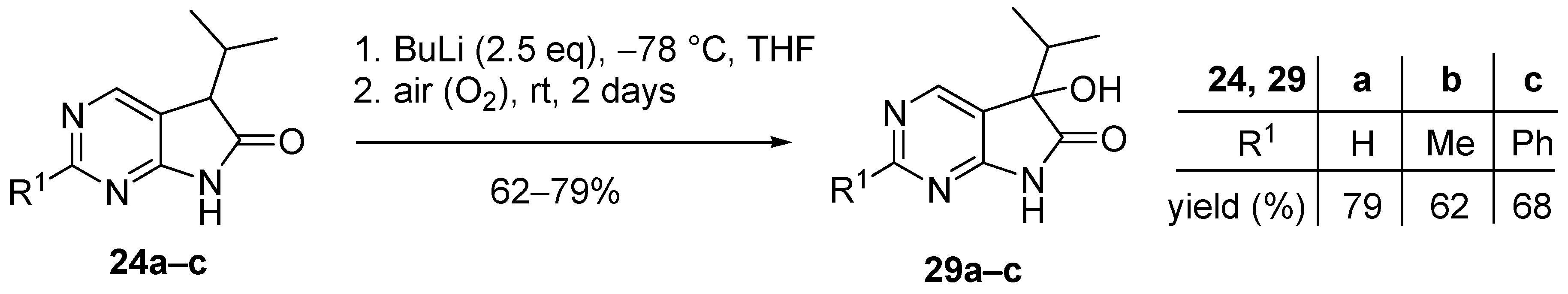
3.6. General Procedure V for the Synthesis of Compounds 29a–c
To a mixture of BuLi in hexane (1.50 mL, 3.75 mmol, 2.5 eq., 2.5 M) and THF (3 mL), the solution of the appropriate 2-substituted 5-isopropyl-1,3-diazaoxindole 24a–c (1.50 mmol) in THF (8 mL) was added dropwise at −78 °C, under argon atmosphere. After the addition was complete, the acetone–dry ice bath was removed, the reaction mixture was allowed to warm to room temperature and the apparatus was opened to the air. The stirring was continued for further 2 days. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the layers were separated and the aqueous layer was extracted with EtOAc (3 × 5 mL). The combined organic layer was dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane/DIPE = 3/1 (7 mL), then it was filtered off, washed with hexane/DIPE = 3/1 (2 × 2 mL) and dried.
5-Hydroxy-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (29a). Method A: This compound was prepared according to General Procedure V. The product 29a was obtained as off-white crystals (228 mg, 79%), m.p. 175–176 °C. IR (KBr) ν 1760, 1159 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 11.42 (br s, 1H), 8.71 (s, 1H), 8.39 (s, 1H), 6.25 (s, 1H), 2.12 (sp, J = 6.9 Hz, 1H), 1.03 (d, J = 6.9 Hz, 3H), 0.64 (d, J = 6.9 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 179.3, 164.1, 158.3, 149.8, 122.4, 78.1, 34.4, 16.3, 16.0. HSQC: 8.71–158.3; 8.39–149.8; 2.12–34.4; 1.03–16.0; 16.3. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 8.71–164.0, 149.8; 8.39–164.0, 158.3, 122.4; 6.25–122.4, 78.1, 34.4; 1.03–78.1, 34.4, 16.3; 0.64–78.1, 34.4, 16.0. HRMS calcd. for C9H11N3O2 [M + H]+ 193.0851; found 193.0850. Method B: To a mixture of BuLi in hexane (3.80 mL, 9.44 mmol, 3.3 eq., 2.5 M) and THF (3 mL), the solution of 5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 500 mg, 2.86 mmol) in THF (8 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to –20 °C and MeI (0.59 mL, 1.33 g, 9.44 mmol, 3.3 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 1.5 h. The mixture was quenched with saturated NH4Cl solution (10 mL), and water (5 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (20 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining yellow oil was purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (2 mL), washed with DIPE (2 × 1 mL) and dried to give 29a (50 mg, 9%) as colorless crystals. Spectral data were identical with those of the product obtained using Method A.
5-Hydroxy-2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (29b). Method A: This compound was prepared according to General Procedure V. The product 29b was obtained as off-white crystals (194 mg, 62%), m.p. 182–184 °C. IR (KBr) ν 1624, 1432 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 11.28 (br s, 1H), 8.26 (s, 1H), 6.16 (s, 1H), 2.49 (s, 3H), 2.10 (sp, J = 6.8 Hz, 1H), 1.03 (d, J = 6.8 Hz, 3H), 0.62 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 179.5, 167.2, 164.3, 149.8, 119.1, 78.0, 34.3, 25.9, 16.4, 16.0. HRMS calcd. for C10H13N3O2 [M˙]+ 207.1008; found 207.1026. Method B: To a mixture of BuLi in hexane (4.13 mL, 6.60 mmol, 2.2 eq., 1.6 M) and THF (5 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b, 574 mg, 3.00 mmol) in THF (14 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then MeI (0.41 mL, 0.94 g, 6.60 mmol, 2.2 eq.) in THF (1 mL) was added dropwise. The temperature was allowed to warm to room temperature and stirring was continued for further 5 h. The mixture was quenched with saturated NH4Cl solution (10 mL), and water (5 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining orange oil was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product was triturated in hexane (2 mL), washed with DIPE (2 × 1 mL) and dried to give 29b (100 mg, 16%) as colorless crystals. Spectral data were identical with those of the product obtained using Method A.
5-Hydroxy-2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (29c). Method A: This compound was prepared according to General Procedure V. The product 29c was obtained as off-white crystals (273 mg, 68%), m.p. 184–185 °C. IR (KBr) ν 1730, 1621, 1167 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 11.49 (br s, 1H), 8.51 (s, 1H), 8.35–8.33 (m, 2H), 7.54–7.51 (m, 3H), 6.24 (s, 1H), 2.16 (sp, J = 6.8 Hz, 1H), 1.08 (d, J = 6.8 Hz, 3H), 0.68 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 179.6, 164.8, 163.2, 150.2, 137.3, 131.1, 128.9, 127.9, 120.5, 78.1, 34.4, 16.4, 16.1. HRMS calcd. for C15H15N3O2 [M˙]+ 269.1164; found 269.1150. Method B: To a mixture of BuLi in hexane (4.13 mL, 6.60 mmol, 2.2 eq., 1.6 M) and THF (15 mL), the solution of 2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24c, 760 mg, 3.00 mmol) in THF (12 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to –20 °C and MeI (0.22 mL, 0.51 g, 3.60 mmol, 1.2 eq.) in THF (1 mL) was added dropwise. The temperature was allowed to warm to 0 °C and stirring was continued for further 4 h. The mixture was quenched with saturated NH4Cl solution (10 mL), and water (5 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining yellow crystals were purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product was triturated in hexane (2 mL), washed with DIPE (2 × 1 mL) and dried to give 29c (64 mg, 8%) as colorless crystals. Spectral data were identical with those of the product obtained using Method A.
3,5-Dibenzyl-5-(1-methylethyl)-3,5-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (30a). To a mixture of BuLi in hexane (3.20 mL, 5.07 mmol, 3.0 eq., 1.6 M) and THF (3 mL), the solution of 5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 300 mg, 1.69 mmol) in THF (6 mL) was added dropwise at −20 °C under Ar atmosphere. The temperature was allowed to warm to room temperature and the mixture was stirred for 30 min, then BnBr (0.60 mL, 0.87 g, 5.07 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 1.5 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining yellow oil was purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 30a (95 mg, 16%) as colorless crystals, m.p. 271–272 °C. IR (KBr) ν 1705, 1656, 1464 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 8.61 (d, J = 2.0 Hz, 1H), 7.97 (d, J = 2.0 Hz, 1H), 7.50 (m, 2H), 7.44 (m, 1H), 7.39 (m, 2H), 6.99 (m, 1H), 6.90 (m, 2H), 6.74 (m, 2H), 5.25 (d, J = 14.8 Hz, 1H), 5.19 (d, J = 14.8 Hz, 1H), 2.99 (d, J = 12.7 Hz, 1H), 2.91 (d, J = 12.7 Hz, 1H), 2.02 (sp, J = 6.8 Hz, 1H), 1.00 (d, J = 6.8 Hz, 3H), 0.67 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 194.5, 182.0, 153.6, 136.8, 136.1, 132.9, 130.0, 129.2, 128.8, 128.0, 127.5, 127.0, 126.2, 58.8, 57.0, 39.4, 34.3, 17.74, 17.73. COSY (characteristic cross-peaks): 7.50–7.44, 7.39; 2.02–1.00, 0.67. HSQC: 8.61–153.6; 7.97–132.9; 7.50–129.2; 7.44–128.8; 7.39–128.0; 6.99–127.0; 6.90–127.5; 6.74–130.0; 5.25–57.0; 5.19–57.0; 2.99–39.5; 2.91–39.5; 2.02–34.3; 1.00, 0.67–17.74, 17.73. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 8.61–182.0, 132.9, 57.0; 7.97–182.0, 153.6, 127.0; 7.39–57.0; 6.74–39.5; 5.25, 5.19–153.6, 136.1, 132.9, 128.0; 2.99–136.8, 130.0; 2.91–136.8, 130.0, 127.0; 2.02–194.5. HRMS calcd. for C23H23N3O [M˙]+ 357.1841; found 357.1851.
3,5-Dibenzyl-2-methyl-5-(1-methylethyl)-3,5-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (30b). To a mixture of BuLi in hexane (1.26 mL, 3.14 mmol, 3.0 eq., 2.5 M) and THF (2 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b, 200 mg, 1.05 mmol) in THF (6 mL) was added dropwise at −20 °C under Ar atmosphere. The temperature was allowed to warm to room temperature and the mixture was stirred for 30 min, then BnBr (0.37 mL, 0.54 g, 3.14 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 2 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then it was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining orange oil was purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 30b (215 mg, 56%) as off-white crystals, m.p. 250–252 °C. IR (KBr) ν 1757, 1653 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 9.11 (br s, 1H), 7.53 (m, 2H), 7.46 (m, 1H), 7.29 (m, 2H), 7.15 (m, 1H), 7.12 (m, 2H), 6.98 (m, 2H), 5.71 (d, J = 16.0 Hz, 1H), 5.64 (d, J = 16.0 Hz, 1H), 3.32 (d, J = 13.1 Hz, 1H), 3.18 (d, J = 13.1 Hz, 1H), 2.61 (s, 3H), 2.27 (sp, J = 6.8 Hz, 1H), 1.10 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 183.4 (br), 170.9 (br), 165.2, 142.8 (br), 135.7, 134.1, 129.9, 129.4, 128.9, 128.2, 127.4, 127.1, 122.8, 58.7, 58.5, 39.4, 34.9, 23.1, 17.6, 17.5. HSQC: 9.11–142.8; 7.53–129.4; 7.46–128.9; 7.29–127.4; 7.15–127.2; 7.12–128.2; 6.98–129.9; 5.71–58.5; 5.64–58.5; 3.32–39.4; 3.18–39.4; 2.61–23.1; 2.27–34.9; 1.10–17.6; 0.85–17.5. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 9.11–170.9, 165.2, 122.8, 58.5; 7.53–134.1; 7.29–58.5; 7.12–135.7; 5.71, 5.64–165.2, 142.8, 134.1; 3.32–183.4; 3.18–183.4, 122.8; 2.61–165.2. HRMS calcd. for C24H25N3O [M˙]+ 371.1998; found 371.1994.
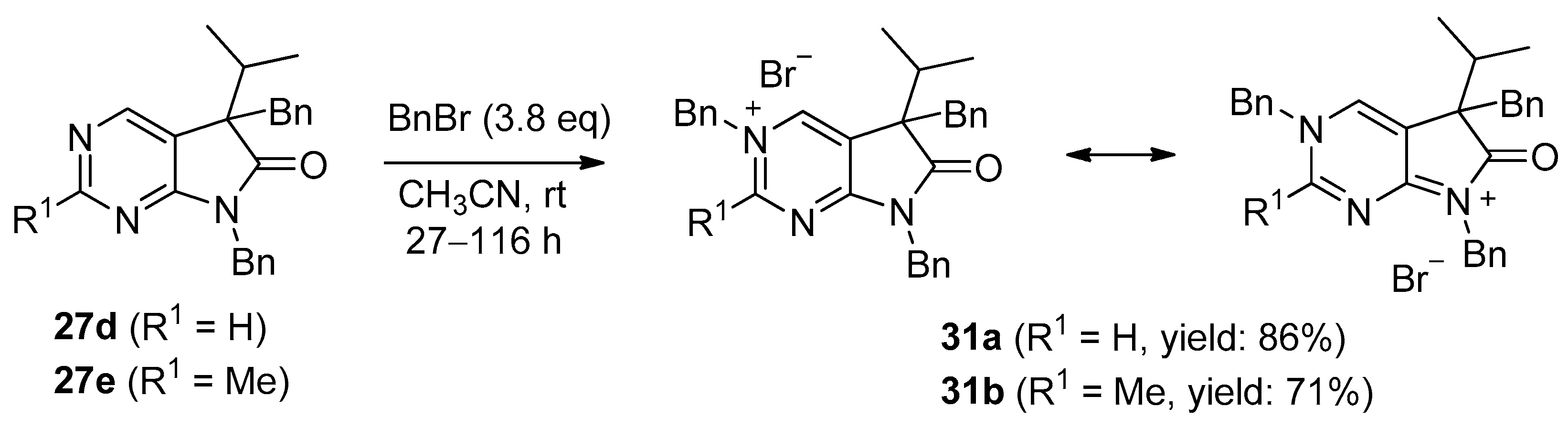
3,5,7-Tribenzyl-5-(1-methylethyl)-6-oxo-6,7-dihydro-5H-pyrrolo[2,3-d]pyrimidin-3-ium bromide (31a). Method A: A mixture of 27d (340 mg, 0.95 mmol), BnBr (0.43 mL, 0.62 g, 3.80 mmol) and acetonitrile (3 mL) was stirred for 27 h at room temperature. The solvent was removed at 40 °C in vacuo and the crystals was triturated with hexane (5 mL), filtered off, washed with DIPE (3 × 2 mL) and dried. The product 31a was obtained as colorless crystals (431 mg, 86%), m.p. 220–222 °C (i-PrOH). IR (KBr) ν 1728, 1625, 1410 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 9.65 (d, J = 1.6 Hz, 1H), 9.36 (d, J = 1.6 Hz, 1H), 7.59 (m, 2H), 7.56 (m, 2H), 7.51 (m, 1H), 7.26 (m, 3H), 7.02 (m, 1H), 6.94 (m, 2H), 6.80 (m, 2H), 6.63 (m, 2H), 5.73 (d, J = 14.4 Hz, 1H), 5.67 (d, J = 14.4 Hz, 1H), 4.85 (d, J = 15.3 Hz, 1H), 4.80 (d, J = 15.3 Hz, 1H), 3.42 (d, J = 13.2 Hz, 1H), 3.25 (d, J = 13.2 Hz, 1H), 2.41 (sp, J = 6.9 Hz, 1H), 1.04 (d, J = 6.9 Hz, 3H), 0.86 (d, J = 6.9 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 178.0, 166.1, 156.3, 143.0, 134.5, 134.4, 134.2, 129.7, 129.6, 129.4, 128.9, 128.8, 128.3, 128.1, 127.7, 127.2, 123.8, 59.9, 58.2, 43.1, 39.2, 35.4, 17.7, 17.2. COSY (characteristic cross-peaks): 7.26–6.94; 7.02–6.80; 6.80–6.63; 3.42–3.25; 2.41–1.04, 0.86. HSQC: 9.65–156.3; 9.36–143.0; 7.59–128.9; 7.56–129.5; 7.51–129.6; 7.26–128.8, 128.1; 7.02–127.2; 6.94–127.7; 6.63–129.7; 5.73–59.9; 5.67–59.9; 4.85–43.1; 4.80–43.1; 3.42–39.2; 3.25–39.2; 2.41–35.4; 1.04–17.7; 0.86–17.2. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 9.65–166.1, 143.0, 59.9; 9.36–166.1, 156.3, 123.8, 59.9; 7.59–59.9; 7.56–134.4; 7.26–134.2; 6.94–43.1; 6.80–134.5; 6.63–39.2; 2.41–123.8, 178.0; 1.04–58.2; 0.86–58.2. COSY: 7.26–6.94; 7.02–6.80; 6.80–6.63; 3.42–3.25; 2.41–1.04, 0.86. HSQC: 9.65–156.3; 9.36–143.0; 7.59–128.9; 7.56–129.5; 7.51–129.6; 7.26–128.8, 128.1; 7.02–127.2; 6.94–127.7; 6.63–129.7; 5.73–59.9; 5.67–59.9; 4.85–43.1; 4.80–43.1; 3.42–39.2; 3.25–39.2; 2.41–35.4; 1.04–17.7; 0.86–17.2. Selective NOESY: 9.36–7.59, 6.63, 5.73, 5.67, 3.42, 1.04; 9.65–7.59, 5.73, 5.67. HRMS calcd. for C30H30N3O [M]+ 448.238; found 448.238. Elementary analysis of C30H30BrN3O calcd. for Br: 15.12%; found: 14.80%. Method B: To a mixture of BuLi in hexane (3.20 mL, 5.07 mmol, 3.0 eq., 1.6 M) and THF (3 mL), the solution of 5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 300 mg, 1.69 mmol) in THF (6 mL) was added dropwise at −20 °C, under Ar atmosphere. The temperature was allowed to warm to room temperature and the mixture was stirred for 30 min, then BnBr (0.60 mL, 0.87 g, 5.07 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 1.5 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then it was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining yellow oil was purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 30a (110 mg, 12%) as colorless crystals. Spectral data were identical with those of the product obtained using Method A.
3,5,7-Tribenzyl-2-methyl-5-(1-methylethyl)-6-oxo-6,7-dihydro-5H-pyrrolo[2,3-d]pyrimidin-3-ium bromide (31b). Method A: A mixture of 27e (371 mg, 1.00 mmol), BnBr (0.45 mL, 0.65 g, 3.80 mmol) and acetonitrile (3 mL) was stirred for 116 h at room temperature. The solvent was removed at 40 °C in vacuo and the crystals was triturated with hexane (5 mL), filtered off, washed with DIPE (3 × 2 mL) and dried. The product 31b was obtained as off-white crystals (386 mg, 71%), m.p. 104–106 °C (i-PrOH). IR (KBr) ν 1756, 1651 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 9.36 (s, 1H), 7.55 (t, J = 7.4 Hz, 2H), 7.48 (t, J = 7.4 Hz, 1H), 7.36 (d, J = 7.4 Hz, 2H), 7.27–7.26 (m, 3H), 7.07 (t, J = 7.4 Hz, 1H), 7.02–6.99 (m, 4H), 6.86 (d, J = 7.2 Hz, 2H), 5.79 (d, J = 15.8 Hz, 1H), 5.71 (d, J = 15.8 Hz, 1H), 4.84 (d, J = 15.2 Hz, 1H), 4.79 (d, J = 15.2 Hz, 1H), 3.44 (d, J = 13.3 Hz, 1H), 3.29 (d, J = 13.3 Hz, 1H), 2.77 (s, 3H), 2.39 (sp, J = 6.8 Hz, 1H), 1.08 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 178.3, 166.6, 165.8, 145.0, 134.8, 134.3, 133.4, 129.7, 129.4, 129.2, 128.8, 128.5, 128.1, 127.9, 127.8, 127.4, 121.5, 59.3, 58.0, 43.0, 39.2, 35.6, 23.5, 17.6, 17.3. HRMS calcd. for C31H32N3O [M]+ 462.254; found 462.253. Elementary analysis of C31H32BrN3O calcd. for Br: 14.73%; found: 14.80%. Method B: To a mixture of BuLi in hexane (1.26 mL, 3.14 mmol, 3.0 eq., 2.5 M) and THF (2 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b, 200 mg, 1.05 mmol) in THF (6 mL) was added dropwise at −20 °C, under Ar atmosphere. The temperature was allowed to warm to room temperature and the mixture was stirred for 30 min, then BnBr (0.37 mL, 0.54 g, 3.14 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 2 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then it was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining orange oil was purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 31b (108 mg, 19%) as off-white crystals. Spectral data were identical with those of the product obtained using Method A.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}