4.3. Synthesis and Characterization
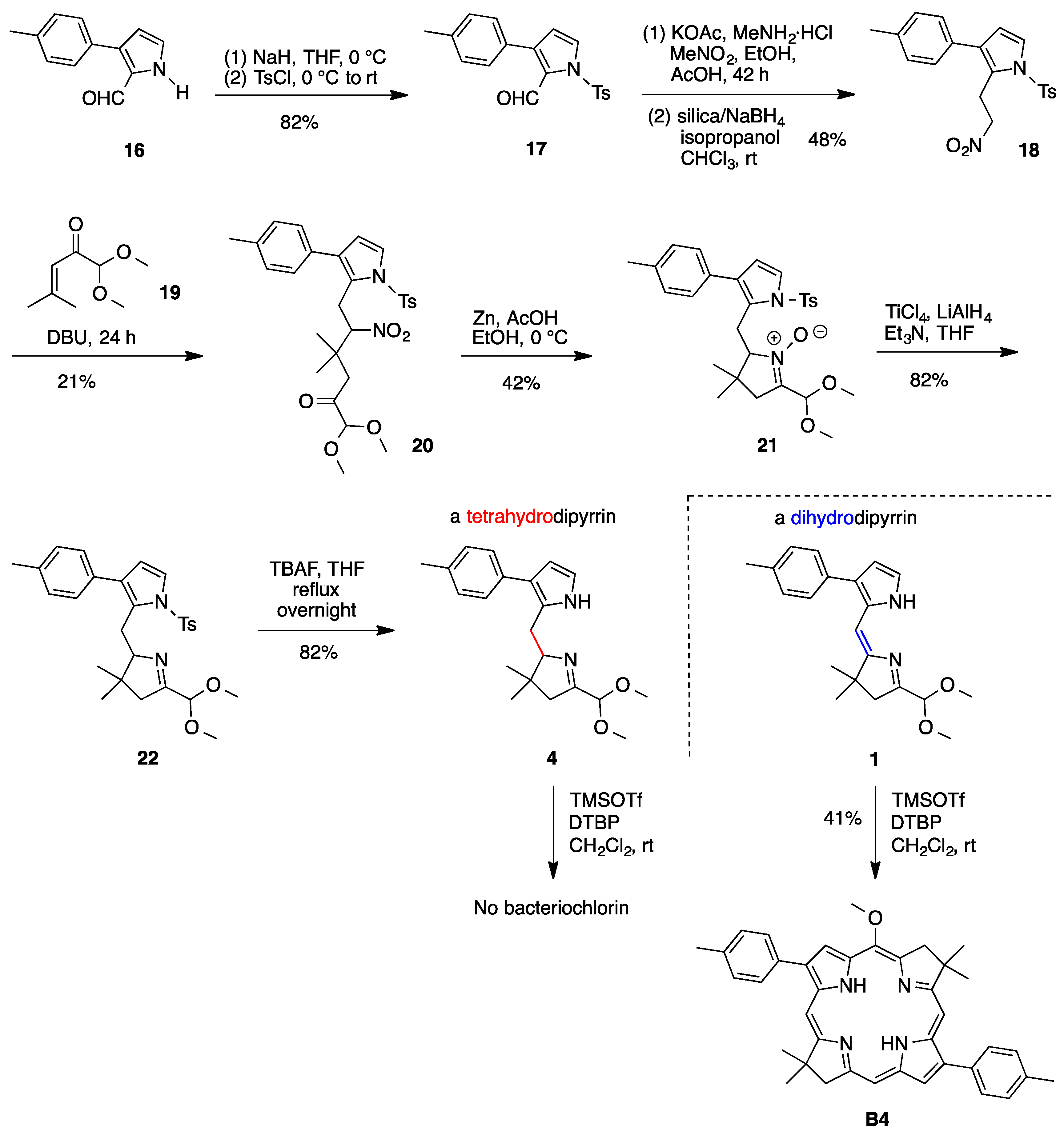
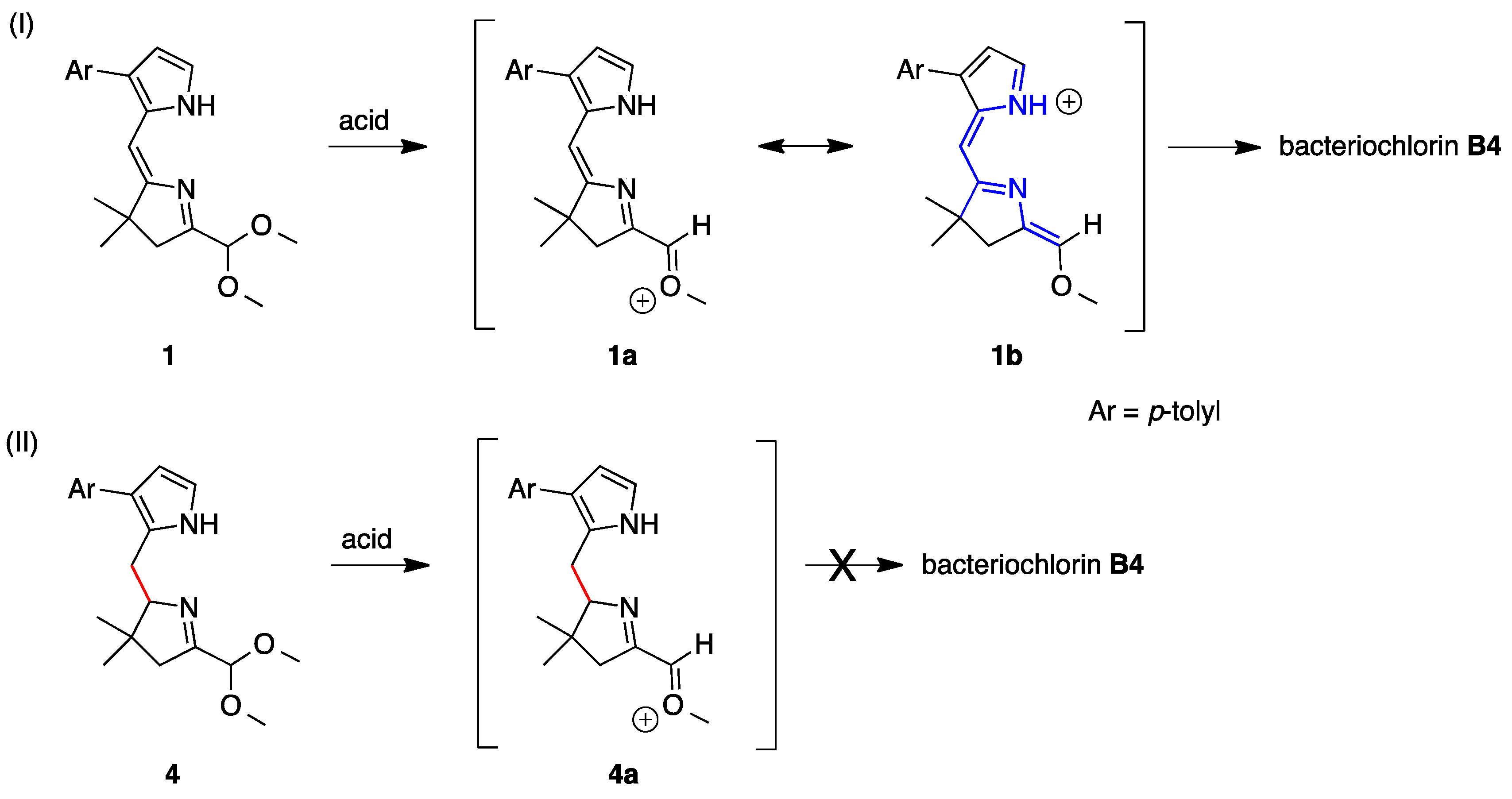
2,3,4,5-Tetrahydro-1-(dimethoxymethyl)-3,3-dimethyl-7-(4-methylphenyl)dipyrrin (
4). Following a reported procedure [
42] with some modification, a sample of
22 (200 mg, 0.426 mmol) was treated with TBAF (1.3 mL, 1.3 mmol, 1.0 M in THF) under argon. The reaction mixture was stirred overnight at reflux. Saturated aqueous NaHCO
3 was added, and the mixture was extracted with ethyl acetate. The organic extract was washed (brine), dried (Na
2SO
4), concentrated and chromatographed (silica, CH
2Cl
2/ethyl acetate (4:1)) to afford a yellow oil (118 mg, 82%):
1H-NMR δ 0.95 (s, 3H), 1.12 (s, 3H), 2.37 (s, 3H), 2.43, 2.51 (AB,
J = 2.2 Hz,
J = 17.4 Hz, 2H), 2.64 (ABX,
J = 11.7 Hz,
J = 15.2 Hz, 1H), 3.05 (ABX,
J = 2.9 Hz,
J = 15.2 Hz, 1H), 3.45 (s, 6H), 3.76–3.79 (m, 1H), 4.87 (s, 1H), 6.28–6.30 (m, 1H), 6.75–6.77 (m, 1H), 7.18 (d,
J = 8.1 Hz, 2H), 7.31 (d,
J = 8.1 Hz, 2H), 9.95 (br, 1H);
13C-NMR (100 MHz) δ 21.3, 22.9, 26.5, 27.3, 41.5, 48.7, 54.9, 80.4, 103.1, 108.3, 116.3, 121.2, 127.5, 128.2, 129.3, 134.7, 174.3; ESI-MS obsd 341.2225, calcd. 341.2224 [(M + H)
+, M = C
21H
28N
2O
2].
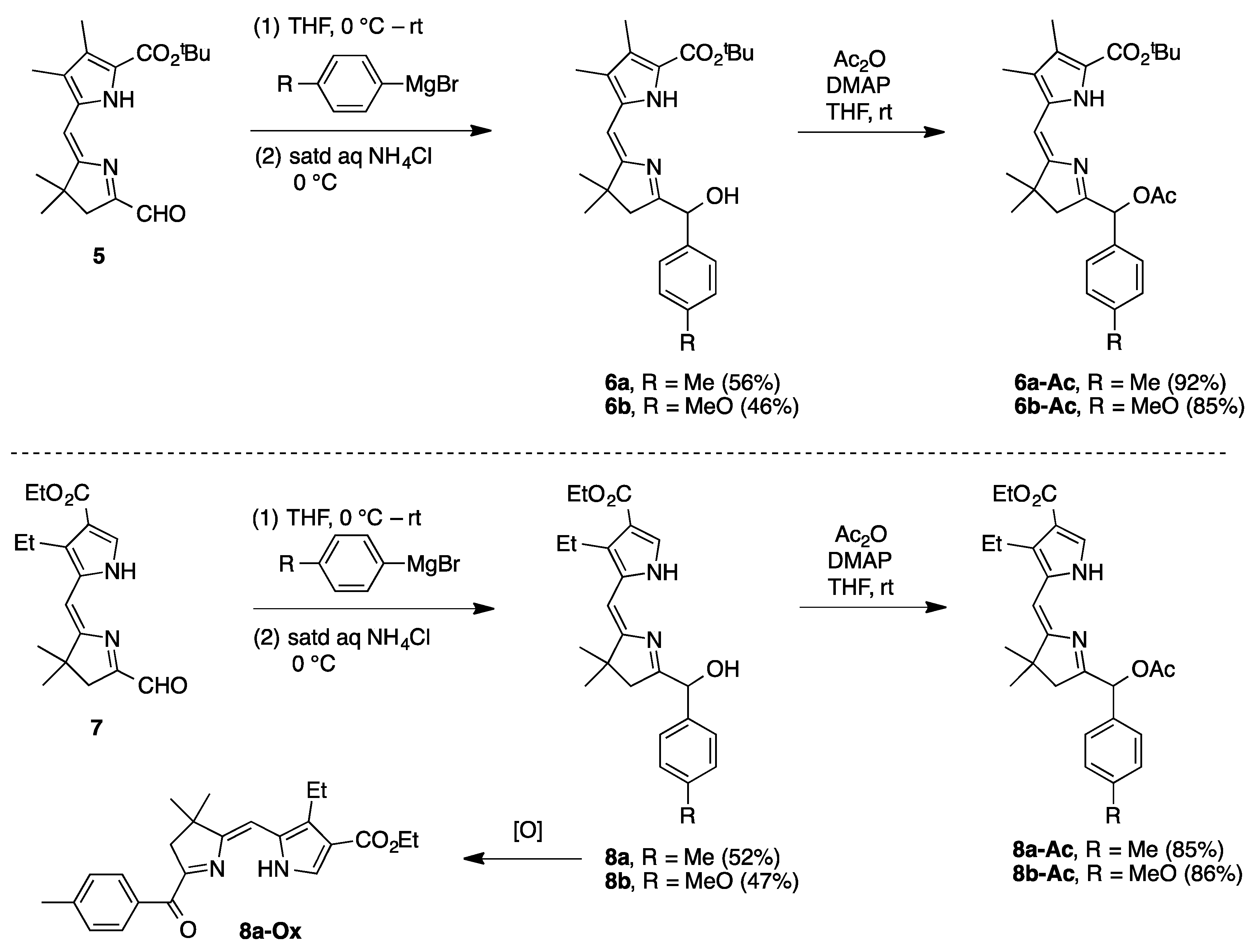
9-tert-Butoxycarbonyl-2,3-dihydro-1-[(hydroxy)(4-methylphenyl)methyl]-3,3,7,8-tetramethyldipyrrin (
6a). Following a literature procedure ([
55]; see
Supporting Information therein) with slight modification, a solution of
5 (97 mg, 0.29 mmol) in THF (1.9 mL) at 0 °C was treated with
p-tolylmagnesium bromide (1.0 mL, 1.0 mmol, 3.5 equiv, 1.0 M in THF) over the course of 2 min. After 5 min further, the ice bath was removed, and the reaction mixture was stirred for 2 h. The reaction mixture was then cooled to 0 °C, whereupon saturated aqueous NH
4Cl (4.0 mL) was added. The mixture was extracted with ethyl acetate. The organic extract was washed with water, dried (Na
2SO
4), and chromatographed (silica, ethyl acetate/hexanes (1:3)) to give a light brown solid (69 mg, 56%): m.p. 130–132 °C;
1H-NMR δ 1.11 (s, 3H), 1.20 (s, 3H), 1.59 (s, 9H), 2.05 (s, 3H), 2.29 (s, 3H), 2.36 (s, 3H), 2.30, 2.45 (AB,
2J =18.6 Hz, 2H), 3.86 (d,
J = 4.2 Hz, 1H), 5.43 (d,
J = 4.2 Hz, 1H), 5.81 (s, 1H), 7.18 (d,
J = 8.0 Hz, 2H), 7.27 (d,
J = 8.0 Hz, 2H), 10.65–10.72 (br, 1H);
13C-NMR δ 8.8, 10.2, 21.2, 28.5, 28.85, 28.94, 41.9, 48.8, 74.5, 80.0, 103.3, 119.2, 119.9, 126.7, 126.8, 129.4, 129.5, 136.5, 138.2, 160.7, 160.9, 181.2; ESI-MS obsd 423.2637, calcd. 423.2642 [(M + H)
+, M = C
26H
34N
2O
3]; λ
abs (toluene) 360 nm.
1-[(Acetoxy)(4-methylphenyl)methyl]-9-tert-butoxycarbonyl-2,3-dihydro-3,3,7,8-tetramethyldipyrrin (
6a-Ac). Following a procedure [
48] with slight modification, a solution of
6a (22 mg, 52 µmol) in THF (2.1 mL) at room temperature was treated with acetic anhydride (11 mg, 0.11 mmol) and DMAP (13 mg, 0.11 mmol). The reaction mixture was stirred for 2 h. Saturated aqueous NaHCO
3 (2 mL) was added, and the mixture was extracted with ethyl acetate. The organic extract was washed with water, dried (Na
2SO
4), and chromatographed (silica, ethyl acetate/hexanes (1:4)) to give a light brown solid (22 mg, 92%): m.p. 159–161 °C;
1H-NMR δ 1.14 (s, 3H), 1.16 (s, 3H), 1.58 (s, 9H), 2.02 (s, 3H), 2.250 (s, 3H), 2.252 (s, 3H), 2.35 (s, 3H), 2.41, 2.55 (AB,
2J =18.3 Hz, 2H), 5.77 (s, 1H), 6.41 (s, 1H), 7.18 (d,
J = 8.1 Hz, 2H), 7.33 (d,
J = 8.1 Hz, 2H), 10.90–10.98 (br, 1H);
13C-NMR δ 8.8, 10.4, 20.0, 21.2, 28.6, 28.8, 41.1, 49.3, 75.7, 79.7, 103.5, 118.9, 120.3, 125.7, 127.4, 129.5, 130.0, 132.6, 138.8, 161.1, 161.9, 170.3, 176.8; ESI-MS obsd 487.2567, calcd. 487.2567 [(M + Na)
+, M = C
28H
36N
2O
4]; λ
abs (toluene) 369 nm.
9-tert-Butoxycarbonyl-2,3-dihydro-1-[(hydroxy)(4-methoxyphenyl)methyl]-3,3,7,8-tetramethyldipyrrin (
6b). Following a literature procedure ([
55]; see
Supporting Information therein) with slight modification, a solution of
5 (80 mg, 0.24 mmol) in THF (1.6 mL) at 0 °C was treated with
p-anisylmagnesium bromide (0.85 mL, 0.85 mmol, 3.5 equiv, 1.0 M in THF) over the course of 5 min. After an additional 5 min, the ice bath was removed, and the reaction mixture was stirred for 2 h. The reaction mixture was then cooled to 0 °C, whereupon saturated aqueous NH
4Cl (4.0 mL) was added. The mixture was extracted with ethyl acetate. The organic extract was washed with water, dried (Na
2SO
4), and chromatographed (silica, ethyl acetate/hexanes (1:3)) to give a light brown solid (48 mg, 46%): m.p. 52–54 °C;
1H-NMR δ 1.11 (s, 3H), 1.20 (s, 3H), 1.58 (s, 9H), 2.04 (s, 3H), 2.29 (s, 3H), 2.30, 2.44 (AB,
2J = 18.6 Hz, 2H), 3.81 (s, 3H), 3.91 (br, 1H), 5.42 (s, 1H), 5.80 (s, 1H), 6.90 (d,
J = 8.0 Hz, 2H), 7.29 (d,
J = 8.0 Hz, 2H), 10.69 (br, 1H);
13C-NMR δ 8.89, 10.3, 28.6, 28.98, 29.0, 41.9, 48.9, 55.4, 74.3, 80.1, 103.3, 114.3, 119.3, 119.9, 126.8, 128.2, 129.6, 131.7, 159.8, 160.8, 161.1, 181.5; ESI-MS obsd 439.2599, calcd. 439.2591 [(M + H)
+, M = C
26H
34N
2O
4].
1-[(Acetoxy)(4-methoxyphenyl)methyl]-9-tert-butoxycarbonyl-2,3-dihydro-3,3,7,8-tetramethyldipyrrin (
6b-Ac). Following a procedure [
48] with slight modification, a solution of
6b (20 mg, 46 µmol) in THF (1.8 mL) at room temperature was treated with acetic anhydride (9.3 mg, 92 µmol) and DMAP (11 mg, 92 µmol). The reaction mixture was stirred for 2 h. Saturated aqueous NaHCO
3 (2 mL) was added, and the mixture was extracted with ethyl acetate. The organic extract was washed with water, dried (Na
2SO
4), and chromatographed (silica, ethyl acetate/hexanes (1:4)) to give a light brown oil (19 mg, 85%):
1H-NMR δ 1.14 (s, 3H), 1.17 (s, 3H), 1.57 (s, 9H), 2.02 (s, 3H), 2.25 (s, 6H), 2.41, 2.55 (AB,
2J = 18.0 Hz, 2H), 3.81 (s, 3H), 5.77 (s, 1H), 6.38 (s, 1H), 6.90 (dd,
J = 7.2, 2.1 Hz, 2H), 7.37 (dd,
J = 7.2, 2.1 Hz, 2H), 10.95 (br, 1H);
13C-NMR δ 8.9, 10.5, 21.1, 28.7, 28.88, 28.92, 41.2, 49.5, 55.4, 75.3, 79.8, 103.5, 114.3, 119.0, 120.4, 125.7, 127.7, 129.1, 130.1, 160.1, 161.2, 162.0, 170.4, 176.9; ESI-MS obsd 503.2517, calcd. 503.2516 [(M + Na)
+, M = C
28H
36N
2O
5].
8-Carbethoxy-7-ethyl-2,3-dihydro-1-[(hydroxy)(4-methylphenyl)methyl]-3,3-dimethyldipyrrin (
8a). Following a literature procedure ([
55]; see
Supporting Information therein) with slight modification, a solution of
7 (128 mg, 0.424 mmol) in THF (4.24 mL) was cooled to 0 °C and treated with
p-tolylmagnesium bromide (1.48 mL, 1.48 mmol, 3.5 equiv, 1.0 M in THF) over the course of 5 min. After 10 min further, the ice bath was removed, and the reaction mixture was stirred for 2 h. The reaction mixture was cooled to 0 °C, whereupon saturated aqueous NH
4Cl (15 mL) was added. The reaction mixture was extracted with ethyl acetate. The organic extract was washed with water, dried (Na
2SO
4), and chromatographed (silica, hexanes/ethyl acetate (3:1)) to give a pale yellow oil (75 mg, 45%):
1H-NMR δ 1.13 (s, 3H), 1.17 (t,
J = 7.2 Hz, 3H), 1.19 (s, 3H), 1.34 (t,
J = 7.2 Hz, 3H), 2.35 (s, 3H), 2.34, 2.45 (AB,
J = 18.6 Hz, 2H), 2.82 (q,
J = 7.2 Hz, 2H), 3.69–3.71 (br, 1H), 4.27 (q,
J = 7.2 Hz, 2H), 5.44 (s, 1H), 5.82 (s, 1H), 7.19 (d,
J = 8.4 Hz, 2H), 7.26 (d,
J = 8.4 Hz, 2H), 7.42 (d,
J = 3.0 Hz, 1H), 10. 72 (brs, 1H);
13C-NMR δ 14.5, 16.4, 18.0, 21.3, 29.0, 29.1, 41.6, 48.8, 59.3, 74.6, 103.4, 114.2, 125.1, 126.2, 126.8, 127.9, 129.6, 136.6, 138.4, 159.2, 165.5, 180.1; ESI-MS obsd 395.2328, calcd. 395.2329 [(M + H)
+, M = C
24H
30N
2O
3]; λ
abs (CH
2Cl
2) 340 nm.
8-Carbethoxy-7-ethyl-2,3-dihydro-1-(4-methylbenzoyl)-3,3-dimethyldipyrrin (8a-Ox). A solution of 8a in CDCl3 in an NMR tube changed from yellow to orange on standing for 7 days; chromatography (silica, hexanes/ethyl acetate (4:1)) gave a yellow oil: 1H-NMR δ 1.20 (t, J = 6.9 Hz, 3H), 1.31 (s, 6H), 1.35 (t, J = 7.2 Hz, 3H), 2.47 (s, 3H), 2.86 (q, J = 6.9 Hz, 2H), 2.98 (s, 2H), 4.27 (q, J = 7.2 Hz, 2H), 6.14 (s, 1H), 7.31 (d, J = 8.1 Hz, 2H), 7.38 (d, J = 3.6 Hz, 1H), 8.03 (d, J = 8.1 Hz, 2H), 10.65 (br, 1H); ESI-MS obsd 393.2168, calcd. 393.2173 [(M + H)+, M = C24H28N2O3]; λabs (CH2Cl2) 451 nm.
1-[(Acetoxy)(4-methylphenyl)methyl]-8-carbethoxy-7-ethyl-2,3-dihydro-3,3-dimethyldipyrrin (
8a-Ac). Following a procedure [
48] with slight modification, a solution of
8a (70 mg, 0.18 mmol) in THF (7.2 mL) at room temperature was treated with acetic anhydride (38 mg, 0.36 mmol) and DMAP (44 mg, 0.361 mmol). The reaction mixture was stirred for 2 h. The reaction mixture was treated with saturated aqueous NaHCO
3 (15 mL) and extracted with ethyl acetate. The organic extract was washed with water, dried (Na
2SO
4), and chromatographed [silica, hexanes/ethyl acetate (4:1)] to give a light brown oil (67 mg, 85%):
1H-NMR δ 1.16 (t,
J = 7.2 Hz, 3H), 1.18 (s, 6H), 1.34 (t,
J = 7.2 Hz, 3H), 2.19 (s, 3H), 2.37 (s, 3H), 2.45, 2.57 (AB,
2J =18.6 Hz, 2H), 2.80 (q,
J = 7.2 Hz, 2H), 4.27 (q,
J = 7.2 Hz, 2H), 5.82 (s, 1H), 6.49 (s, 1H), 7.20 (d,
J = 8.4 Hz, 2H), 7.29 (d,
J = 8.4 Hz, 2H), 7.37 (d,
J = 3.0 Hz, 1H), 10.95 (brs, 1H);
13C-NMR δ 14.5, 16.4, 18.0, 21.0, 21.3, 28.9, 29.0, 41.0, 49.3, 59.2, 75.5, 103.8, 114.1, 124.9, 126.1, 127.3, 128.3, 129.6, 133.0, 138.9, 159.8, 165.4, 169.8, 175.5; ESI-MS obsd 437.2423, calcd. 437.2435 [(M + H)
+, M = C
26H
32N
2O
4]; λ
abs (CH
2Cl
2) 343 nm.
8-Carbethoxy-7-ethyl-2,3-dihydro-1-[(hydroxy)(4-methoxyphenyl)methyl]-3,3-dimethyldipyrrin (
8b). Following a literature procedure ([
55]; see
Supporting Information therein) with slight modification, a solution of
7 (52 mg, 0.17 mmol) in THF (1.1 mL) at 0 °C was treated with
p-anisylmagnesium bromide (0.60 mL, 0.60 mmol, 3.5 equiv, 1.0 M in THF) over the course of 5 min. After 10 min further, the ice bath was removed, and the reaction mixture was stirred for 2 h. The reaction mixture was then cooled to 0 °C, whereupon saturated aqueous NH
4Cl (4.0 mL) was added. The reaction mixture was extracted with ethyl acetate. The organic extract was washed with water, dried (Na
2SO
4), and chromatographed (silica, hexanes/ethyl acetate (3:2)) to give a red oil (33 mg, 47%):
1H-NMR δ 1.13 (s, 3H), 1.17 (t,
J = 7.2 Hz, 3H), 1.20 (s, 3H), 1.35 (t,
J = 7.2 Hz, 3H), 2.34, 2.46 (AB,
J = 18.6 Hz, 2H), 2.82 (q,
J = 7.2 Hz, 2H), 3.71 (br, 1H), 3.81 (s, 3H), 4.28 (q,
J = 7.2 Hz, 2H), 5.43 (s, 1H), 5.82 (s, 1H), 6.90 (d,
J = 8.7 Hz, 2H), 7.29 (d,
J = 8.7 Hz, 2H), 7.43 (d,
J = 3.0 Hz, 1H), 10.71 (br, 1H); ESI-MS obsd 411.2271, calcd. 411.2278 [(M + H)
+, M = C
24H
30N
2O
4].
1-[(Acetoxy)(4-methoxyphenyl)methyl]-8-carbethoxy-7-ethyl-2,3-dihydro-3,3-dimethyldipyrrin (
8b-Ac). Following a procedure [
48] with slight modification, a solution of
8b (20 mg, 49 µmol) in THF (1.9 mL) at room temperature was treated with acetic anhydride (9.5 µL, 98 µmol) and DMAP (12 mg, 98 µmol). The reaction mixture was stirred for 2 h. The reaction mixture was treated with saturated aqueous NaHCO
3 (10 mL) and extracted with ethyl acetate. The organic extract was washed with water, dried (Na
2SO
4), and chromatographed (silica, hexanes/ethyl acetate (4:1)) to give a light brown oil (19 mg, 86%):
1H-NMR δ 1.13–1.18 (m, 9H), 1.34 (t,
J = 6.9 Hz, 3H), 2.19 (s, 3H), 2.45, 2.57 (AB,
2J = 18.6 Hz, 2H), 2.80 (q,
J = 7.2 Hz, 2H), 3.82 (s, 3H), 4.27 (q,
J = 6.9 Hz, 2H), 5.82 (s, 1H), 6.47 (s, 1H), 6.92 (d,
J = 8.2 Hz, 2H), 7.33 (d,
J = 8.2 Hz, 2H), 7.38 (d,
J = 3.0 Hz, 1H), 10.95 (br, 1H);
13C-NMR δ 14.5, 16.4, 18.0, 21.1, 28.9, 29.0, 41.1, 49.4, 55.4, 59.3, 75.2, 103.8, 114.1, 114.3, 124.9, 126.1, 127.9, 128.3, 128.9, 159.8, 160.1, 165.5, 170.0, 175.6; ESI-MS obsd 475.2203, calcd. 475.2203 [(M + Na)
+, M = C
26H
32N
2O
5]; λ
abs (CH
2Cl
2) 343 nm.
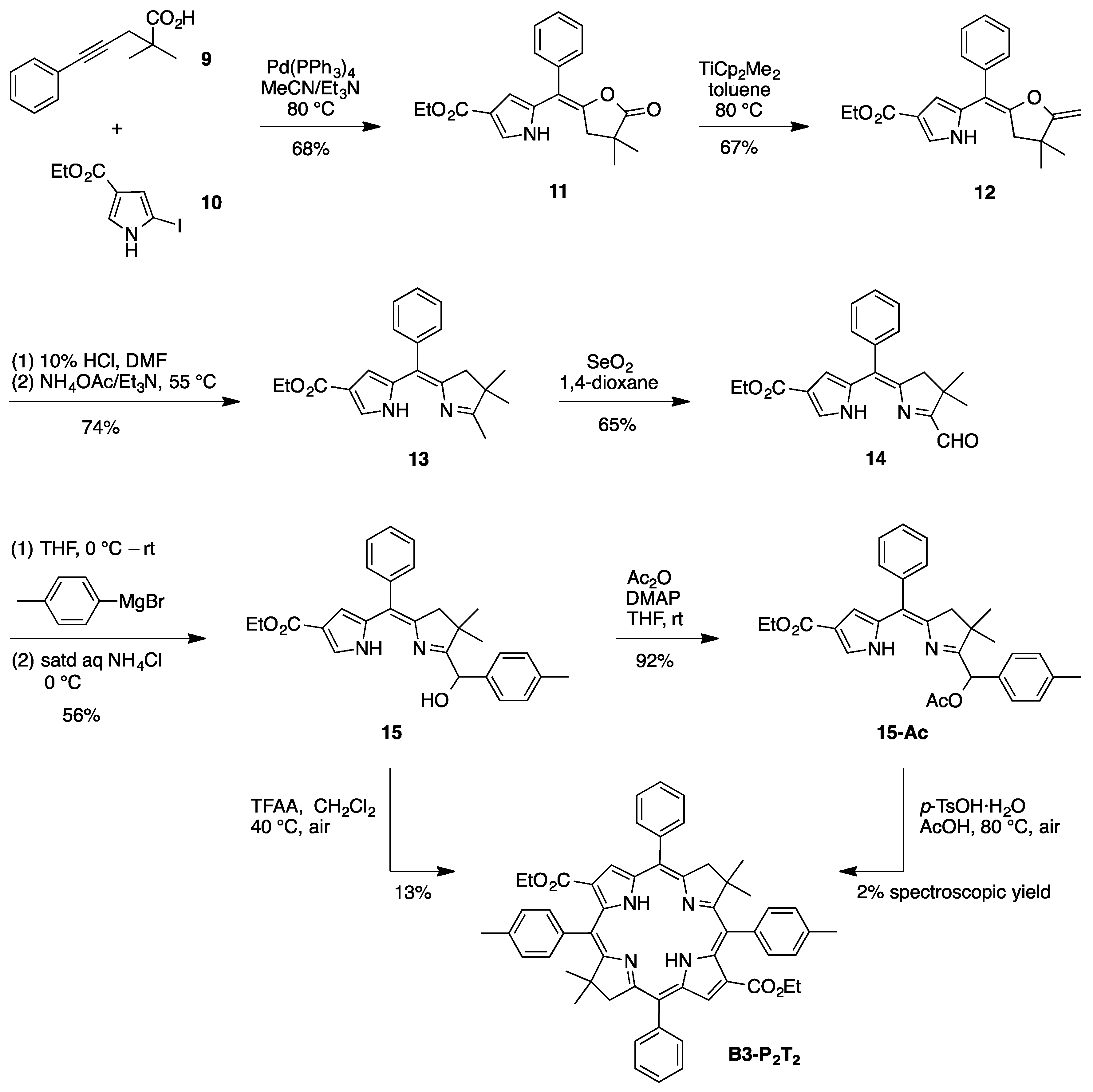
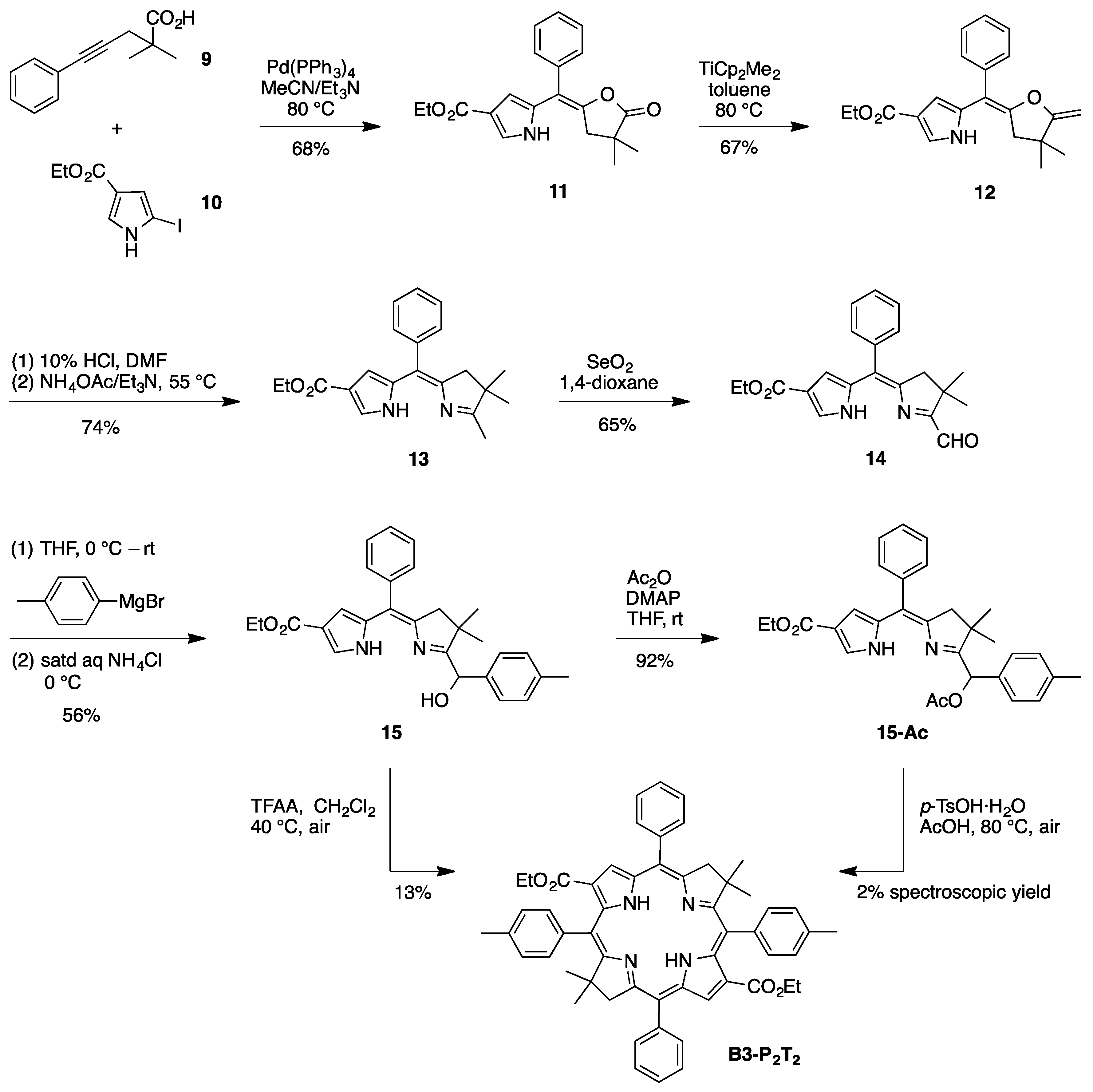
(E)-Ethyl 5-[(4,4-dimethyl-5-oxodihydrofuran-2(3H)-ylidene)(phenyl)methyl]-1H-pyrrole-3-carboxylate (
11). Following a literature procedure [
19], in a Schlenk flask, a solution of
9 (2.5 g, 10 mmol) in dry acetonitrile (16.5 mL) was treated with
10 (2.1 g, 8.2 mmol), BnNEt
3Cl (2.3 g, 8.2 mmol), and Et
3N (10 mL). The mixture was deaerated by three freeze–pump–thaw cycles. A sample of Pd(PPh
3)
4 (0.47 g, 0.41 mmol) was then added, and the resulting mixture was further deaerated. The reaction mixture was heated at 80 °C for 16 h, and upon allowing to cool to room temperature, CH
2Cl
2 and water were added. The organic layer was dried (Na
2SO
4), concentrated, and chromatographed (silica, hexanes/ethyl acetate (7:3)) to afford a light yellow solid (1.9 g, 68%): m.p. 115–116 °C;
1H-NMR δ 1.35 (t,
J = 6.9 Hz, 3H), 1.37 (s, 6H), 3.06 (s, 2H), 4.29 (q,
J = 6.9 Hz, 2H), 6.56–6.57 (m, 1H), 7.27–7.37 (m, 6H), 8.17 (brs, 1H);
13C-NMR δ 14.5, 25.1, 39.9, 41.8, 59.9, 109.1, 111.0, 117.3, 123.3, 127.9, 128.6, 129.5, 129.7, 135.1, 145.1, 164.9, 179.6; ESI-MS obsd 340.1548, calcd. 340.1543 [(M + H)
+, M = C
20H
21NO
4].
(E)-Ethyl 5-[(4,4-dimethyl-5-methylenedihydrofuran-2(3H)-ylidene)(phenyl)methyl]-1H-pyrrole-3-carboxy-late (
12). Following a general procedure [
19], a solution of TiCp
2Cl
2 (6.2 g, 25 mmol) in toluene (100 mL) was treated dropwise with a solution of MeLi (1.6 M, 32 mL in Et
2O, 50 mmol) at 0 °C under argon. The resulting mixture was stirred at 0 °C for 1 h, whereupon saturated aqueous NH
4Cl solution was added. The organic layer was washed (water and brine), dried (Na
2SO
4) and filtered. The filtrate was treated with lactone
11 (1.7 g, 5.0 mmol) and TiCp
2Cl
2 (50 mg) in a Schlenk flask under argon. The resulting solution was heated at 80 °C for 16 h. The resulting mixture was allowed to cool to room temperature whereupon MeOH (1.8 mL), NaHCO
3 (50 mg) and water (1.0 mL) were added. The resulting mixture was kept at 40 °C for 2 h with stirring and then filtered through Celite. The filtrate was concentrated and chromatographed (silica, hexanes/ethyl acetate (4:1)) to afford a gummy oil (1.1 g, 67%):
1H-NMR δ 1.27 (s, 6H), 1.34 (t,
J = 7.2 Hz, 3H), 2.76 (s, 2H), 4.02 (d,
J = 2.4 Hz, 1H), 4.24 (q,
J = 7.2 Hz, 2H), 4.37 (d,
J = 2.4 Hz, 1H), 6.46–6.48 (m, 1H), 7.21–7.36 (m, 6H), 8.53 (brs, 1H);
13C-NMR δ 14.5, 27.5, 39.8, 43.9, 59.8, 81.3, 105.7, 108.7, 116.5, 123.0, 126.8, 128.3, 129.6, 131.5, 136.8, 152.0, 165.3, 169.8; ESI-MS obsd 338.1756, calcd. 338.1751 [(M + H)
+, M = C
21H
23NO
3].
8-Carbethoxy-2,3-dihydro-1,2,2-trimethyl-5-phenyldipyrrin (
13). Following a general procedure [
19], a solution of
12 (544 mg, 1.60 mmol) in DMF (26.0 mL) was treated with 10% aqueous HCl (2.0 mL). After 30 min, NH
4OAc (2.46 g, 32 mmol) and Et
3N (4.5 mL, 32 mmol) were added, and the resulting mixture was stirred at 55 °C for 16 h. Then, the reaction mixture was treated with saturated aqueous KH
2PO
4 solution. Ethyl acetate (100 mL) was added, and the organic layer was washed (water), dried (Na
2SO
4), concentrated and chromatographed (silica, hexanes/ethyl acetate (4:1)) to afford a yellow solid (401 mg, 74%): m.p. 165–167 °C;
1H-NMR δ 1.13 (s, 6H), 1.28 (t,
J = 6.9 Hz, 3H), 2.17 (s, 3H), 2.36 (s, 2H), 4.22 (q,
J = 6.9 Hz, 2H), 6.01 (d,
J = 2.4 Hz, 1H), 7.28–7.49 (m, 6H), 11.83 (brs, 1H);
13C-NMR δ 14.5, 15.7, 25.7, 43.9, 48.0, 59.5, 109.4, 116.0, 120.3, 124.2, 127.2, 128.4, 129.7, 134.7, 139.0, 147.9, 165.4, 186.9; ESI-MS obsd 337.1912, calcd. 337.1911 [(M + H)
+, M = C
21H
24N
2O
2]; λ
abs (CH
2Cl
2) 330 nm.
8-Carbethoxy-1-formyl-2,3-dihydro-2,2-dimethyl-5-phenyldipyrrin (
14). Following a general procedure [
19], a solution of
13 (0.35 g, 1.0 mmol) in 1,4-dioxane (20 mL) was treated with SeO
2 (0.33 g, 3.0 mmol) and stirred at room temperature for 60 min. Ethyl acetate and saturated NaHCO
3 were then added, and the organic layer was washed (brine), dried, concentrated and chromatographed (silica, hexanes/ethyl acetate (4:1)) to afford a yellow solid (234 mg, 65%): m.p. 157–159 °C;
1H-NMR δ 1.29 (t,
J = 7.2 Hz, 3H), 1.31 (s, 6H), 2.49 (s, 2H), 4.23 (q,
J = 7.2 Hz, 2H), 6.23 (s, 1H), 7.29–7.60 (m, 6H), 10.0 (s, 1H), 11.42 (brs, 1H);
13C-NMR δ 14.5, 25.7, 45.8, 46.1, 59.9, 113.9, 117.0, 126.7, 128.1, 128.7, 129.1, 130.3, 133.8, 137.9, 148.4, 164.8, 177.0, 190.0; ESI-MS obsd 351.1701 calcd. 351.1703 [(M + H)
+, M = C
21H
22N
2O
3]; λ
abs (CH
2Cl
2) 438 nm.
8-Carbethoxy-1-[(hydroxy)(4-methylphenyl)methyl]-2,3-dihydro-2,2-dimethyl-5-phenyldipyrrin (
15). Following a literature procedure ([
55]; see
Supporting Information therein) with slight modification, a solution of
14 (97 mg, 0.29 mmol) in THF (1.9 mL) at 0 °C was treated with
p-tolylmagnesium bromide (1.0 mL, 1.0 mmol, 3.5 equiv, 1.0 M in THF) over the course of 5 min. After 5 min further, the ice bath was removed, and the mixture was stirred for 2 h. The reaction mixture was then cooled to 0 °C whereupon saturated aqueous NH
4Cl (10.0 mL) was added. The mixture was then extracted with ethyl acetate. The organic extract was washed with water, dried (Na
2SO
4), and chromatographed (silica, hexanes/ethyl acetate (3:1)) to give a light yellow solid (72 mg, 56%): m.p. 146–148 °C;
1H-NMR δ 0.75 (s, 3H), 1.24 (s, 3H), 1.29 (t,
J = 6.9 Hz, 3H), 2.37 (s, 3H), 2.34, 2.46 (AB,
2J = 17.1 Hz, 2H), 3.49 (d,
J = 4.8 Hz, 1H), 4.22 (q,
J = 6.9 Hz, 2H), 5.52 (d,
J = 4.8 Hz, 1H), 6.06 (s, 1H), 7.19–7.46 (m, 10H), 11.25 (brs, 1H);
13C-NMR δ 14.6, 21.3, 26.2, 26.5, 46.1, 47.6, 59.7, 72.0, 110.6, 116.2, 122.5, 124.8, 127.5, 127.6, 128.5, 129.6, 134.1, 137.2, 138.6, 138.7, 146.1, 165.3, 188.5; ESI-MS obsd 443.2341, calcd. 443.2329 [(M + H)
+, M = C
28H
30N
2O
3]; λ
abs (CH
2Cl
2) 343 nm.
1-[(Acetoxy)(4-methylphenyl)methyl]-8-carbethoxy-2,3-dihydro-2,2-dimethyl-5-phenyldipyrrin (
15-Ac). Following a procedure [
48] with slight modification, a solution of
15 (22 mg, 52 µmol) in THF (2.1 mL) at room temperature was treated with acetic anhydride (11 mg, 0.11 mmol) and DMAP (13 mg, 0.11 mmol). The reaction mixture was stirred for 2 h, and then treated with saturated aqueous NaHCO
3 (5 mL). The reaction mixture was extracted with ethyl acetate. The organic extract was washed with water, dried (Na
2SO
4), and chromatographed (silica, hexanes/ethyl acetate (4:1)) to give a light yellow solid (22 mg, 92%): m.p. 65–67 °C;
1H-NMR δ 0.91 (s, 3H), 1.28 (s, 3H), 1.29 (t,
J = 6.9 Hz, 3H), 2.18 (s, 3H), 2.39 (s, 3H), 2.35, 2.45 (AB,
2J = 16.8 Hz, 2H), 4.22 (q,
J = 6.9 Hz, 2H), 6.06 (d,
J = 1.8 Hz, 1H), 6.53 (s, 1H), 7.23–7.43 (m, 10H), 11.58 (brs, 1H);
13C-NMR δ 14.5, 21.1, 21.3, 26.2, 26.3, 45.3, 48.3, 59.6, 72.6, 110.4, 116.1, 123.0, 124.6, 127.5, 128.3, 128.5, 129.5, 133.4, 134.4, 138.4, 139.2, 146.8, 165.2, 169.9, 183.8; ESI-MS obsd 485.2445, calcd. 485.2435 [(M + H)
+, M = C
30H
32N
2O
4]; λ
abs (CH
2Cl
2) 343 nm.
2-Formyl-3-(4-methylphenyl)-N-p-tosylpyrrole (
17). Following a reported procedure [
42], a mixture of NaH (1.17 g, 60 wt % in oil, 29 mmol) in THF (90 mL) was treated portionwise with pyrrole
16 (4.20 g, 22.7 mmol) at 0 °C under argon. The reaction mixture was stirred for 20 min at 0 °C, whereupon
p-tosyl chloride (6.50 g, 34.1 mmol) was added. The resulting heterogeneous mixture was stirred for 3 h at room temperature. Water and CH
2Cl
2 were added. The organic phase was washed (brine), dried (Na
2SO
4) and concentrated to a pale brown solid. Column chromatography (silica, hexanes/CH
2Cl
2 (1:3)) afforded a pale grey solid (6.34 g, 82%): m.p. 178–180 °C;
1H-NMR (400 MHz) δ 2.38 (s, 3H), 2.43 (s, 3H), 6.47 (d,
J = 3.3 Hz, 1H), 7.21 (d,
J = 7.9 Hz, 2H), 7.30 (d,
J = 7.9 Hz, 2H), 7.33 (d,
J = 8.2 Hz, 2H), 7.82–7.83 (m, 1H), 7.94 (d,
J = 8.2 Hz, 2H), 9.62 (s, 1H);
13C-NMR (100 MHz) δ 21.5, 21.9, 113.1, 128.1, 128.6, 129.5, 129.6, 129.7, 129.8, 129.9, 135.4, 139.0, 144.0, 145.6, 178.5; ESI-MS obsd 340.1007, calcd. 340.1002 [(M + H)
+, M = C
19H
17NO
3S].
3-(4-Methylphenyl)-2-(2-nitroethyl)-N-p-tosylpyrrole (
18). Following a reported procedure [
43], a mixture of
17 (6.30 g, 18.6 mmol), potassium acetate (2.00 g, 20.4 mmol), methylamine hydrochloride (1.51 g, 22.3 mmol), nitromethane (2.50 mL, 46.6 mmol) and acetic acid (106 µL, 1.86 mmol) in ethanol (6.5 mL) was sonicated (benchtop sonication bath) for 10 min, and then stirred at room temperature for 42 h. Water was added. The resulting yellow precipitate was washed (water, cold ethanol), and then dried under high vacuum for 4 h. The crude product was dissolved in CHCl
3/2-propanol (133 mL, 3:1), treated with silica (22.3 g), and vigorously stirred upon addition of NaBH
4 (1.41 g, 37.1 mmol). The mixture was vigorously stirred for 2 h at room temperature. The progress of the reaction was monitored by
1H-NMR spectroscopy. The reaction mixture was filtered. The filtrate was washed (water, brine), dried (Na
2SO
4), and concentrated to a brown oil. The oil was chromatographed (silica, hexanes/ethyl acetate (3:1)). The product was recrystallized from hot EtOH and cooled overnight at 1 °C to afford a white solid (3.45 g, 48%): m.p. 115–117 °C;
1H-NMR δ 2.37 (s, 3H), 2.44 (s, 3H), 3.46 (t,
J = 8.0 Hz, 2H), 4.55 (t,
J = 8.0 Hz, 2H), 6.35–6.37 (m, 1H), 7.11 (d,
J = 7.9 Hz, 2H), 7.18 (d,
J = 7.9 Hz, 2H), 7.33 (d,
J = 8.1 Hz, 2H), 7.37–7.39 (m, 1H), 7.69 (d,
J = 8.1 Hz, 2H);
13C-NMR (100 MHz) δ 21.4, 21.9, 24.0, 74.3, 113.9, 123.4, 126.9, 128.4, 129.7, 130.5, 130.9, 131.2, 136.1, 137.6, 145.7; Anal. Calcd. for C
20H
20N
2O
4S: C, 62.48; H, 5.24; N, 7.29; S, 8.34. Found: C, 62.40; H, 5.10; N, 7.21; S, 8.24.
1,1-Dimethoxy-4,4-dimethyl-6-[3-(4-methylphenyl)-N-p-tosylpyrrol-2-yl]-5-nitro-2-hexanone (20). A mixture of 18 (3.42 g, 8.90 mmol) and 19 (3.19 g, 20.2 mmol) was treated with DBU (5.20 mL, 26.7 mmol) at room temperature. The reaction mixture was stirred for 24 h at room temperature. Saturated aqueous NH4Cl was added, and the mixture was extracted with ethyl acetate. The organic phase was washed (water, brine), dried (Na2SO4), and concentrated to a brown oil. A first chromatography (silica, hexanes/ethyl acetate (1:3)) and a second chromatography (silica, CH2Cl2) afforded a slightly yellow oil (1.02 g, 21%): 1H-NMR δ 1.05 (s, 3H), 1.12 (s, 3H), 2.37 (s, 3H), 2.43 (s, 3H), 2.49, 2.59 (AB, J = 18.4 Hz, 2H), 3.28 (ABX, J = 2.9 Hz, J = 15.3 Hz, 1H), 3.34 (s, 3H), 3.38 (s, 3H), 3.62 (ABX, J = 10.9 Hz, J = 15.3 Hz, 1H), 4.31 (s, 1H), 5.10 (ABX, J = 2.9 Hz, J = 10.9 Hz, 1H), 6.26 (d, J = 3.3 Hz, 1H), 7.07 (d, J = 7.8 Hz, 2H), 7.16 (d, J = 7.8 Hz, 2H), 7.30–7.34 (m, 3H), 7.65 (d, J = 8.5 Hz, 2H); 13C-NMR (100 MHz) δ 21.4, 21.9, 23.6, 23.9, 25.3, 29.9, 36.6, 44.3, 55.0, 94.3, 104.5, 114.9, 124.2, 124.6, 126.6, 128.9, 129.3, 130.3, 131.5, 132.2, 136.3, 137.2, 145.4, 203.2; ESI-MS obsd 565.1994, calcd. 565.1979 [(M + Na)+, M = C28H34N2O7S].
2,3,4,5-Tetrahydro-1-(dimethoxymethyl)-3,3-dimethyl-7-(4-methylphenyl)-N11-p-tosyldipyrrin N10-oxide (
21). Following a general procedure [
42], a solution of
20 (1.02 g, 1.88 mmol) in AcOH/EtOH (19 mL, 1:1) was treated with zinc dust (3.09 g, 47.6 mmol) at 0 °C. The reaction mixture was vigorously stirred for 1 h at 0 °C, then diluted with ethyl acetate and filtered through Celite. The filtrate was concentrated. The resulting oil was purified by column chromatography (silica, hexanes/ethyl acetate (2:3)) to afford a white solid (400 mg, 42%): m.p. 160–163 °C;
1H-NMR (400 MHz) δ 0.57 (s, 3H), 0.78 (s, 3H), 2.32 (s, 2H), 2.34 (s, 3H), 2.41 (s, 3H), 3.23 (ABX,
J = 11.0 Hz,
J = 15.3 Hz, 1H), 3.40 (s, 3H), 3.43 (s, 3H), 3.70 (ABX,
J = 4.0 Hz,
J = 15.3 Hz, 1H), 4.33–4.37 (m, 1H), 5.43 (s, 1H), 6.33 (d,
J = 3.3 Hz, 1H), 7.11–7.15 (m, 4H), 7.27 (d,
J = 8.4 Hz, 2H), 7.38 (d,
J = 3.3 Hz, 1H), 7.75 (d,
J = 8.4 Hz, 2H);
13C-NMR (100 MHz) δ 21.4, 21.9, 22.6, 23.0, 26.7, 38.1, 41.9, 55.3, 55.8, 79.9, 97.9, 114.6, 124.1, 125.9, 127.1, 128.9, 129.6, 130.3, 131.0, 132.2, 137.2, 145.3. Anal. Calcd. for C
28H
34N
2O
5S: C, 65.86; H, 6.71; N, 5.49. Found: C, 65.94; H, 6.70; N, 5.38.
2,3,4,5-Tetrahydro-1-(dimethoxymethyl)-3,3-dimethyl-7-(4-methylphenyl)-N11-p-tosyldipyrrin (
22). Following a general procedure [
42], TiCl
4 (5.35 mL, 5.35 mmol, 1.0 M in CH
2Cl
2) was slowly added with stirring to THF (15.3 mL) under argon at 0 °C. The resulting yellow solution was slowly treated with LiAlH
4 (3.82 mL, 3.82 mmol, 1.0 M in THF). The resulting black mixture was stirred at room temperature for 15 min. TEA (4.78 mL, 34.4 mmol) was added. The resulting black mixture was stirred for 2 min at room temperature. The black mixture was slowly poured into a solution of
21 (378 mg, 0.740 mmol) in THF (12.7 mL) at 0 °C. The mixture was stirred for 1 h at room temperature. Water and ethyl acetate were added. The organic extract was washed (brine), dried (Na
2SO
4), concentrated and chromatographed (silica, CH
2Cl
2/ethyl acetate (5:1)) to afford a white solid (300 mg, 82%): m.p. 105–107 °C;
1H-NMR (400 MHz) δ 0.79 (s, 3H), 1.05 (s, 3H), 2.32 (s, 3H), 2.35 (s, 2H), 2.40 (s, 3H), 2.94–2.98 (m, 2H), 3.30 (s, 3H), 3.35 (s, 3H), 4.07–4.12 (m, 1H), 4.65 (s, 1H), 6.36 (d,
J = 3.3 Hz, 1H), 7.09 (d,
J = 8.1 Hz, 2H), 7.27 (d,
J = 8.1 Hz, 2H), 7.32–7.36 (m, 3H), 7.64 (d,
J = 8.1 Hz, 2H);
13C-NMR (100 MHz) δ 21.6, 22.1, 23.3, 27.1, 27.4, 42.0, 49.1, 54.9, 55.2, 79.7, 94.8, 103.7, 115.0, 123.6, 127.0, 129.3, 129.4, 130.5, 133.1, 136.7, 137.2, 145.2, 173.6; ESI-MS obsd 495.2307, calcd. 495.2313 [(M + H)
+, M = C
28H
34N
2O
4S].
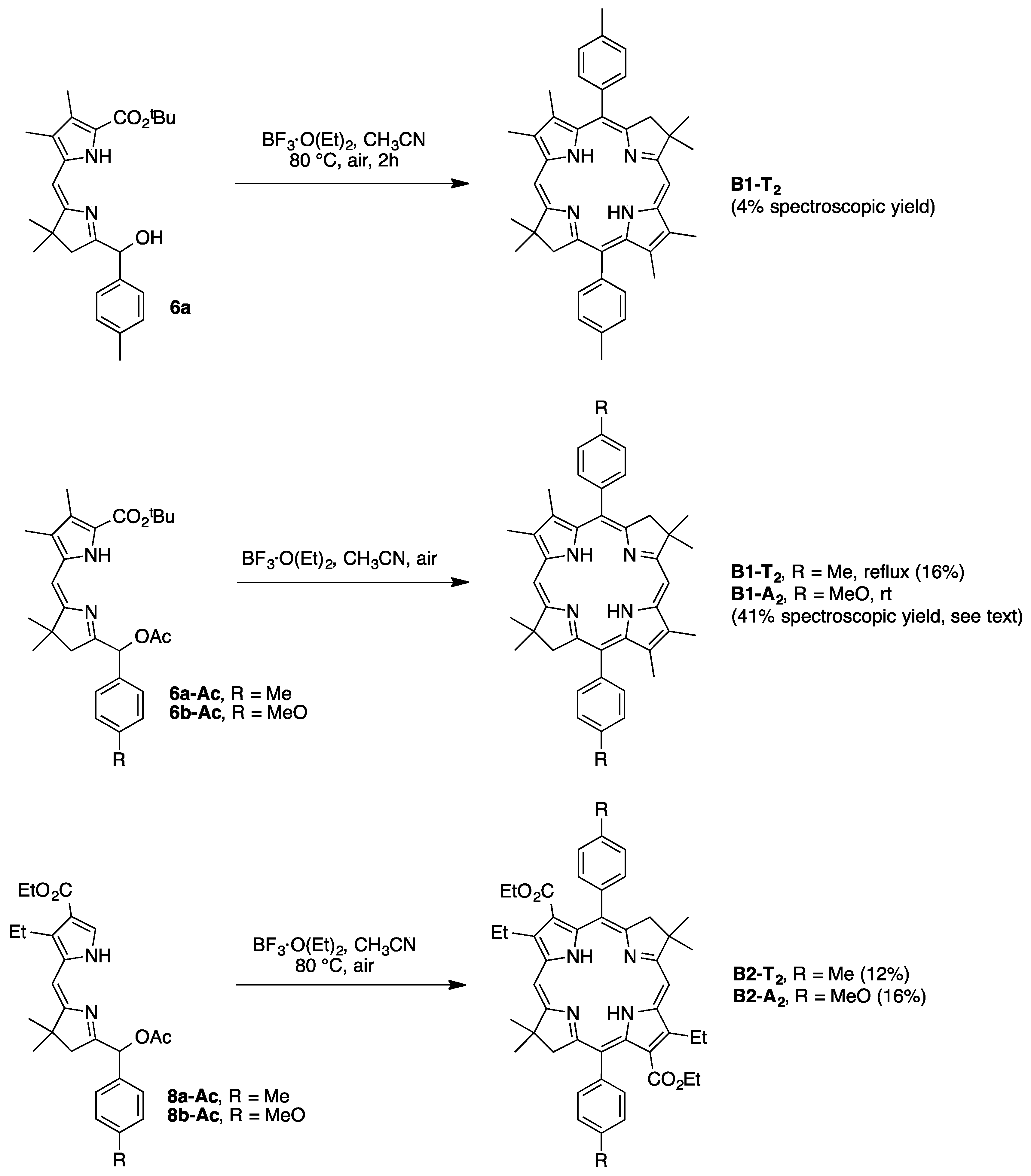
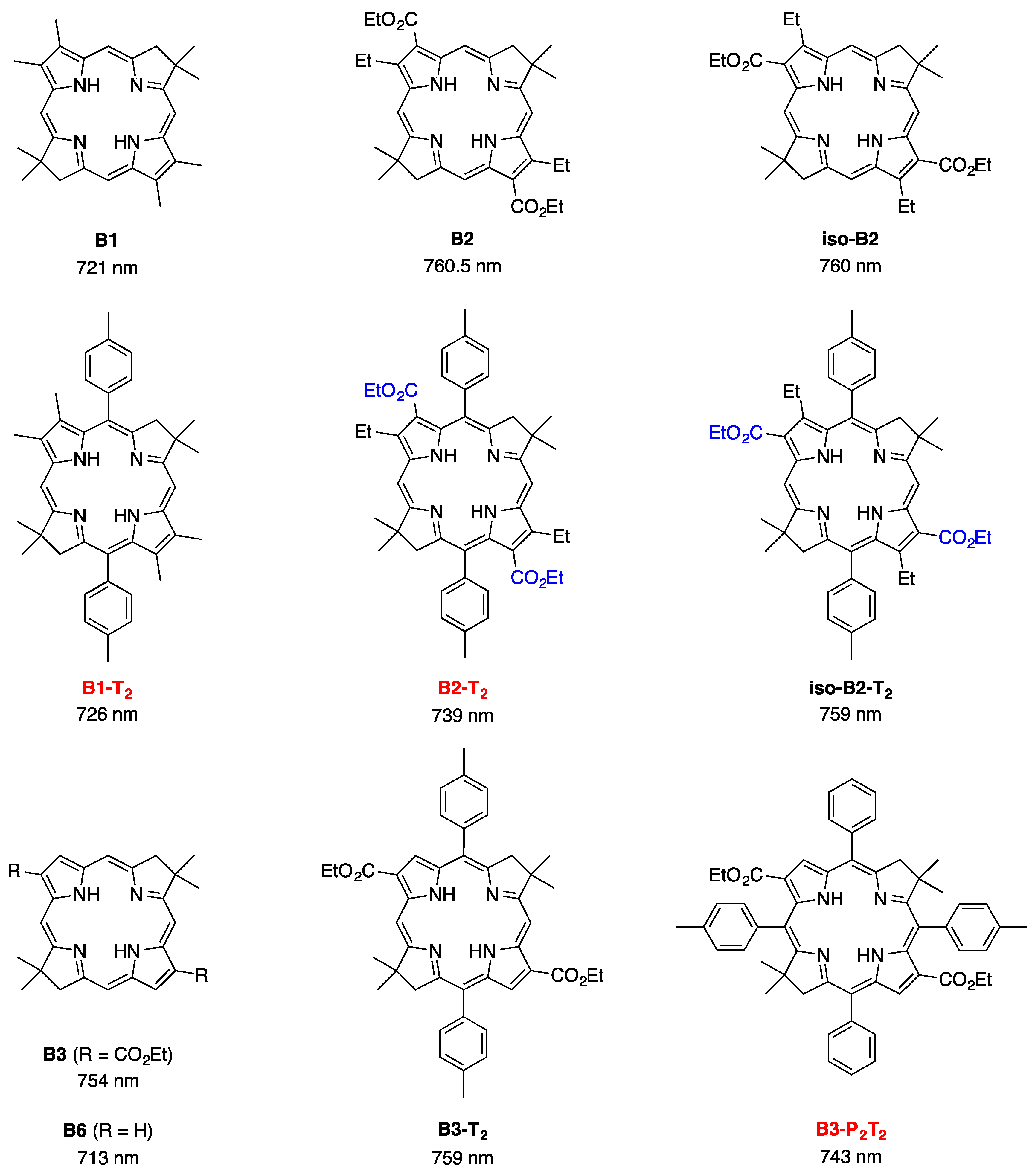
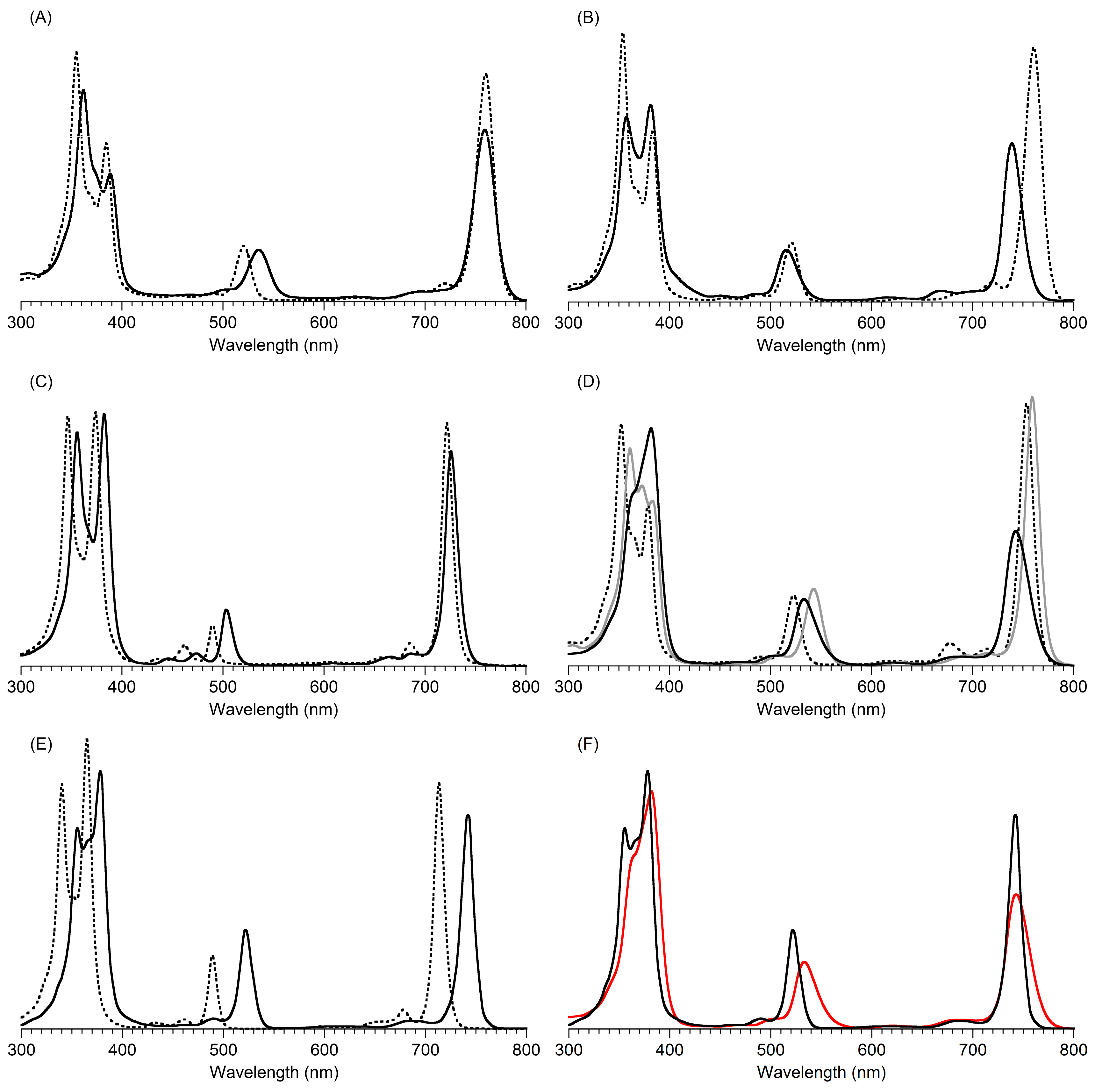
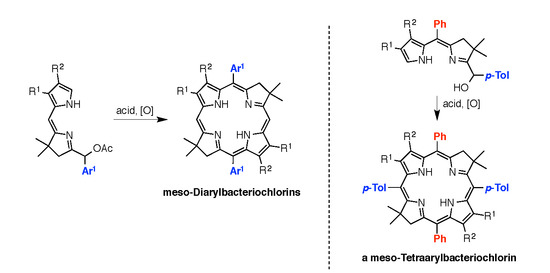
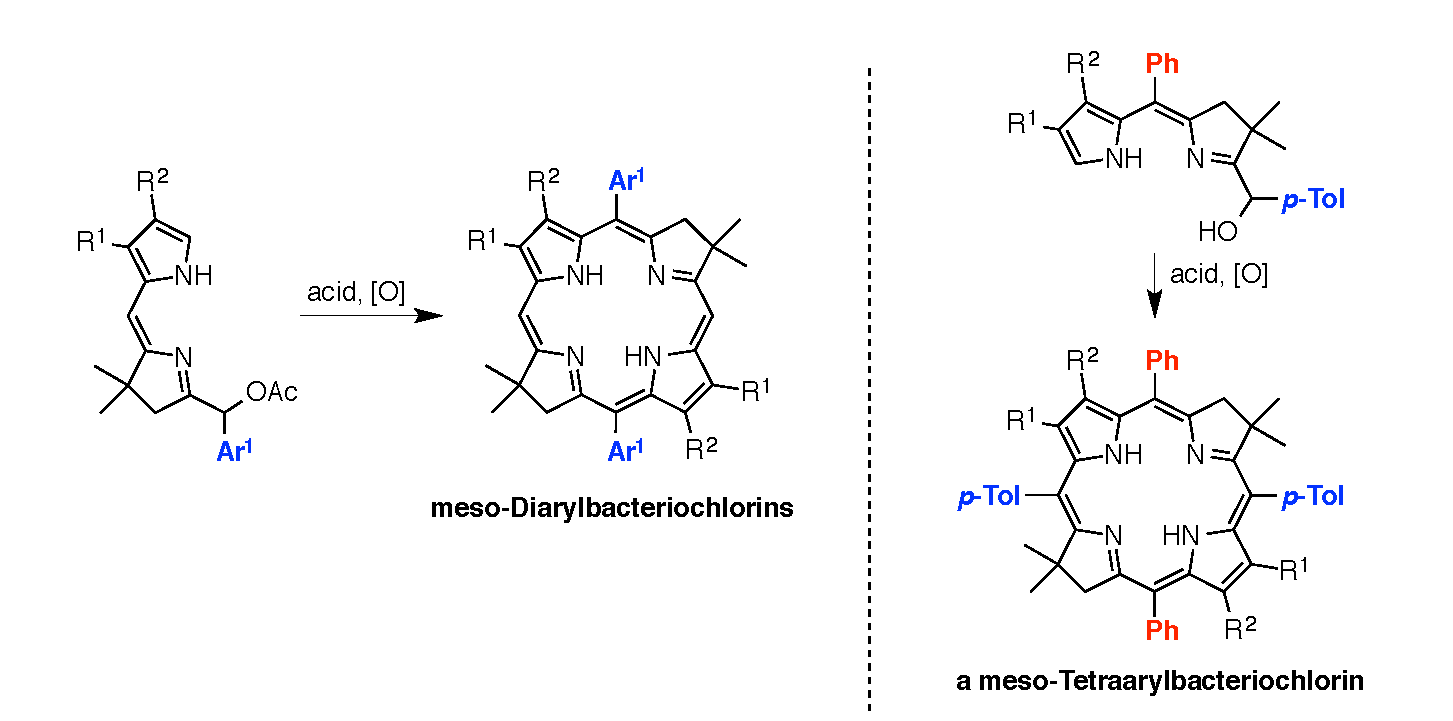
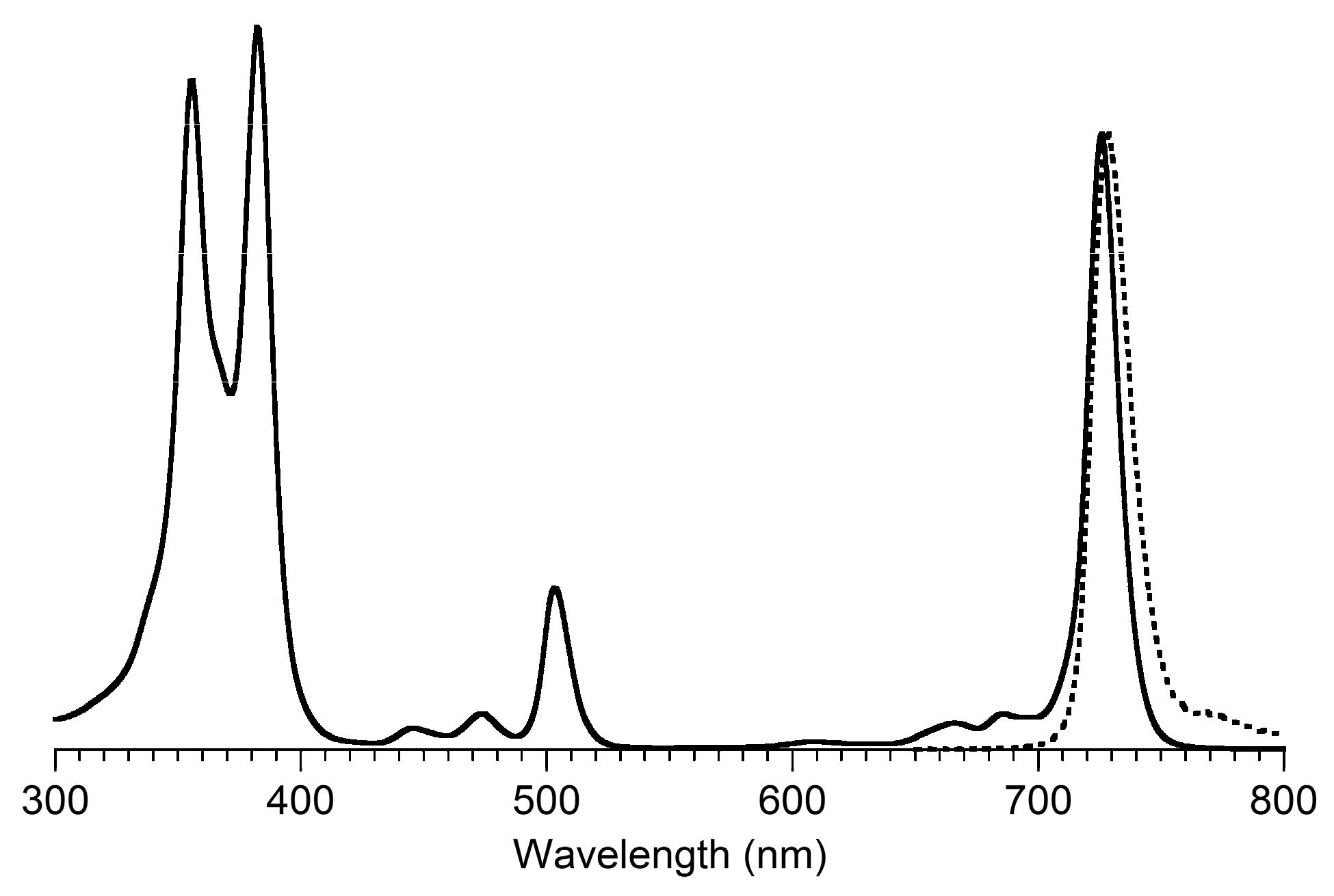
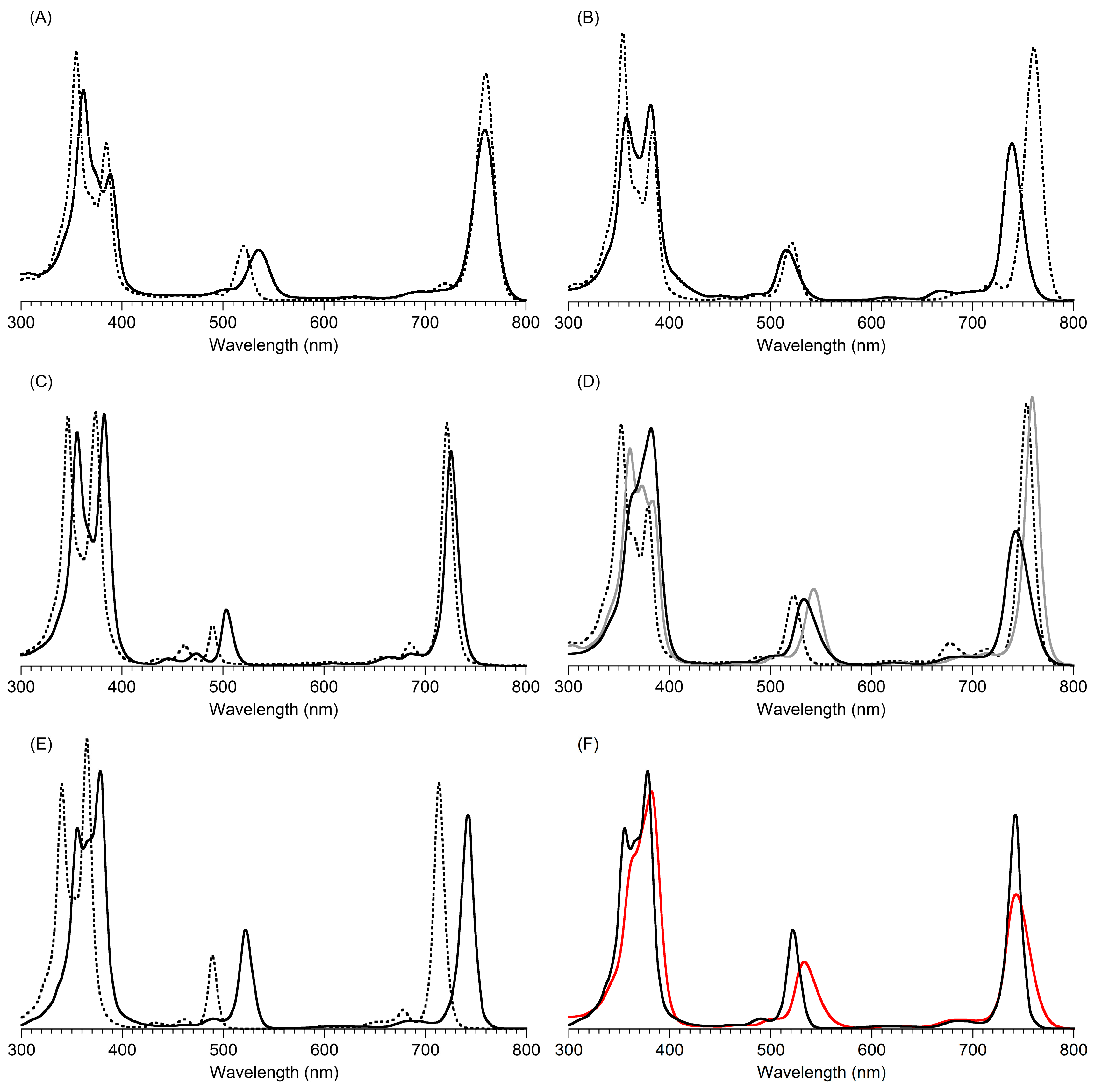
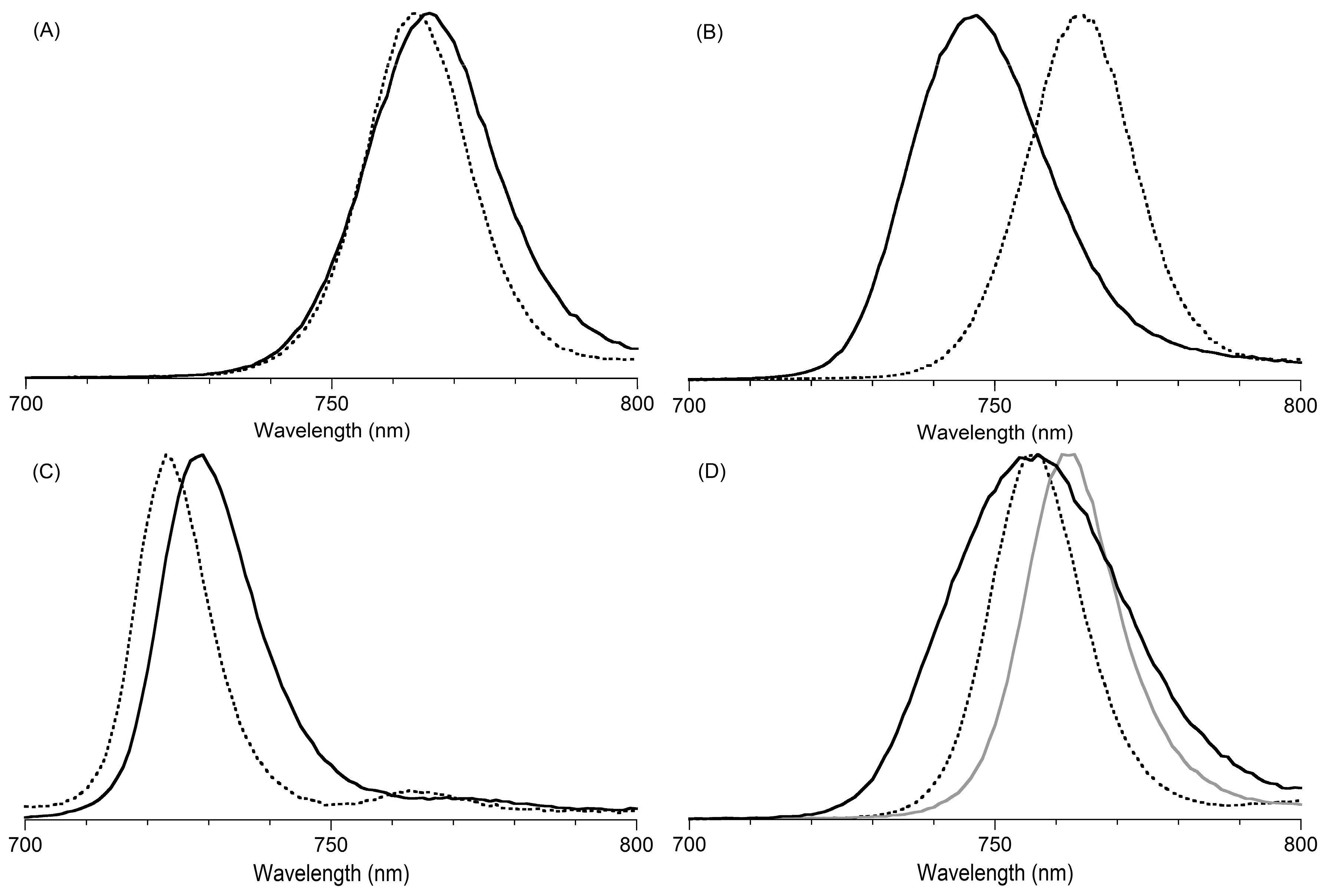
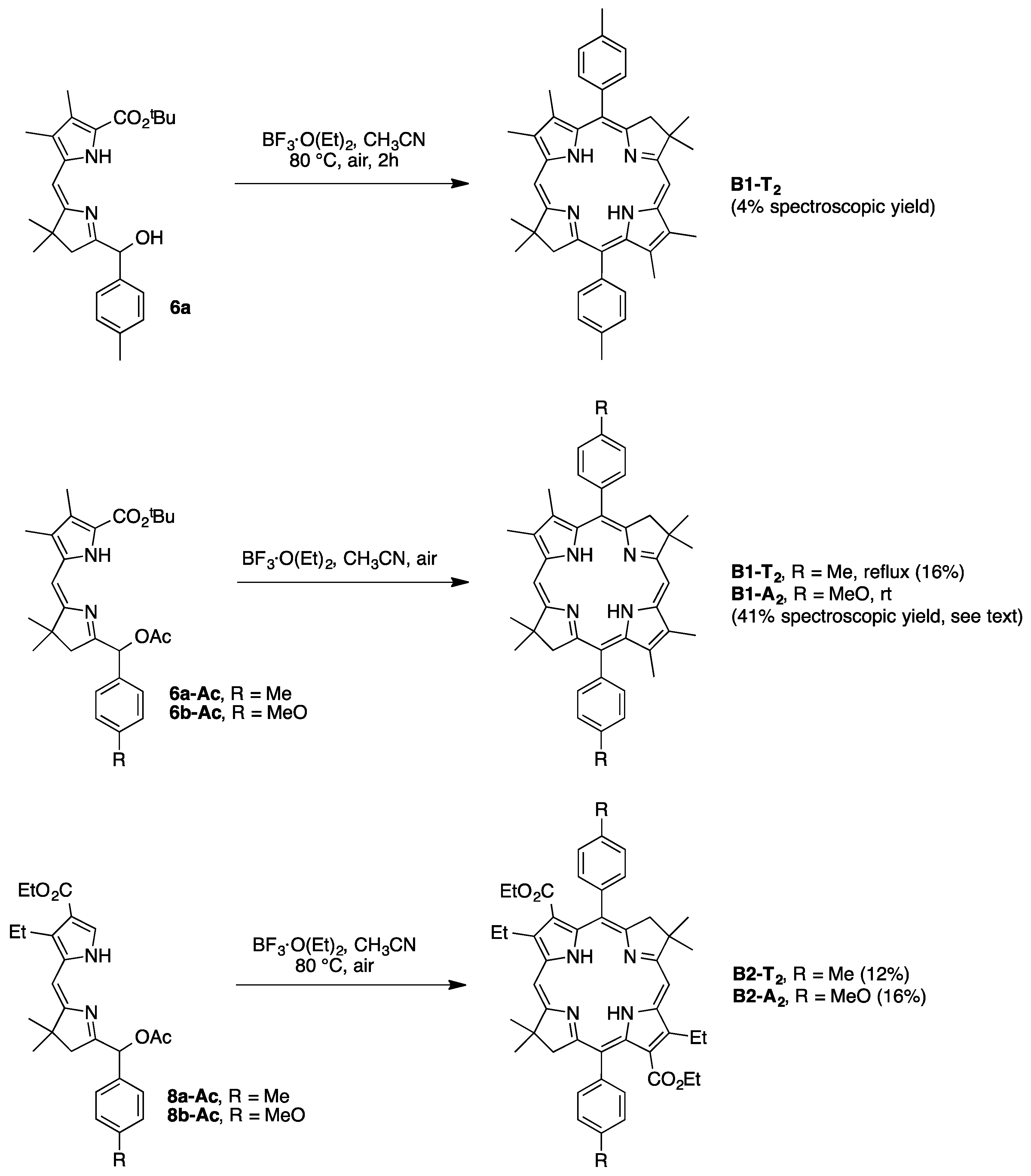
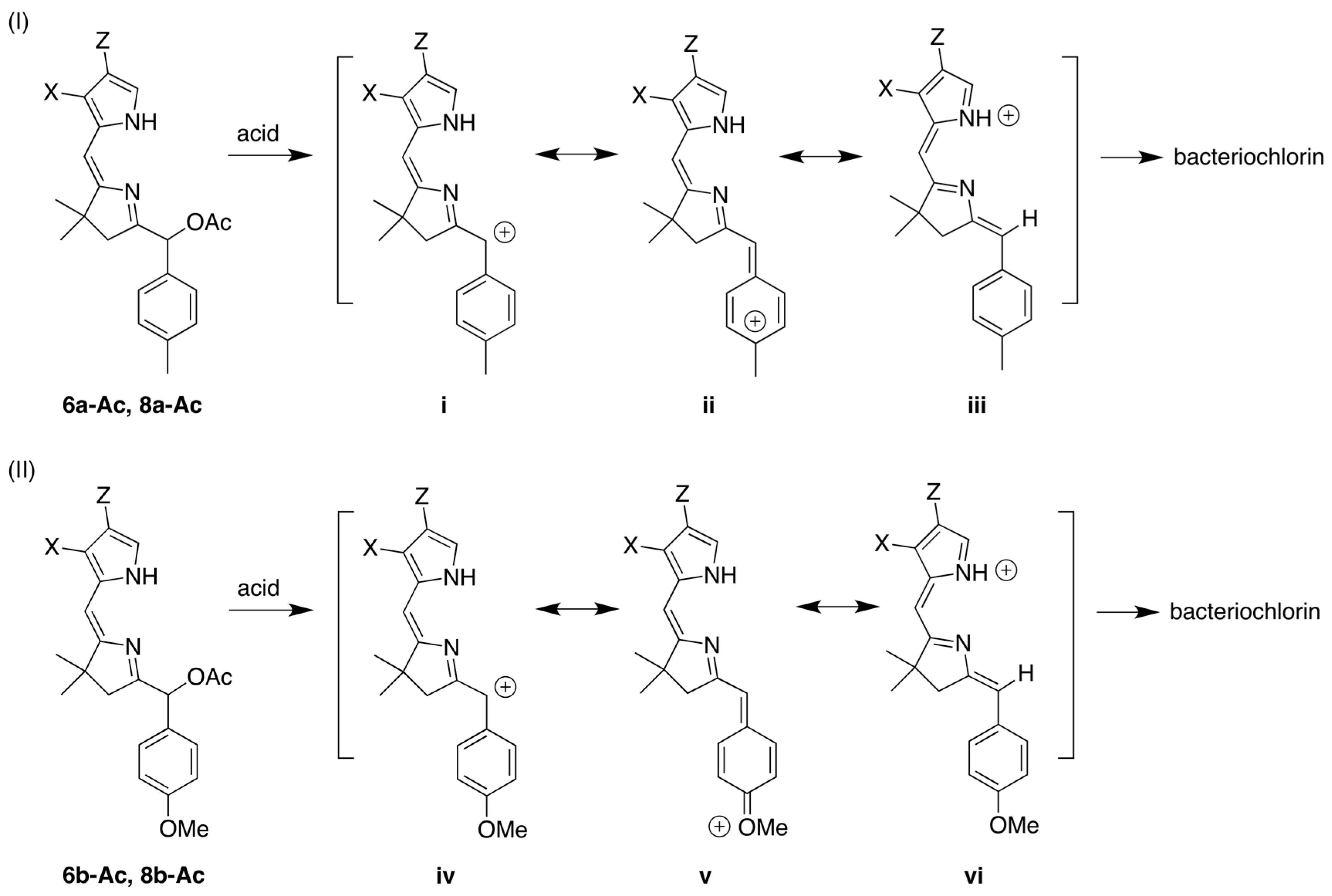
2,3,8,8,12,13,18,18-Octamethyl-5,15-bis(4-methylphenyl)bacteriochlorin (B1-T2). A solution of 6a-Ac (32.0 mg, 0.0689 mmol) in CH3CN (3.83 mL) was treated with BF3·O(Et)2 (67.9 µL, 0.536 mmol). The reaction mixture was heated to 80 °C for 2 h in a round-bottomed flask (air atmosphere) fitted with a rubber stopper, which itself was pierced with a syringe needle. The reaction mixture was allowed to cool to room temperature and then treated with TEA (89.6 µL, 0.643 mmol). The resulting mixture was diluted with ethyl acetate (~15 mL), washed with water, dried (Na2SO4), and concentrated. Column chromatography (silica, hexanes/CH2Cl2 (1:1)) afforded a greenish solid (3.8 mg). Trituration with CH2Cl2/hexanes gave a green solid (3.3 mg, 16%): 1H-NMR δ −1.64–1.58 (brs, 2H), 1.85 (s, 12H), 2.35 (s, 6H), 2.63 (s, 6H), 3.24 (s, 6H), 3.93 (s, 4H), 7.46 (d, J = 8.0 Hz, 4H), 7.65 (d, J = 8.0 Hz, 4H), 8.59 (s, 2H); MALDI-MS obsd 607.7; ESI-MS obsd 606.3714, calcd. 606.3717 [(M)+, M = C42H46N4]; λabs(toluene) 356, 382, 504, 726 nm.
5,15-Bis(4-methoxyphenyl)-2,3,8,8,12,13,18,18-octamethylbacteriochlorin (B1-A2). A solution of 6b-Ac (2.2 mg, 4.6 µmol) in CH3CN (0.25 mL) was treated with BF3·O(Et)2 (4.4 µL, 36 µmol) in a glass vial (air atmosphere) fitted with a rubber stopper, which itself was pierced with a syringe needle. The reaction mixture was stirred at room temperature for 24 h. The yield of bacteriochlorin was 41% upon absorption spectroscopic examination. (The weight of the vial was checked to confirm the absence of any significant solvent loss, the occurrence of which would inflate the spectroscopic yield determination; no correction was required.) The reaction mixture was treated with TEA (11 µL, 72 µmol). The resulting mixture was diluted with ethyl acetate (5 mL) and washed with water, dried (Na2SO4), and concentrated. Silica chromatography afforded a greenish brown solid, which exhibited limited solubility in diverse organic solvents (hexanes, ethyl acetate, dichloromethane, methanol): MALDI-MS obsd 639.4, calcd. 638.36 (C42H46N4O2); λabs (CH2Cl2) 355, 382, 503, 726 nm.
3,13-Dicarbethoxy-2,12-diethyl-8,8,18,18-tetramethyl-5,15-bis(4-methylphenyl)bacteriochlorin (B2-T2). A solution of 8a-Ac (31 mg, 71 µmol) in CH3CN (3.9 mL) was treated with BF3·O(Et)2 (68 µL, 0.55 mmol) for 4 h following the procedure for A2-BC1. The reaction mixture was treated with TEA (77 µL, 0.55 mmol) and diluted with ethyl acetate (20 mL), and then washed with water, dried (Na2SO4), and concentrated. Column chromatography (silica, hexanes/ethyl acetate (4:1)) afforded a green solid (3.1 mg, 12%): 1H-NMR δ −1.28 (br, 2H), 1.29 (t, J = 7.2 Hz, 6H), 1.67 (t, J = 7.2 Hz, 6H), 1.83 (s, 12H), 2.59 (s, 6H), 3.78 (q, J = 7.2 Hz, 4H), 3.87 (q, J = 7.2 Hz, 4H), 3.95 (s, 4H), 7.44 (d, J = 7.5 Hz, 4H), 7.71 (d, J = 7.5 Hz, 4H), 8.62 (s, 2H); 13C-NMR δ 14.2, 17.7, 20.1, 21.6, 31.2, 45.5, 51.6, 61.2, 94.5, 113.5, 125.6, 128.3, 131.9, 132.0, 133.0, 137.1, 137.5, 138.7, 159.9, 168.1, 169.0; MALDI-MS obsd 751.3; ESI-MS obsd 751.41805, calcd. 751.42178 [(M + H)+, M = C48H54N4O4]; λabs (toluene) 358, 382, 516, 739 nm.
3,13-Dicarbethoxy-2,12-diethyl-5,15-bis(4-methoxyphenyl)-8,8,18,18-tetramethylbacteriochlorin (B2-A2). A solution of 8b-Ac (14 mg, 31 µmol) in CH3CN (1.7 mL) was treated with BF3·O(Et)2 (30 µL, 0.24 mmol) for 2 h following the procedure for A2-BC1. The reaction mixture was treated with TEA (33 µL, 0.24 mmol). The resulting mixture was diluted with ethyl acetate (20 mL) and then washed with water, dried (Na2SO4), and concentrated. Column chromatography (silica, CH2Cl2) afforded a green solid (1.9 mg, 16%): 1H-NMR δ −1.31 (br, 2H), 1.30 (t, J = 7.5 Hz, 6H), 1.67 (t, J = 7.5 Hz, 6H), 1.84 (s, 12H), 3.77 (br, 4H), 3.95 (q, J = 7.5 Hz, 8H), 4.01 (s, 6H), 7.16 (d, J = 8.7 Hz, 4H), 7.73 (d, J = 8.7 Hz, 4H), 8.62 (s, 2H); MALDI–MS obsd 783.8; ESI-MS obsd 783.41449, calcd. 783.41161 [(M + H)+, M = C48H54N4O6]; λabs(toluene) 358, 382, 517, 739 nm. In this reaction one additional bacteriochlorin was observed in trace quantities upon chromatography (following the title compound), although not isolated in pure form (LD-MS m/z = 739.1; λabs = 382, 549, 759 nm).
3,13-Dicarbethoxy-8,8,18,18-tetramethyl-5,15-bis(4-methylphenyl)-10,20-diphenylbacteriochlorin (B3-P2T2). A solution of 15 (10 mg, 23 µmol) in CH2Cl2 (1.3 mL) was treated with trifluoroacetic anhydride (16 µL, 0.12 mmol) in a glass vial. The reaction flask was sealed with a rubber stopper and heated at 40 °C for 24 h. Upon cooling to room temperature, the reaction mixture was treated with TEA (33 µL, 0.24 mmol). The resulting mixture was concentrated and chromatographed (silica, CH2Cl2)) to afford a pink solid (1.3 mg, 13%): 1H-NMR δ −0.46 (br, 2H), 1.18 (t, J = 7.2 Hz, 6H), 1.30 (s, 12H), 2.54 (s, 6H), 3.67 (s, 4H), 3.77 (q, J = 7.2 Hz, 4H), 7.30 (d, J = 8.4 Hz, 4H), 7.59–7.61 (m, 7H), 7.71–7.73 (m. 7H), 8.03 (d, J = 2.1 Hz, 2H); MALDI-MS obsd 847.6; ESI-MS obsd 847.41996, calcd. 847.42178 [(M + H)+, M = C56H54N4O4]; λabs(toluene) 382, 533, 743 nm.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}