
Constituents of the Roots of Dichapetalum pallidum and Their Anti-Proliferative Activity

Abstract
:1. Introduction
2. Results
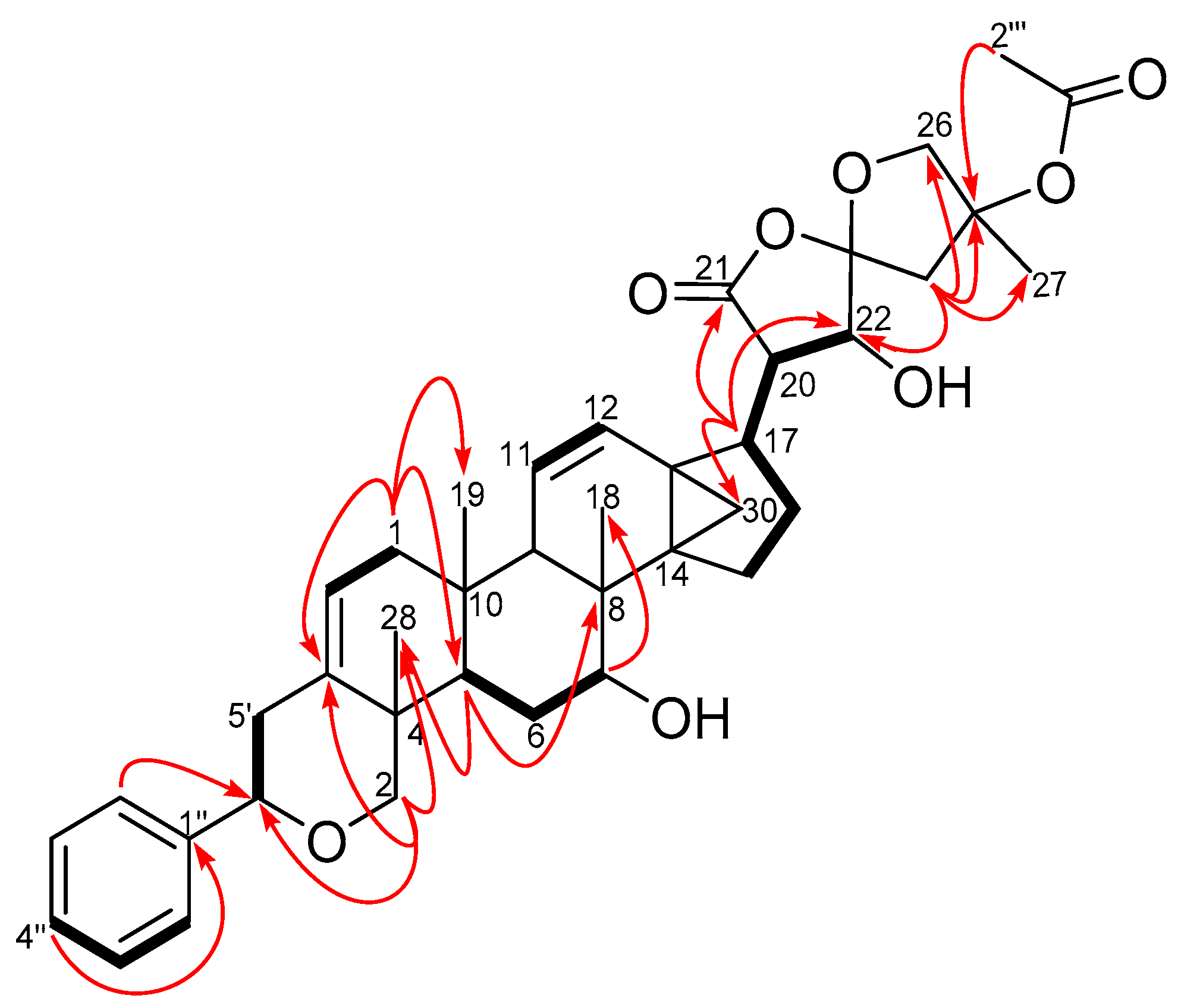
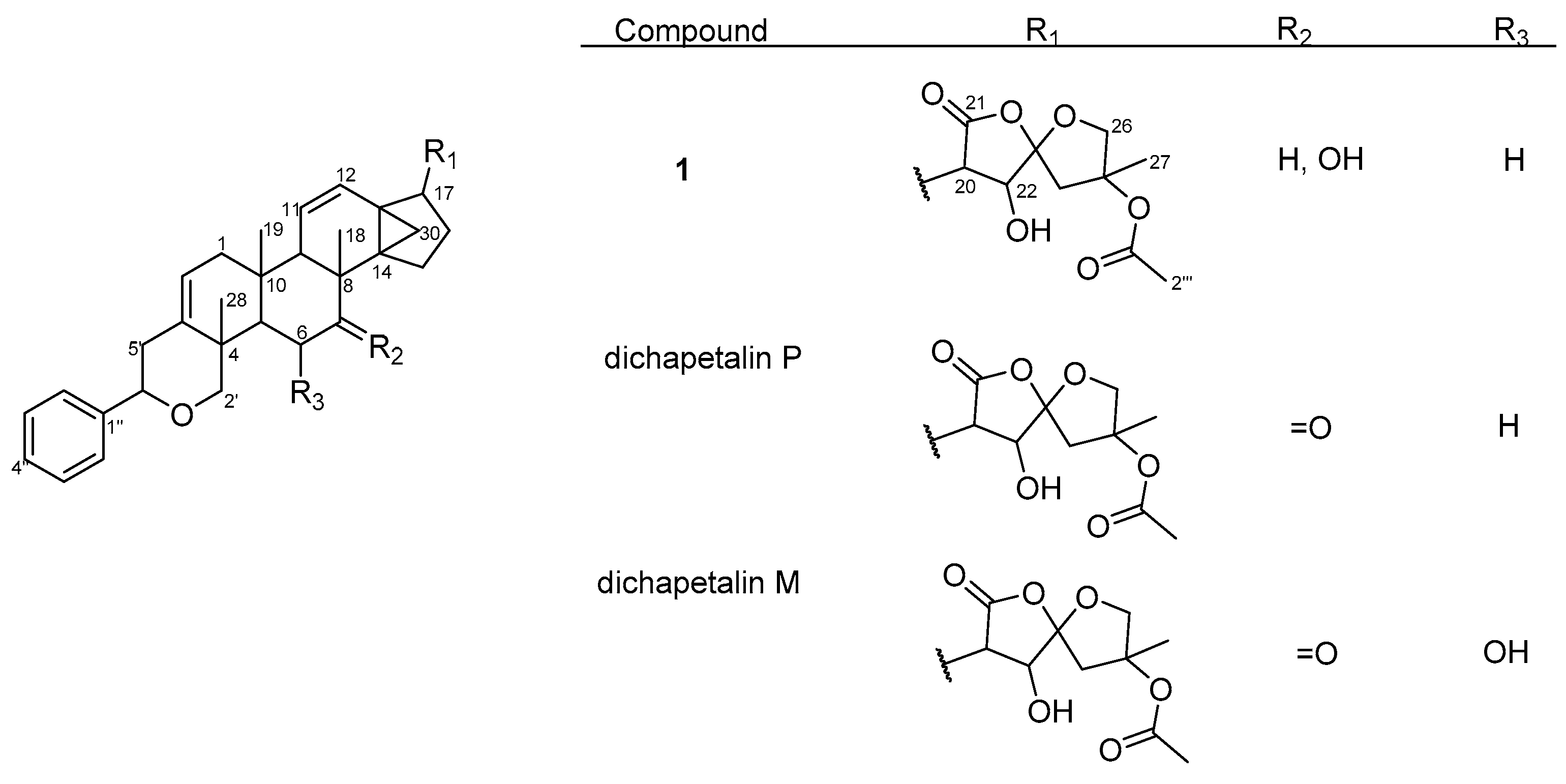
2.1. Characterization of Compounds
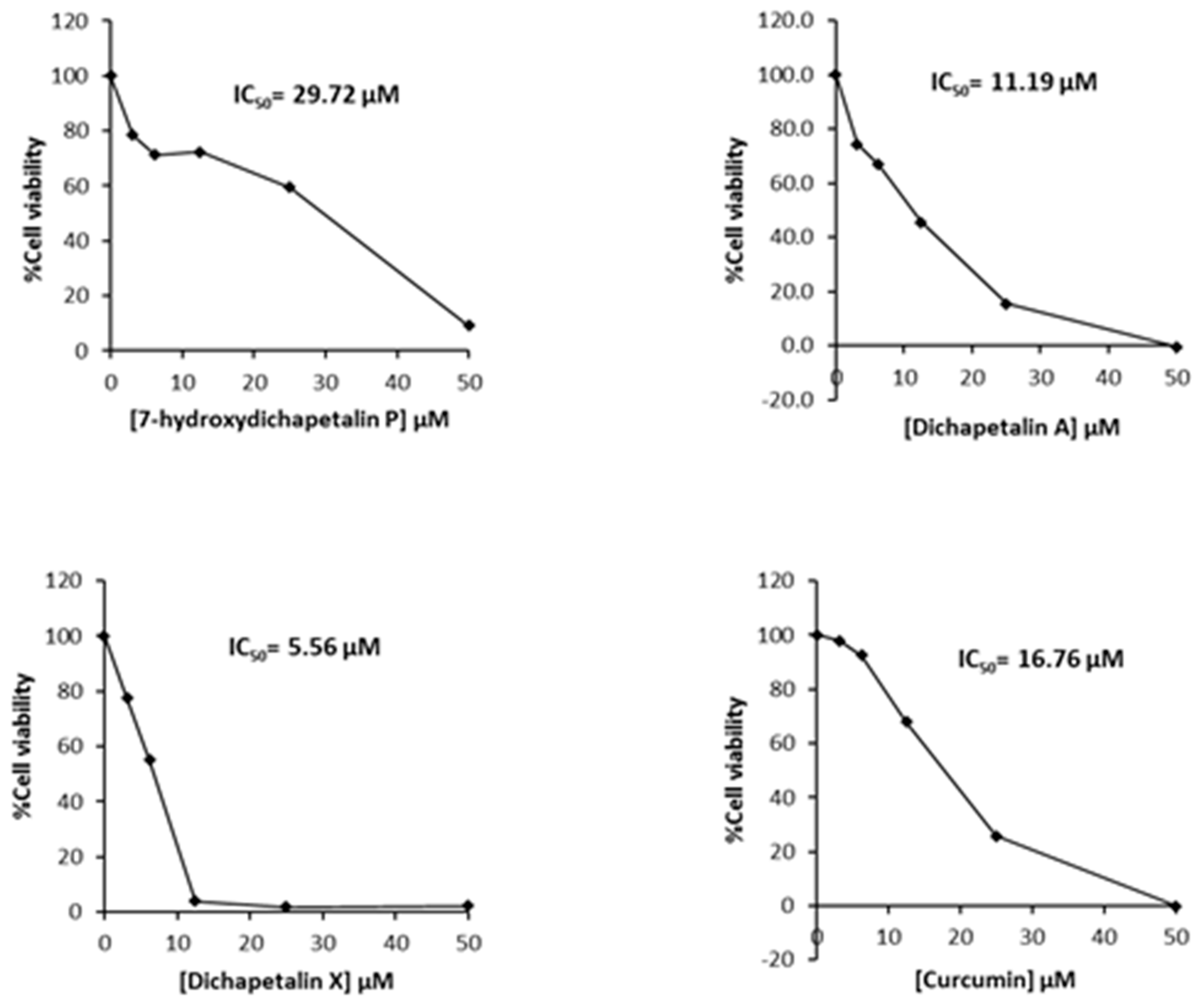
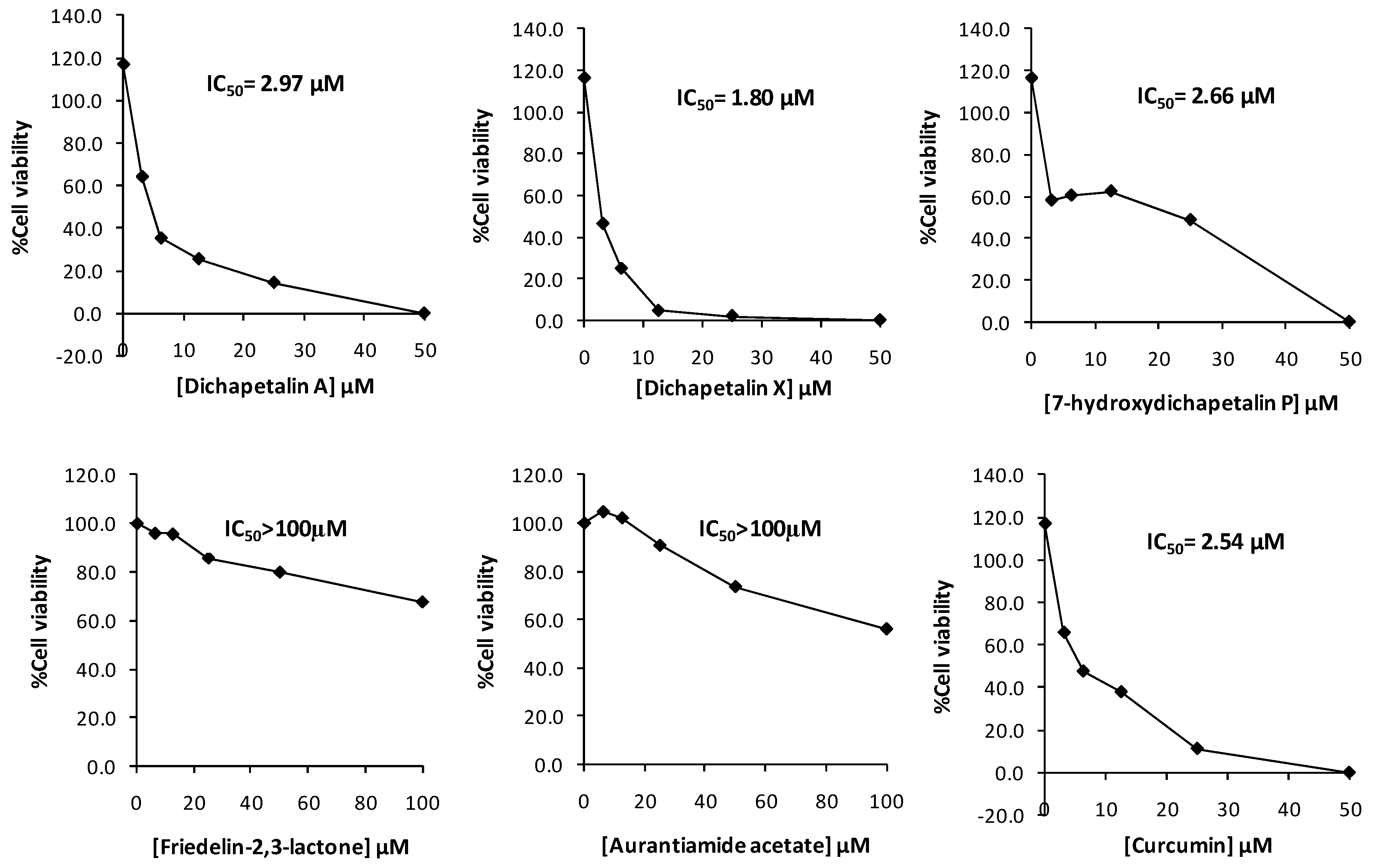
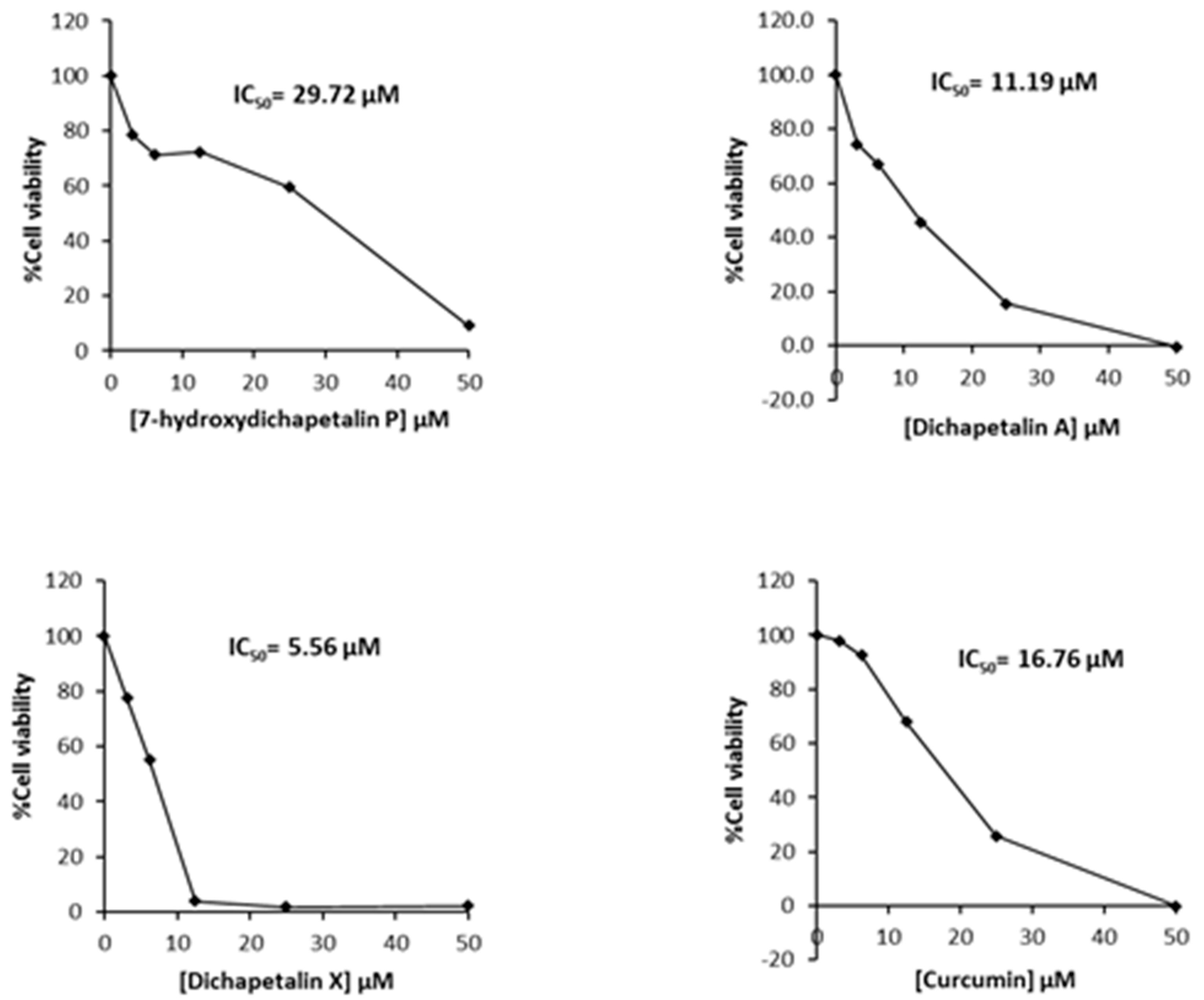
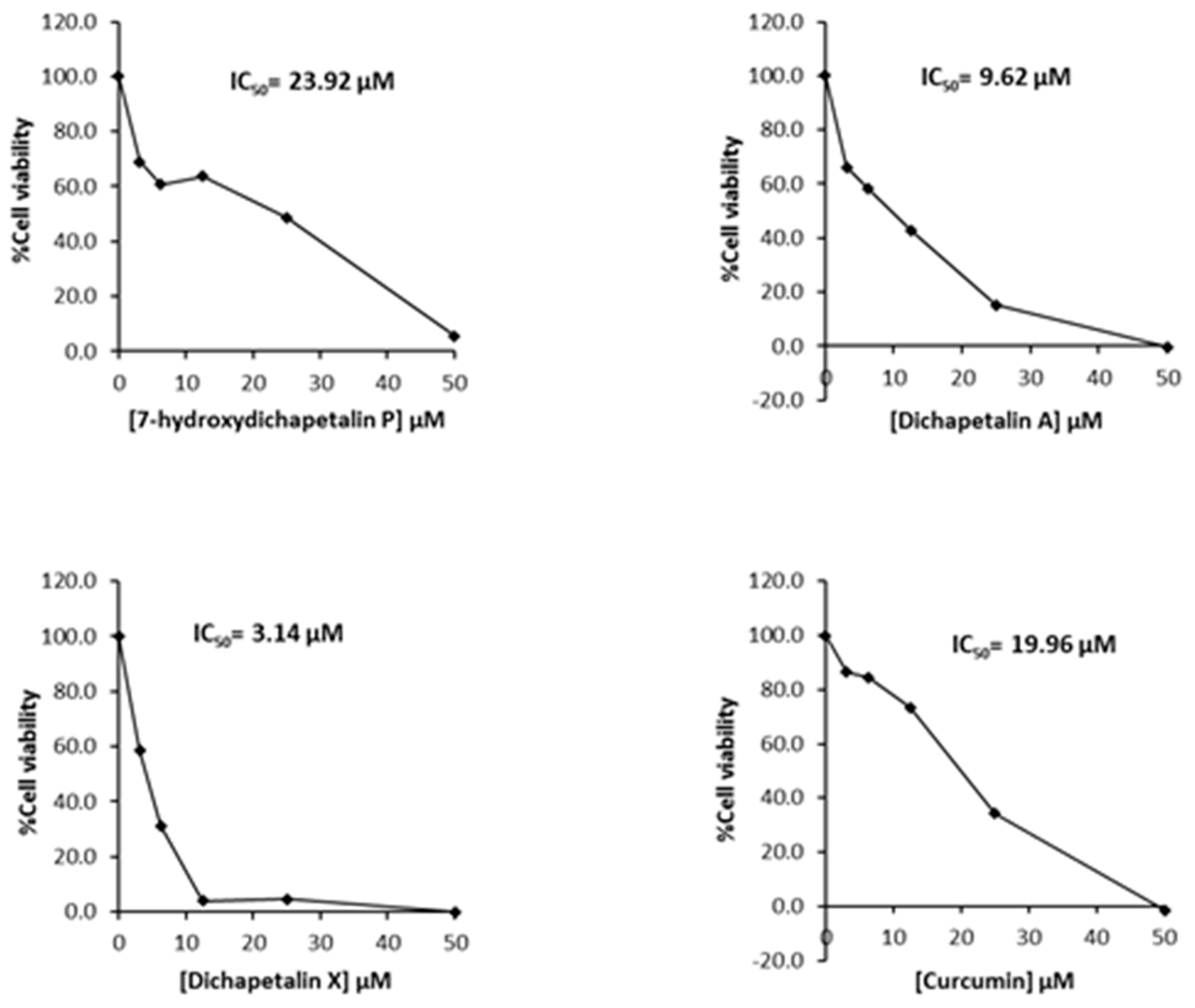
2.2. Effect of Compounds 1–5 on Cell Proliferation of Leukemia Cancer Cell Lines
3. Conclusions
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Plant Material
4.3. Extraction and Isolation
4.4. Cytotoxicity Assay
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Achenbach, H.; Asunka, S.A.; Waibel, R.; Addae-Mensah, I.; Oppong, I.V. Dichapetalin A, A novel plant constituent from Dichapetalum madagascariense. Nat. Prod. Lett. 1995, 7, 93–100. [Google Scholar] [CrossRef]
- Addae-Mensah, I.; Waibel, R.; Asunka, S.A.; Oppong, I.V.; Achenbach, H. The Dichapetalins—A new class of Triterpenes. Phytochemistry 1996, 43, 649–656. [Google Scholar] [CrossRef]
- Fang, L.; Ito, A.; Chai, H.-B.; Mi, Q.; Jones, W.P.; Madulid, D.R.; Oliveros, M.B.; Gao, Q.; Orjala, J.; Farnsworth, N.R.; et al. Cytotoxic constituents from the stem bark of D. gelonioides collected in the Philippines. J. Nat. Prod. 2006, 69, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Osei-Safo, D.; Chama, M.A.; Addae-Mensah, I.; Waibel, R.; Asomaning, W.A.; Oppong, I.V. Dichapetalin M from Dichapetalum madagascariensis. Phytochem. Lett. 2008, 1, 147–150. [Google Scholar] [CrossRef]
- Long, C.; Aussagues, Y.; Molinier, N.; Marcourt, L.; Vendier, L.; Samson, A.; Poughon, V.; Patrick, B.; Mutiso, C.; Ausseil, F.; et al. Dichapetalins from Dichapetalum species and their cytotoxic properties. Phytochemistry 2013, 94, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Jing, S.-X.; Luo, S.-H.; Li, C.-H.; Hua, J.; Wang, Y.-L.; Niu, X.-M.; Li, X.-N.; Liu, Y.; Huang, C.-S.; Wang, Y.; et al. Biologically active dichapetalins from Dichapetalum gelonioides. J. Nat. Prod. 2014, 77, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Chama, M.A.; Dziwornu, G.A.; Waibel, R.; Osei-Safo, D.; Addae-Mensah, I.; Otchere, J.; Wilson, M. Isolation, characterization and anthelminthic activity of a novel dichapetalin and other constituents of Dichapetalum filicaule. Pharm. Biol. 2015, 54, 1179–1188. [Google Scholar] [PubMed]
- Irvine, F.R. Woody Plants of Ghana; Oxford University Press: London, UK, 1961; Volume 1, p. 268. [Google Scholar]
- Annan, K.; Adu, F.; Gbedema, S.Y. Friedelin: A bacterial resistance modulator from Paullinia pinnata L. J. Sci. Technol. 2009, 29, 152–159. [Google Scholar] [CrossRef]
- Li, X.J.; Liu, Z.Z.; Kim, K.-W.; Li, Z.; Kim, Y.-C.; Yook, C.S.; Liu, X.Q. Chemical Constituents from Leaves of Pileostegia viburnoides Hook.f.et Thoms. Nat. Prod. Sci. 2016, 22, 154–161. [Google Scholar] [CrossRef]
- Moiteiro, C.; Marcelo Curto, M.J.; Mohamed, N.; Bailén, M.; Martínez-Díaz, R.; González Coloma, A. Biovalorization of friedelane triterpenes derived from cork processing industry byproducts. J. Agric. Food Chem. 2006, 54, 3566–3571. [Google Scholar] [CrossRef]
- Wahidulla, S.; D’Souza, L.; Patel, J. 5α-Cholestane-3,6-dione from the red alga Acantophora spicifera. Phytochemistry 1987, 26, 2864–2865. [Google Scholar] [CrossRef]
- Songue, J.L.; Kouam, D.E.; Mpondo, T.N.; White, R.L. Chemical constituents from stem bark and roots of Clausena anisata. Molecules 2012, 17, 13673–13686. [Google Scholar] [CrossRef] [PubMed]
- Appiah-Opong, R.; Asante, I.K.; Osei-Safo, D.; Tuffour, I.; Ofori-Attah, E.; Uto, T.; Nyarko, A.K. Cytotoxic effects of Albizia zygia (DC) J.F. Macbr, a Ghanaian medicinal plant against T-lymphoblast-like leukemia, prostate and breast cancer cell lines. Int. J. Pharm. Pharm. Sci. 2016, 8, 392–396. [Google Scholar]
Sample Availability: Samples of the compounds 2–8 are available from the authors. |
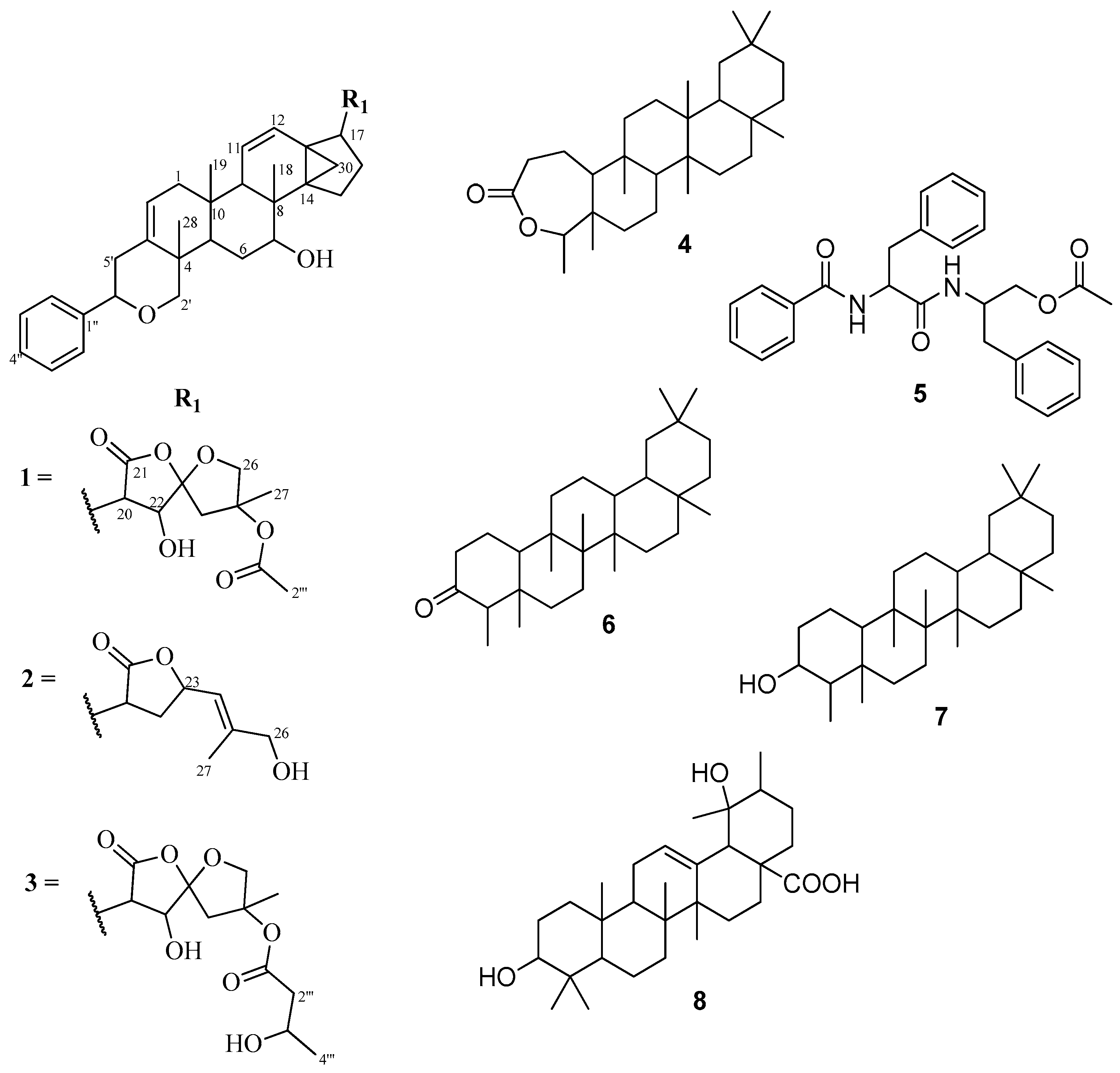





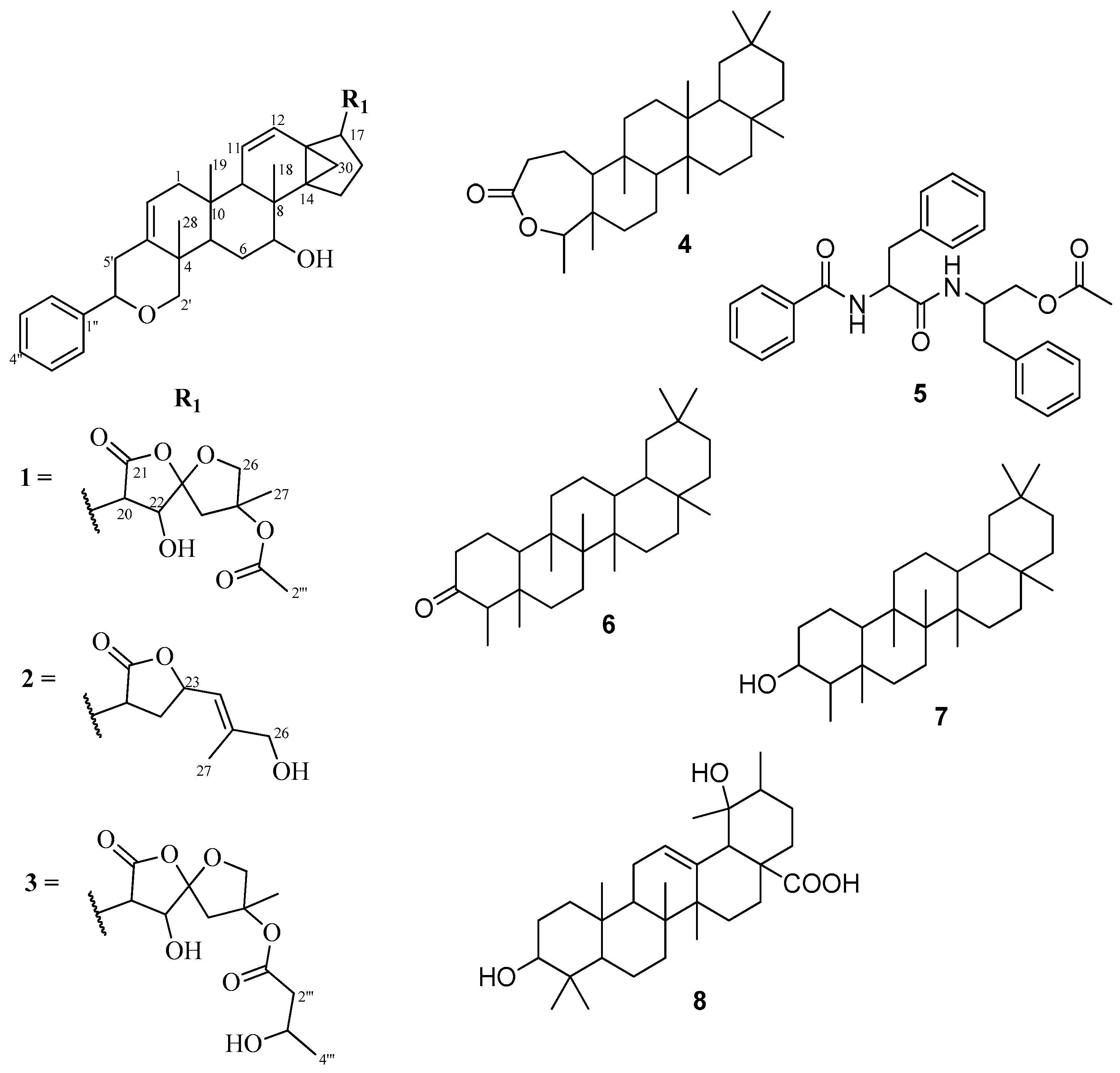{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 (in CDCl3, 600 MHz) | 2 (in CDCl3, 600 MHz) | 3 (in CDCl3, 500 MHz) | |||
|---|---|---|---|---|---|---|
| δc, Type | δH (J/Hz) | δc, Type | δH (J/Hz) | δc, Type | δH (J/Hz) | |
| 1a | 1.68, m | 1.66, m | 1.69, m | |||
| 1b | 40.1, CH2 | 2.03, dd (7.1, 16.1) | 40.1, CH2 | 2.10, dd (7.0, 16.2) | 40.1, CH2 | 2.11, dd (7.1, 16.2) |
| 2 | 117.8, CH | 5.42, d (7.1) | 117.7, CH | 5.40, d (6.9) | 117.8, CH | 5.43, ddd (1.8, 1.8, 7.1) |
| 3 | 140.0, C | 140.1, C | 140.0, C | |||
| 4 | 38.3, C | 38.3, C | 38.3, C | |||
| 5 | 43.7, CH | 2.01, m | 43.8, CH | 2.00, m | 43.8, CH | 2.04, m |
| 6a | 1.62, ddd (2.4, 13.8, 13.8) | 1.62, m | 1.67, m | |||
| 6b | 24.1, CH2 | 1.85, ddd (3.0, 3.0, 13.8) | 24.2, CH2 | 1.85, dd (10.5, 12.7) | 24.2, CH2 | 1.86, ddd (3.0, 3.0, 13.8) |
| 7 | 72.3, CH | 3.95, dd (2.4, 3.0) | 72.3, CH | 3.94, m | 72.3, CH | 3.97, dd (2.4, 3.0) |
| 8 | 36.4, C | 36.4, C | 36.4, C | |||
| 9 | 45.7, CH | 2.01, m | 45.7, CH | 1.98, m | 45.7, CH | 2.02, m |
| 10 | 35.7, C | 35.2, C | 35.7, C | |||
| 11 | 124.1, CH | 5.46, dd (2.5, 10.2) | 124.1, CH | 5.45, dd (2.1, 10.0) | 124.0, CH | 5.48, dd (2.6, 10.2) |
| 12 | 129.1, CH | 6.20, dd (3.1, 10.2) | 129.0, CH | 6.14, dd (2.9, 10.0) | 129.1, CH | 6.21, dd (3.1, 10.2) |
| 13 | 29.9, C | 30.1, C | 30.0, C | |||
| 14 | 36.2, C | 36.2, C | 36.3, C | |||
| 15a | 1.73, m | |||||
| 15b | 24.8, CH2 | 2.05, m | 24.9, CH2 | 2.05, d (12.2) | 24.8, CH2 | 2.08, m |
| 16a | 1.27, m | |||||
| 16b | 23.2, CH2 | 1.78, ddd (9.6, 15.6, 19.2) | 22.8, CH2 | 1.12, m | 23.2, CH2 | 1.78, ddd (9.6, 15.6, 19.2) |
| 17 | 40.3, CH | 2.63, m | 41.0, CH | 2.62, m | 40.3, CH | 2.64, m |
| 18 | 17.5, CH3 | 0.92, s | 17.4, CH3 | 0.90, s | 17.5, CH3 | 0.92, s |
| 19 | 18.2, CH3 | 1.08, s | 18.1, CH3 | 1.08, s | 18.2, CH3 | 1.11, s |
| 20 | 47.1, CH | 3.01, dd (5.1, 10.0) | 42.1, CH | 3.08, ddd (4.9, 8.3, 12.9) | 47.0, CH | 3.05, dd (5.1, 10.0) |
| 21 | 173.9, C | 178.2, C | 173.9, C | |||
| 22 | 71.9, CH | 4.18, t (9.6) | 31.4, CH2 | 2.37, m | 72.1, CH | 4.19, d (9.5) |
| 23 | 111.3, C | 75.1, CH | 5.12, m | 111.4, C | ||
| 24a | 2.51, d (15.1) | 2.53, d (15.2) | ||||
| 24b | 45.7, CH2 | 2.84, d (15.1) | 122.0, CH | 5.52, dd (8.3, 1.0) | 45.8, CH2 | 2.91, d (15.2) |
| 25 | 85.0, C | 141.7, C | 86.0, C | |||
| 26a | 4.09, d (10.2) | 4.09, d (10.2) | ||||
| 26b | 78.9, CH2 | 4.32, d (10.2) | 67.2, CH2 | 4.05, s | 78.3, CH2 | 4.37, d (10.2) |
| 27 | 22.0, CH3 | 1.70, s | 14.1, CH3 | 1.74, s | 21.6, CH3 | 1.74, s |
| 28 | 23.8, CH3 | 1.33, s | 23.8, CH3 | 1.32, s | 23.8, CH3 | 1.36, s |
| 30a | 0.93, d (5.3) | 0.78, d (5.2) | 0.96, d (5.3) | |||
| 30b | 15.4, CH2 | 1.29, m | 15.0, CH2 | 1.18, d (4.6) | 15.5, CH2 | |
| 2′a | 3.62, d (10.5) | 3.59, d (10.6) | 3.63, d (10.5) | |||
| 2′b | 72.5, CH2 | 3.80, d (10.5) | 72.5, CH2 | 3.76, d (10.6) | 72.5, CH2 | 3.80, d (10.5) |
| 5′a | 2.20, dd | 2.19, dd (2.4, 13.4) | 2.23, dd (2.7, 13.7) | |||
| 5′b | 40.7, CH2 | 2.63, m | 40.7, CH2 | 2.62, m | 40.8, CH2 | |
| 6′ | 81.8, CH | 4.28, dd (2.5, 9.1) | 81.8, CH | 4.26, dd (2.3, 11.6) | 81.8, CH | 4.29, dd (3.6, 11.6) |
| 1″ | 142.6, C | 142.6, C | 142.6, C | |||
| 2″,6′′ | 125.8, CH | 7.35, m | 125.8, CH | 7.37, m | 125.8, CH | 7.36, m |
| 3″,5″ | 128.4, CH | 7.33, m | 128.3, CH | 7.33, m | 128.4, CH | 7.40, m |
| 4″ | 127.5, CH | 7.24, m | 127.5, CH | 7.25, m | 127.5, CH | 7.28, m |
| 1′′′ | 170.4, C | 172.1, C | ||||
| 2′′′a | 21.8, CH3 | 2.03, s | 2.40, dd (9.2, 16.2) | |||
| 2′′′b | 43.7, CH2 | 2.47, dd (3.6, 16.2) | ||||
| 3′′′ | 64.6, CH | 4.20, m | ||||
| 4′′′ | 22.7, CH3 | 1.26, d (6.1) | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osei-Safo, D.; Dziwornu, G.A.; Appiah-Opong, R.; Chama, M.A.; Tuffour, I.; Waibel, R.; Amewu, R.; Addae-Mensah, I. Constituents of the Roots of Dichapetalum pallidum and Their Anti-Proliferative Activity. Molecules 2017, 22, 532. https://doi.org/10.3390/molecules22040532
Osei-Safo D, Dziwornu GA, Appiah-Opong R, Chama MA, Tuffour I, Waibel R, Amewu R, Addae-Mensah I. Constituents of the Roots of Dichapetalum pallidum and Their Anti-Proliferative Activity. Molecules. 2017; 22(4):532. https://doi.org/10.3390/molecules22040532
Chicago/Turabian StyleOsei-Safo, Dorcas, Godwin Akpeko Dziwornu, Regina Appiah-Opong, Mary Anti Chama, Isaac Tuffour, Reiner Waibel, Richard Amewu, and Ivan Addae-Mensah. 2017. "Constituents of the Roots of Dichapetalum pallidum and Their Anti-Proliferative Activity" Molecules 22, no. 4: 532. https://doi.org/10.3390/molecules22040532