
Metal-Based PSMA Radioligands

1
Institute of Basic Medical Sciences, University of Oslo, Oslo 0372, Norway
2
Norwegian Medical Cyclotron Centre Ltd., P.O. Box 4950 Nydalen, Oslo 0424, Norway
3
Institute of Physics, University of Oslo, Oslo 0317, Norway
*
Author to whom correspondence should be addressed.
Molecules 2017, 22(4), 523; https://doi.org/10.3390/molecules22040523
Submission received: 30 January 2017
/
Revised: 13 March 2017
/
Accepted: 18 March 2017
/
Published: 24 March 2017
(This article belongs to the Special Issue Metal Based Drugs: Opportunities and Challenges)
Abstract
:Prostate cancer is one of the most common malignancies for which great progress has been made in identifying appropriate molecular targets that would enable efficient in vivo targeting for imaging and therapy. The type II integral membrane protein, prostate specific membrane antigen (PSMA) is overexpressed on prostate cancer cells in proportion to the stage and grade of the tumor progression, especially in androgen-independent, advanced and metastatic disease, rendering it a promising diagnostic and/or therapeutic target. From the perspective of nuclear medicine, PSMA-based radioligands may significantly impact the management of patients who suffer from prostate cancer. For that purpose, chelating-based PSMA-specific ligands have been labeled with various diagnostic and/or therapeutic radiometals for single-photon-emission tomography (SPECT), positron-emission-tomography (PET), radionuclide targeted therapy as well as intraoperative applications. This review focuses on the development and further applications of metal-based PSMA radioligands.
1. Introduction
Prostate cancer is the most common malignancy found in men and the second leading cause of cancer death in the Western world. Even though prostate cancer is one of the few slow-growing cancers, it becomes potentially lethal upon metastasis. Consequently, early detection is particularly important for effective treatment. Functional imaging methods to detect prostate cancer include magnetic resonance imaging (MRI), computed tomography (CT), single-photon-emission tomography (SPECT) and positron emission tomography (PET). Prostate cancer is a complex and biologically heterogeneous disease and cannot be fully assessed with conventional imaging alone. MRI has shown high sensitivity for localizing prostate cancer, although it’s diagnostic performance and impact on the treatment regimen and varies in the patient populations studied so far. MRI is also not sensitive in detecting cancer in regions other than the peripheral zone of the prostate [1]. Both conventional imaging modalities, MRI and CT, focus on morphological changes, and their contribution to primary staging of prostate cancer is limited. Therefore, efforts to discover and evaluate new diagnostic and therapeutic biomarkers for prostate cancer continue. Radionuclide molecular imaging with SPECT or PET is poised to fill this unmet need through noninvasive detection of the multiple molecular and cellular processes that are active in prostate cancer patients [2]. Furthermore, the hybrid imaging techniques such as SPECT/CT and PET/CT combine functional and morphological information leading to high diagnostic accuracy. Nowadays, both hybrid imaging techniques are used in the clinical routine, not only as the primary staging tool in prostate cancer, but also in patients with suspected disease recurrence. The basis for the successful application of either SPECT or PET relies on the over-expression of specific receptors in various tumors [3,4]. In particular, prostate specific membrane antigen (PSMA) is a well-established enzyme/target for diagnostic and potential therapeutic applications via its targeting with suitable radiolabeled anti-PSMA tracers [5,6].
PSMA, with a molecular weight of 100 kDa, consists of 750 amino acids and is a type II integral membrane glycoprotein. It has a unique structure containing three distinct parts; a 707-amino acid extracellular region, a cell membrane part of 24 amino acids and a cytoplasmic tail which contains 19 amino acids. PSMA is a key player in prostate carcinogenesis and disease progression, glutamatergic neurotransmission, and folate absorption [7]. These various functions and the tissue distribution of the protein result in different designations. The name which is also frequently used for this enzyme is glutamate carboxypeptidase II, or GCPII. Furthermore, in central nervous system, it metabolizes the brain neurotransmitter N-acetyl aspartyl glutamate (NAAG), and is named NAALADase. In the proximal small intestine, it removes γ-linked glutamates from poly-g-glutamated folate, which is reflected in its name, folate hydrolase FOLH1 (Figure 1) [5,7,8].
The NAALDase activity of PSMA has been extensively investigated for the development of PSMA-specific based ligands with the potential to be used for prostate cancer diagnosis and/or therapy. Briefly, the substrate, NAAG binds extracellularly to PSMA followed by hydrolysis of NAAG to glutamate and N-acetyl aspartate. Studies of the enzyme structure have shown the presence of a deep tunnel with a length of ~20 Å which connects the extracellular surface of PSMA with the active site of the enzyme, called the NAAG binding pocket. This binding pocket is also the binding site for the binding of the newly developed anti-PSMA ligands [9]. The proposed pharmacophore for PSMA active site can be divided into three parts: (i) three carboxylate groups; (ii) a carbonyl oxygen as part of the zinc complexation unit and (iii) nearby aromatic residues. More specifically, the crystal structure reveals two zinc atoms separated by approximately 3.2 Å. Each zinc atom in the PSMA active site is coordinated by three endogenous amino acids: a histidine (His-553 or -377), a terminal aspartate (Asp-453) or glutamate (Glu-425), and a bridged aspartate (Asp-387). The formed complex is finally surrounded by water molecules including conservative points [9,10]. Human PSMA has structural homology to another type II transmembrane glycoprotein, the transferrin receptor, which consists of 760 amino acids with a molecular weight of 190 kDa. Part of the extracellular domain of PSMA shows sequence similarity to the transferrin receptor (~54% similarity). The transferrin receptor however exists in dimeric form, whilst PSMA exists in dimeric/monomeric form although it is only enzymatically active as a dimer [11]. Both PSMA and transferrin receptor bind their ligands followed by internalization of the PSMA-bound ligand complex through the clathrin-coated pits [11,12,13]. PSMA and transferrin receptor are expressed on various tumor cells and inhibiting their functions may lead to implications for cancer therapeutics.
In pathological situations, PSMA is primarily expressed in the human prostate epithelium, salivary and lacrimal glands and kidneys with enhanced expression by almost all prostate cancers and further up-regulation in poorly differentiated, metastatic and hormone-refractory carcinomas without being shed into the circulation [5,14]. These characteristics render PSMA a promising target for imaging and therapy of prostate cancer. In particular, it is overexpressed by almost all prostate cancers with an increased expression by a factor of about 1000 in poorly differentiated, metastatic, and hormone-refractory cases [6,15,16]. Given the overexpression of PSMA in advanced prostate cancer, several efforts have been made especially during the last decade for the development of anti-PSMA-based imaging agents for SPECT and PET imaging.
2. PSMA-Based Radioligands
As part of the ongoing efforts of the scientific community to develop new anti-PSMA-specific ligands, various radiotracers have been reported, with some of them having shown great promise not only preclinically, but also in the clinical assessment. The dual nature of PSMA to act not only as a receptor protein, but also as an enzyme, has paved the way for the establishment of several approaches for its targeting via radiolabeled molecules. Firstly, based on the macromolecular protein structure of PSMA, specific monoclonal antibodies and smaller molecules called aptamers have been developed. These molecules bind tightly, selectively and specifically to PSMA [17,18]. Secondly, the enzymatic activity of PSMA served as the trigger for the synthesis and the further evaluation of a variety of anti-PSMA inhibitors of low molecular weight, with the potential to be used as nuclear imaging probes [19].
2.1. Anti-PSMA Monoclonal Antibodies as Radiotracers for Prostate Cancer Imaging and Therapy
PSMA was originally targeted by the monoclonal antibody 7E11-C5 (capromab) functionalized with the chelator diethylenetriaminepentaacetic acid (DTPA) and labeled with 111In to obtain 111In-7E11-C5 (ProstaScint®; Cytogen Corporation, Princeton, NJ, USA) [20,21,22,23]. ProstaScint® binds to the intracellular site of PSMA (the amino terminus) which is accessible on necrotic tumors only [24,25]. Therefore, this tracer fails to gain wide acceptance in the field of nuclear medicine for the detection of prostate cancer. Hence, the need for the development of monoclonal antibodies which bind to the extracellular site of PSMA was evident. Consequently, several monoclonal antibodies, such as the humanized monoclonal antibody J591 [26,27] and three others murine monoclonal antibodies, named 3/A12, 3/E7 and 3/F11, were generated. These antibodies exhibit high and specific binding to cell-adherent PSMA [28,29].
Contrary to 7E11-C5, J591 can recognize PSMA that is present on the surface of nearly all prostate cancer tumors and circulating tumor cells. J591 is the first monoclonal antibody targeting the extracellular domain of PSMA as well as the first humanized monoclonal antibody to PSMA to be tested in humans. J591 has been evaluated preclinically for SPECT, PET as well as radioimmunotherapy. Its functionalization has been performed using a variety of chelators, such as 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and deferoxamine (DFO) and was further labeled with 99mTc, 111In, 89Zn, 64Cu, 177Lu, 90Y [30,31,32,33,34,35,36,37,38,39,40,41]. PSMA SPECT imaging using 99mTc-J591 depicts the presence of prostate cancer, and it can be also considered as a sensitive tool for the delineation of micro-invasion of the capsule, seminal vesicles or bladder neck. Although 99mTc-J591 is of value in locating recurrence of the disease after radical prostatectomy, the results of the study indicated that extraprostatic cancer was not reliably detected. This difficulty may be due to the high activity of 99mTc degradation products in the bladder [30]. 111In-J591 was also evaluated for SPECT imaging preclinicaly and in clinical trials. In the preclinical settings, the pharmacokinetics, biodistribution, and tumor uptake of 111In-J591 were compared with those of 111In-7E11 in nude mice with PSMA-positive LNCaP tumors [41]. The most important findings were that the blood clearance of 111In-J591 was relatively faster (8.98 ± 2.10, 4.78 ± 0.85, 2.52 ± 0.56 % IA/g at 2, 4 and 6 days p.i.) than that of 111In-7E11 (7.22 ± 0.46, 5.69 ± 1.00, 4.16 ± 0.41 % IA/g at 2, 4 and 6 days p.i.), leading to higher tumor-to-blood ratios (111In-J591: 1.62 ± 0.72, 3.35 ± 0.39, 6.07 ± 1.79, 111In-7E11: 1.34 ± 0.24, 2.83 ± 0.51, 4.57 ± 0.31 at 2, 4 and 6 days p.i. respectively). Furthermore, in clinical trials, 111In-J591 was found superior to ProstaScint®, revealing most soft-tissue and bone metastases [31,39,40]. J591 has also been radiolabeled with 89Zr, after its functionalization with 3.9 ± 0.3 accessible DFO chelators [34]. 89Zr-DFO-J591 was produced in high radiochemical yield (>77%) with radiochemical purity >99% and a specific activity of 181.7 ± 1.1 MBq/mg. The radiolabeled immunoconjugate remained active for up to 7 days at 37 °C. In vivo biodistribution studies revealed high uptake of 89Zr-DFO-J591 in LNCaP tumors after 24, 48, 96 and 144 h (34.4 ± 3.2 % IA/g; 38.0 ± 6.2 % IA/g; 40.4 ± 4.8 % IA/g; and 45.8 ± 3.2 % IA/g, respectively). The immunoPET imaging indicated that 89Zr-DFO-J591 shows high potential as a radiotracer for specific, non-invasive delineation of PSMA positive prostate cancers in vivo. In localized human prostate cancer cases, 89Zr-J591 bound to tumor foci in situ and PET identified primarily Gleason score 7 or greater and larger tumors. High grade tumors were generally better visualized with 89Zr-J591. These clinical data provide a basis on which to investigate the various roles of 89Zr-J591 PET in prostate cancer diagnosis, management and treatment [42]. The coupling of the chelator DOTA to J591 and the subsequent labeling with 64Cu led to 64Cu-J519, which was also evaluated for PET imaging preclinically [35]. 89Zr-labeled rather than 64Cu-labeled J591 was proved to successfully image PSMA-expression in preclinical prostate cancer models. J591 has also been adapted for radioimmunotherapy, and Ab–drug conjugates and therapeutic doses are well tolerated in patients. Multiple (two or three) administrations of 177Lu-J591 (30–60 mCi/m2) or 90Y-J591 (17.5 mCi/m2) over a 4- to 6-month period were tolerated by the patients with manageable thrombocytopenia. Furthermore, the clinical trials showed that even though a single large dose may deliver optimal radiation dose to kill a larger fraction of tumor cells, fractionated therapy offers the advantage of lower myelotoxicity and prolonged tumor response [33,36].
For the development of antibodies with a high cell binding activity which may have an important impact on antibody-based imaging and therapy, a series of monoclonal antibodies against PSMA, named 3/F11, 3/A12 and 3/E7, generated [28,29]. All three antibodies were able to bind efficiently to cell surface expressed PSMA. In addition, the in vivo behavior and tumor uptake of the 64Cu-labeled monoclonal antibodies 3/A12, 3/F11 and 3/E7 were also investigated. Radiolabeling of 3/A12, 3/F11 and 3/E7 with 64Cu through a conjugated DOTA moiety was accomplished without loss of immunoreactivity [43]. Biodistribution as well as PET imaging studies of the three 64Cu-labeled antibodies showed specific tumor uptake in PSMA-positive C4-2 tumors, whereas the uptake of tracers in PSMA-negative DU 145 tumors was found to be at the background level. In the same study, the pharmacokinetics of the 64Cu-DOTA-3/A12 Fab and 64Cu-DOTA-3/A12 F(ab′)2 fragments compared to that of 64Cu-DOTA-3/A12 were also evaluated at 3, 24 and 48 h after the injection of the radiotracers. Only a faint uptake of 64Cu-DOTA-3/A12 Fab and 64Cu-DOTA-3/A12 F(ab′)2 fragments in the PSMA-positive tumors was observed at all time points tested, indicating that the rate of clearance from blood was too fast to allow for sufficient tracer binding at the receptors. Furthermore, high kidney retention was also reported for both 64Cu-labeled fragments [43]. Behe et al. was published the labeling of 3/F11 through a conjugated DOTA moiety with 177Lu along with the in vitro and in vivo evaluation of 177Lu-DOTA-3/F11 [44]. The biodistribution data showed that the tumor uptake of 177Lu-DOTA-3/F11 gradually increased with time up to 72 h after the injection of the radiotracer. The high and persistent tumor uptake may be caused by a rapid internalization of the 177Lu DOTA-3/F11-PSMA complex into the target cells after binding [45], followed by metabolism and trapping of the radioactivity at the tumor site [46]. The same study also revealed that the treatment of C4-2 bearing prostate cancer xenografts with a single dose of 1 MBq 177Lu-DOTA-3/F11 led to a more than two-fold enhanced mean survival and a delay in tumor growth. However, this therapeutic window is small since mice treated with 2 MBq 177Lu-DOTA-3/F11 apparently died of myelotoxicity, which is the predominant dose-limiting factor in radioimmunotherapeutic approaches with 177Lu [36,47,48].
Despite the improved targeting of J591 and the subsequent generation of anti-PSMA monoclonal antibodies, the major impediment to the use of antibodies for diagnostic purposes continues to be the slow clearance from the non-target tissues. This, in most of the cases, is mitigated by introducing a time interval of several days between radiotracer administration and imaging. An alternative approach is to optimize the pharmacokinetics of the monoclonal antibodies by the generation of antibody fragments, such as single domain antibodies (VHH), called nanobodies. The comparatively low molecular mass of nanobodies leads both to a better permeability in tissues, and a shorter plasma half-life [49,50]. Recently, the anti-PSMA nanobodies, JVZ-007-c-myc-his and JVZ-007-cys, were developed and the nanobody-DTPA conjugates were labeled with 111In to obtain 111In-JVZ-007-c-myc-his and 111In-JVZ-007-cys [51]. 111In-JVZ-007-c-myc-his was evaluated in mice bearing PC-310 (PSMA+) and PC-3 (PSMA-) tumors, showing good and specific tumor targeting already at 4 h after injection, with low background intensity. Only kidney tracer uptake was high (110.89 ± 7.35 % IA/g at 4 h p.i.) which was reduced by about 75% after the administration of gelofusin and lysine [52,53]. The introduction of a cysteine at the C terminus for site-specific coupling to maleimide-DTPA and the labeling with 111In led to 111In-JVZ-007-cys. The in vivo evaluation of 111In-JVZ-007-cys in the same tumor model as described above revealed a further drop in renal uptake without loss of tumor targeting. These optimal characteristics of 111In-JVZ-007-cys may pave the way for its applicability to radionuclide therapy. The engineering of the new nanobody construct, based on JVZ-007, in which the c-myc-his tag was substituted with Cys tag, may have a considerable impact on the clinical implementation of a wide range of nanobodies in the field of nuclear medicine.
In addition to nanobodies, a variety of small, genetically engineered immunological constructs have been developed for a broader range of in vivo applications. Examples of these constructs include Fab (fragment antigen-binding), F(ab′)2, single-chain (sc) Fv, bis-scFV, diabodies and minibodies. As described above, their small size potentially gives them access to tissues that are poorly accessible by intact antibodies; rapid clearance from blood and non-targeted tissues, and at the same time they exhibit high affinity properties to antigens. As far the PSMA targeting concerns, an anti-PSMA minibody named IAB2M was genetically engineered from the parent antibody J591 and labeled with 89Zr [54]. Preclinical studies demonstrated faster clearance and rapid in vivo biokinetics, with efficient target penetration, allowing for high-contrast images within a few hours after the injection [46,55,56]. 89Zr-IAB2M [54] showed properties similar to that of 89Zr-J591 [34], coupled with a significantly faster blood clearance and lower uptake at 24 h after the injection in PSMA-positive tissue compared to 89Zr-J591, as shown by the PET images [57]. The latter finding can be attributed to the extended blood circulation of the intact antibody rather than loss of the affinity of IAB2M. Nevertheless, the longer circulation may benefit immunotherapies, but a key element for the successful diagnosis of cancer is a fast blood clearance in combination with high and retained tumor uptake, leading to high tumor-to-background ratios over time. The first-in-human imaging with 89Zr-IAB2M in metastatic prostate patients showed that PET imaging is feasible. Furthermore, favorable biodistribution profile in patients with advanced prostate cancer was also revealed. In addition to these characteristics, targeting of both bone and soft tissue lesions was observed after the administration of 89Zr-IAB2M [54].
Although immunoPET holds a very important role in the field of diagnostic imaging, the long blood circulation in combination with the nonspecific accumulation in normal tissues may lead to radiotoxicity of several vital organs such as the liver or the spleen. These obstacles could be overcome by generating smaller molecules which show fast blood clearance and retained radiotracer target specificity. The development of the genetically engineered immunological constructs properly modified in order to be used as nuclear probes can be considered as alternative imaging tools for cancer imaging.
2.2. Aptamers as Radiotracers for Prostate Cancer Imaging and Therapy
Aptamers are synthetic small molecules of 8–15 kDa is size which can be readily modified in a site-specific manner [58]. In particular, they are single-chain oligonucleotides that are selected from high-complexity RNA (or DNA) pools [58,59]. Their characteristics are described by tight binding to and inhibition of molecular targets. They can be chemically modified in such a way that they exhibit low immunogenicity and toxicity, in addition to an increased half-life in the circulation, rendering them attractive molecules either for diagnostic or therapeutic purposes. In fact, a variety of aptamer-based conjugates accumulate in tumors, suggesting that with specific modifications they may be used for imaging and potentially therapeutic applications [18,60,61,62]. Within this concept, two anti-PSMA aptamers, A9 and A10, have been developed and evaluated by Lupold et al. for their potential as specific targeting agents of prostate cancer [63]. They both exhibit specific binding to PSMA-positive cells in vitro. Additionally, A9g and A10-3.2 are the truncated versions of A9 and A10 respectively, which show enhanced binding affinity to PSMA and, importantly, enable inhibition of NAALADase/glutamate carboxypeptidase II activity. Rockey et al. published the functionalization of A10-3.2 with the chelators 1,4,7,10-tetraazacyclododecane-1,4,7-triaceticacid mono-N-hydroxy-succinimide (DOTA-NHS), S-2-(4-isothiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid (p-SCN-Bn-NOTA), p-SCN-Bn-3,6,9,15-tetraazabicyclo [9.3.1]pentadeca-1(15),11,13-triene-3,6,9-triacetic acid (p-SCN-Bn-PCTA) and 1,8-diamino-3,6,10,13,16,19-hexaazabicyclo(6,6,6)eicosane (diAmSar) and their subsequent labeling with 64Cu [64]. The NOTA and PCTA constructs appear to be the most promising for further studies of the chelator-aptamer derivatives; however, in vivo images or biodistribution data of these 64Cu-labeled agents have so far not been reported.
2.3. PSMA Inhibitors of Low Molecular Weight
Since 2-PMPA (compound 1, Figure 2) was firstly reported as a potent PSMA inhibitor in 1996 [65], thorough efforts have been devoted to generating further molecules with inhibitory action towards PSMA. The main strategy for the discovery of those inhibitors was to find zinc-binding groups which are linked to a glutamate moiety. Three functionalities with affinity for zinc, including phosphonates (phosphates, phosphoramidates), thiols and ureas (compounds 2–6, Figure 2) have been identified [66,67,68,69,70,71]. In the field of radiopharmaceutical development, a great number of phosphoramidates and urea inhibitors have been synthesized and modified accordingly in order to be labeled with a variety of radiometals. In the following chapters we will have a closer look on the derived radiotracers from these two categories.
3. Phosphoramidate-Metal Based PSMA Inhibitors
Misra et al. reported the development of a series of irreversible phosphoramidate inhibitors (Figure 3) that also target PSMA with high affinity and specificity [72]. The starting molecule for the synthesis of the new PSMA inhibitors was GPI (compound 7, Figure 3). The phosphoramidate-based PSMA inhibitors were shown to rapidly internalize into subcellular organelles of PSMA-positive (PSMA+) prostate tumor cells, presumably through the enzyme-inhibitor complex [73]. 99mTc-labeling of the derived inhibitors was obtained using a simple, cartridge-based, solid phase prelabeled 2-mercaptoacetyl-Ser-Ser-Ser- (MAS3) chelator. However, no in vivo imaging studies have been reported.
The modification of another irreversible phosphoramidate inhibitor, named CTT-54 (compound 11, Figure 4), for the delivery of 99mTc(CO)3-DTPA-CTT-54, was reported by Nedrow-Byers et al. in 2012. 99mTc(CO)3-DTPA-CTT-54 was evaluated in vitro and in vivo for its potential as a SPECT imaging nuclear probe for PSMA targeting [74]. Although DTPA may not be the ideal chelator for [99mTc(CO)3]+, the results from the study justify a proof-of-concept evidence for the development of a next-generation of phosphoramidate-based PSMA inhibitors as SPECT imaging agents.
A PET tracer, 64Cu-ABN-1 (compound 12, Figure 5) was generated from the irreversible phosphoramidate inhibitor of PSMA CTT-1297 after its conjugation to the phosphonate-pendant-armed cross-bridged chelator, 1,4,8,11-tetraazacyclotetradecane-1-(methanephosphonic acid)-8-(methanecarboxylic acid) (CB-TE1A1P) [75], and the incorporation of 64Cu [74]. 64Cu-ABN-1 was evaluated for its selective uptake both in vitro and in vivo with PSMA-positive cells. A particular focus was given to assess the ability of this 64Cu-labeled radiotracer to detect and distinguish elevated levels of PSMA in a panel of prostate tumor-bearing mouse models. The study showed that 64Cu-ABN-1 demonstrated selective uptake in PSMA-positive cells and tumors, which was also correlated to the level of PSMA expression. 64Cu-ABN-1 may serve as a non-invasive method to follow the progression of prostate cancer in the future.
The irreversible binding profile of the phosphoramidate-based PSMA inhibitors is desirable because it induces a higher rate of internalization compared to the slowly released, but reversiblebinding of phosphate-based inhibitors and the reversible profile urea- and phosphonic-based inhibitors. The phosphoramidate-based PSMA inhibitors are effective in delivering a therapeutically relevant dose from a relatively low dose due to the higher internalization rate which reduces radiation exposure of patients. The further optimization of this class of PSMA targeting agents has the potential to enable efficient in vivo targeting of PSMA positive tumors either for radiodiagnostic or radiotherapeutic applications.
4. Urea-Based PSMA Inhibitors
In 2001 the Kozikowski group was the first to first synthesize and evaluate urea-based PSMA inhibitors [70]. In 2002, the Pomper group at John Hopkins School of Medicine (Baltimore, MD, USA) published the synthesis and preclinical evaluation of the first radiolabeled urea-based PSMA inhibitor, (N-(N-[(S)-1,3-dicarboxypropyl]carbomoyl)-S-11C-methyl-l-cysteine) (11C-DCMC), also referred to as 11C-MeCys-C(O)-Glu (11C-MCG) [75]. To date, a variety of urea-based PSMA inhibitors have been developed and labeled with different SPECT (99mTc, 111In), PET (68Ga, 64Cu), and therapeutic (177Lu) radiometals using several chelators and showed great promise not only preclinicaly but also in the clinical assessment. Apart from the radiometals to be used as the cytotoxic units, a considerable amount of urea-based PSMA inhibitors labeled with 11C, 125I, 124I, 131I and 18F, have also been reported [19,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96]. These radiotracers were also able to successfully image PSMA-expressing xenografted mice, but since this is beyond the focus of this review no further information will be given here with regard to this topic.
The development of urea-based PSMA precursors for subsequent labeling with radiometals requires the attachment of a relatively bulky chelator to the peptidomimetic structure of the PSMA inhibitors. As it has been noticed earlier in this review, a ~20 Å deep tunnel connects the extracellular surface of PSMA with the active site of the enzyme, rendering difficult the entry of the radiometal-bearing chelator into the enzymatic binding site. Therefore, the insertion of a spacer between the urea-based motif and the chelator is essential. Although the binding affinity can be considered as the most crucial parameter which affects the tumor uptake, the overall pharmacokinetic performance of a radiotracer is determined by many other factors which certainly need to be taken into consideration during the development of potential radiopharmaceuticals. In particular, parameters such as the nature of the spacer, lipophilicity, charge, plasma protein binding and molecular weight also influence the pharmacokinetic performance of a radiotracer [97]. Indeed, the presence of the spacer not only greatly influences the affinity of the derived radiotracers towards PSMA but also the pharmacokinetic properties of the chelator-conjugated PSMA inhibitors [97].
4.1. 99mTc-Labeled Urea-Based PSMA Inhibitors
99mTc remains the radionuclide of choice for SPECT applications and a variety of PSMA inhibitors have been modified to facilitate efficient labeling with 99mTc. Banerjee et al. have published a series of PSMA inhibitors suitable for labeling with 99mTc [98,99]. The influence of the spacer length as well as the nature of different chelators was thoroughly investigated with respect to the overall performance of the derived 99mTc-labeled Lys-urea-Glu-based PSMA inhibitors (compounds 13–25, Figure 6, Table 1). In particular, the PSMA inhibitors are attached to hydrophilic ligands for radiolabeling with the 99mTc(I)-tricarbonyl-labeled ([99mTc(CO)3]+) core, traditional NxSy-based chelator for the 99mTc-oxo ([99mTcO]3+) core, and a 99mTc-organohydrazine (99mTc-hynic) core. It was proved that parameters such the charge, polarity and hydrophilicity are crucial and contribute not only to the efficient targeting of PSMA by the 99mTc-labeled agent but also to its pharmacokinetics.
The above tested low-molecular-weight, urea-based, 99mTc-labeled, PSMA-targeted radiotracers, differ in the chelator and in the lipophilicity. Except for HYNIC-labeled 99mTc-25 irrespective of charge and lipophilicity the radioligands 99mTc-(13-14) enabled visualization of PSMA positive tumors (>20 % IA/g at 2 h p.i.) and kidneys. More specifically, the study showed that the degree of lipophilicity had a considerable effect on binding affinity as well as on in vivo performance. The ligands which showed higher lipophilic character revealed higher inhibitory activity, lower tumor and higher background uptake. Indeed, compounds 13 to 15, which possess different degree of lipophilicity due to different number of Phe moieties (from zero (13) to three (15)) on the spacer, displayed considerable differences in tumor and background uptake as measured by SPECT/CT images from male SCID mice bearing PSMA+ PC3 PIP and PSMA− PC3 flu tumors at 2 h p.i. High gallbladder, liver, and gastrointestinal uptake were observed for 99mTc-15 as compared to that of 99mTc-13. Between the radioligands bearing the tricarbonyl core, 99mTc-21 and 99mTc-22, the neutral complex 99mTc-22, exhibited favorable pharmacokinetics and promising SPECT/CT images, presumably because of the comparatively low background within the gastrointestinal tract relative to the positively charged 99mTc-21 (99mTc-21: small intestine: 1.29 ± 0.76 % IA/g, large intestine: 16.02 ± 12.39 % IA/g at 1 h p.i., 99mTc-22: small intestine: 1.35 ± 0.19 % IA/g, large intestine: 0.29 ± 0.06 % IA/g at 1 h p.i.). In addition, 99mTc-22, the most hydrophilic radioligand in the tricarbonyl series, had high, specific and retained tumor uptake in the PSMA+ PC3 PIP tumor (28.31 ± 4.38, 28.05 ± 2.04, 26.29 ± 7.45, 23.22 ± 6.02 % IA/g, at 0.5, 1, 2 and 5 h p.i. respectively). Another point which also needs to be pointed out is that the 99mTc-oxo radioliagnds showed high and prolonged spleen uptake, which was not observed with the tricarbonyl-based radioligands. 99mTc-22 proved superior with respect to tumor-to background ratios over time. Despite high tumor uptake, other compounds had either high gastrointestinal or spleen uptake, which cannot be easily attributed on their charge and lipophilicity. No clinical studies with of these 99mTc compounds have been reported.
Six targeted radioimaging agents labeled with 99mTc derived from the Lys-urea-Glu peptidomimetic structure functionalized with the chelator Dap-Asp-Cys through spacers of different lengths (compounds 26–31, Figure 7, Table 2) were investigated by Kularante et al. [100].
This study demonstrates the importance of the spacer between the urea-based PSMA inhibitors and the chelator which have been used for their functionalization. Docking studies revealed that coupling the urea analogue to an 8-aminooctanoic acid moiety (to avoid steric overlap within the narrow regions at the base of the tunnel) followed by two phenylalanine residues (for maximal interaction with hydrophobic pockets near the mouth of the tunnel) generated an agent (compound 28) with high affinity and specificity. Also the most lipophilic compound was found to be the most affine towards PSMA. The in vivo data were in accordance with the findings from in vitro studies, with remarkable tumor uptake upon injection in nu/nu mice xenografted with LNCaP tumor cells (average 9.8 ± 2.4 % IA/g at 1 h p.i.) was observed for radiotracers 27, 28, and 29 with little accumulation in other tissues except the kidneys. These ligands have so far not been transferred to clinical evaluation.
Robu et al. recently published the synthesis of two 99mTc-labeled anti-PSMA agents [101]. In this study the Lys-urea-Glu motif was coupled to the spacer d-Tyr-d-2-Nal-d-Lys-suberoyl (y-nal-k-Sub) to enhance interaction of the peptidic spacer with a remote arene binding site and the functionalization of the precursor took place through the conjugation of the chelators MAS3 (2-mercaptoacetyl-Ser-Ser-Ser-) (compound 32, Figure 8) and mas3 (2-mercaptoacetyl-d-Ser-d-Ser-d-Ser-) (compound 33, Figure 8). The derived conjugates labeled with 99mTc (Table 3).
The main focus of the study was to investigate whether or not the all-L-amino acid chelator MAS3 is susceptible towards proteolytic degradation. As anticipated, no detectable influence of chelator stereochemistry on the outcome of the 99mTc-labeling reaction was observed, since the formation of the 99mTcO-MAS3/mas3-complex should be independent from the spatial orientation of the serine side chains. On the other hand, the MAS3-chelating 99mTc-labeled tracer showed substantially decreased in vivo stability compared to the mas3-derivative 99mTc-PSMA-I&S (PSMA-I&S for Imaging and Surgery), for which only intact tracer was detected in blood, urine and kidney at 1 h p.i. Furthermore, 99mTc-PSMA-I&S uptake in kidney and LNCaP-tumor was found to be high (186 ± 23, 8.28 ± 3.27 % IA/g at 1 h p.i., respectively) and specific, as shown by the blocking studies. The straightforward and reliable kit-production and the initial patient data indicate the potentiality of 99mTc-PSMA-I&S as a SPECT imaging agent.
In another study, Kimura et al. [102] synthesized and evaluated a novel anionic 99mTc-tricarbonyl complex (99mTc-TMCE) with high hydrophilicity due to strong polarity and electric charges with the intent to further decrease non-target tissue uptake and enhance renal clearance (compound 34, Figure 9, Table 4). 99mTc-TMCE was obtained in very low radiochemical yield (12–17%), introducing the implementation of a HPLC for purification of the radioligand. Compared to the neutral and positively charged tricarbonyl core-bearing radioligands published by Banerjee et al. the in vivo performance of the anionic 99mTc-tricarbonyl complex 99mTc-TMCE is characterized by a relatively long blood circulation and fast washout from the tumor (blood: 11.4 ± 2.3, 3.2 ± 0.5, 0.6 ± 0.6 ± 0.1 % IA/g and LNCaT-tumor: 4.0 ± 1.2, 12.8 ± 2.2 and 5.0 ± 2.7 % IA/g at 5, 30 and 120 min p.i. respectively) The outcome of the study demonstrated the effect of affinity, hydrophilicity, and electric charge in the pharmacokinetics of PSMA imaging probes. In particular, the hydrophilicity of both cationic and anionic charges led to rapid hepatobiliary clearance, although anionic charges might enhance renal clearance to a greater extent (kidney: 124.9 ± 26.2, 136 ± 6.4 and 56.8 ± 20.6 % IA/g at 5, 30 and 120 min p.i.). The implementation of this 99mTc-labeled anti-PSMA radiotracer into the clinical pathway has not taken place yet.
Four small-molecule inhibitors for PSMA labeled with 99mTc via technetium tricarbonyl chemistry were reported by Hillier et al. [103]. The 99mTc-labeled PSMA inhibitors derived from the glutamate-urea-glutamate or glutamate-urea-lysine pharmacophores after the incorporation of the chelators 2,2′-(2,2′-(azanediylbis(methylene))bis(1H-imidazole-2,1-diyl))diacetic acid (CIM) or 2,2′,2′′,2′′′-((2,2′-(2,2′-(azanediylbis(methylene))bis(1H-imidazole-2,1-diyl))bis(acetyl))bis(azanetriyl))tetraacetic acid (TIM) and the labeling with 99mTc (compounds 35–38, Figure 10) were evaluated in vitro and in vivo (Table 5).
The introduction of the chelators CIM and TIM to glutamate-urea-glutamate or glutamate-urea-lysine pharmacophores and the subsequent labeling with 99mTc did not influence the affinity of the derived radioligands towards PSMA (Table 5), as their KD values were found to be in the low nanomolar range. Furthermore, a side-by-side comparison of the affinity reveals a slightly higher affinity for PSMA of the glutamate-urea-glutamate-containing compounds, 99mTc-MIP-1404 and 99mTc-MIP-1427. The formal addition of an extra carboxylic acid group to the CIM chelator, yielding the TIM chelator, resulted in improved pharmacokinetics of the derived radioligands. Indeed, 99mTc-MIP-1404 and 99mTc-MIP-1428 displayed lower uptake in and more rapid clearance from blood, liver, kidneys (99mTc-MIP-1404: blood: 0.13 ± 0.03 and 0.02 ± 0, liver: 0.14 ± 0.03 and 0.07 ± 0.01, kindey: 105 ± 37 and 12 ± 7 % IA/g at 1 and 4 h p.i. respectively, 99mTc-MIP-1428: blood: 0.44 ± 0.07 and 0.14 ± 0.03, liver: 0.27 ± 0.05 and 0.14 ± 0.04, kidney: 136 ± 6 and 63 ± 27 % IA/g at 1 and 4 h p.i. respectively), and most other nontarget tissues than did 99mTc-MIP-1405 and 99mTc-MIP-1427 (99mTc-MIP-1405: blood: 0.72 ± 0.19 and 0.26 ± 0.03, liver: 0.43 ± 0.14 and 0.19 ± 0.05, kidney: 157 ± 69 and 14 ± 6 % IA/g at 1 and 4 h p.i. respectively, 99mTc-MIP-1427: blood: 0.55 ± 0.08 and 0.28 ± 0.03, liver: 0.83 ± 0.24 and 0.76 ± 0.26, kidney: 149 ± 24 and 142 ± 39 % IA/g at 1 and 4 h p.i. respectively). The chelator TIM did not diminish uptake or retention in the LNCaP xenograft tumor, as there was no statistical difference between the tumor uptake of any of the compounds at either the 1 h or the 4 h time points. More specifically, uptake in LNCaP xenografts ranged from 9.3% to 12.4% injected dose per gram at 1 h after injection and from 7.2% to 11.0% at 4 h, with tumor-to-blood ratios ranging from 29:1 to 550:1 and tumor–to–muscle ratios ranging from 31:1 to 157:1 at 4 h. 99mTc-MIP-1404 is currently under clinical investigation in a phase II trial.
4.2. 68Ga-Labeled Urea-Based PSMA Inhibitors
Because of its availability from the 68Ge/68Ga generator systems and the relative ease of labeling chemistry, the positron emitter 68Ga (t½ = 68 min, β+max = 1899 keV) has gained increasing interest in the field of molecular imaging [104] and can considered a PET-applicable counterpart of 99mTc.
Many efforts have been made with respect to the development of 68Ga-PSMA-based imaging agents in the hope of developing a cyclotron-independent nuclear probe for prostate cancer imaging. A variety of PSMA-based inhibitors have been generated where several chelators suitable for labeling with 68Ga as well as spacers have been investigated. In solution, gallium is most stable in the +3-oxidation state and forms stable complexes with several chelating agents. The HBED chelator (N,N′-bis[2-hydroxybenzyl] ethylenediamine-N,N′-diacetic acid) which contains an amine-phenol backbone which renders it suitable for complexation with +3 metal ions, and it forms a thermodynamically stable complex with 68Ga even at room temperature [105]. A representative example of PSMA-inhibitors associated with the chelator HBED, is the peptidomimetic structure Lys-urea-NH-Glu, published by Eder et al., when coupled to the spacer 6-aminohexanoic acid (Ahx) and functionalized with the chelator N,N′-dis[2-hydroxy-5-(carboxyethyl)benzyl] ethylenediamine-N,N′-diacetic acid (HBED-CC) to obtain HBED-CC-Ahx-Lys-urea-Glu (HBED-CC-PSMA or PSMA-11) (compound 39, Figure 11) [106]. HBED-CC has a dual nature, it also acts as a lipophilic moiety due to the presence of the two phenolic rings and therefore an aliphatic spacer such as Aca is adequate for providing the required lipophilicity and retaining the affinity of the derived ligand towards PSMA (Ki = 12.1 ± 2.1 nM). Radiolabeling with 68Ga was performed at pH 4.2 by incubating PSMA-11 in a mixture consisting of 50–100 MBq [68Ga]Ga3+ and HEPES. An amount of 0.1 nmol of PSMA-11 at a final concentration of 1.7 μM led to a radiochemical yield of more than 99% in less than 1 min at room temperature, with specific activities in the range og 500–1000 GBq/μmol. These are the highest specific activities which have been reported for 68Ga-lebeled PSMA inhibitors. In order to achieve comparable high radiochemical yields with the DOTA-conjugated PSMA inhibitors, the compounds were incubated for 2 min at 80 °C using 1 nmol of precursor [106]. 68Ga-PSMA-11 exhibited fast blood clearance, relatively low liver uptake (0.87 ± 0.05 % IA/g at 1 h p.i.), and high specific uptake in PSMA-expressing tissues and tumor (tumor uptake 7.7 ± 1.5 % IA/g at 1 h p.i.). The ability to image PSMA using 68Ga-HBED-CC-PSMA showed great promise not only preclinically but also clinically [106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129]. The dimerization of the same pharmacophore, Lys-Urea-Glu, coupled to the same spacer Ahx, through the chelator HBED-CC (PSMA-10) (compound 40, Figure 11) was also published by Schäfer et al. [130]. The dimer was also labeled with 68Ga to obtain 68Ga-PSMA-10 and evaluated in vitro and in vivo compared to 68Ga-PSMA-11. Despite the fact that the preclinical evaluation revealed that 68Ga-PSMA-10 was superior to 68Ga-PSMA-11 in terms of binding affinity and tumor to background ratios (IC50 = 3.9 ± 1.8 nM with a target to non-target ratio of 26.5 at 1 h p.i. as compared to a value of 9.2 for the monomer at the same time point), most of the clinical studies, so far, have been conducted using 68Ga-PSMA-11.
In the field of radiopharmaceutical chemistry, the cyclic chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-triacetic acid (DOTA) has been used for complexation of +3 radiometal ions such as 68Ga, 90Y and 177Lu. Banerjee et al. described two DOTA-conjugated PSMA inhibitors based on the Lys-Urea-Glu construct that confers PSMA specificity (compounds 41 and 42, Figure 12) and are suitable for labeling with 68Ga [131]. The difference between those two tracers is confined to the spacer unit. More specifically, a benzyl group was inserted into compound 41 (Figure 12) in order to provide a chromophore that facilitates purification. On the other hand, two phenylalanines were added to compound 42 (Figure 12) in order to offset its high hydrophilicity and enable sustained retention and potentially higher absolute uptake in tumor [98]. Furthermore, in a recent study from the same group [132], a new radiotracer containing the macrocyclic chelator NOTA was prepared, since NOTA has been shown to be an effective chelator for 68Ga (compound 43, Figure 13,). A head-to-head preclinical comparison of 68Ga-42 with 68Ga-43 (Figure 12) was conducted. 68Ga-PSMA-11, the radioligand which has been used throughout Europe in clinical trials, was also included in the study for comparison 68Ga-41, 68Ga-42 and 68Ga-43 were directly labeled with the eluted 68Ga from the 68Ge/68Ga generator without the use of a buffer solution within 10 min at 90–95 °C. The yields ranged from 60% to 70% with HPLC purification and the reported radiochemical purities were >99% with specific activities >168 GBq/μmol. Both 68Ga-41 (−3.9) and 68Ga-43 (−4.01 ± 0.16) were found to be 1 order of magnitude more hydrophilic than 68Ga-42 (−3.0 ± 0.1). It is evident that 68Ga-42 is more lipophilic than 68Ga-41 and 68Ga-43, which is reasonable because of the presence of two phenylalanine residues in the spacer. The corresponding metal-labeled compounds demonstrated high binding affinity to PSMA, with Ki values ranging from 0.33 to 29 nM (compound 41 was the less affine towards PSMA).
Both compounds, 68Ga-41 and 68Ga-42, exhibited PSMA specific tumor imaging in vivo. Nevertheless, the improved tumor-to-background ratios of 68Ga-42 at later time points after injection renders it a more promising candidate for clinical translation. The PSMA+ PC3 PIP tumors as well as PSMA positive organs such as, kidney and urinary bladder were clearly delineated already at 15 min p.i. More specifically, the NOTA-conjugated radioligand 68Ga-43 exhibited the highest tumor uptake with 42.2 ± 6.7 % IA/g at 1 h p.i. and the fastest background clearance. The PSMA+ PC3 PIP-to-PSMA− PC3 flu tumor ratios were 110 ± 22 at 1 h, 232 ± 26 at 2 h, and 182 ± 15 at 3 h p.i. Renal uptake for 68Ga-43 was highest at 1 h, 106 ± 23 % IA/g, much higher than that seen for the DOTA-conjugated radioligand 68Ga-42 (26.5 ± 6.9 % IA/g), and showed faster renal clearance, which decreased to 34.7 ± 5.7 % IA/g by 2 h p.i. There was no significant difference in PSMA+ PIP tumor uptake between 68Ga-42 and 68Ga-PSMA-11 or between 68Ga-43 and 68Ga-PSMA-11 (p > 0.05) at any time-point. 68Ga-PSMA-11 demonstrated the highest and retained uptake in normal tissues, including kidney, blood, spleen, salivary glands and PSMA-negative PC3 flu tumors up to 3 h post-injection. This preclinical evaluation showed that 68Ga-43 was superior for PSMA-targeted PET imaging in clinical settings.
Benešová et al. [133], published the synthesis and preclinical evaluation a novel theranostic compound termed PSMA-617 (compound 44, Figure 13). In this case, the chelator DOTA, was conjugated to the pharmacophore Lys-urea-Glu via a naphthylic spacer. PSMA-617 was labeled with [68Ga]Ga3+ eluate in HEPES buffer, pH 4.0, within 15 min at 95 °C, with a radiochemical yield of more than 90% and a specific activity in the range of 14–140 GBq/μmol. The study showed that the presence of the naphthylic linker has a significant impact on the tumor-targeting as well as on the pharmacokinetics and the resulting imaging contrast. In fact, 68Ga-PSMA-617 (Ki = 6.40 ± 1.02 nM) was superior to 68Ga-PSMA-11 (Ki = 12.1 ± 2.1 nM) as far as the affinity towards PSMA and efficacy of internalization (up to 17.67 6 4.34 percentage injected activity/106 LNCaP cells) into the cancer cells concerns. Biodistibution studies upon injection of 68Ga-PSMA-617 on LNCaP tumor bearing mice 1 h p.i. revealed a high specific uptake in LNCaP tumors (8.47 ± 4.09 % IA/g; 0.98 ± 0.32 % IA/g by coinjection of 2-PMPA) and in the kidneys (113.3 ± 24.4 % IA/g). Other organs such as the liver (1.17 ± 0.10 % IA/g), lung (1.41 ± 0.41 % IA/g), and spleen (2.13 ± 0.16 % IA/g) showed rather low uptake. Furthermore, 68Ga-PSMA-617 dynamic PET imaging showed that the maximum kidney uptake was reached within 15 min after injection and decreased substantially as early as 20 min p.i. High and sustained tumor uptake was observed. The fast kidney clearance emboldened clinical translation of this compound.
When Lys-Urea-Glu (KuE) was coupled to the spacer Phe-Phe-Lysine-suberoyl (l-amino acid spacer, FFK-Sub) and functionalized with the chelator (1-(1,3-carboxypropyl)-4,7,10(carboxymethyl)-1,4,7,10 tetraazacyclododecane (DOTAGA) DOTAGA-FFK(Sub-KuE) was obtained [134]. HEPES buffer was used for the labeling with 68Ga (pH 4.5). The reaction completed within 5 min at 95 °C and resulted in a radioligand with a specific activity of 250–300 GBq/μmol. However, a rapid in vivo metabolysis of the 68Ga-labeled radiovector was demonstrated. In the same report by Weineisen et al. the inclusion of a D-amino acids spacer led to an in vivo metabolic stable radiotracer (DOTAGA-ffk(Sub-KuE)) (compounds 45–47, Figure 14).
In an attempt from the same group to further optimize this second-generation of the PSMA inhibitor with respect to its affinity towards the PSMA enzyme, additional modifications on the spacer unit were undertaken. [135]. For that purpose, d-Phe (f) was substituted with d-I-Tyr (I-y) and DOTAGA-(I-y)fk(Sub-KuE) was synthesized (Figure 14). DOTAGA-(I-y)fk(Sub-KuE) is also termed PSMA I&T (PSMA I&T for Imaging &Therapy), since it can be labeled with both the diagnostic radionuclide 68Ga and the therapeutic radionuclide 177Lu. The necessary modifications for the generation of a metabolically stable radioligand did not alter the affinity of the derived ligands towards PSMA, since all the precursors as well as the corresponding metalloconjugates exhibited IC50 values within the range of 8 to 16 nM. 68Ga-PSMA I&T is characterized by rapid tumor targeting (4.95 ± 1.57 % IA/g at 1 h p.i.) and pharmacokinetics with high uptake in PSMA-positive organs such as the tumor and the kidneys (53.26 ± 9.02 % IA/g at 1 h p.i.). 68Ga-PSMA I&T is currently under clinical investigation.
The enantiomerically pure prochelator (R)-1-(1-carboxy-3-carbotertbutoxypropyl)-4,7-carbotartbutoxymethyl)-1,4,7-triazacyclononane ((R)-NODAGA(tBu)3) was reported by Gourni et al. for the functionalization of urea-based PSMA inhibitor derived from the Lys-urea-Glu peptidomimetic structure. The spacer Phe-Phe-d-Lys(suberoyl) was included to obtain (R)-NODAGA-Phe-Phe-d-Lys(suberoyl)-Lys-urea-Glu (CC34) (compound 48, Figure 15) [136]. 68Ga-CC34 was obtained in high specific activity (75–80 MBq/nmol) and evaluated in vitro and in vivo in LNCaP tumor xenografts by biodistribution and PET imaging studies. 68Ga-PSMA-11 was also evaluated for comparison. 68/natGa-CC34 exhibited high affinity for the LNCaP cells, with a KD value of 19.3 ± 2.5 nM. Tumor uptake of 68Ga-CC34 (14.5 ± 2.9 % IA/g) in LNCaP xenografts at 1 h p.i. was comparable to 68Ga-PSMA-11 (15.8 ± 1.4 % IA/g) (p = 0.67). The tumor-to-normal tissue ratios at 1 and 2 h p.i of 68Ga-CC34 were also comparable to that of 68Ga-PSMA-11 (p > 0.05).
In another recent preclinical study, the cyclohexyl-diethylene triamine pentaacetic acid (CHX-A″-DTPA) was used as the chelator for the Glu-urea-GLu-based peptide (Pep) bearing a 2-[3-(1,3-dicarboxypropyl)-ureido]pentanedioic acid (DUPA) moiety to obtain CHX-A″-DTPA-DUPA-Pep (compound 49, Figure 16). The study describes the efficient labeling of CHX-A″-DTPA-DUPA-Pep with 68Ga, 90Y and 177Lu and the first in vitro characterization which shows high affinity of the tested radiotracers towards PSMA with KD values of ≤14.67 ± 1.95 nM. Labelling with 68Ga was performed at room temperature under neutral conditions (HEPES, pH 7.4). Significant differences in radiochemical yields were observed. Radio labelling with 68Ga succeeded in 10 min with high radiochemical yields; ≥95% when 50 μg (36 nM) of the peptide was used. However, in vivo images or biodistribution data of these agents have so far not been published [137].
4.3. 64Cu-Labeled Urea-Based PSMA Inhibitors
Amongst the PET-radioisotopes, 64Cu has gained particular attention because of its decay characteristics (t1/2 = 12.7 h; β+, Emax = 0.653 MeV [17.8%]; β−, Emax = 0.579 MeV [38.4%]) and the well-established coordination chemistry with a variety of chelators. The longer half-life of 64Cu relative to 68Ga (t1/2 = 67.8 min) and 18F (t1/2 = 109.8 min) is particularly attractive, as it allows PET-images at later time points with improved tumor-to-background ratios [138]. Additionally, 64Cu-labeled radiopharmaceuticals can be produced at a central facility and distributed to remote hospitals. The successful application of 64Cu as a diagnostic agent may also lead to potential targeted radionuclide therapy with 67Cu (t1/2 = 61.9 h; β−, Emax = 0.141 MeV [100%]) providing a promising theranostic pair.
Banerjee et al. reported the synthesis and preclinical evaluation of a series of five PSMA inhibitors labeled with 64Cu [139]. The main focus of the study was given on the effect of various chelators on PSMA-targeted ureas with respect to their pharmacokinetics for in vivo PET imaging. The investigated chelators are 1,4,7,10-tetraazacyclodoadecane-N,N′,N″,N′′′-tetraacetic acid, (DOTA), oxa-4,7,1-tetraazacyclododecane-4,7,10-triacetic acid, (Oxo-DO3A), 1,4,8,11-tetraazabicyclo[6.6.2]-hexadecane-4,11-diacetic acid, (CB-TE2A), 3,6,9,15-tetraazabicyclo[9.3.1]-pentadeca-1(15),11,13-triene)-3,6,9-triacetic acid (PCTA) and 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) (compounds 50–54, Figure 17). The radiolabeling with 64Cu was performed in acetate buffer and the conditions differed based on the chelator (Table 6). The metal-free CB-TE2A-conjugated ligand 53 contained two nearly inseparable isomeric peaks by HPLC. This is most probably caused by the presence of an asymmetric center at the Phe moiety of the spacer, located very close to CB-TE2A. Radiolabeling of 53 resulted in two HPLC separable products, named 64Cu-53A and 64Cu-53B. All radioligands were obtained in specific activities in the range 2.9–9.1 GBq/μmol. 64Cu-53A, 64Cu-53B and 64Cu-54 were found to be more hydrophilic despite the presence of two Phe moieties at the spacer. The diastereomeric metalloconjuagtes 53A and 53B were the ones with highest affinity for PSMA. PET imaging as well as ex vivo biodistribution studies showed that 64Cu-50 and 64Cu-53A/B (NOTA and CB-TE2A chelators, respectively) exhibited high in vivo stability as evidenced by their lower liver uptake than that of the other three conjugates (blood: 1.06 ± 0.29, 1.87 ± 0.6, 0.20 ± 0.03 and 0.20 ± 0.07 % IA/g; liver: 8.63 ± 0.92, 17.04 ± 1.44, 1.68 ± 0.38 and 1.63 ± 0.72 % IA/g for 64Cu-50, 64Cu-51, 64Cu-53A and 64Cu-53B at 2 h p.i.). An additional finding of this study was that liver uptake and blood concentration were much lower at all time points for 64Cu-53A/B compared to that of 64Cu-50, suggesting a higher in vivo stability of CB-TE2A-conjugated 64Cu-53A/B than of NOTA-conjugated 64Cu-50. That might also be related to the higher hydrophilicity of 64Cu-53A/B compared to 64Cu-50. On the other hand, the high liver uptake and the slow blood clearance for the 64Cu-DOTA-conjugated radiotracer were indicative of free Cu(II), which is accumulated in liver [140,141]. Noteworthy is also the fact that negatively charged 64Cu-labeled NOTA-conjugated PSMA inhibitor, 64Cu-50, similarly to 99mTc-oxo compounds [99], exhibited higher kidney and spleen uptake compared to 64Cu-53A/B (kidney: 125 ± 42, 26 ± 9 and 25 ± 11 % IA/g; spleen: 13.42 ± 1.18, 0.39 ± 0.30 and 0.90 ± 0.23 % IA/g for 64Cu-50, 64Cu-53A and 64Cu-53B at 2 h p.i.) Furthermore, although the PSMA-mediated renal uptake has been shown to be specific by several groups, it is interesting that variations are observed not only in regard to the absolute kidney uptake, but also with regard to the renal elimination.
The NODAGA-conjugated PSMA inhibitor CC34 (Figure 15), was also radiolabeled with 64Cu in ammonium acetate buffer, pH 5.4, to completion within 30 min at 95 °C, and was used without further purification. The specific activity of 64Cu-CC34 was 40 MBq/nmol and a logP of −3.01 ± 0.06. When CC34 was labeled with 64Cu, the resulting tracer exhibited a slightly higher lipophilicity than did 68Ga-CC34 (LogP = −3.54 ± 0.06) [136]. 64Cu-CC34 was preclinically evaluated in LNCaP xenografted mice. A high affinity towards PSMA on LNCaP cells was shown (KD = 27.5 ± 2.7 nM). Furthermore, the versatility of NODAGA was exploited for efficient labeling also with 64Cu, which allows for the conduction of biodistribution/imaging studies at later time points compared to 68Ga-CC34, and thus a complete assessment of the pharmacokinetics of the radiotracer. 64Cu-CC34 is specifically taken up by the PSMA positive organs at early time points and washed out faster from the PSMA positive organs compared to uptake in tumor, leading to improved tumor to background ratios (tumor/blood: 41 ± 10 and 114 ± 17, tumor/muscles: 61 ± 18 and 103 ± 21 at 1 and 4 h p.i., respectively). The very low liver uptake of 64Cu-CC34 at all time points in combination with the short blood circulation, comparable to what was also reported for the CB-2A-conjugated PSMA inhibitor from Banerjee et al. [139] is a strong indication of the excellent in vivo stability of the 64Cu-NODAGA complex.
4.4. 111In-Labeled Urea-Based PSMA Inhibitors
111In has suitable nuclear characteristics (t½ = 2.8 d, E(γ) = 173, 245 keV) for use as an important SPECT label. Additionally, intraoperative gamma probes are now an important, well-established technology in the management of cancer, particularly in the detection of sentinel lymph nodes [140]. So far two already established PSMA inhibitors, DOTAGA-(I-y)fk(Sub-KuE) (also named PSMA-I&T) and PSMA-617, have been labeled with 111In, as companion nuclear probes for radioguided surgery and SPECT imaging [135,136,142].
111In-PSMA-I&T exhibited high PSMA-affinity (natIn-PSMA-I&T: IC50: 7.5 ± 1.5 nM) and enhanced internalization (104 ± 7%) compared to 177Lu-PSMA-I&T (76 ± 2%) into LNCaP cells. Biodistribution studies in LNCaP tumor-bearing mice (1 h p.i.) revealed slightly reduced background accumulation of 111In-PSMA-I&T compared to 177Lu-PSMA-I&T and comparable tumor uptake of both compounds. These findings led to a somehow improved tumor/background ratios for 111In-PSMAI&T compared to 177Lu-PSMA-I&T at 1 h p.i. (tumor/blood-, tumor/liver-, tumor/intestines-, and tumor/muscle-ratios of 34 ± 8, 32 ± 6, 53 ± 8, and 43 ± 6, respectively, versus 18 ± 9, 7 ± 3, 12 ± 3, and 14 ± 9 for 177Lu-PSMA-I&T) [135,142].
111In-PSMA-617 was prepared within 30 min at 95 °C in ammonium acetate buffer pH 5.4 and were used without any further purification with a specific activity of 10 MBq/nmol and a KD value of approximately 5 nM The pharmacokinetics of 111In labeled naphthyl-containing DOTA-conjugated PSMA inhibitor, PSMA-617were excellent, with impressive tumor to background ratios over time (i.e., tumor/blood-, tumor/kidney-, and tumor-muscle ratios of 72 ± 19, 0.2 ± 0.01, 148 ± 61; 772 ± 153, 0.4 ± 0.04, 582 ± 24; 4768 ± 110, 9 ± 2, 2819 ± 864; 5343 ± 1033, 16 ± 2, 1987 ± 177 at 1-, 4-, 24-, and 48-h p.i. respectively) [136].
4.5. 177Lu-Labeled Urea-Based PSMA Inhibitors
The development of new radiolabeled specific biomarkers is mainly focused on the early diagnosis and prognostic prediction continuing with the adaptation of their therapeutic interventions that are individually optimized. Taking into account the theranostic approach, an ideal radiopharmaceutical should be able to combine the ability to be used for both diagnosis and therapy when labeled with a diagnostic or a suitable therapeutic radionuclide respectively. The beta emitter 177Lu can be considered as an appropriate therapeutic unit during the construction of a therapeutic nuclear probe due to its favorable characteristics (t1/2 = 6.73 days, Eβmax = 497 keV, Eγ = 113 keV (6.4%) and 208 keV (11%)).
The tailor-made DOTA-conjugated PSMA inhibitor, PSMA-617, has also been reported to be efficiently labeled with 177Lu [133] in addition to 68Ga. Radiolabeling with 177Lu reached a radiochemical yield of greater than 99% at low amount of precursor (0.5 mg, 0.5 nmol) in sodium acetate buffer, pH 5. The specific activity was in the range of 4–40 GBq/mmol. 177Lu-PSMA-617 exhibited high stability for at least 72 h, high binding affinity towards PSMA (Ki = 6.91 ± 1.32 nM for natLu-PSMA-617) along with enhanced internalization rate into the LNCaP prostate cancer cells. 68Ga-labeled PSMA-617 was specifically internalized up to 17.67 ± 4.34% injected activity/106 LNCaP cells and 177Lu-labeled PSMA-617 up to 17.51 ± 3.99% injected activity/106 LNCaP cells, both at 37 °C. The in vivo performance of the 177Lu-PSMA-617 was characterized by high and retained tumor uptake combined with a rapid clearance from the kidneys within 24 h p.i., which led to improved tumor to background ratios over time (1058 (tumor to blood) and 529 (tumor to muscle), 24 h p.i.). It is worth noting that 177Lu-PSMA-617 exhibited similar pharmacokinetics as 111In-PSMA-617 [131]. 177Lu-PSMA-617 seems attractive for endoradiotherapy due to its higher tumor uptake at later time points, lower spleen accumulation, and the highly efficient clearance from the kidneys. The clinical evaluation of 177Lu-PSMA-617 is under way.
Under the same concept of the theranostic approach, the PSMA inhibitors DOTAGA-ffk(Sub-KuE) and DOTAGA-(I-y)fk(Sub-KuE) (also named PSMA I&T) were also labeled with 177Lu and evaluated preclinically [135]. Radiolabeling with 177Lu performed in ammonium acetate buffer, pH 5, and the radiolabeled products were achieved in a specific activity of more than 38 GBq/μmol. Both cold lutetium analogues exhibited high affinity towards PSMA (IC50: natLu-DOTAGA-ffk(Sub-KuE): 13.1 ± 2.2 nM, natLu-PSMA I&T: 7.9 ± 2.4 nM). Compared with 177Lu-DOTAGA-ffk(Sub-KuE), tumor targeting of 177Lu-PSMA I&T was fast, with the highest uptake in tumor (7.96 ± 1.76 % IA/g at 1 h p.i.) xenografts and kidneys (107 ± 16 % IA/g at 1 h p.i.) (both PSMA-specific). 177Lu-PSMA I&T exhibits suitable targeting and retention characteristics and an assessment of this compound for application in endoradiotherapy is currently ongoing.
5. Clinical Assessment of Radiolabeled PSMA Inhibitors: Current Status
5.1. SPECT Imaging with 99mTc-Labeled PSMA Inhibitors
An exploratory investigational new drug application (IND) was implemented to bring the compounds MIP-1404 and MIP-1405 which been developed by the group at Molecular Insight Pharmaceuticals (MIP), and labeled with 99mTc, into the clinic. Vallabhajosula et al. [143], reported the comparison of the pharmacokinetics, biodistribution, and tumor uptake of 99mTc-MIP-1404 and 99mTc-MIP-1405 in 6 healthy men and 6 men with radiographic evidence of metastatic prostate cancer. SPECT imaging was performed between 3 and 4 h after injection of the radiotracers (Figure 18). Both radiotracers showed fast blood clearance persistent uptake in the salivary, lacrimal, and parotid glands. They localized in bone and lymph node lesions as early as 1 h. Because of the lower urinary activity of 99mTc-MIP-1404 (7%) compared to 99mTc-MIP-1405 (26%), a clear advantage for detecting prostate cancer in the gland and pelvis at early stages of the disease is indicated for this compound. 99mTc-MIP-1404 was selected for phase II studies to determine sensitivity and specificity to detect prostate cancer in high-risk patients. Furthermore, the development and optimization of a “cold” kit for the preparation of 99mTc-MIP-1404 using generator eluted pertechnetate has also been reported [144].
5.2. PET Imaging with 68Ga-Labeled PSMA Inhibitors
PET imaging of prostate cancer using PSMA-ligands has gained great attention during the last years. The numerous clinical trials evaluating 68Ga-PSMA-11 PET-CT imaging in prostate cancer patients have proved that this tracer can be used for lesion characterization, staging, planning and monitoring of therapy as well as evaluation of recurrence. 68Ga-PSMA-11 PET-CT imaging was firstly introduced in 2013 by Afshar-Oromieh et al. in 37 patients with relapsing prostate cancer (Figure 19) [107,145]. Within a relatively short time frame, 68Ga-PSMA-11 has been thoroughly clinically assessed by several research groups, indicative of the large number of publications the last four years [107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129]. Although the background uptake of 68Ga-PSMA-11 in salivary and lacrimal glands was abnormally high, PET-CT imaging was obtained leading to excellent detection of the metastatic lesions of prostate cancer.
Sterzing et al. [111] reported that PET-CT imaging with 68Ga-PSMA-11 may greatly contribute to the therapy planning of prostate cancer. The results of the study showed that 68Ga-PSMA-11 PET-CT had impact on the therapy regimen in 50.8% of the cases.
In a retrospective study, Afshar-Oromieh et al. [110] demonstrated that tumor detection mediated by 68Ga-PSMA-11 uptake was positively associated with prostate specific antigen (PSA) level and androgen deprivation therapy (ADT). Furthermore, 68Ga-PSMA-11 PET-CT imaging in patients with biochemical recurrence was proved to be crucial for recurrence evaluation, restaging as well as early treatment [112].
The ability of 68Ga-PSMA-11 to detect lesions of prostate cancer in patients with biochemical recurrence was also evaluated versus [18F]fluoromethylcholine [108,111]. In both studies 68Ga-PSMA-11 was found to be superior in detecting metastatic lesions associated with prostate cancer and displayed higher SUVmax values and superior tumor to background ratios. Another study by Dietlein et al. [113], PSMA-PET-CT imaging was conducting by applying 68Ga-PSMA-11 and 18F-DCFPyl in 14 patients with biochemical recurrence. 18F-DCFPyl was found to be somehow superior compared to 68Ga-PSMA-11 and could be considered as an alternative for PSMA-PET-CT imaging in relapsed prostate cancer.
The PSMA inhibitor PSMA-617 labeled with 68Ga was also clinically assessed in the diagnosis of prostate cancer by PET-CT [146]. 68Ga-PSMA-617 shows lesions of prostate cancer with high contrast, especially in late images (Figure 20). Maximum contrast of tumor lesions was achieved between 3 and 4 h after injection.
First-in-human 68Ga-PSMA I&T PET imaging allowed high-contrast detection of bone lesions, lymph node, and liver metastases [135]. In another study [143], 68Ga-PSMA I&T PET delayed images were also systematically evaluated after forced diuresis in 66 prostate cancer patients. The study explored the rather complex influence of delayed imaging and timing of forced diuresis on image quality. The authors suggest diuretic administration at 60 min p.i. in combination with delayed imaging at 180 min p.i. in order to obtain the maximum information possible, at least for 68Ga-PSMA I&T. However, the approach is time consuming [147].
5.3. PET Imaging with 64Cu-Labeled PSMA Inhibitors
The first clinical use of the 64Cu-labeled ligand PSMA-617 for PET imaging was conducted at two different centers in Austria and Germany [148]. 29 patients with either a high suspicion of recurrent disease were imaged in the context of therapy planning including surgery or PSMA radioligand therapy. The PET images showed an excellent resolution of the detected lesions with very high lesion to background contrast. An additional advantage of the 64Cu-labeled radiopharmaceuticals is the long half-life of 64Cu which allows distribution of the radiotracer to clinical PET centers where 68Ga-PSMA ligands are not available.
5.4. PSMA Radionuclide Therapy Using 177Lu-Labeled PSMA Inhibitors
177Lu-labeled monoclonal antibody J591, which binds to the extracellular domain of PSMA, was firstly used for radionuclide therapy of prostate cancer [149]. Although the response to therapy was encouraging, the dose-limiting toxicity which is transient myelosuppression was the major reason of abandoning this therapeutic approach. In contrast, the development and preclinical evaluation of new PSMA ligands which could be labeled with therapeutic radionuclides such as 177Lu, paved the way for the reinvestigation of the 177Lu-based radionuclide therapy in patients with advanced prostate cancer. The radiotracers which have been used of the conduction of those studies are 177Lu-PSMA-617 and 177Lu-PSMA I&T (Figure 21). The clinical trials reported so far demonstrate promising results [150,151,152,153,154,155,156] with a mean tumor dose 6 to 12 fold higher compared to other critical organs such as kidneys and salivary glands. The PSA levels were found to be reduced in 60–90% of the patients after a single dose of both 177Lu-labeled PSMA-based radiotracers. The patients tolerated the therapy well, with no significant nephrotoxicity and just rarely occurring other severe side effects were observed. Although PSMA-directed radionuclide therapy bears a potential for implementation in routine clinical practice, the limited number of studies so far requires further controlled clinical trials for the establishment of the clinical value of 177Lu-PSMA-617 and 177Lu-PSMA I&T. 177Lu-PSMA-617 recently received FDA approval for phase II clinical trials in the U.S., (“First U.S. multi-center investigational clinical trial of 177Lu-PSMA-617 targeted radioligand therapy in metastatic castration resistant prostate cancer receives FDA clearance”. Daily Tribune, SyndiGate Media Inc. (Houston, TX, USA); 7 February, 2017, as found on http://www.pharmacychoice.com/News/article.cfm?Article_ID= 1690538).
5.5. Image-Guided Surgery of Prostate Cancer Lesions Using 111In-Labeled PSMA Inhibitors
The promising results of the diagnostic and endoradiotherapeutic approaches which have been successfully applied into the clinical setting using PSMA-based radiotracers rendered several research groups to make use of suitable PSMA-based radiotracers for other therapeutic strategies, such as the image-guided surgery of prostate cancer lesions. 111In-PSMA I&T 111In-PSMA-617 are the probes which have been used in two different clinical trials for radio-guided surgery for the detection of lymph node metastasis [117,142,157]. Both trials showed that all PET-positive lesions were also detected by PSMA-radio-guided surgery. Furthermore, tracer uptake and median gamma probe counts were significantly different between regions with lymph node metastases and other normal tissues. Overall, these studies showed that the gamma probes measurements could be considered as an indicator for lymph node metastasis and that radio-guided surgery may consequently lead to improvements with respect to the resection of prostate cancer.
6. Conclusions
Targeting the integral membrane glycoprotein PSMA by using radiometal-based PSMA inhibitors shows a great promise for diagnosis, (re)staging and therapy of prostate cancer. Specific PSMA-based ligands show high potential for initial staging, lymph node staging, restaging and therapy of prostate cancer. Between the different groups of PSMA inhibitors which have been developed so far, the peptidomimetic structure Lys-urea-Glu is the most successful biomolecule for utilization in radiopharmaceutical development. Furthermore, the choice of the chelator significantly affects the stability, affinity, lipophilicity and ultimately pharmacokinetics of the final radiolabeled PSMA inhibitor.
At the moment, 99mTc-MIP-1404 seems to be the tracer of choice for SPECT imaging of prostate cancer, while 68Ga-PSMA-11 is one of the most promising tracers for PET imaging of patients with this disease. Due to the fact that 68Ga-PSMA-11 can be used only for diagnostic purposes, two modified PSMA inhibitors, PSMA-617 and PSMA I&T, have been developed. The superiority of PSMA-617 and PSMA I&T compared to PSMA-11 is that they can be efficiently labeled with both 68Ga for PET, as well as, 177Lu for radionuclide therapy. Therefore, both have the potential to be used as theranostic pairs. Although PSMA-directed radionuclide therapy shows great promise for implementation in routine clinical practice, the limited number of studies so far emphasizes the need for further controlled clinical trials for assessing the clinical value of 177Lu-PSMA-617 and 177Lu-PSMA I&T. Finally, image-guided surgery of prostate cancer lesions using 111In-labeled PSMA inhibitors seems to be an appealing approach for detection of lymph node metastasis.
Acknowledgments
Financial support by the Scientia Fellows Programme (Marie-Curie fellowship) managed by the University of Oslo. F.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hricak, H.; Choyke, P.L.; Eberhardt, S.C.; Leibel, S.A.; Scardino, P.T. Imaging prostate cancer: A multidisciplinary perspective. Radiology 2007, 243, 28–53. [Google Scholar] [CrossRef] [PubMed]
- Beer, A.J.; Eiber, M.; Souvatzoglou, M.; Schwaiger, M.; Krause, B.J. Radionuclide and hybrid imaging of recurrent prostate cancer. Lancet Oncol. 2011, 12, 181–191. [Google Scholar] [CrossRef]
- Fani, M.; Maecke, H.R. Radiopharmaceuticals development of radiolabelled peptides. Eur. J Nucl. Med. Mol. Imaging 2012, 1, S11–S30. [Google Scholar] [CrossRef] [PubMed]
- Schottelius, M.; Wester, H.J. Molecular imaging targeting peptide receptors. Methods 2009, 48, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S. Overview of prostate-specific membrane antigen. Rev. Urol. 2004, 6 (Suppl. 10), S13–S18. [Google Scholar] [PubMed]
- Perner, S.; Hofer, M.D.; Kim, R.; Shah, R.B.; Li, H.; Möller, P.; Hautmann, R.E.; Gschwend, J.E.; Kuefer, R.; Rubin, M.A. Prostate-specific membrane antigen expression as a predictor of prostate cancer progression. Hum. Pathol. 2007, 38, 696–701. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, D.S.; Bachich, D.; Heston, W.D.W. Prostate specific membrane antigen. In Prostate Cancer, Biology, Genetics, and the New Therapeutics, 2nd ed.; Chung, L.W.K., Issacs, W.B., Simons, J.W., Eds.; Humana Press: Totowa, NJ, USA, 2001; pp. 307–326. [Google Scholar]
- Navrátil, M.; Ptáček, J.; Šacha, P.; Starková, J.; Lubkowski, J.; Bařinka, C.; Konvalinka, J. Structural and biochemical characterization of the folyl-poly-γ-l-glutamate hydrolyzing activity of human glutamate carboxypeptidase II. FEBS. J. 2014, 281, 3228–3242. [Google Scholar] [CrossRef] [PubMed]
- Bařinka, C.; Rojas, C.; Slusher, B.; Pomper, M. Glutamate carboxypeptidase II in diagnosis and treatment of neurologic disorders and prostate cancer. Curr. Med. Chem. 2012, 19, 856–870. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Toriyabe, Y.; Kazak, M.; Berkman, C.E. Pseudoirreversible inhibition of prostate-specific membrane antigen by phosphoramidate peptidomimetics. Biochemistry 2008, 47, 12658–12660. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Heston, W.D. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J. Cell Biochem. 2004, 91, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Dautry-Varsat, A. Receptor-mediated endocytosis: The intracellular journey of transferrin and its receptor. Biochimie 1986, 68, 375–381. [Google Scholar] [CrossRef]
- Schulke, N.; Varlamova, O.A.; Donovan, G.P.; Ma, D.; Gardner, J.P.; Morrissey, D.M.; Arrigale, R.R.; Zhan, C.; Chodera, A.J.; Surowitz, K.G.; et al. The homodimer of prostatespecific membrane antigen is a functional target for cancer therapy. Proc. Natl. Acad. Sci. USA 2003, 100, 12590–12595. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar] [PubMed]
- Chang, S.S.; Reuter, V.E.; Heston, W.D.; Gaudin, P.B. Comparison of anti-prostate-spacific membrane antigen antibodies and other immunomarkers in metastatic prostate carcinoma. Urology 2001, 57, 1179–1183. [Google Scholar] [CrossRef]
- Wright, G.L.J.; Grob, B.M.; Haley, C.; Grossman, K.; Newhall, K.; Petrylak, D.; Troyer, J.; Konchuba, A.; Schellhammer, P.F.; Moriarty, R. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology 1996, 48, 326–334. [Google Scholar] [CrossRef]
- Yao, D.; Trabulsi, E.J.; Kostakoglu, L.; Vallabhajosula, S.; Joyce, M.A.; Nanus, D.M.; Milowsky, M.; Liu, H.; Goldsmith, S.J. The utility of monoclonal antibodies in the imaging of prostate cancer. Semin. Urol. Oncol. 2002, 20, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.C.; Missalidis, S. Radiolabelled aptamers for tumor imaging and therapy. Q. J. Nucl. Med. Mol. Imaging 2007, 51, 292–296. [Google Scholar] [PubMed]
- Pomper, M.G.; Musachio, J.L.; Zhang, J.; Scheffel, U.; Zhou, Y.; Hilton, J.; Maini, A.; Dannals, R.F.; Wong, D.F.; Kozikowski, A.P. 11C-MCG: Synthesis, uptake selectivity, and primate PET of a probe for glutamate carboxypeptidase II (NAALADase). Mol. Imaging 2002, 2, 96–101. [Google Scholar] [CrossRef]
- Taneja, S.S. ProstaScint® scan: Contemporary use in clinical practice. Rev. Urol. 2004, 6, S19–S28. [Google Scholar] [PubMed]
- Kahn, D.; Williams, R.D.; Haseman, M.K.; Reed, N.L.; Miller, S.J.; Gerstbrein, J. Radioimmunoscintigraphy with In-111-labeled capromab pendetide predicts prostate cancer response to salvage radiotherapy after failed radical prostatectomy. J. Clin. Oncol. 1998, 16, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Kahn, D.; Williams, R.D.; Manyak, M.J.; Haseman, M.K.; Seldin, D.W.; Libertino, J.A.; Libertino, J.A.; Maguire, R.T. 111Indium-capromab pendetide in the evaluation of patients with residual or recurrent prostate cancer after radical prostatectomy. J. Urol. 1998, 159, 2041–2047. [Google Scholar] [CrossRef]
- Sodee, D.B.; Malguria, N.; Faulhaber, P.; Resnick, MI.; Albert, J.; Bakale, G. Multi-center ProstaScint® imaging findings in 2154 patients with prostate cancer. The ProstaScint imaging centers. Urology 2000, 56, 988–993. [Google Scholar] [CrossRef]
- Troyer, J.K.; Becket, M.L.; Wright, G.L. Location of prostate-specific membrane antigen in the LNCaP prostate carcinoma cell line. Prostate 1997, 30, 232–242. [Google Scholar] [CrossRef]
- Zhou, J.; Neale, J.H.; Pomper, M.G.; Kozikowski, A.P. NAAG peptidase inhibitors and their potential potency for diagnosis and therapy. Nat. Rev. Drug Discov. 2005, 4, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Reuter, V.E.; Heston, W.D.; Bander, N.H.; Grauer, L.S.; Gaudin, P.B. Five different prostate membrane antigen (PSMA) antibodies confirm PSMA expression in tumor associated neovasculature. Cancer Res. 1999, 59, 3192–3198. [Google Scholar] [PubMed]
- Liu, H.; Moy, P.; Kim, S.; Xia, Y.; Rajasekaran, A.; Navarro, V.; Knudsen, B.; Bander, N.H. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997, 57, 3629–3634. [Google Scholar] [PubMed]
- Elsaesser-Beile, U.; Wolf, P.; Gierschner, D.; Buehler, P.; Schultze-Seemann, W.; Wetterauer, U. A new generation of monoclonal and recombinant antibodies against cell-adherent prostatespecific membrane antigen for diagnostic and therapeutic targeting of prostate cancer. Prostate 2006, 66, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Freudenberg, N.; Buehler, P.; Alt, K.; Schultze-Seemann, W.; Wetterauer, U.; Elsaesser-Beile, U. Three conformational antibodies specific for different PSMA epitopes are promising diagnostic and therapeutic tools for prostate cancer. Prostate 2010, 70, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Nargund, V.; Al Hashmi, D.; Kumar, P.; Gordon, S.; Otitie, U.; Ellison, D.; Carroll, M.; Baithun, S.; Britton, K.E. Imaging with radiolabelled monoclonal antibody (MUJ591) to prostate-specific membrane antigen in staging of clinically localized prostatic carcinoma: Comparison with clinical, surgical and histological staging. BJU Int. 2005, 95, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Vallabhajosula, S.; Kuji, I.; Mamacher, K.A.; Konishi, S.; Kostakoglu, L.; Kothari, P.A.; Milowski, M.L.; Nanus, D.M.; Bander, N.H.; Goldsmith, S.J. Pharmacokinetics and biodistribution of 111In- and 177Lu-labeled J591 antibody specific for prostate-specific membrane antigen: Prediction of 90Y-J591 radiation dosimetry based on 111In or 177Lu? J. Nucl. Med. 2005, 46, 634–641. [Google Scholar] [PubMed]
- Bander, N.H.; Trabulsi, E.J.; Kostakoglu, L.; Yao, D.; Vallabhajosula, S.; Smith-Jones, P.; Joyce, M.A.; Milowsky, M.; Nanus, D.M.; Goldsmith, S.J. Targeting metastatic prostate cancer with radiolabeled monoclonal antibody J591 to the extracellular domain of prostate specific membrane antigen. J. Urol. 2003, 170, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Bander, N.H.; Milowsky, M.I.; Nanus, D.M.; Kostakoglu, L.; Vallabhajosula, S.; Goldsmith, S.J. Phase I trial of 177Lu-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J. Clin. Oncol. 2005, 23, 4591–4601. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.P.; Divilov, V.; Bander, N.H.; Smith-Jones, P.M.; Larson, S.M.; Lewis, J.S. 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J. Nucl. Med. 2010, 51, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Smith-Jones, P.M.; Wongvipat, J.; Navarro, V.; Kim, S.; Bander, N.H.; Larson, S.M.; Sawyers, C.L. Noninvasive measurement of androgen receptor signaling with a positron-emitting radiopharmaceutical that targets prostate-specific membrane antigen. Proc. Natl. Acad. Sci. USA 2011, 108, 9578–9582. [Google Scholar] [CrossRef] [PubMed]
- Vallabhajosula, S.; Goldsmith, S.J.; Kostakoglou, L.; Milowsky, M.I.; Nanus, D.M.; Bander, N.H. Radioimmunotherapy of prostate cancer using 90Y- and 177Lu-labeled J591 monoclonal antibodies: Effect of multiple treatments on myelotoxicity. Clin. Cancer Res. 2005, 11, 7195s–7200s. [Google Scholar] [CrossRef] [PubMed]
- Kampmeier, F.; Williams, J.D.; Maher, J.; Mullen, G.E.; Blower, P.J. Design and preclinical evaluation of a 99mTc-labelled diabody of mAb J591 for SPECT imaging of prostate-specific membrane antigen (PSMA). Eur. J. Nucl. Med. Mol. Imaging Res. 2014, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Milowsky, M.I.; Nanus, D.N.; Kostakoglou, L.; Sheehan, C.E.; Vallabhajosula, S.; Goldsmith, S.J.; Ross, J.S.; Bander, N.H. Vascular targeted therapy with anti-prostate-specific membrane antigen monoclonal antibody J591 in advanced solid tumors. J. Clin. Oncol. 2007, 25, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Milowsky, M.I.; Nanus, D.M.; Kostakoglu, L.; Vallabhajosula, S.; Goldsmith, S.J.; Bander, N.H. Phase I trial of yttrium-90-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for androgen-independent prostate cancer. J. Clin. Oncol. 2004, 22, 2522–2531. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Beltran, H.; Vallabhajosula, S.; Goldsmith, S.J.; Osborne, J.; Matulich, D.; Petrillo, K.; Parmar, S.; Nanus, D.M.; Bander, N.H. Anti-prostate-specific membrane antigen-based radioimmunotherapy for prostate cancer. Cancer 2010, 116, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Smith-Jones, P.M.; Vallabhajosula, S.; Navarro, V.; Bastidas, D.; Goldsmith, S.J.; Bander, N.H. Radiolabeled Monoclonal Antibodies Specific to the Extracellular Domain of Prostate-Specific Membrane Antigen: Preclinical Studies in Nude Mice Bearing LNCaP Human Prostate Tumor. J. Nucl. Med. 2003, 44, 610–617. [Google Scholar] [PubMed]
- Osborne, J.R.; Green, D.A.; Spratt, D.E.; Lyashchenko, S.; Fareedy, S.B.; Robinson, B.D.; Beattie, B.J.; Jain, M.; Lewis, J.S.; Christos, P.; et al. A prospective pilot study of 89Zr-J591/prostate specific membrane antigen positron emission tomography in men with localized prostate cancer undergoing radical prostatectomy. J. Urol. 2014, 191, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Alt, K.; Wiehr, S.; Ehrlichmann, W.; Reischl, G.; Wolf, P.; Pichler, B.J.; Elsaesser-Beile, U.; Buehler, P. High-resolution animal PET imaging of prostate cancer xenografts with three different (64)Cu-labeled antibodies against native cell-adherent PSMA. Prostate 2010, 70, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Behe, M.; Alt, K.; Deininger, F.; Buehler, P.; Wetterauer, U.; Weber, W.A.; Elsaesser-Beile, U.; Wolf, P. In Vivo testing of 177Lu-Labelled Anti-PSMA antibody as a new radioimmunotherapeutic agent against prostate cancer. In Vivo 2011, 25, 55–60. [Google Scholar] [PubMed]
- Liu, J.; Kopeckova, P.; Buehler, P.; Wolf, P.; Pan, H.; Bauer, H.; Elsaesser-Beile, U.; Kopecek, J. Biorecognition and Subcellular Trafficking of HPMA Copolymer-Anti-PSMA Antibody Conjugates by Prostate Cancer Cells. Mol. Pharm. 2009, 6, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.M.; Senter, P.D. Arming antibodies: Prospects and challenges for immunoconjugates. Nat. Biotechnol. 2005, 23, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Schlom, J.; Siler, K.; Milenic, D.E.; Eggensperger, D.; Colcher, D.; Miller, L.S.; Houchens, D.; Cheng, R.; Kaplan, D.; Goeckeler, W. Monoclonal antibody-based therapy of a human tumor xenograft with a 177lutetium-labeled immunoconjugate. Cancer Res. 1991, 51, 2889–2896. [Google Scholar] [PubMed]
- Vallabhajosula, S.; Goldsmith, S.J.; Hamacher, K.A.; Kostakoglu, L.; Konishi, S.; Milowski, M.I.; Nanus, D.M.; Bander, N.H. Prediction of myelotoxicity based on bone marrow radiation-absorbed dose: Radioimmunotherapy studies using 90Y- and 177Lu-labeled J591 antibodies specific for prostate-specific membrane antigen. J. Nucl. Med. 2005, 46, 850–858. [Google Scholar] [PubMed]
- Cortez-Retamozo, V.; Lauwereys, M.; Hassanzadeh, G.G.; Gobert, M.; Conrath, K.; Muyldermans, S.; De Baetselier, P.; Revets, H. Efficient tumor targeting by single-domain antibody fragments of camels. Int. J. Cancer 2002, 98, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Evazalipour, M.; D’Huyvetter, M.; Tehrani, B.S.; Abolhassani, M.; Omidfar, K.; Abdoli, S.; Arezumand, R.; Morovvati, H.; Lahoutte, T.; Muyldermans, S.; et al. Generation and characterization of nanobodies targeting PSMA for molecular imaging of prostate cancer. Contrast Med. Mol. Imaging 2014, 9, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Chatalic, K.L.S.; Veldhoven-Zweistra, J.; Bolkestein, M.; Hoeben, S.; Koning, G.A.; Boerman, O.C.; de Jong, M.; van Weerden, W.M. A novel 111In-labeled anti-prostate-specific membrane antigen nanobody for targeted SPECT/CT imaging of prostate cancer. J. Nucl. Med. 2015, 56, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Melis, M.; Bijster, M.; de Visser, M.; Konijnenberg, M.W.; de Swart, J.; Rolleman, E.J.; Boerman, O.C.; Krenning, E.P.; de Jong, M. Dose-response effect of Gelofusine on renal uptake and retention of radiolabelled octreotate in rats with CA20948 tumours. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Bernard, B.F.; Krenning, E.P.; Breeman, W.A.; Rolleman, E.J.; Bakker, W.H.; Visser, T.J.; Mäcke, H.R.; de Jong, M. d-Lysine reduction of indium-111 octreotide and yttrium-90 octreotide renal uptake. J. Nucl. Med. 1997, 38, 1929–1933. [Google Scholar] [PubMed]
- Pandit-Taskar, N.; O’Donoghue, J.A.; Ruan, S.; Lyashchenko, S.K.; Carrasquillo, J.A.; Heller, G.; Martinez, D.F.; Cheal, S.M.; Lewis, J.S.; Fleisher, M.; et al. First-in-human imaging with 89Zr-Df-IAB2M Anti-PSMA minibody in patients with metastatic prostate cancer: Pharmacokinetics, biodistribution, dosimetry, and lesion uptake. J. Nucl. Med. 2016, 57, 1858–1864. [Google Scholar] [CrossRef] [PubMed]
- Leyton, J.V.; Olafsen, T.; Lepin, E.J.; Hahm, S.; Bauer, K.B.; Reiter, R.E.; Wu, A.M. Humanized radioiodinated minibody for imaging of prostate stem cell antigen-expressing tumors. Clin. Cancer Res. 2008, 14, 7488–7496. [Google Scholar] [CrossRef] [PubMed]
- Olafsen, T.; Betting, D.; Kenanova, V.E.; Salazar, F.B.; Clarke, P.; Said, J.; Raubitschek, A.A.; Timmerman, J.M.; Wu, A.M. Recombinant anti-CD20 antibody fragments for small-animal PET imaging of B-cell lymphomas. J. Nucl. Med. 2009, 50, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Viola-Villegas, N.T.; Sevak, K.K.; Carlin, S.D.; Doran, M.G.; Evans, H.W.; Bartlett, D.W.; Wu, A.M.; Lewis, J.S. Noninvasive imaging of PSMA in prostate tumors with 89Zr-labeled huJ591 engineered antibody fragments: The faster alternatives. Mol. Pharm. 2014, 11, 3965–3973. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Cerchia, L.; de Franciscis, V. Targeting cancer cells with nucleic acid aptamers. Trends Biotechnol. 2010, 28, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Dassie, J.P.; Liu, X.Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O.; Giangrande, P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009, 27, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhu, X.; Lu, P.Y.; Rosato, R.R.; Tan, W.; Zu, Y. Oligonucleotide aptamers: New tools for targeted cancer therapy. Mol. Ther. Nucleic Acids 2014, 3, e182. [Google Scholar] [CrossRef] [PubMed]
- De Franciscis, V. A Theranostic “SMART” Aptamer for Targeted Therapy of Prostate Cancer. Mol. Ther. 2014, 22, 1886–1888. [Google Scholar] [CrossRef] [PubMed]
- Lupold, S.E.; Hicke, B.J.; Lin, Y.; Coffey, D.S. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002, 62, 4029–4033. [Google Scholar] [PubMed]
- Rockey, W.M.; Huang, L.; Kloepping, K.C.; Baumhover, N.J.; Giangrande, P.H.; Schultz, M.K. Synthesis and radiolabeling of chelator-RNA aptamer bioconjugates with copper-64 for targeted molecular imaging. Bioorg. Med. Chem. 2011, 19, 4080–4090. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.F.; Cole, D.C.; Slusher, B.S.; Stetz, S.L.; Ross, L.E.; Donzanti, B.A.; Trainor, D.A. Design, synthesis, and biological activity of a potent inhibitor of the neuropeptidase N-acetylated alphalinked acidic dipeptidase. J. Med. Chem. 1996, 39, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Majer, P.; Jackson, P.F.; Delahanty, G.; Grella, B.S.; Ko, Y.S.; Li, W.; Liu, Q.; Maclin, K.M.; Polakova, J.; Shaffer, K.A.; et al. Synthesis and biological evaluation of thiol-based inhibitors of glutamate carborypeptidase II: Discovery of an orally active GCP II inhibitor. J. Med. Chem. 2003, 46, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.O.; Wu, L.Y.; Santiago, N.M.; Moser, J.M.; Rowley, J.A.; Bolstad, E.S.; Berkman, C.E. Substrate specificity of prostate-specific membrane antigen. Bioorg. Med. Chen. 2007, 15, 6678–6686. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wu, L.Y.; Kazak, M.; Berkman, C.E. Cell-Surface labeling and internalization by a fluorescent inhibitor of prostate-specific membrane antigen. Prostate 2008, 68, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Maung, J.; Mallari, J.P.; Girtsman, T.A.; Wu, L.Y.; Rowley, J.A.; Santiago, N.M.; Brunelle, A.N.; Berkman, C.E. Probing for a hydrophobic a binding register in prostate-specific membrane antigen with phenylalkylphosphonamidates. Bioorg. Med. Chem. 2004, 12, 4969–4979. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Nan, F.; Conti, P.; Zhang, J.; Ramadan, E.; Bzdega, T.; Wroblewska, B.; Neale, J.H.; Pshenichkin, S.; Wroblewski, J.T. Design of remarkably simple, yet potent urea-based inhibitors of glutamate carboxypeptidase II (NAALADase). J. Med. Chem. 2001, 44, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Zhang, J.; Nan, F.; Petukhov, P.A.; Grajkowska, E.; Wroblewski, J.T.; Yamamoto, T.; Bzdega, T.; Wroblewska, B.; Neale, J.H. Synthesis of urea-based inhibitors as active site probes of glutamate carboxypeptidase II: Efficacy as analgesic agents. J. Med. Chem. 2004, 47, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Misra, P.; Humblet, V.; Pannier, N.; Maison, W.; Frangioni, J.V. Production of multimeric prostate-specific membrane antigen small molecule radiotracers using a solid-phase 99mTc pre-loading strategy. J. Nucl. Med. 2007, 48, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Smith-Jones, P.M.; Vallabahajosula, S.; Goldsmith, S.J.; Navarro, V.; Hunter, C.J.; Bastidas, D.; Bander, N.H. In Vitro characterization of radiolabeled monoclonal antibodies specific for the extracellular domain of prostate-specific membrane antigen. Cancer Res. 2000, 60, 5237–5243. [Google Scholar] [PubMed]
- Nedrow-Byers, J.R.; Jabbes, M.; Jewett, C.; Ganguly, T.; He, H.; Liu, T.; Benny, P.; Bryan, J.N.; Berkman, C.E. A phosphoramidate-based prostate-specific membrane antigen-targeted SPECT agent. Prostate 2012, 72, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Zenga, D.; Ouyangb, Q.; Caia, Z.; Xieb, X.-Q.; Anderson, C.J. New cross-bridged cyclam derivative CB-TE1K1P, an improved bifunctional chelator for copper radionuclides. Chem. Commun. 2014, 50, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Nedrow, J.R.; Latoche, J.D.; Day, K.E.; Modi, J.; Ganguly, T.; Zeng, D.; Kurland, B.F.; Berkman, C.E.; Anderson, C.J. Targeting PSMA with a Cu-64 labeled phosphoramidate inhibitor for PET/CT imaging of variant PSMA-expressing xenografts in mouse models of prostate cancer. Mol. Imaging Biol. 2016, 18, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Foss, C.A.; Mease, R.C.; Fan, H.; Wang, Y.; Ravert, H.T.; Dannals, R.F.; Olszewski, R.T.; Heston, W.D.; Kozikowski, A.P.; Pomper, M.G. Radiolabeled small molecule ligands for prostate-specificmembrane antigen: In Vivo imaging in experimental models of prostate cancer. Clin. Cancer Res. 2005, 11, 4022–4028. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Foss, C.A.; Byun, Y.; Nimmagadda, S.; Pullambhatla, M.; Fox, J.J.; Castanares, M.; Lupold, S.E.; Babich, J.W.; Mease, R.C.; et al. Radiohalogenated prostate-specific membrane antigen (PMSA)-based ureas as imaging agents for prostate cancer. J. Med. Chem. 2008, 51, 7933–7943. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Byun, Y.; Barinka, C.; Pullambhatla, M.; Bhang, H.E.; Fox, J.J.; Lubkowski, J.; Mease, R.C.; Pomper, M.G. Bioisosterismof urea-based GCPII inhibitors: Synthesis and structure-activity relationship studies. Bioorg. Med. Chem. Lett. 2010, 20, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Kiess, A.P.; Minn, I.; Chen, Y.; Hobbs, R.; Sgouros, G.; Mease, R.C.; Pullambhatla, M.; Shen, C.J.; Foss, C.A.; Pomper, M.G. Auger radiopharmaceutical therapy targeting prostate-specific membrane antigen. J. Nucl. Med. 2015, 56, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Maresca, K.P.; Hillier, S.M.; Femia, F.J.; Keith, D.; Barone, C.; Joyal, J.L.; Zimmerman, C.N.; Kozikowski, A.P.; Barrett, J.A.; Eckelman, W.C.; et al. A series of halogenated heterodimeric inhibitors of prostate specific membrane antigen (PSMA) as radiolabeled probes for targeting prostate cancer. J. Med. Chem. 2009, 52, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Hillier, S.M.; Maresca, K.P.; Femia, F.J.; Marquis, J.C.; Foss, C.A.; Nguyen, N.; Zimmerman, C.N.; Barrett, J.A.; Eckelman, W.C.; Pomper, M.G.; et al. Preclinical evaluation of novel glutamate-urea-lysine analogues that target prostate-specific membrane antigen as molecular imaging pharmaceuticals for prostate cancer. Cancer Res. 2009, 69, 6932–6940. [Google Scholar] [CrossRef] [PubMed]
- Hillier, S.M.; Kern, A.M.; Maresca, K.P.; Marquis, J.C.; Eckelman, W.C.; Joyal, J.L.; Babich, J.W. 123I-MIP-1072, a small-molecule inhibitor of prostate-specific membrane antigen, is effective at monitoring tumor response to taxane therapy. J. Nucl. Med. 2011, 52, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Darwish, A.; Blacker, M.; Janzen, N.; Rathmann, S.M.; Czorny, S.; Hillier, S.M.; Joyal, J.L.; Babich, J.W.; Valliant, J.F. Triazole appending agent (TAAG): A newsynthon for preparing iodine-basedmolecular imaging and radiotherapy agents. ACS Med. Chem. Lett. 2012, 8, 313–316. [Google Scholar] [CrossRef] [PubMed]
- El-Zaria, M.E.; Genady, A.R.; Janzen, N.; Petlura, C.I.; Beckford Vera, D.R.; Valliant, J.F. Preparation and evaluation of carborane-derived inhibitors of prostate specific membrane antigen (PSMA). Dalton Trans. 2014, 43, 4950–4961. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.P.; Gage, K.L.; Farai, S.F.; Macura, K.J.; Cornish, T.C.; Gonzalez-Roibon, N.; Guner, G.; Munari, E.; Partin, A.W.; Pavlovich, C.P.; et al. 18F-DCFBC PET/CT for PSMA-based detection and characterization of primary prostate cancer. J. Nucl. Med. 2015, 56, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Z.; Mean, E.; Rowe, S.P.; Plyku, D.; Nidal, R.; Eisenberger, M.A.; Antonarakis, E.S.; Fan, H.; Dannals, R.F.; Chen, Y.; et al. Initial evaluation of 18F-DCFPyl for prostate-specific membrane antigen antigen (PSMA)-targeted PET imaging of prostate cancer. Mol. Imaging Biol. 2015, 17, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Mease, R.C.; Dusich, C.L.; Foss, C.A.; Ravert, H.T.; Dannals, R.F.; Seidel, J.; Seidel, J.; Prideaux, A.; Fox, J.J.; Sgouros, G.; et al. N-[N-[(S)-1,3-Dicarboxypropyl]carbamoyl]-4-[18F]fluorobenzyl-l-cysteine, [18F]DCFBC: A new imaging probe for prostate cancer. Clin. Cancer Res. 2008, 14, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Gage, K.L.; Mease, R.C.; Senthamizhchelvan, S.; Holt, D.P.; Jeffrey-Kwanisai, A.; Endres, C.J.; Dannals, R.F.; Sgouros, G.; Lodge, M.; et al. Biosdistribution, tumor detection, and radiation dosimetry of 18F-DCFBC, a low molecular weight inhibitor of prostate-specific membrane antigen, in patients with metastatic prostate cancer. J. Nucl. Med. 2012, 53, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pullambhatla, M.; Foss, C.A.; Byun, Y.; Nimmagadda, S.; Senthamizhchelvan, S.; Sgouros, G.; Mease, R.C.; Pomper, M.G. 2(3-{1-Carboxy-5[{[6-18F]fluoro-pyridine-3-carbonyl}amino]-pentyl}-ureido)-pentanedioic acid, 18F-DCPyL, a PSMA-based PET imaging agent for prostate cancer. Clin. Cancer Res. 2011, 17, 7645–7653. [Google Scholar] [CrossRef] [PubMed]
- Lapi, S.E.; Wahnishe, H.; Pham, D.; Wu, L.Y.; Nedrow-Byers, J.R.; Liu, T.; Vejdani, K.; VanBrocklin, H.F.; Berkman, C.E.; Jones, E.F. Assessment of an 18F-labeled phosphoramide peptidomimetic as a new prostate-specific membrane antigen-targeted imaging for prostate cancer. J. Nucl. Med. 2009, 50, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Zhang, Z.; Zhu, Z.; Yang, D.; Chen, X.; Wang, J.; Chen, F.; Song, X. Synthesis and positron emission tomography evaluation of 18F-Glu-urea-Lys, a prostate-specific membrane antigen-based imaging agent for prostate cancer. Oncol. Lett. 2015, 10, 2299–2302. [Google Scholar] [PubMed]
- Malik, N.; Machulla, H.J.; Solbach, C.; Winter, G.; Reske, S.N.; Zlatopolskiy, B. Radiosynthesis of a new PSMA targeting ligand ([18F]FPy-DUPA-pep). Appl. Radiat. Isot. 2011, 69, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Malik, N.; Baur, B.; Winter, G.; Reske, S.N.; Beer, A.J.; Solbach, C. Radiofluorination of PSMA-HBED via Al18F2+ chelation and biological evaluations in vitro. Mol. Imaging Biol. 2015, 17, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Ley, C.R.; Beattie, N.R.; Dannoon, S.; Regan, M.; VanBrocklin, H.; Berkman, C.E. Synthesis and evaluation of constrained phosphoramidate inhibitors of prostate-specific membrane antigen. Bioorg. Med. Chem. Lett. 2015, 25, 2536–2539. [Google Scholar] [CrossRef] [PubMed]
- Lesche, R.; Kettschau, G.; Gromov, A.V.; Böhnke, N.; Borkowski, S.; Mönning, U.; Hegele-Hartung, C.; Döhr, O.; Dinkelborg, L.M.; Graham, K. Preclinical evaluation of BAY 1075553, a novel 18F-labelled inhibitor of prostate specific membrane antigen for PET imaging of prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Benešová, M.; Bauder-Wüst, U.; Schäfer, M.; Klika, K.D.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Linker modification strategies to control the prostate-specific membrane antigen (PSMA)-targeting and pharmacokinetic properties of DOTA-conjugated PSMA inhibitors. J. Med. Chem. 2016, 59, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Foss, C.A.; Castanares, M.; Mease, R.C.; Byun, Y.; Fox, J.J.; Hilton, J.; Lupold, S.E.; Kozikowski, A.P.; Pomper, M.G. Synthesis and evaluation of technetium-99m- and rhenium-labeled inhibitors of the prostate-specific membrane antigen (PSMA). J. Med. Chem. 2008, 51, 4504–4517. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Pullambhatla, M.; Foss, C.A.; Falk, A.; Byun, Y.; Nimmagadda, S.; Mease, R.C.; Pomper, M.G. Effect of chelators on the pharmacokinetics of 99mTc-labeled imaging agents for the prostate-specific membrane antigen (PSMA). J. Med. Chem. 2013, 56, 6108–6121. [Google Scholar] [CrossRef] [PubMed]
- Kularatne, S.A.; Zhou, Z.; Yang, J.; Post, C.B.; Low, P.S. Design, synthesis, and preclinical evaluation of prostate-specific membrane antigen targeted 99mTc-radioimaging agents. Mol. Pharm. 2009, 6, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Robu, S.; Schottelius, M.; Eiber, M.; Maurer, T.; Gschwend, J.; Schwaiger, M.; Wester, H.-J. Preclinical evaluation and first patient application of 99mTc-PSMA-I&S for SPECT imaging and radioguided surgery in prostate cancer. J. Nucl. Med. 2017, 58, 235–242. [Google Scholar] [PubMed]
- Kimura, H.; Sampei, S.; Matsuoka, D.; Harada, N.; Watanabe, H.; Arimitsu, K.; Ono, M.; Saji, H. Development of 99mTc-labeled asymmetric urea derivatives that target prostate-specific membrane antigen for single-photon emission computed tomography imaging. Bioorg. Med. Chem. 2016, 24, 2251–2256. [Google Scholar] [CrossRef] [PubMed]
- Hillier, S.M.; Maresca, K.P.; Lu, G.; Merkin, R.D.; Marquis, J.C.; Zimmerman, C.N.; Eckelman, W.C.; Joyal, J.L.; Babich, J.W. 99mTc-labeled small-molecule inhibitors of prostate-specific membrane antigen for molecular imaging of prostate cancer. J. Nucl. Med. 2013, 54, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Maecke, H.R.; Hofmann, M.; Haberkorn, U. 68Ga-labeled peptides in tumor imaging. J. Nucl. Med. 2005, 46, 172S–178S. [Google Scholar] [PubMed]
- Roesch, F.; Riss, P.J. The renaissance of the 68Ge/68Ga radionuclide generator initiates new developments in 68Ga radiopharmaceutical chemistry. Curr. Top. Med. Chem. 2010, 10, 1633–1668. [Google Scholar] [CrossRef] [PubMed]
- Eder, M.; Schäfer, M.; Bauder-Wüst, U.; Hull, W.E.; Wängler, C.; Mier, W.; Haberkorn, U.; Eisenhut, M. 68Ga-Complex lipophilicity and the targeting property of a urea based PSMA inhibitor for PET imaging. Bioconjug. Chem. 2012, 23, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Malcher, A.; Eber, M.; Eisenhut, M.; Linhart, H.G.; Hadaschik, B.A.; Holland-Letz, T.; Giesel, F.L.; Kratochwil, C.; Haufe, S.; et al. PET imaging with [68Ga]gallium labeled PSMA ligand for the diagnosis of prostate cancer: Biodistribution in humans and first evaluation in tumor lesions. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Zechmann, C.M.; Malcher, A.; Eder, M.; Eisenhut, M.; Linhart, H.G.; Holland-Letz, T.; Hadaschik, B.A.; Giesel, F.L.; Debus, J.; et al. Comparison of PET imaging with a 68Ga-labelled PSMA ligand and 18F-choline-based PET/CT for the diagnosis of recurrent prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bluemel, C.; Krebs, M.; Polat, B.; Linke, F.; Eiber, M.; Samnick, S.; Lapa, C.; Lassmann, M.; Riedmiller, H.; Czernin, J.; et al. 68Ga-PSMA-PET/CT in patients with biochemical prostate cancer recurrence and negative 18F-choline-PET/CT. Clin. Nucl. Med. 2016, 41, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Avtzi, E.; Giesel, F.L.; Holland-Letz, T.; Linhart, H.G.; Eder, M.; Eisenhut, M.; Boxler, S.; Hadaschik, B.A.; Kratochwil, C.; et al. The diagnostic value of PET/CT imaging with the 68Ga-labelled PSMA ligand HBED-CC in the diagnosis of recurrent prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Sterzing, F.; Kratochwil, C.; Fiedler, H.; Katayama, S.; Habl, G.; Kopka, K.; Afshar-Oromieh, A.; Debus, J.; Haberkorn, U.; Giesel, F.L. 68Ga-PSMA-11 PET/CT: A new technique with high potential for the radiotherapeuticmanagement of prostate cancer patients. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Giesel, F.L.; Fiedler, H.; Stefanova, M.; Sterzing, F.; Rius, M.; Kopka, K.; Moltz, J.H.; Afshar-Oromieh, A.; Choyke, P.L.; Haberkorn, U.; et al. PSMA PET/CT with Glu-urea-Lys-(Ahx)-[68Ga(HBED-CC)] versus 3D CT volumetric lymph node assessment in recurrent prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Dietlein, M.; Kobe, C.; Kuhnert, G.; Stockter, S.; Fischer, T.; Schomäcker, K.; Schmidt, M.; Dietlein, F.; Zlatopolskiy, B.D.; Krapf, P.; et al. Comparison of [18F]DCFPyL and [68Ga]Ga-PSMA-HBED-CC for PSMA-PET imaging in patients with relapsed prostate cancer. Mol. Imaging Biol. 2015, 17, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Giesel, F.L.; Sterzing, F.; Schlemmer, H.P.; Holland-Letz, T.; Mier, W.; Rius, M.; Afshar-Oromieh, A.; Kopka, K.; Debus, J.; Haberkorn, U.; et al. Intraindividual comparison of 68Ga-PSMA-11-PET/CT and multi-parametric MR for imaging of primary prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Freitag, M.T.; Radtke, J.P.; Hadaschik, B.A.; Kopp-Schneider, A.; Eder, M.; Kopka, K.; Haberkorn, U.; Roethke, M.; Schlemmer, H.P.; Afshar-Oromieh, A. Comparison of hybrid 68Ga-PSMA PET/MRI and 68Ga-PSMA PET/CT in the evaluation of lymph node and bone metastases of prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Herlemann, A.; Wenter, V.; Kretschmer, A.; Thierfelder, K.M.; Bartenstein, P.; Faber, C.; Gildehaus, F.J.; Stief, C.G.; Gratzke, C.; Fendler, W.P. 68Ga-PSMA positron emission tomography/computed tomography provides accurate staging of lymph node regions prior to lymph node dissection in patients with prostate cancer. Eur. Urol. 2016, 40, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Maurer, T.; Weirich, G.; Schottelius, M.; Weineisen, M.; Frisch, B.; Okur, A.; Kübler, H.; Thalgott, M.; Navab, N.; Schwaiger, M.; et al. Prostate specific membrane antigen-radioguided surgery for metastatic lymph nodes in prostate cancer. Eur. Urol. 2015, 68, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Lawal, I.; Vorster, M.; Boshomane, T.; Ololade, K.; Ebenhan, T.; Sathekge, M. Metastatic prostate carcinoma presenting as a Superscan on 68Ga-PSMA PET/CT. Clin. Nucl. Med. 2015, 40, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Ceci, F.; Uprimmy, C.; Nilica, B.; Geraldo, L.; Kendler, D.; Kroiss, A.; Bektic, J.; Horninger, W.; Lukas, P.; Decristoforo, C.; et al. 68Ga-PSMA PET/CT for restaging recurrent prostate cancer: Which factors are associated with PET/CT detection rate? Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Schlenkhoff, C.D.; Gaertner, F.; Essler, M.; Hauser, S.; Ahmadzadehfar, H. 68Ga-labeled anti-prostate-specific membrane antigen peptide as marker for androgen deprivation therapy response in prostate cancer. Clin. Nucl. Med. 2016, 41, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Rai, B.P.; Baum, R.; Patel, A.; Hughes, R.; Alonzi, R.; Lane, T.; Adshead, J.; Vasdev, N. The role PET with 68Galabelledbprostate-specific membrane antigen (PSMA) in the management of patient with organ confined and locally advanced prostate cancer prior to radical treatment and after radical prostatectomy. Urology 2016, 95, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Sasikumar, A.; Joy, A.; Nanabala, R.; Pillai, M.R.A.; Thomas, B. 68Ga-PSMA PET/CT imaging and 153Sm-EDTMP bone pain palliation therapy. Clin. Nucl. Med. 2016, 41, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Eber, M.; Maurer, T.; Souvatzoglou, M.; Beer, A.J.; Ruffani, A.; Haller, B.; Graner, F.P.; Kübler, H.; Haberhorn, U.; Eisenhut, M.; et al. Evaluation of hybrid 68Ga-PMSA ligand PET/CT in 248 patients with biochemical recurrence after radical prostatectomy. J. Nucl. Med. 2015, 56, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Verburg, F.A.; Pfister, D.; Heidenreich, A.; Vogg, A.; Drude, N.I.; Vöö, S.; Mottaghy, F.M.; Behrendt, F.F. Extent of disease in recurrent prostate cancer determined by [68Ga]PSMA-HBED-CC PET/CT in relation to PSA levels, PSA doubling time and Gleason score. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Sachpekidis, C.; Eder, M.; Kopka, K.; Mier, W.; Hadaschik, B.A.; Haberkorn, U.; Dimitrakopoulou-Strauss, A. 68Ga-PSMA-11 dynamic PET/CT imaging in biochemical relapse of prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Minamimoto, M.; Hancock, S.; Schneider, B.; Chin, F.T.; Jamali, M.; Loening, A.; Vasanawala, S.; Gambhir, S.S.; Iagaru, A. Pilot comparison of 68Ga-RM2 and 68Ga-PSMA PET in patients with biochemically recurrent prostate cancer. J. Nucl. Med. 2016, 57, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Soydal, C.; Ozkan, E.; Yerlikaya, H.; Utkan, G.; Kucuk, O.N. Widespread metastatic prostate carcinoma shown by 68Ga-PSMA PET/CT. Clin. Nucl. Med. 2016, 41, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Morigi, J.J.; Stricker, P.D.; van Leeuwen, P.J.; Tang, R.; Ho, B.; Nguyen, Q.; Hruby, G.; Fogarty, G.; Jagavkar, R.; Kneebone, A.; et al. Prospective comparison of 18F-fluoromethylcholine versus 68Ga-PSMA PET/CT in prostate cancer patients who have rising PSA after curative treatment and are being considered for targeted therapy. J. Nucl. Med. 2016, 57, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, L.; Fattoni, F.; Rossi, E.; Karnes, R.J.; Lowe, V. Early detection of prostate cancer relapse by biochemistry and diagnostic imaging. Q. J. Nucl. Med. Mol. Imaging 2015, 59, 359–373. [Google Scholar] [PubMed]
- Schäfer, M.; Bauder-Wüst, U.; Leotta, K.A.; Zoller, F.; Mier, W.; Haberkorn, U.; Eisenhut, M.; Eder, M. Dimerized urea-based inhibitor of the prostate-specific membrane antigen for 68Ga-PET imaging of prostate cancer. Eur. J. Nucl. Med. Mol. Imaging Res. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Pullambhatla, M.; Byun, Y.; Nimmagadda, S.; Green, G.; Fox, J.J.; Horti, A.; Mease, R.C.; Pomper, M.G. 68Galabeled inhibitors of prostate-specificmembrane antigen (PSMA) for imaging prostate cancer. J. Med. Chem. 2010, 53, 5333–5341. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Chen, Z.; Pullambhatla, M.; Lisok, A.; Chen, J.; Mease, R.C.; Pomper, M.G. Preclinical Comparative Study of 68Ga-Labeled DOTA, NOTA, and HBED-CC Chelated Radiotracers for Targeting PSMA. Bioconjug. Chem. 2016, 27, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Benešová, M.; Schäfer, M.; Bauder-Wüst, U.; Afshar-Oromieh, A.; Kratochwil, C.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Preclinical evaluation of a tailor-made DOTA-conjugated PSMA inhibitor with optimized linker moiety for imaging and endoradiotherapy of prostate cancer. J. Nucl. Med. 2015, 56, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Weineisen, M.; Simecek, J.; Schottelius, M.; Schwaiger, M.; Wester, H.J. Synthesis and preclinical evaluation of DOTAGA-conjugated PSMA ligands for functional imaging and endoradiotherapy of prostate cancer. Eur. J. Nucl. Med. Mol. Imaging Res. 2014, 4, 63. [Google Scholar] [CrossRef] [PubMed]
- Weineisen, M.; Schottelius, M.; Simecek, J.; Baum, R.P.; Yildiz, A.; Beykan, S.; Kulkarni, H.R.; Lassmann, M.; Klette, I.; Eiber, M.; et al. 68Ga- and 177Lu-labeled PMSA I&T: Optimization of a PMSA-targeted theranostic concept first proof-of-concept in human studies. J. Nucl. Med. 2015, 56, 1169–1176. [Google Scholar] [PubMed]
- Gourni, E.; Canovas, C.; Goncalves, V.; Denat, F.; Meyer, P.T.; Maecke, H.R. (R)-NODAGAPSMA: A versatile precursor for radiometal Leling and nuclear imaging of PSMA positive tumors. PLoS ONE 2015, 10, e0145755. [Google Scholar] [CrossRef] [PubMed]
- Baur, B.; Solbach, C.; Andreolli, E.; Winter, G.; Machulla, H.J.; Reske, S.N. Synthesis, Radiolabelling and in vitro characterization of the gallium-68-, yttrium-90- and lutetium-177-labelled PSMA ligand, CHX-A″-DTPA-DUPA-Pep. Pharmaceuticals 2014, 7, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Ferdani, R. Copper-64 radiopharmaceuticals for PET imaging of cancer: Advances in preclinical and clinical research. Cancer Biother. Radiopharm. 2009, 24, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Pullambhatla, M.; Foss, C.A.; Nimmagadda, S.; Ferdani, R.; Anderson, C.J.; Mease, R.C.; Pomper, M.G. 64Cu-labeled inhibitors of prostate-specific membrane antigen for PET imaging of prostate cancer. J. Med. Chem. 2014, 57, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Voss, S.T.; Smith, S.V.; DiBartolo, N.; McIntosh, L.J.; Cyr, E.M.; Bonab, A.A.; Dearling, J.L.J.; Carter, E.A.; Fishman, A.J.; Treves, S.T.; et al. Positron emission tomography (PET) imaging of neuroblastoma and melanoma with 64Cu-SarAr immunoconjugates. Proc. Natl. Acad. Sci. USA 2007, 104, 17489–17493. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.C.; Frier, M. Radionuclide imaging in drug development. Curr. Pharm. Des. 2004, 10, 2907–2921. [Google Scholar] [CrossRef] [PubMed]
- Schottelius, M.; Wirtz, M.; Eiber, M.; Maurer, T.; Wester, H.J. [111In]PSMA-I&T: Expanding the spectrum of PSMA-I&T applications towards SPECT and radioguided surgery. Eur. J. Nucl. Med. Mol. Imaging Res. 2015, 5, 68. [Google Scholar] [CrossRef]
- Vallabhajosula, S.; Nikolopoulou, A.; Babich, J.W.; Osborne, J.R.; Tagawa, S.T.; Lipai, I.; Solnes, L.; Maresca, K.P.; Armor, T.; Joyal, J.L.; et al. 99mTc-labeled small-molecule inhibitors of prostate-specific membrane antigen: Pharmacokinetics and biodistribution studies in healthy subjects and patients with metastatic prostate cancer. J. Nucl. Med. 2014, 55, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Maresca, K.; Wang, J.C.; Hillier, S.; Femia, F.; Lu, G.; Zimmermann, C.; Kronauge, J.; Eckelman, W.; Joyal, J.; Babich, J. Development of a simple kit for Tc-99m-MIP-1404, a single amino acid chelate (SAAC II) derived small molecule inhibitor of prostate specific membrane antigen (PSMA) for imaging prostate cancer. J. Nucl. Med. 2012, 53, S523. [Google Scholar]
- Lütje, S.; Heskamp, S.; Cornelissen, A.S.; Poeppel, T.D.; van den Broek, S.A.M.W.; Rosenbaum-Krumme, S.; Bochisch, A.; Gotthardt, M.; Rijpkema, M.; Boerman, O.C. PSMA ligands for radionuclide imaging and therapy of prostate cancer: Clinical status. Theranostics 2015, 5, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Hetzheim, H.; Kratochwil, C.; Benesova, M.; Eder, M.; Neels, O.C.; Eisenhut, M.; Kübler, W.; Holland-Letz, T.; Giesel, F.L.; et al. The Theranostic PSMA ligand PSMA-617 in the diagnosis of prostate cancer by PET/CT: Biodistribution in humans, radiation dosimetry, and first evaluation of tumor lesions. J. Nucl. Med. 2015, 56, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Derlin, T.; Weiberg, D.; von Klot, C.; Wester, H.J.; Henkenberens, C.; Ross, T.L.; Christiansen, H.; Merseburger, A.S.; Bengel, F.M. 68Ga-PSMA I&T PET/CT for assessment of prostate cancer: Evaluation of image quality after forced diuresis and delayed imaging. Eur. Radiol. 2016, 26, 4345–4353. [Google Scholar] [PubMed]
- Grubmüller, B.; Baum, R.P.; Capasso, E.; Singh, A.; Ahmadi, Y.; Knoll, P.; Floth, A.; Righi, S.; Zandieh, S.; Meleddu, C.; et al. 64Cu-PSMA-617 PET/CT imaging of prostate adenocarcinoma: First in-human studies. Cancer Biother. Radiopharm. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Milowsky, M.I.; Morris, M.; Vallabhajosula, S.; Christos, P.; Akhtar, N.H.; Osborne, J.; Goldsmith, S.J.; Larson, S.; Taskar, N.P.; et al. Phase II study of Lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody j591 for metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 5182–5191. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadehfar, H.; Eppard, E.; Kürpig, S.; Fimmers, R.; Yordanova, A.; Schlenkhoff, C.D.; Gärtner, F.; Rogenhofer, S.; Essler, M. Therapeutic response and side effects of repeated radioligand therapy with 177Lu-PSMA-DKFZ-617 of castrate-resistant metastatic prostate cancer. Oncotarget 2016, 7, 12477–12488. [Google Scholar] [PubMed]
- Rahbar, K.; Bode, A.; Weckesser, M.; Avramovic, N.; Claesener, M.; Stegger, L.; Bögemann, M. Radioligand therapy with 177Lu-PSMA-617 as a novel therapeutic option in patients with metastatic castration resistant prostate cancer. Clin. Nucl. Med. 2016, 41, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadehfar, H.; Rahbar, K.; Kürpig, S.; Bögemann, M.; Claesener, M.; Eppard, E.; Gärtner, F.; Rogenhofer, S.; Schäfers, M.; Essler, M. Early side effects and first results of radioligand therapy with 177Lu-DKFZ-617 PSMA of castrate-resistant metastatic prostate cancer: A two-centre study. Eur. J. Nucl. Med. Mol. Imaging Res. 2015, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Baum, R.P.; Kulkarni, H.R.; Schuchardt, C.; Singh, A.; Wirtz, M.; Wiessalla, S.; Schottelius, M.; Mueller, D.; Klette, I.; Wester, H.J. Lutetium-177 PSMA radioligand therapy of metastatic castration-resistant prostate cancer: Safety and efficacy. J. Nucl. Med. 2016, 57, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Heck, M.M.; Retz, M.; D’Alessandria, C.; Rauscher, I.; Scheidhauer, K.; Maurer, T.; Storz, E.; Janssen, F.; Schottelius, M.; Wester, H.J.; et al. Systemic radioligand therapy with 177Lu Labeled prostate specific membrane antigen ligand for imaging and therapy in patients with metastatic castration resistant prostate cancer. J. Urol. 2016, 196, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Giesel, F.L.; Stefanova, M.; Benešová, M.; Bronzel, M.; Afshar-Oromieh, A.; Mier, W.; Eder, M.; Kopka, K.; Haberkorn, U. PSMA-targeted radionuclide therapy of metastatic castration-resistant prostate cancer with lu-177 labeled psma-617. J. Nucl. Med. 2016, 57, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, K.; Schmidt, M.; Heinzel, A.; Eppard, E.; Bode, A.; Yordanova, A.; Claesener, M.; Ahmadzadehfar, H. Response and tolerability of a single dose of 177Lu-PSMA-617 in patients with metastatic castration-resistant prostate cancer: A multicenter retrospective analysis. J. Nucl. Med. 2016, 57, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Schaal, K.; Stoykow, C.; Mix, M.; Bartholomä, M.; Drendel, V.; Mäcke, H.R.; Gourni, E.; Wetterauer, U.; Schultze-Seemann, W.; Meyer, P.T.; et al. Feasibility of 111In-PSMA-guided surgery for treatment of nodal prostate cancer relapse. Eur. Urol. Suppl. 2016, 15, e551. [Google Scholar] [CrossRef]
Figure 1.
The enzyme actions of PSMA. (a) Glutamic acid is released from folate polyglutamate; (b) N-Acetyl-l-aspartyl-l-glutamate (NAAG) is hydrolyzed to aspartate (NAA) and glutamate (l-Glu).
Figure 1.
The enzyme actions of PSMA. (a) Glutamic acid is released from folate polyglutamate; (b) N-Acetyl-l-aspartyl-l-glutamate (NAAG) is hydrolyzed to aspartate (NAA) and glutamate (l-Glu).
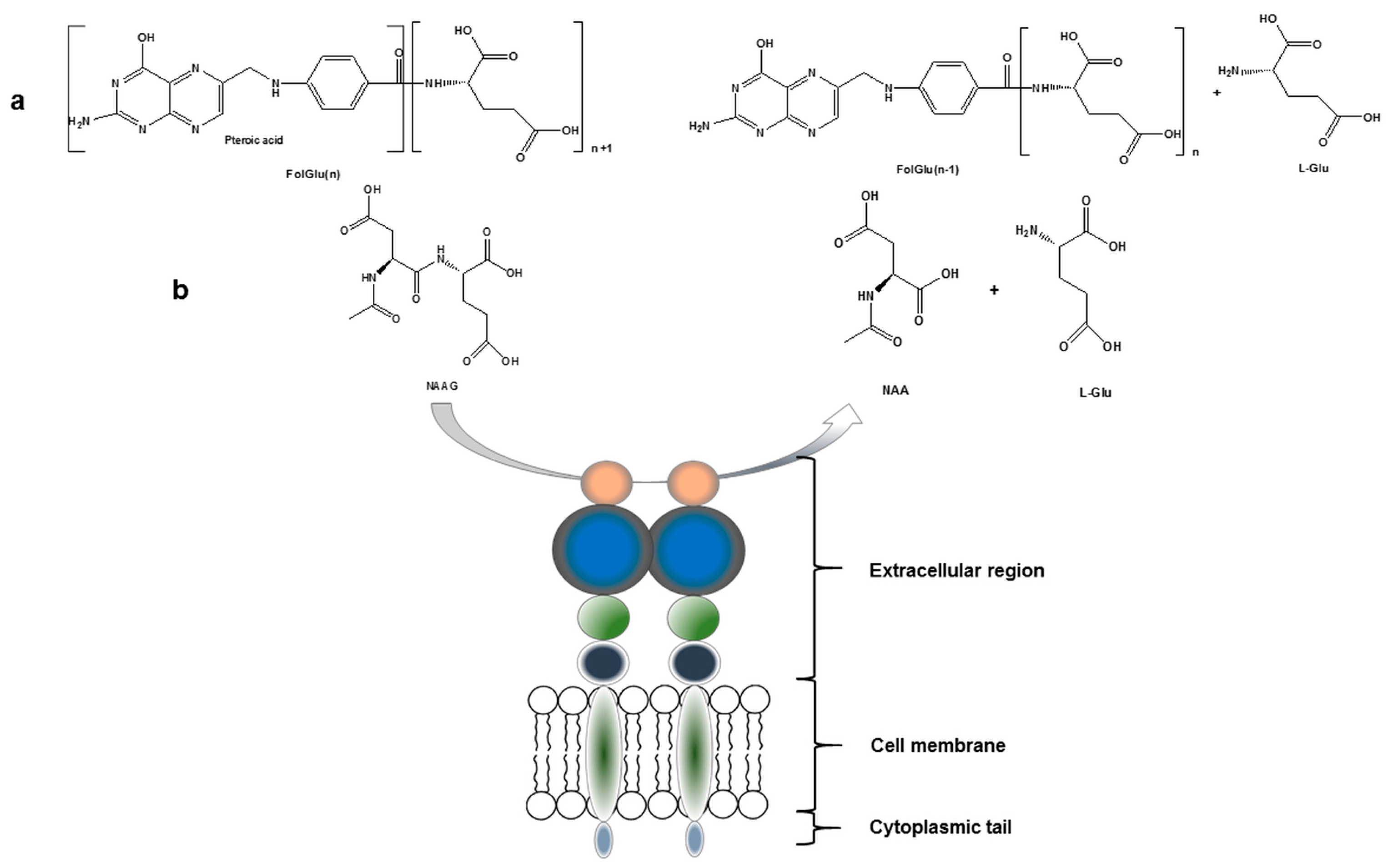
Figure 2.
PSMA inhibitors. 2-PMPA (1) is the first potent PSMA inhibitor. The three groups of PSMA inhibitors include: thiols 2 and 3, phosphonates 4 and ureas 5 and 6.
Figure 2.
PSMA inhibitors. 2-PMPA (1) is the first potent PSMA inhibitor. The three groups of PSMA inhibitors include: thiols 2 and 3, phosphonates 4 and ureas 5 and 6.
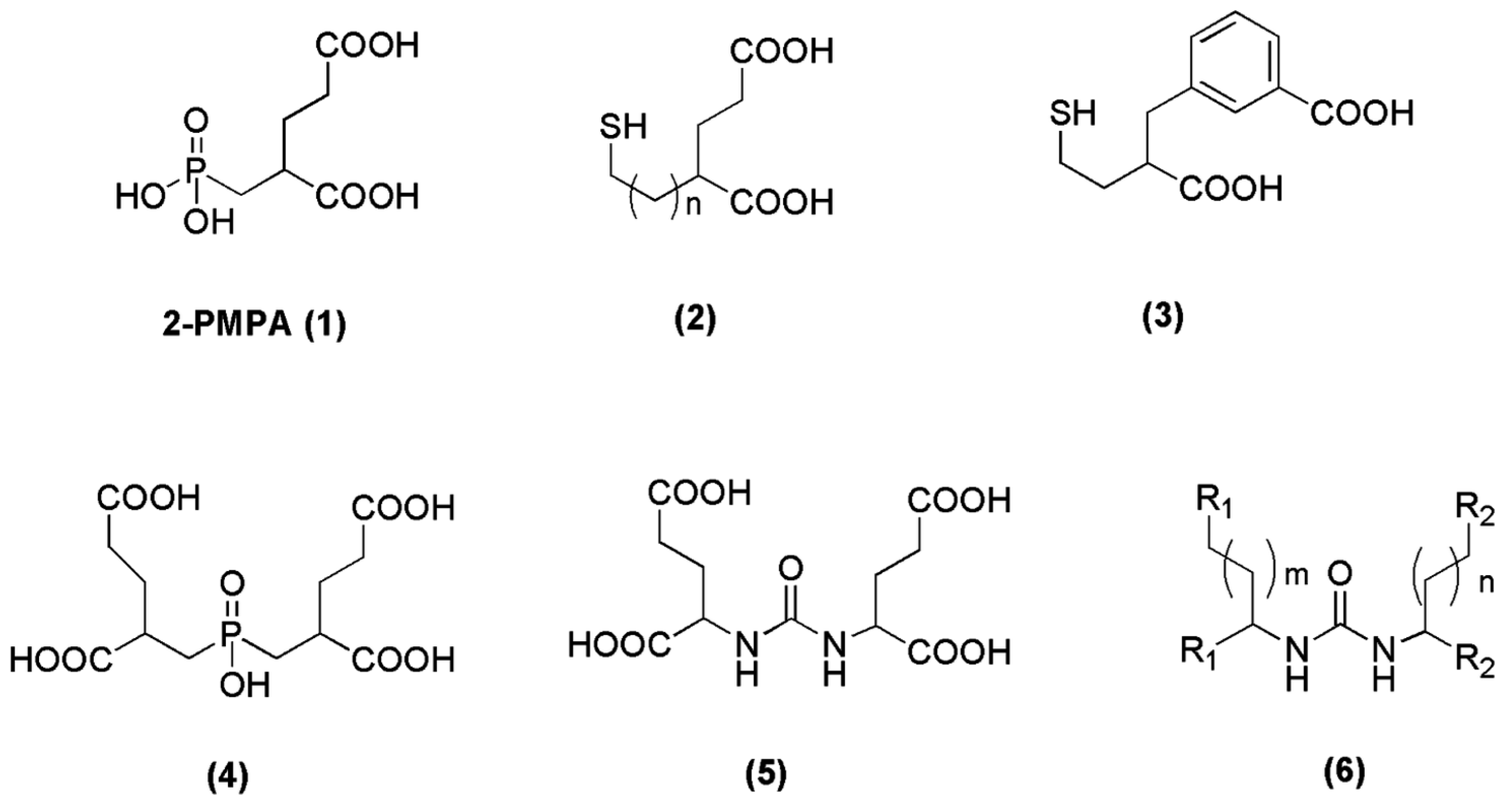
Figure 3.
Chemical structures of the 99mTc-labeled GPI (7) GPI monomer (8), GPI dimer (9) and GPI trimer (10) [72,73].

Figure 4.
Chemical structure of 99mTc(CO)3-DTPA-CTT-54 (11) [74].
Figure 4.
Chemical structure of 99mTc(CO)3-DTPA-CTT-54 (11) [74].
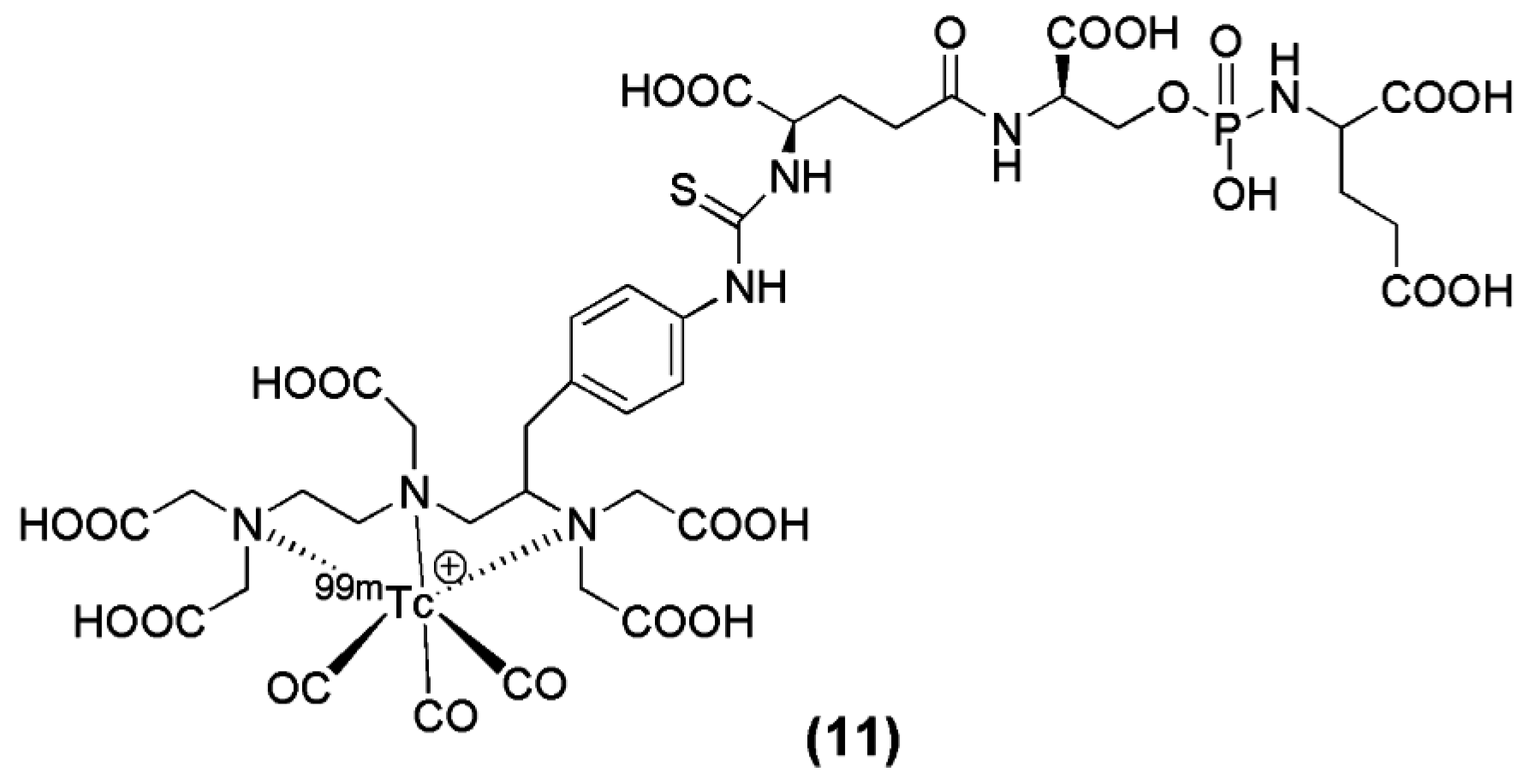
Figure 5.
Chemical structure of 64Cu-ABN-1 (12) [75].
Figure 5.
Chemical structure of 64Cu-ABN-1 (12) [75].

Figure 6.
PSMA-inhibitors labeled via the ([99mTcO]3+) (13–20), ([99mTc(CO)3]+) (21–24) and (99mTc-hynic) (25) core [98,99].
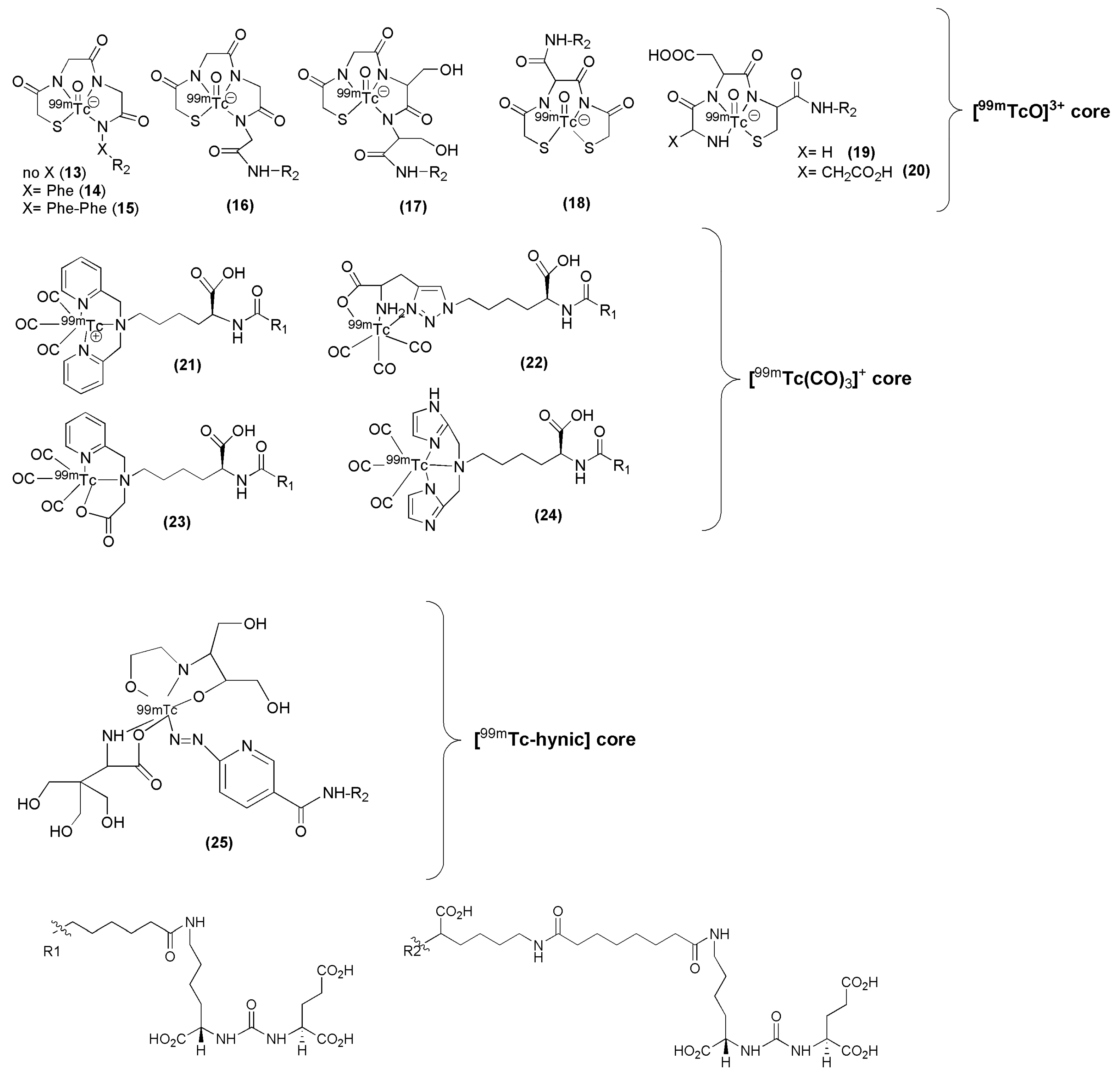
Figure 7.
99mTc-labeled PSMA inhibitors (26–31) [100].
Figure 7.
99mTc-labeled PSMA inhibitors (26–31) [100].
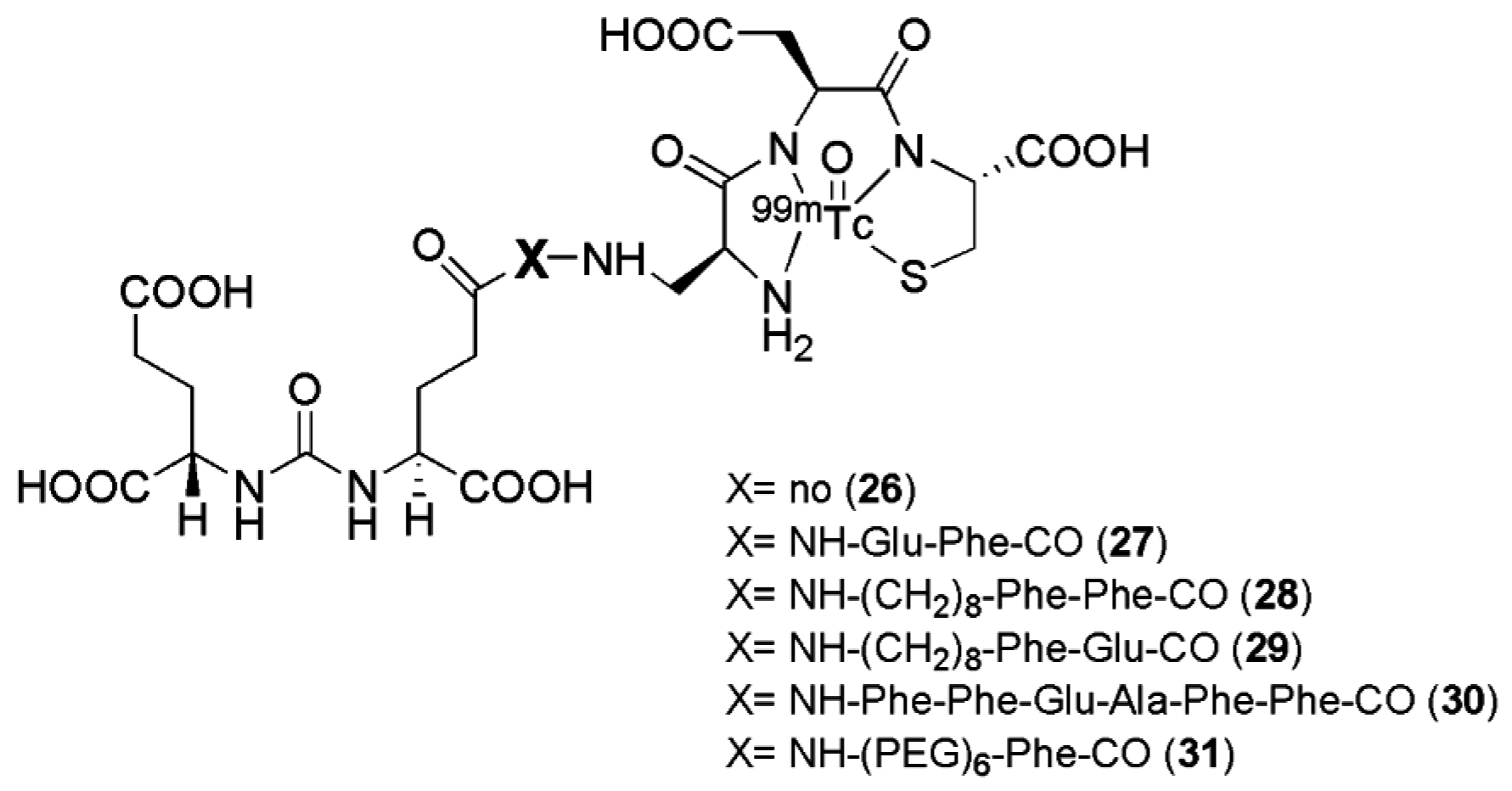
Figure 8.
Chemical structure of 99mTc-MAS3 (32)/mas3 (33)-y-nal-k-Sub-KuE (MAS3/mas3-D-Tyr-D-2-Nal-D-Lys-suberoyl-Lys-urea-Glu) [101].
Figure 8.
Chemical structure of 99mTc-MAS3 (32)/mas3 (33)-y-nal-k-Sub-KuE (MAS3/mas3-D-Tyr-D-2-Nal-D-Lys-suberoyl-Lys-urea-Glu) [101].
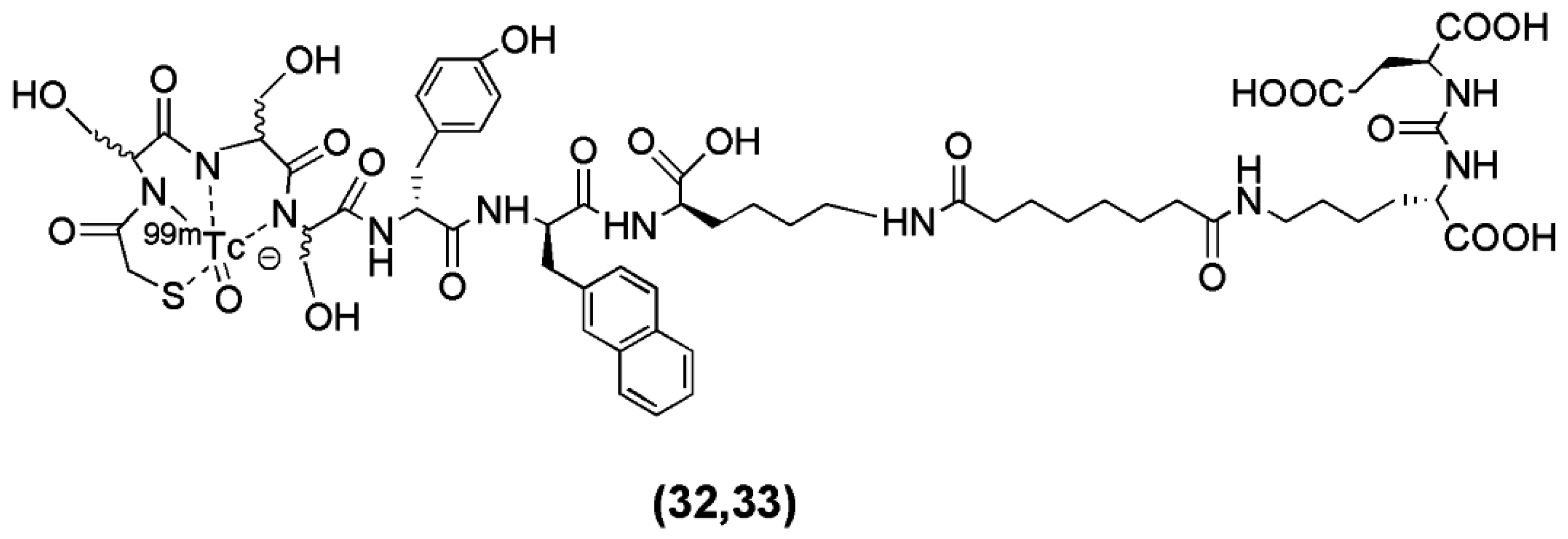
Figure 9.
Chemical structure of 99mTc-TMCE (34) [102].
Figure 9.
Chemical structure of 99mTc-TMCE (34) [102].
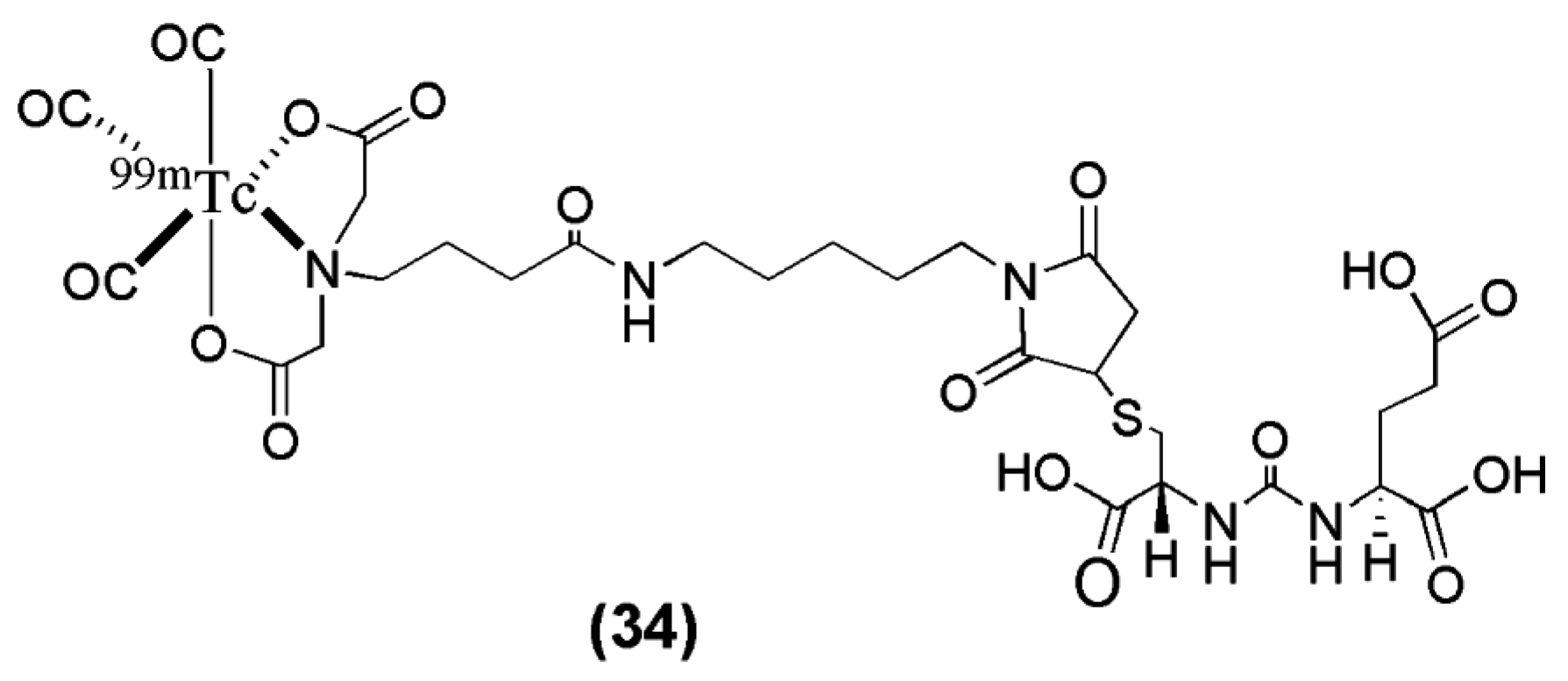
Figure 10.
Chemical structures of 99mTc-MIP-1428 (35), 99mTc-MIP-1405 (36), 99mTc-MIP-1404 (37) and 99mTc-MIP-1427 (38) [103].
Figure 10.
Chemical structures of 99mTc-MIP-1428 (35), 99mTc-MIP-1405 (36), 99mTc-MIP-1404 (37) and 99mTc-MIP-1427 (38) [103].

Figure 11.
Chemical structures of the monomer 68Ga-PSMA-11 (39) and the dimer 68Ga-PSMA-10 (40) [106,130].

Figure 12.
Chemical structures of the two DOTA- (41 and 42) and the NOTA-conjugated (43) PSMA inhibitors labeled with 68Ga [131,132].
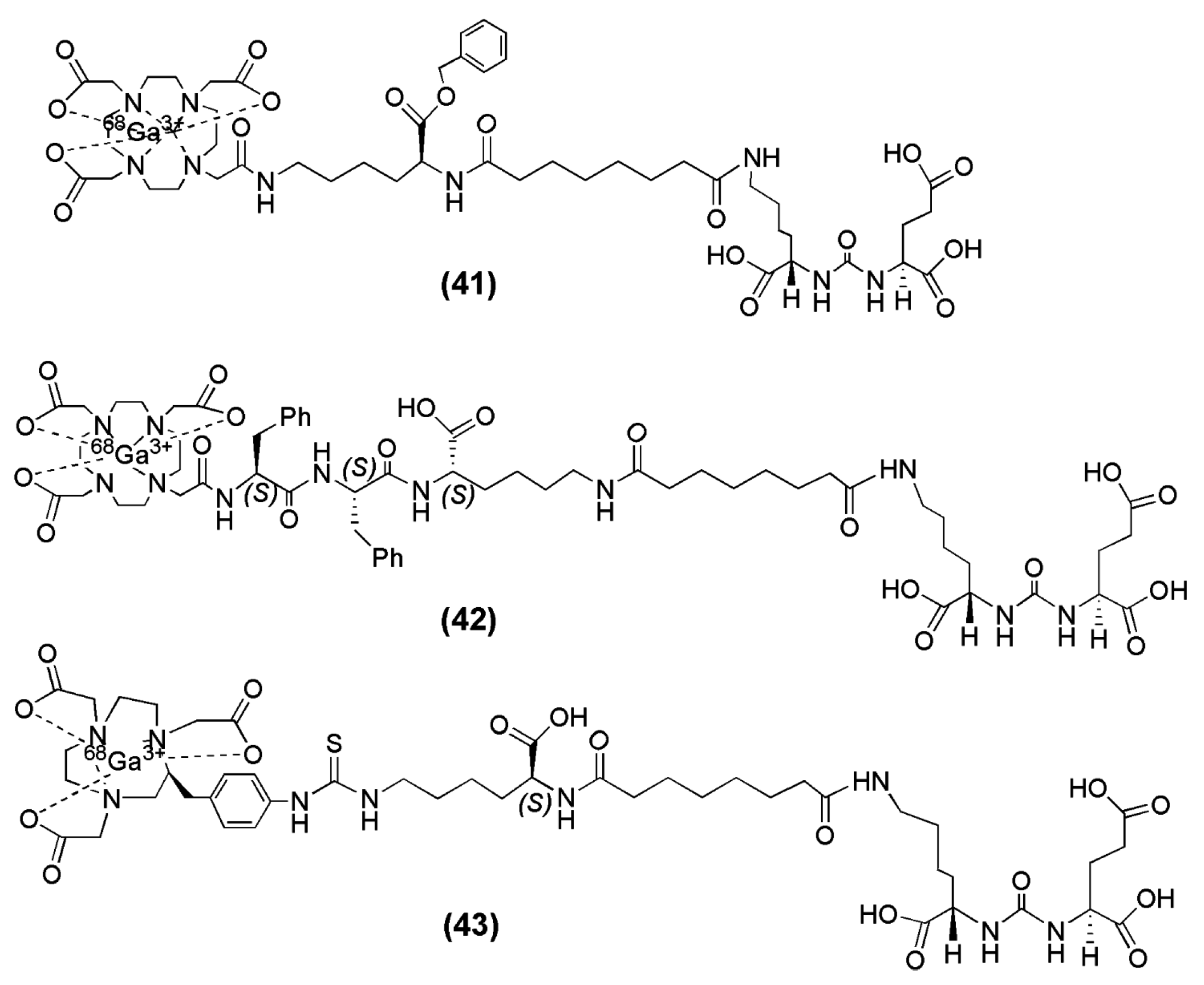
Figure 13.
Chemical structure of 68Ga-PSMA-617 (44) [133].
Figure 13.
Chemical structure of 68Ga-PSMA-617 (44) [133].
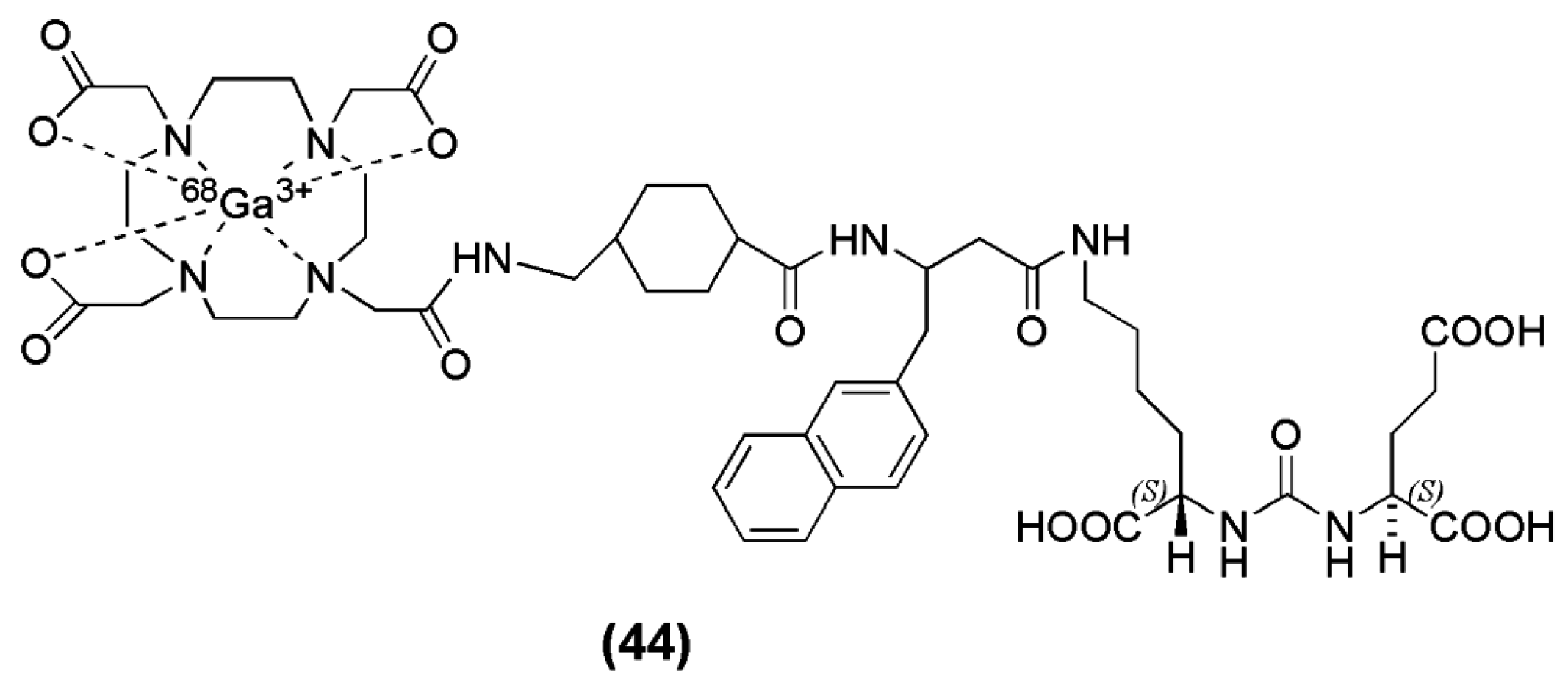
Figure 14.
Chemical structures of the three generations (DOTAGA-FFK(Sub-KuE) (45), DOTAGA-ffk(Sub-KuE) (46) and of DOTAGA-(I-y)fk(Sub-KuE) (47)) PSMA inhibitors labeled with 68Ga [134,135].
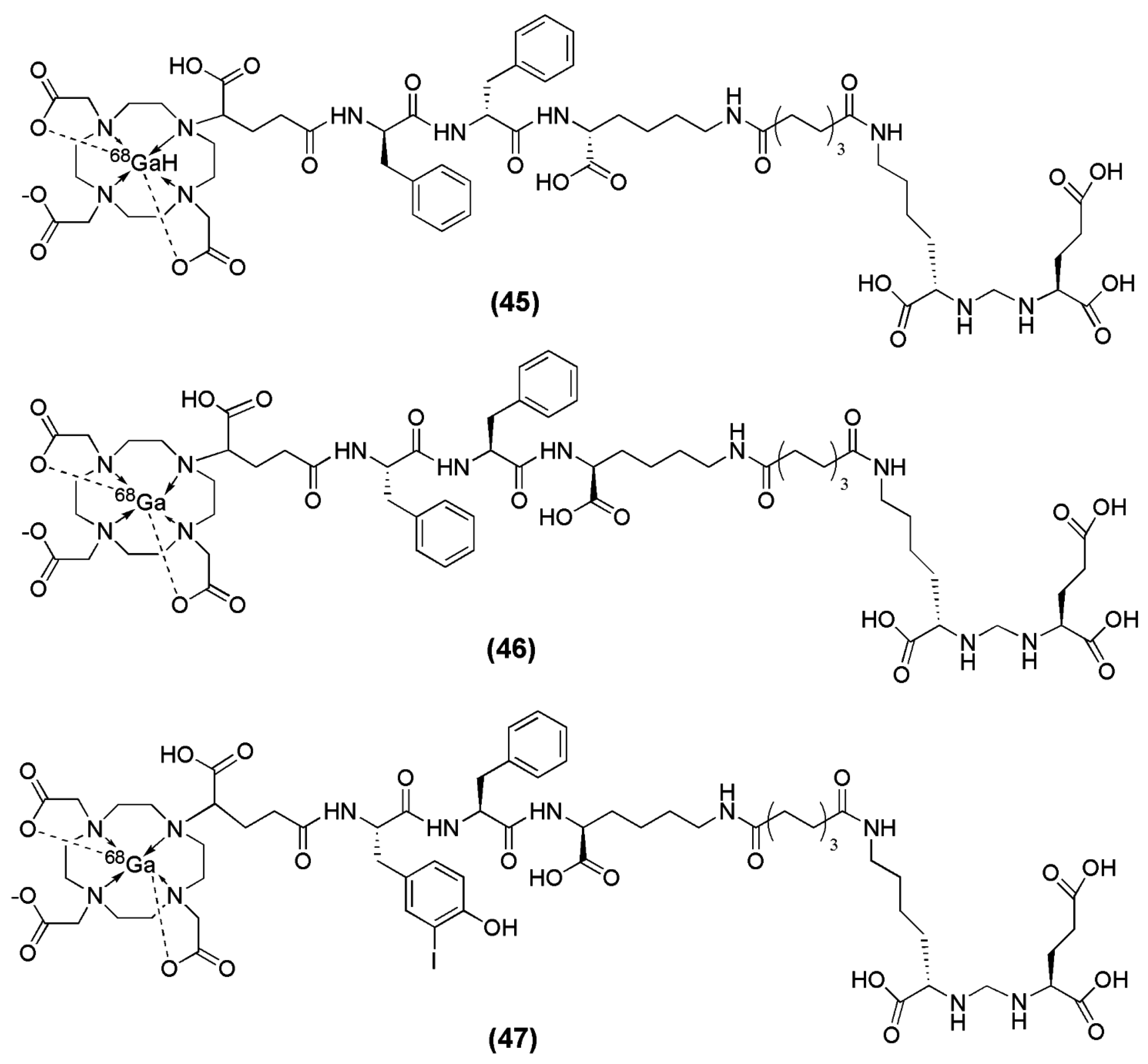
Figure 15.
Chemical structure of 68Ga-(R)-NODAGA-Phe-Phe-d-Lys(suberoyl)-Lys-urea-Glu (68Ga-CC34) (48) [136].
Figure 15.
Chemical structure of 68Ga-(R)-NODAGA-Phe-Phe-d-Lys(suberoyl)-Lys-urea-Glu (68Ga-CC34) (48) [136].
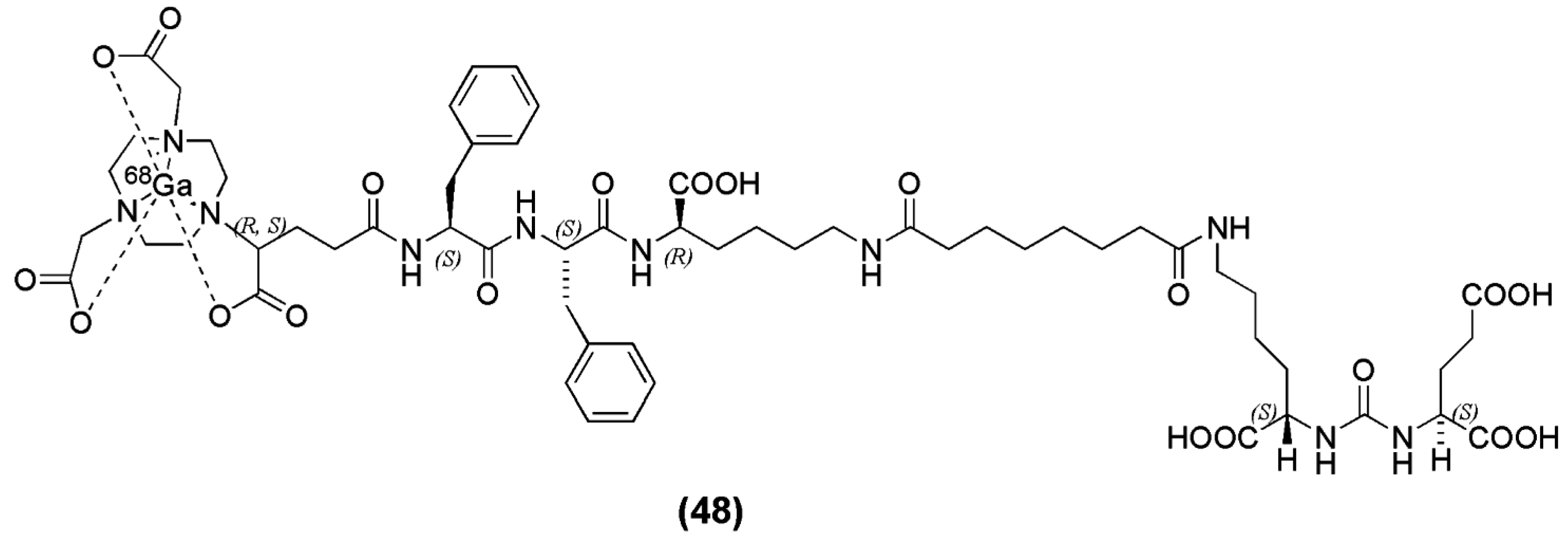
Figure 16.
Chemical structure of 68Ga-CHX-A″-DTPA-DUPA-Pep (49) [137].
Figure 16.
Chemical structure of 68Ga-CHX-A″-DTPA-DUPA-Pep (49) [137].

Figure 17.
Chemical structures of the PSMA-based inhibitors (50–54) labeled with 64Cu [139].
Figure 17.
Chemical structures of the PSMA-based inhibitors (50–54) labeled with 64Cu [139].

Figure 18.
Tumor uptake of 99mTc-MIP-1404 (A) or 99mTc-MIP-1405 (B) at 4 h in patient with metastatic prostate cancer, compared with that of standard bone scan (C). Images also show uptake of radiotracer in normal parotid and salivary glands. Ant = anterior; Post = posterior (Reprinted with permission of [143]).
Figure 18.
Tumor uptake of 99mTc-MIP-1404 (A) or 99mTc-MIP-1405 (B) at 4 h in patient with metastatic prostate cancer, compared with that of standard bone scan (C). Images also show uptake of radiotracer in normal parotid and salivary glands. Ant = anterior; Post = posterior (Reprinted with permission of [143]).

Figure 19.
68Ga-PSMA-11 PET/CT image of a patient with locally recurrent prostate cancer (PSA 3.7 ng/mL) after radical prostatectomy (SUVmax 12.4) who received 140 MBq of the 68Ga-labeled tracer molecule and was scanned at 1 h p.i.; (a) CT image; (b) PET image; (c) PET/CT fusion image; (d) MIP (Reprinted with permission of [145]).
Figure 19.
68Ga-PSMA-11 PET/CT image of a patient with locally recurrent prostate cancer (PSA 3.7 ng/mL) after radical prostatectomy (SUVmax 12.4) who received 140 MBq of the 68Ga-labeled tracer molecule and was scanned at 1 h p.i.; (a) CT image; (b) PET image; (c) PET/CT fusion image; (d) MIP (Reprinted with permission of [145]).
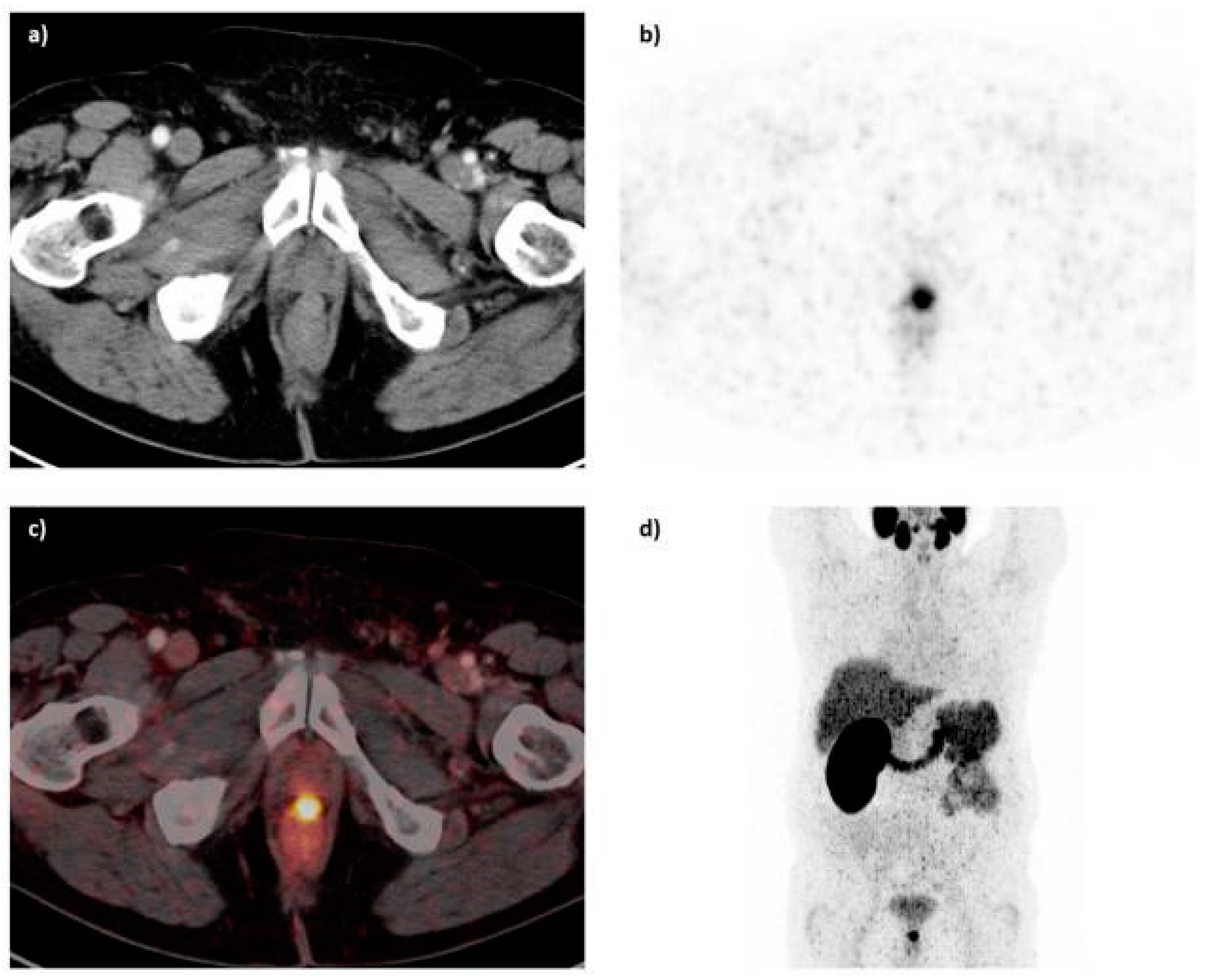
Figure 20.
(A–C) 68Ga-PSMA-617 PET/CT of a patient at 1 h after injection. Red arrows point to a bone metastasis with SUVmax of 21.7 at 1 h and 32.6 at 3 h after injection. (A) Low dose CT; (B) Fusion of PET and CT; (C) MIP of PET/CT. MIP = maximum-intensity projection (Reprinted with permission of [146]).
Figure 20.
(A–C) 68Ga-PSMA-617 PET/CT of a patient at 1 h after injection. Red arrows point to a bone metastasis with SUVmax of 21.7 at 1 h and 32.6 at 3 h after injection. (A) Low dose CT; (B) Fusion of PET and CT; (C) MIP of PET/CT. MIP = maximum-intensity projection (Reprinted with permission of [146]).
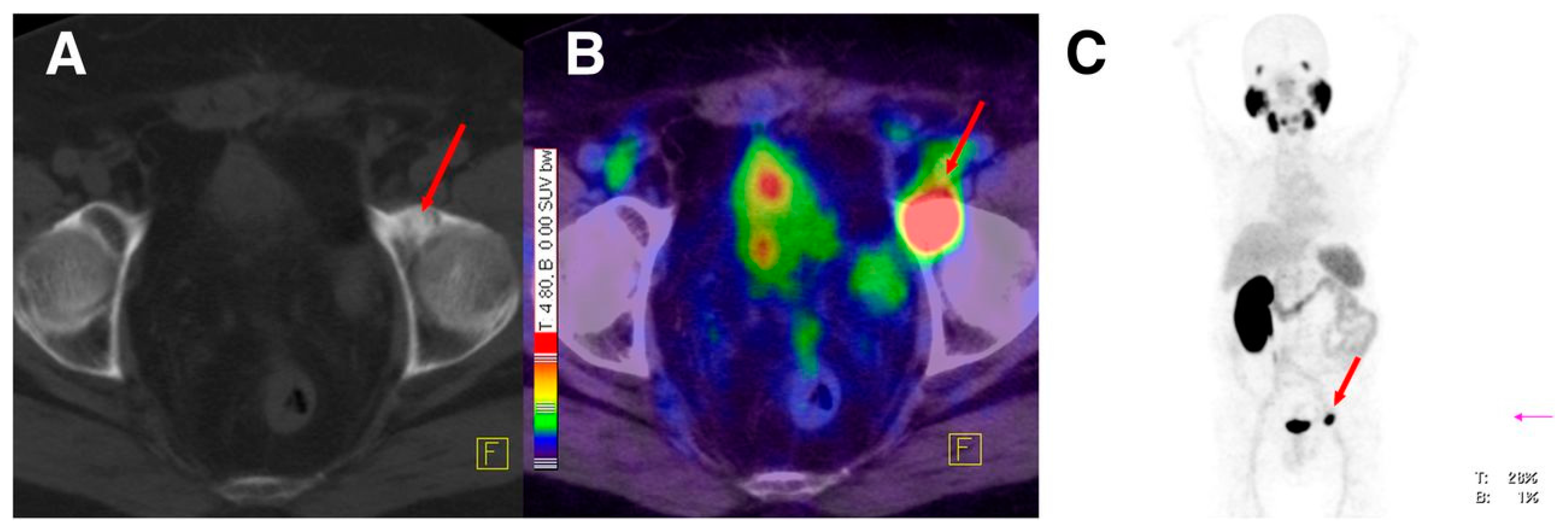
Figure 21.
70-year-old patient with PSMA-avid lymph node metastases on 68Ga-PSMA PET/CT before therapy (A) and on 177Lu-PSMA I&T scintigraphy after first PSMA radionuclide therapy (B); with remarkable reduction in uptake after second PSMA RLT (C). Results were consistent with excellent therapy response projection (Reprinted with permission of [153]).
Figure 21.
70-year-old patient with PSMA-avid lymph node metastases on 68Ga-PSMA PET/CT before therapy (A) and on 177Lu-PSMA I&T scintigraphy after first PSMA radionuclide therapy (B); with remarkable reduction in uptake after second PSMA RLT (C). Results were consistent with excellent therapy response projection (Reprinted with permission of [153]).
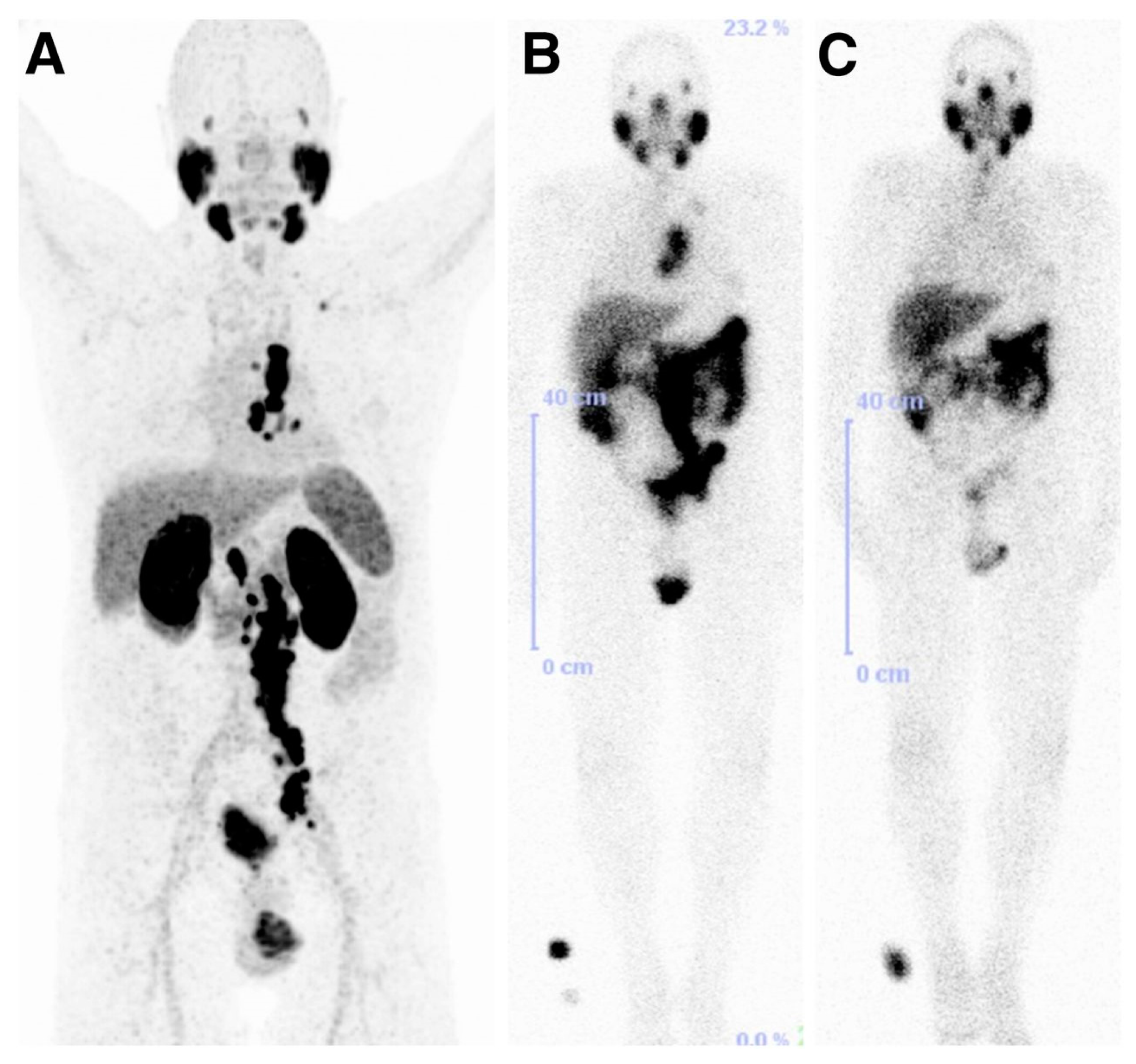

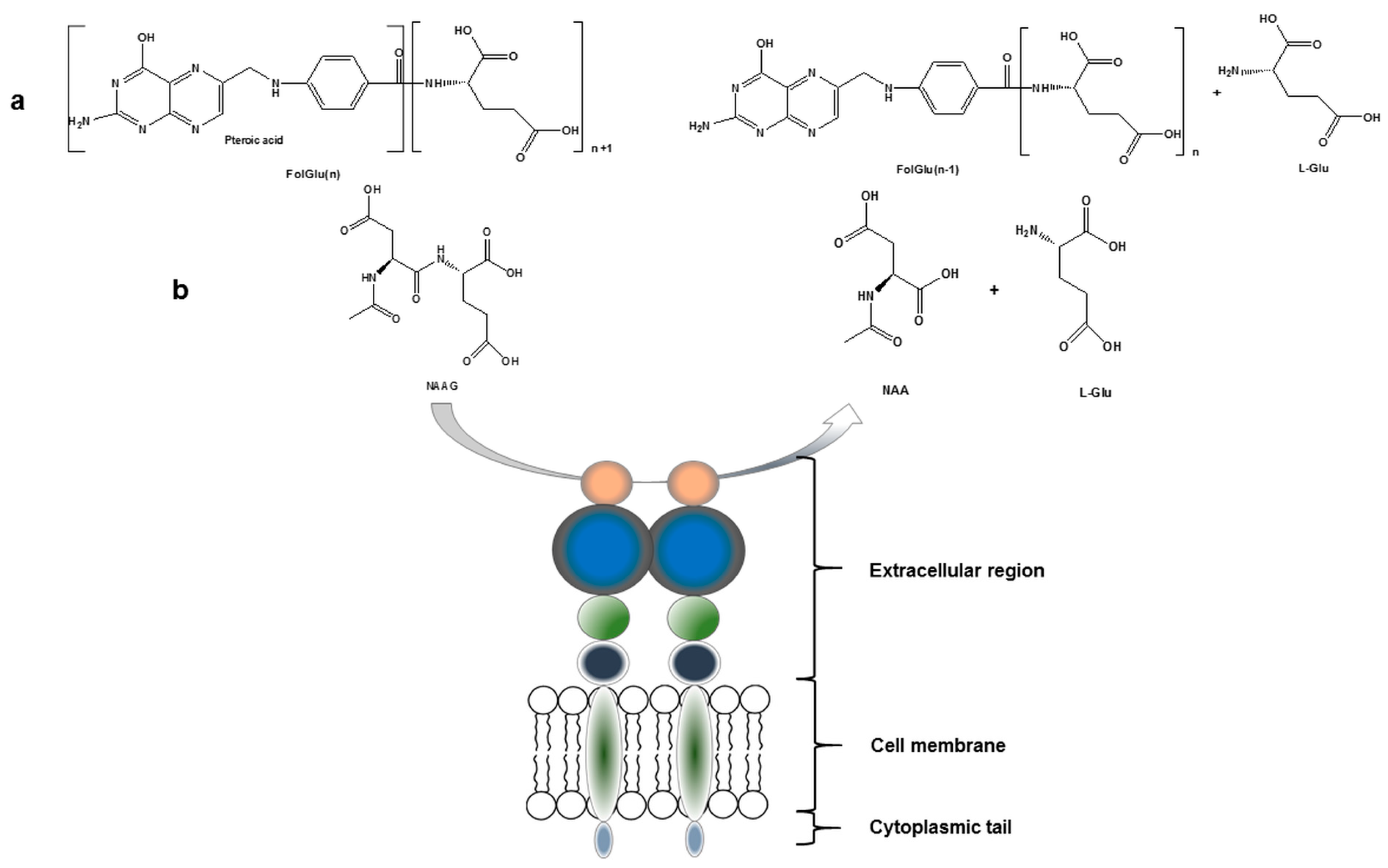{kind=link}
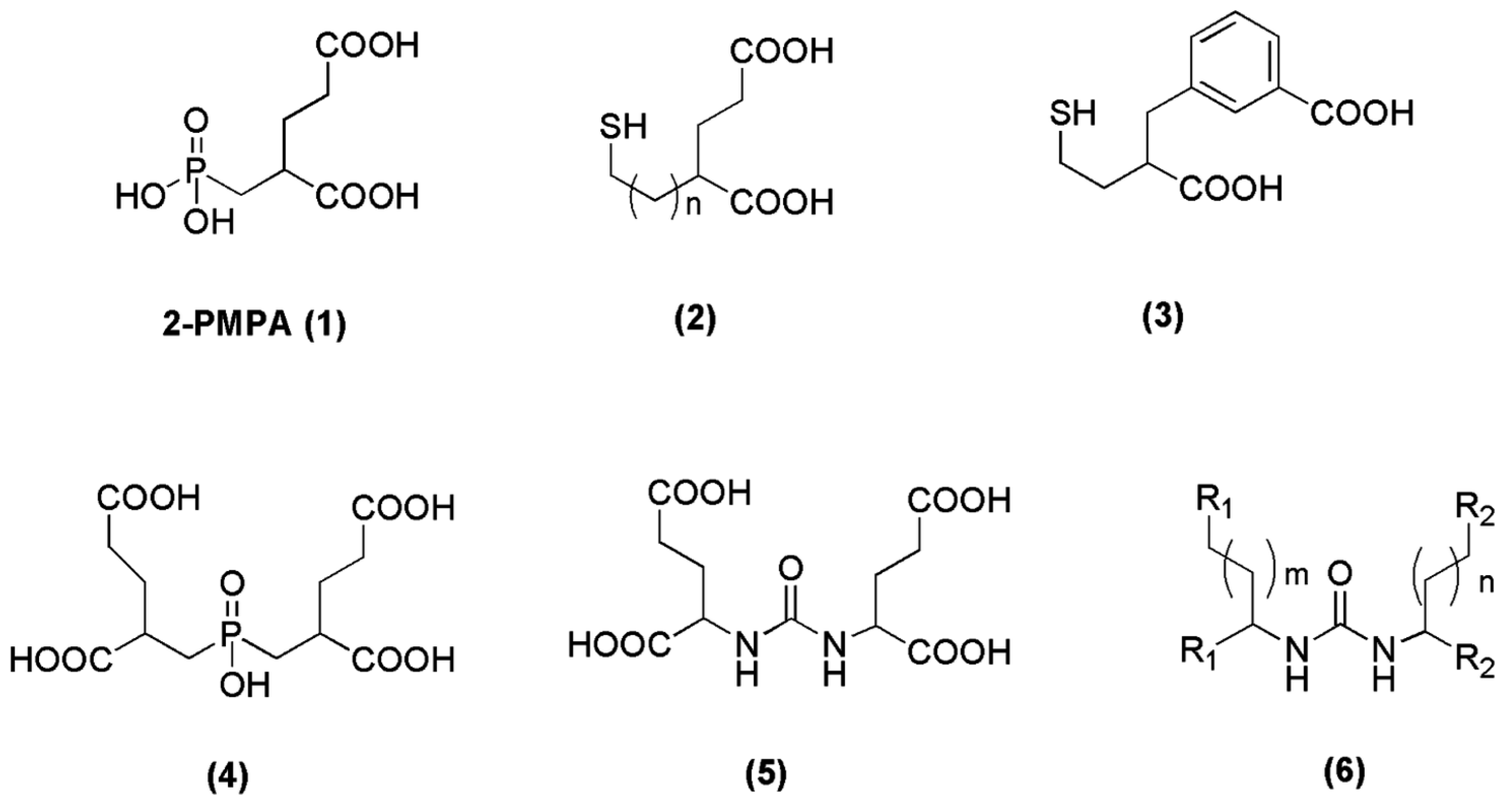{kind=link}
{kind=link}
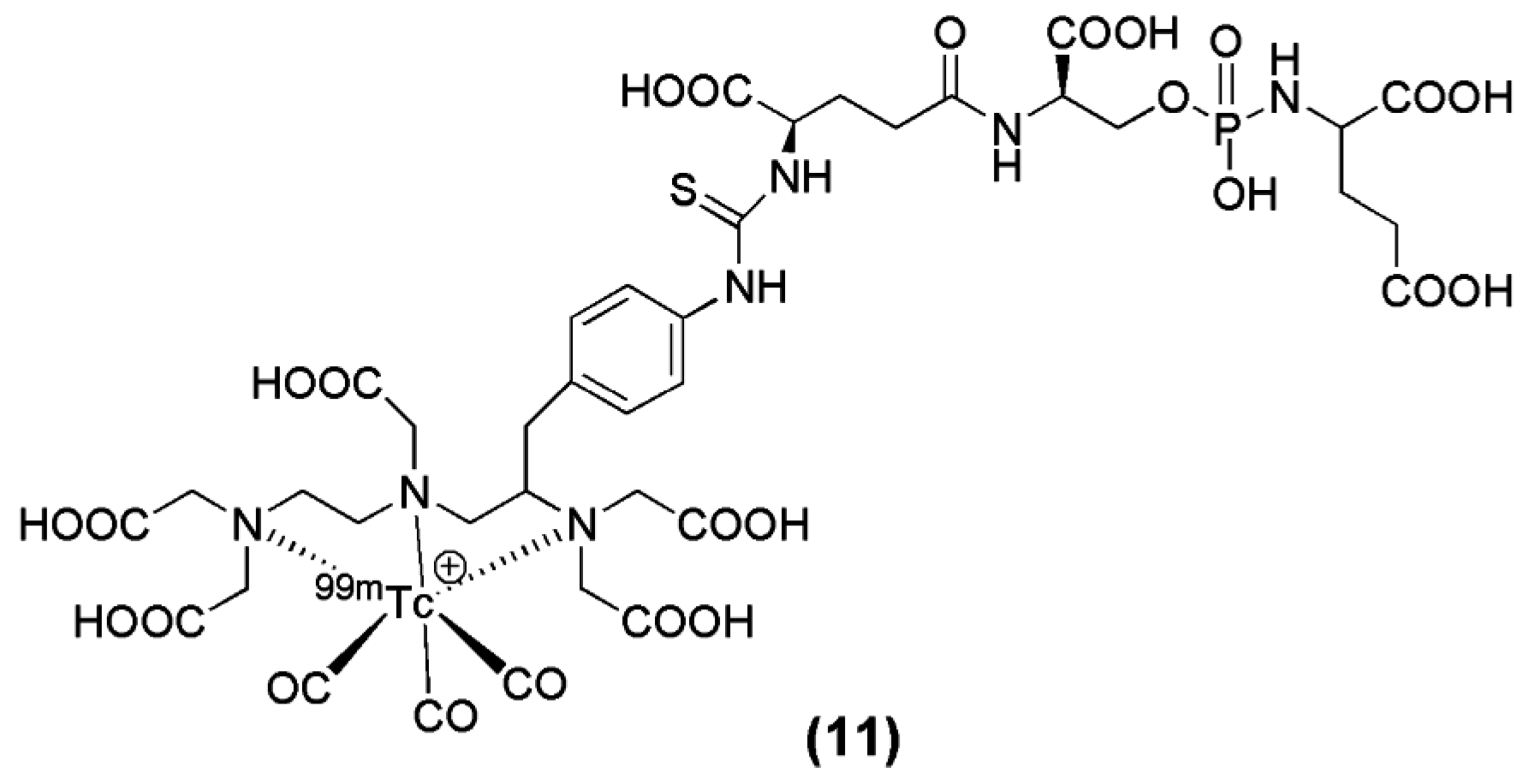{kind=link}
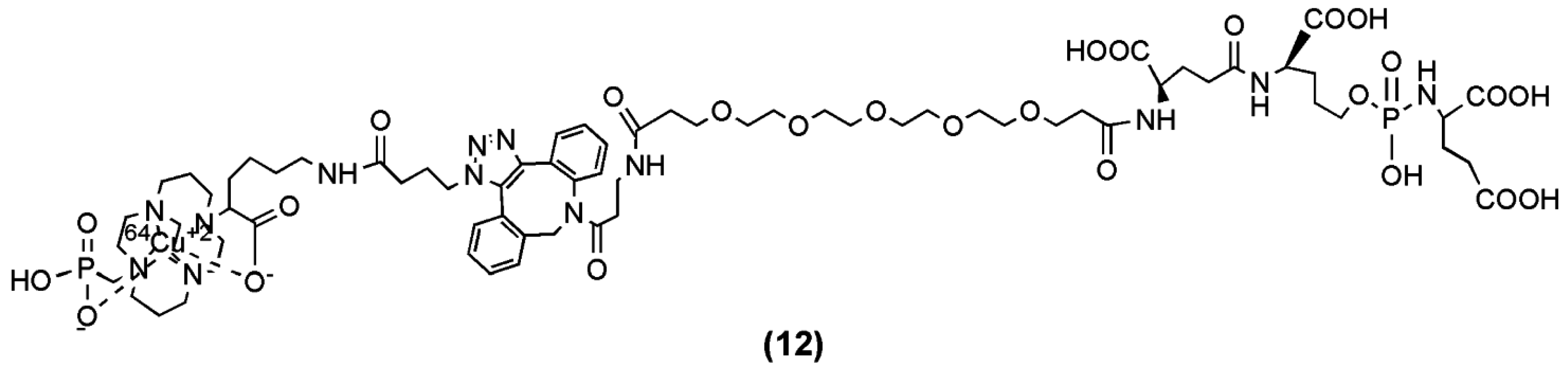{kind=link}
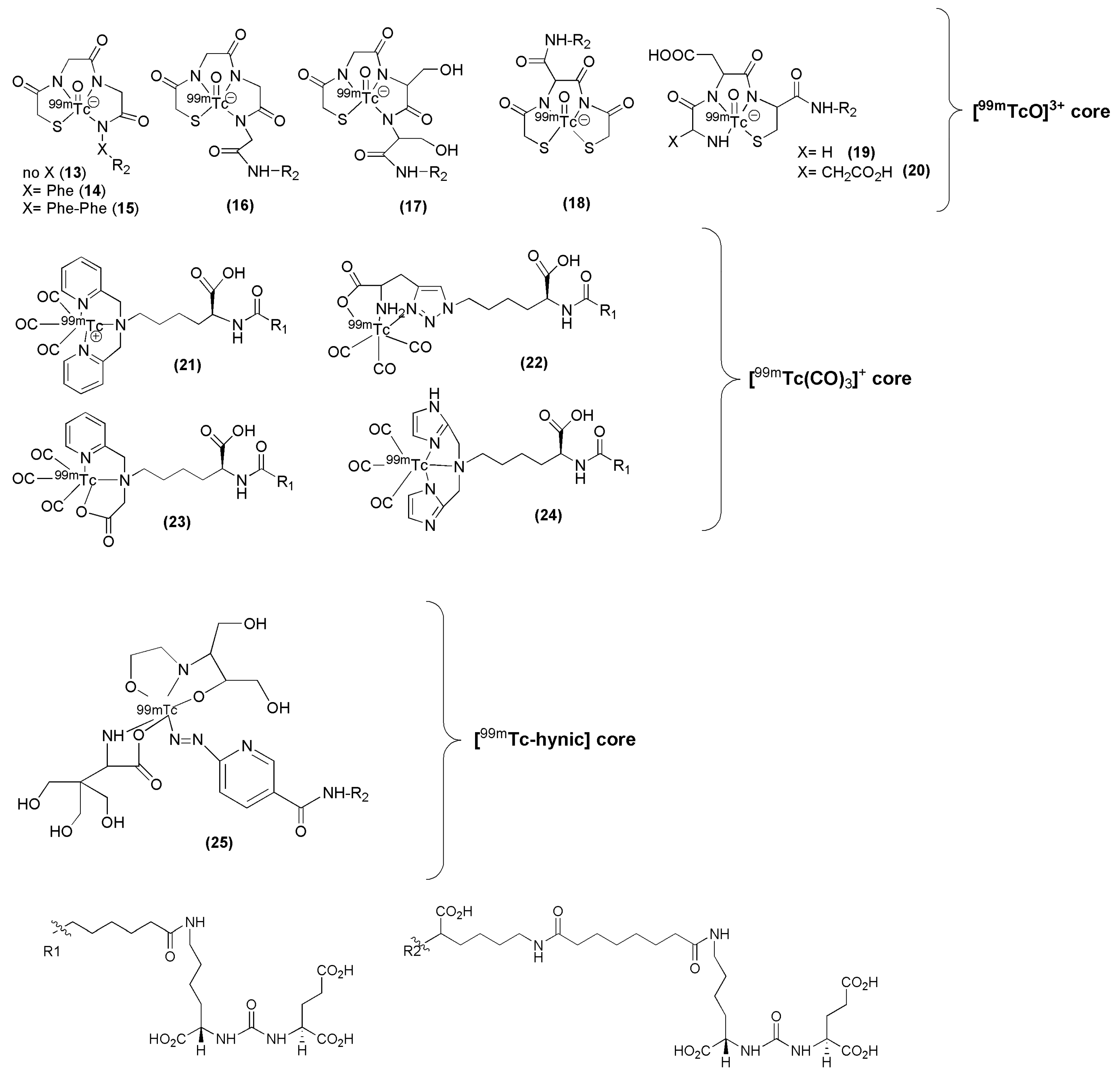{kind=link}
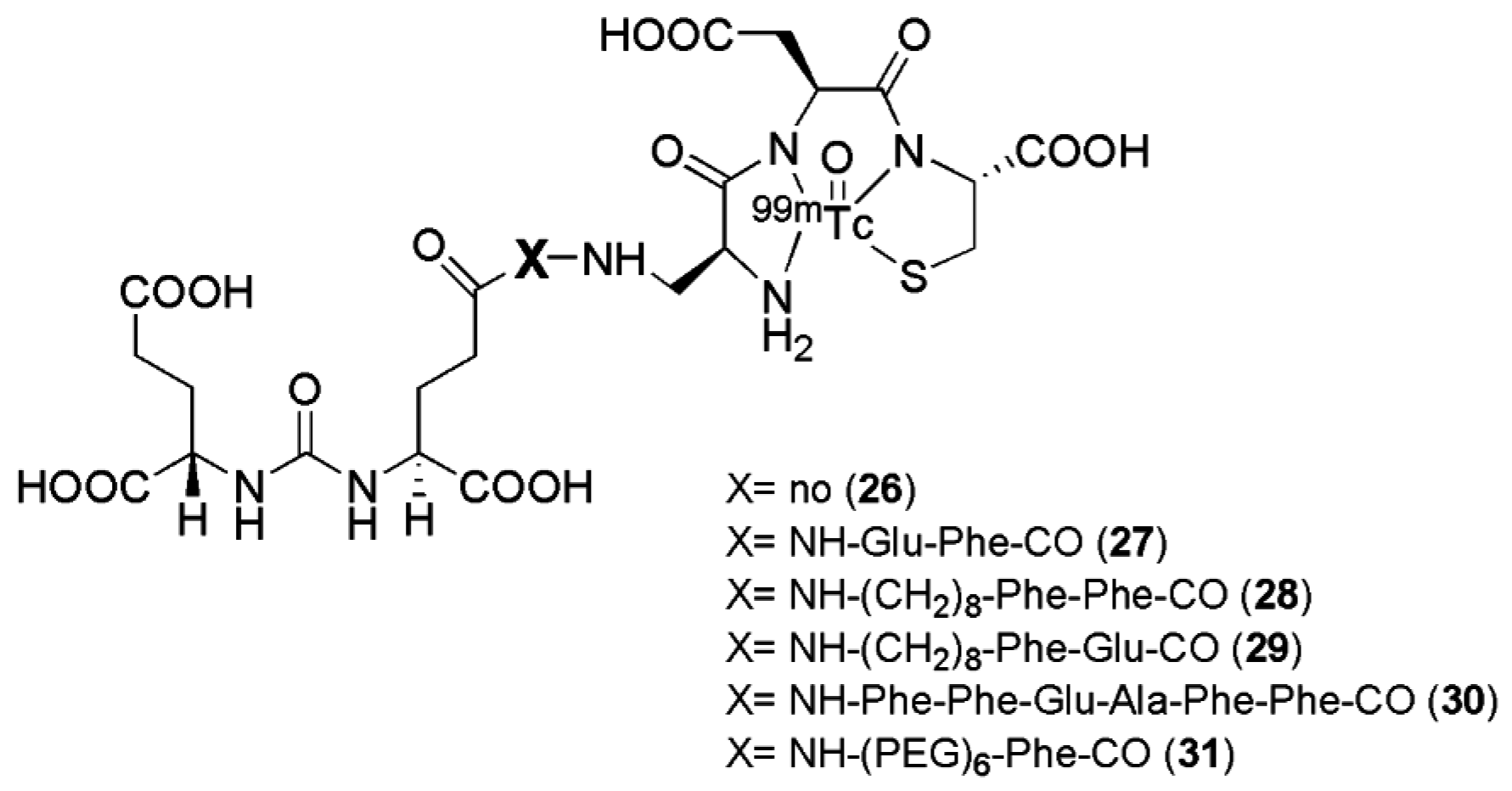{kind=link}
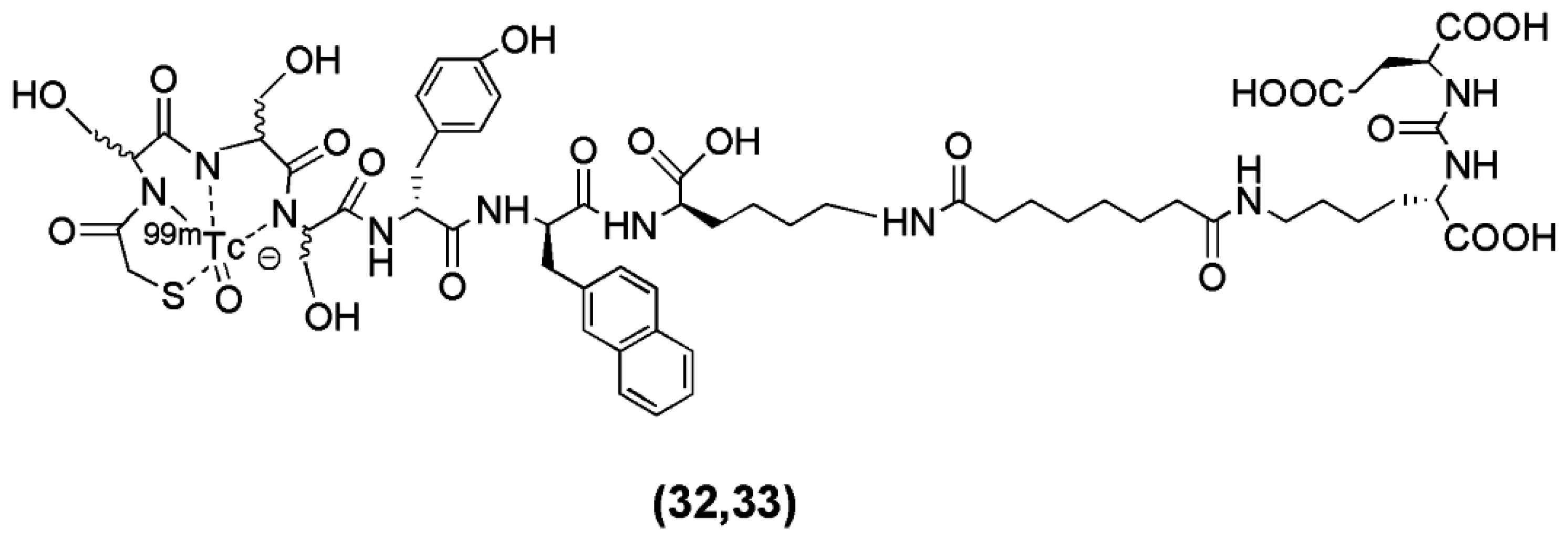{kind=link}
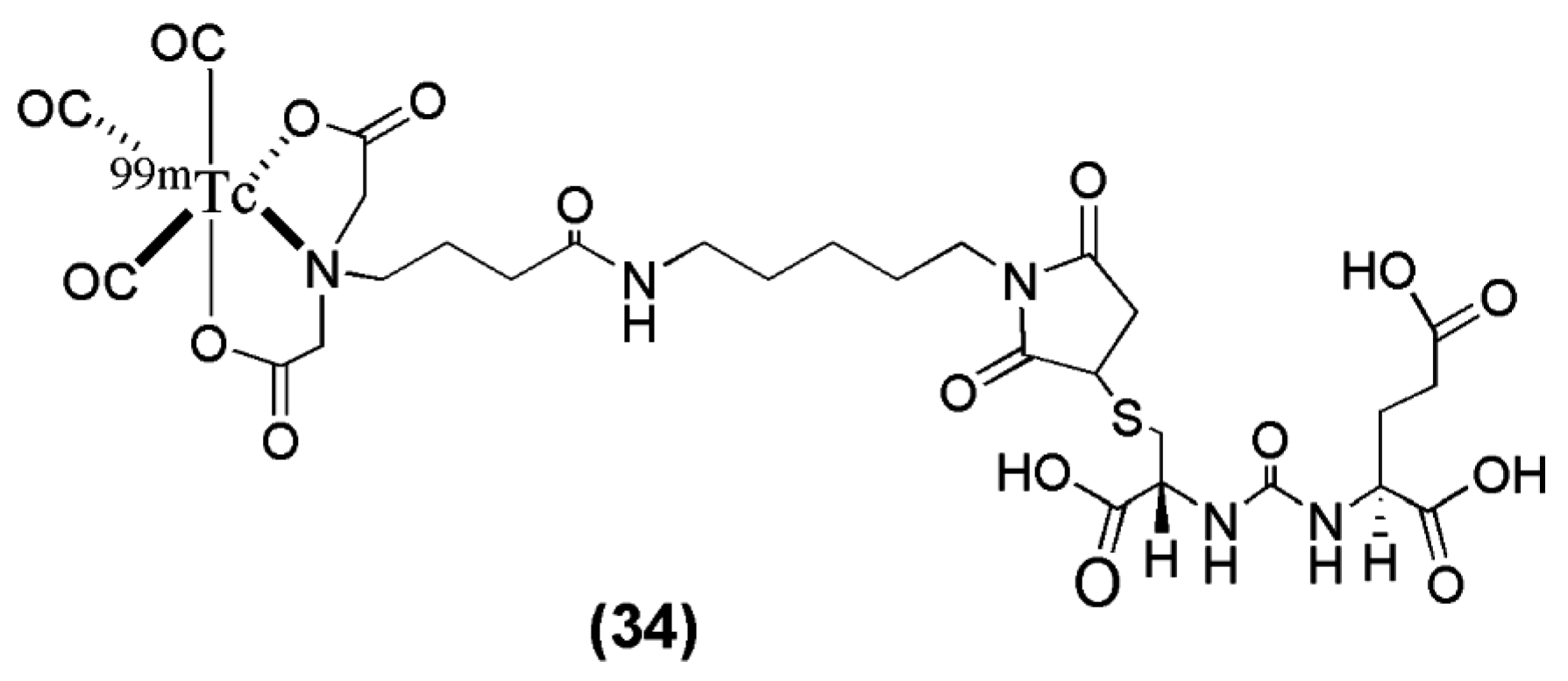{kind=link}
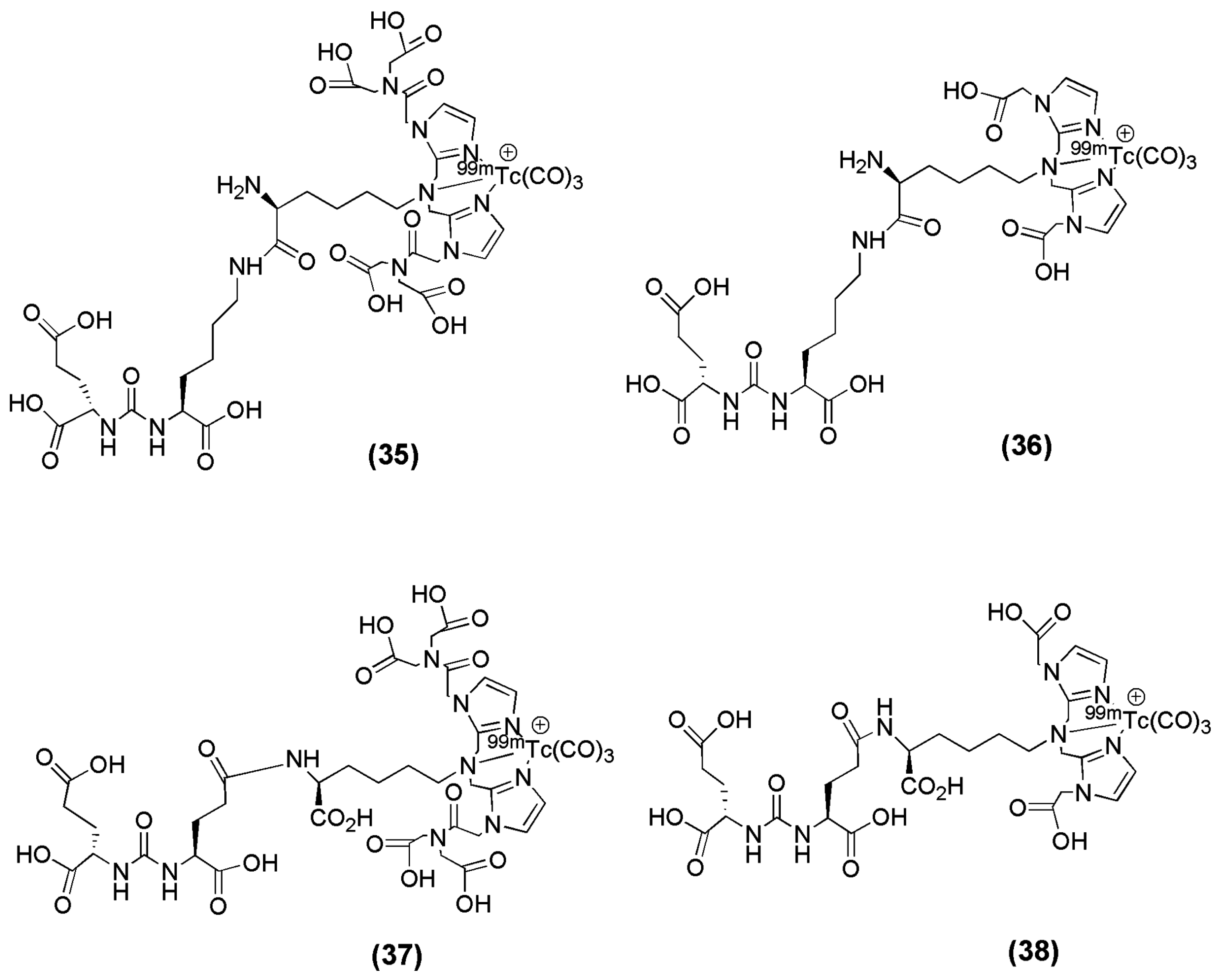{kind=link}
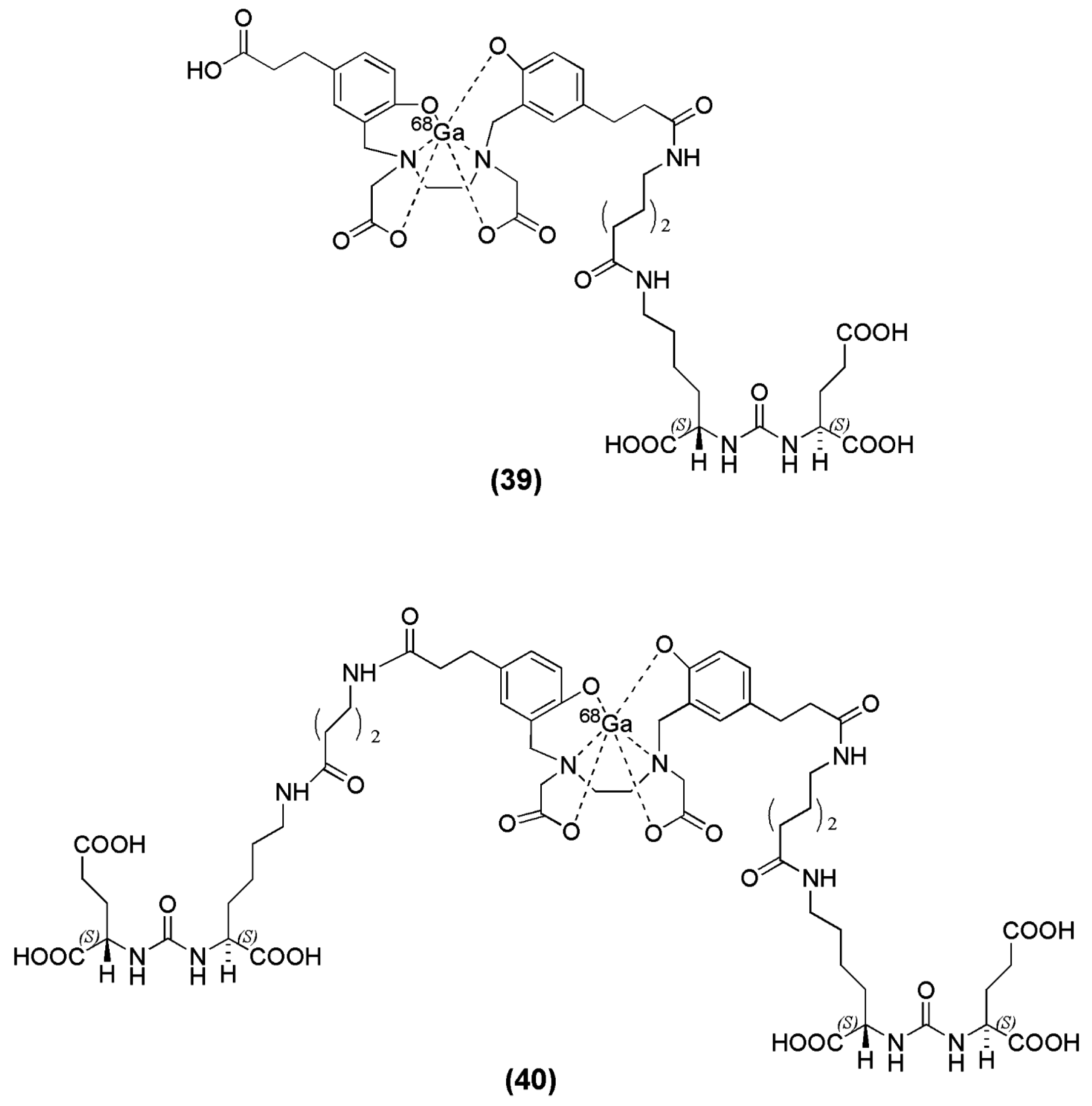{kind=link}
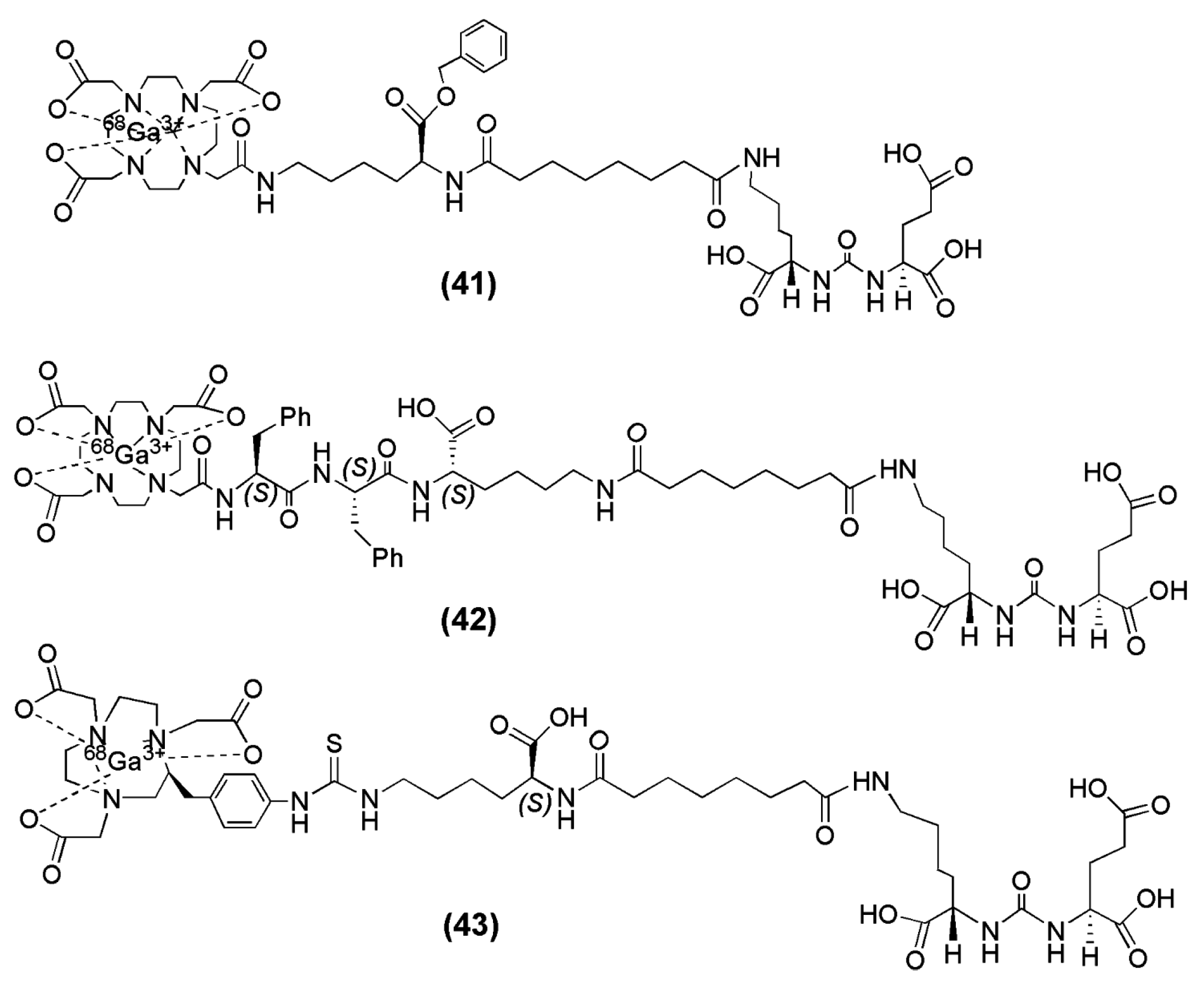{kind=link}
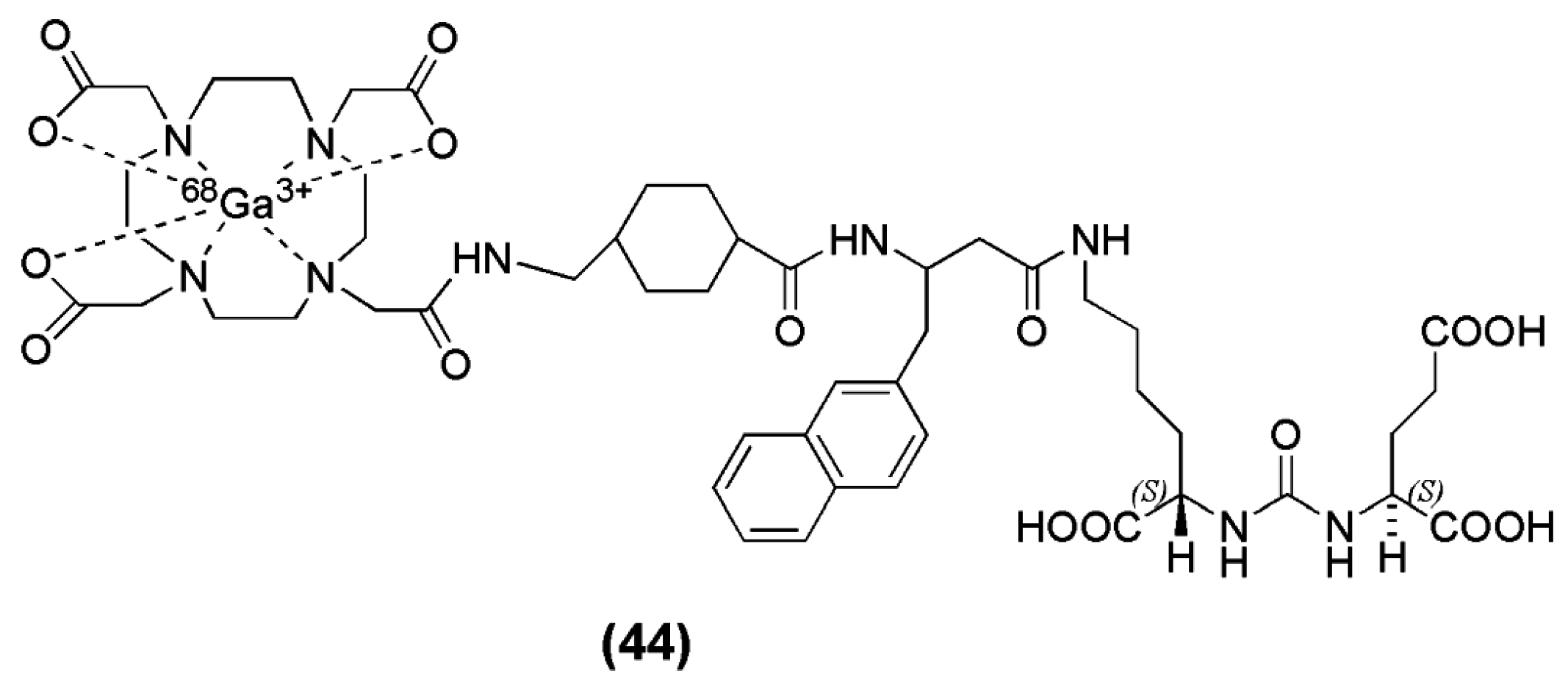{kind=link}
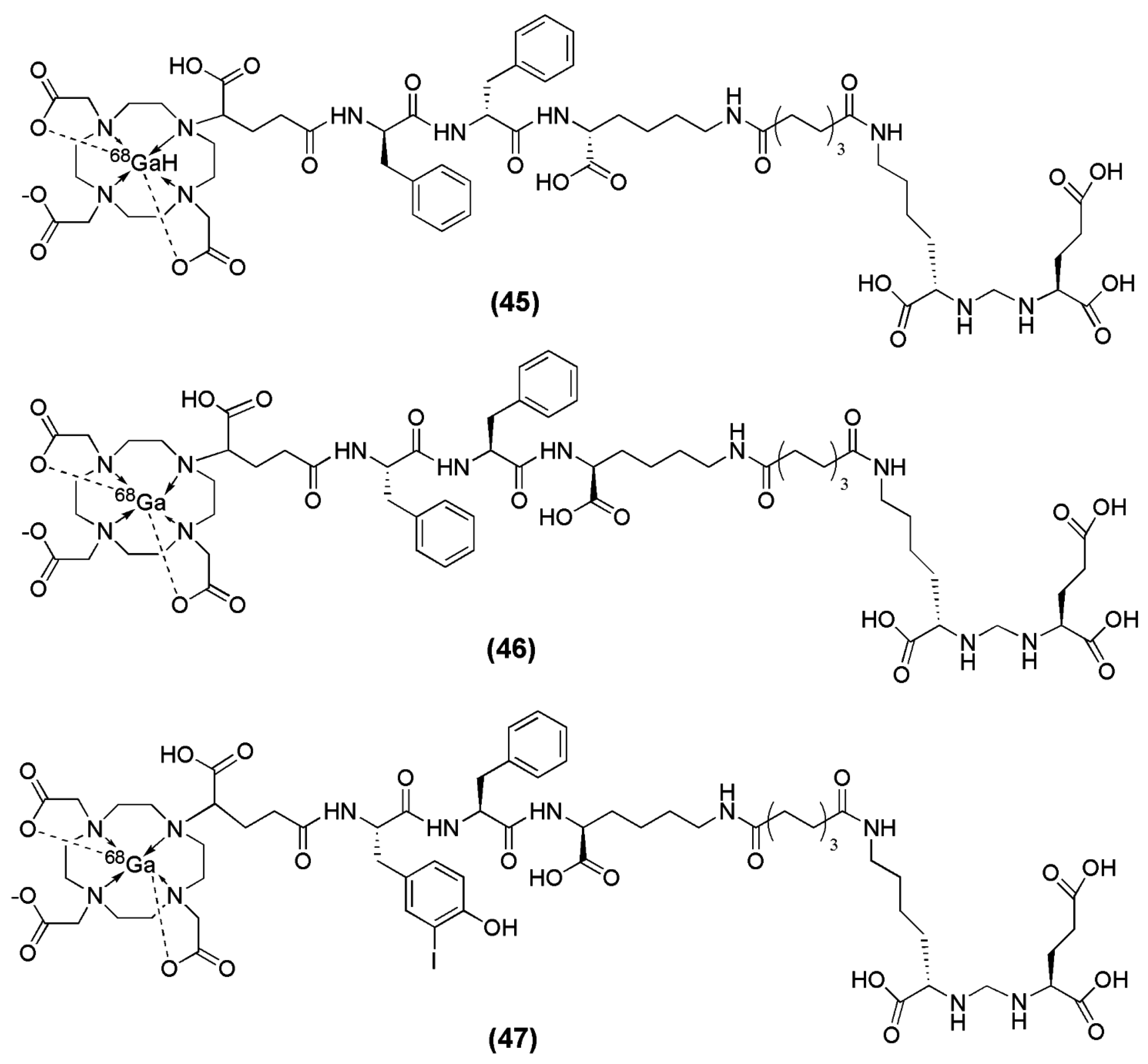{kind=link}
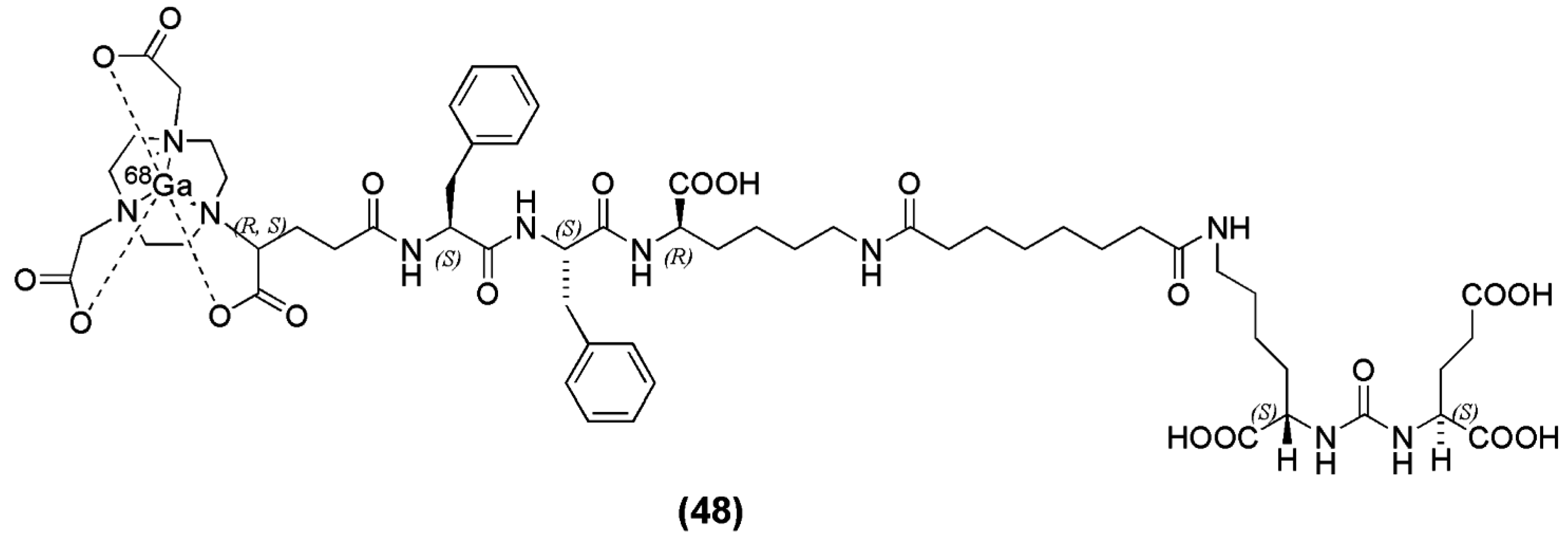{kind=link}
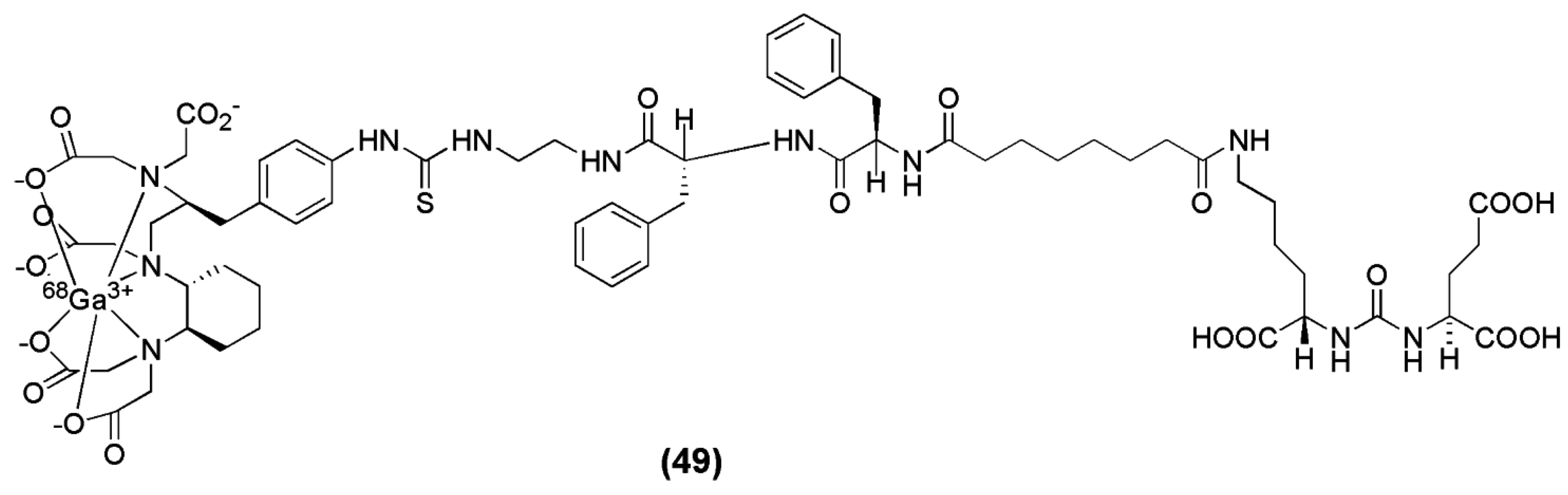{kind=link}
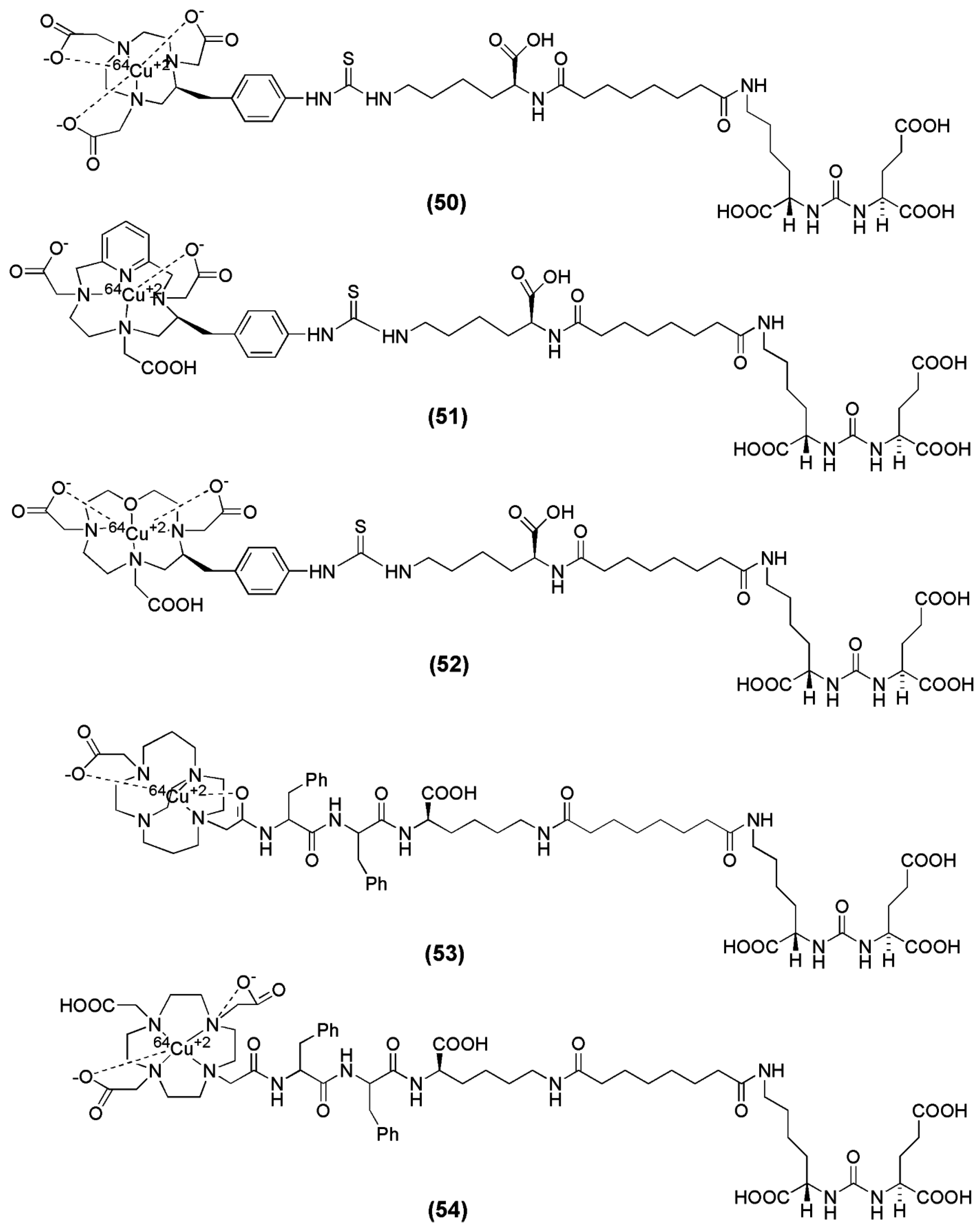{kind=link}
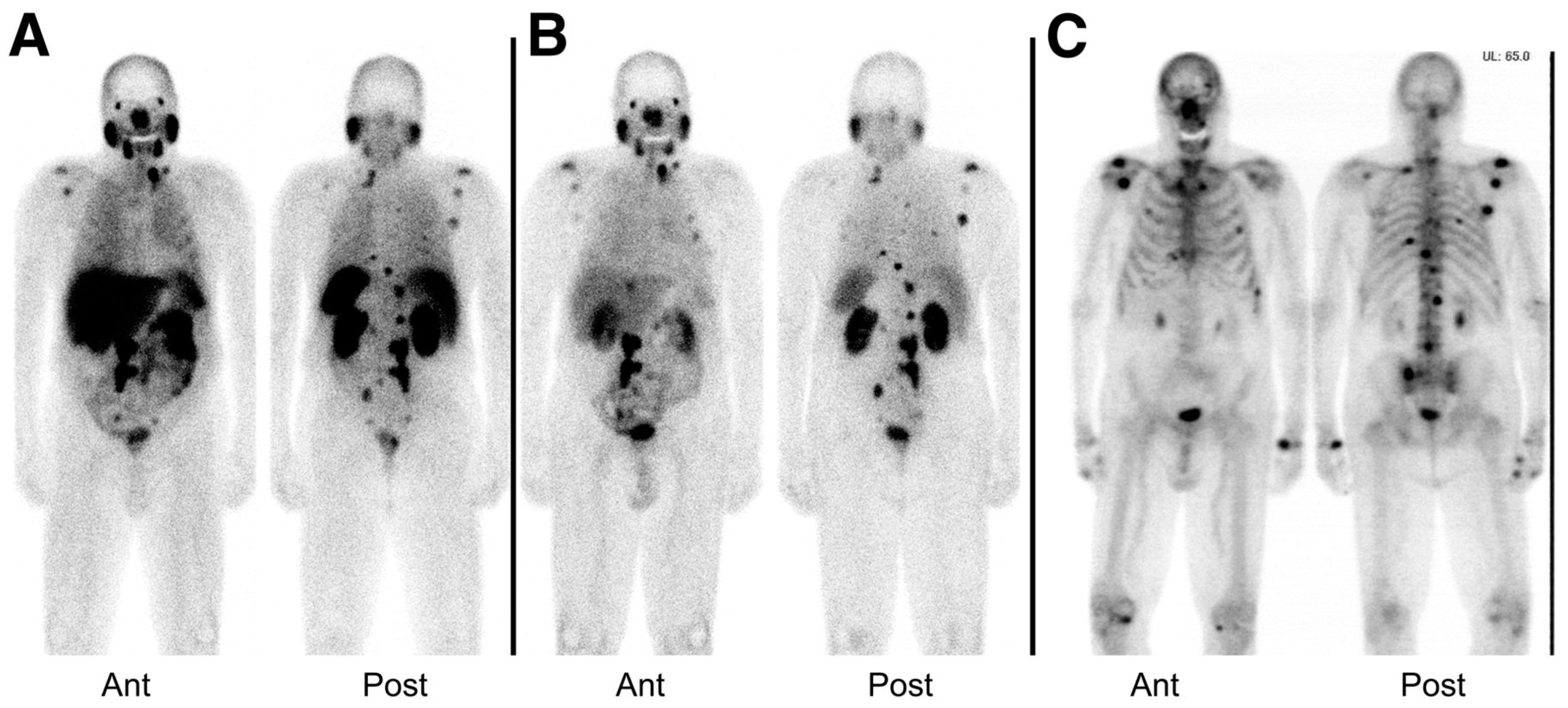{kind=link}
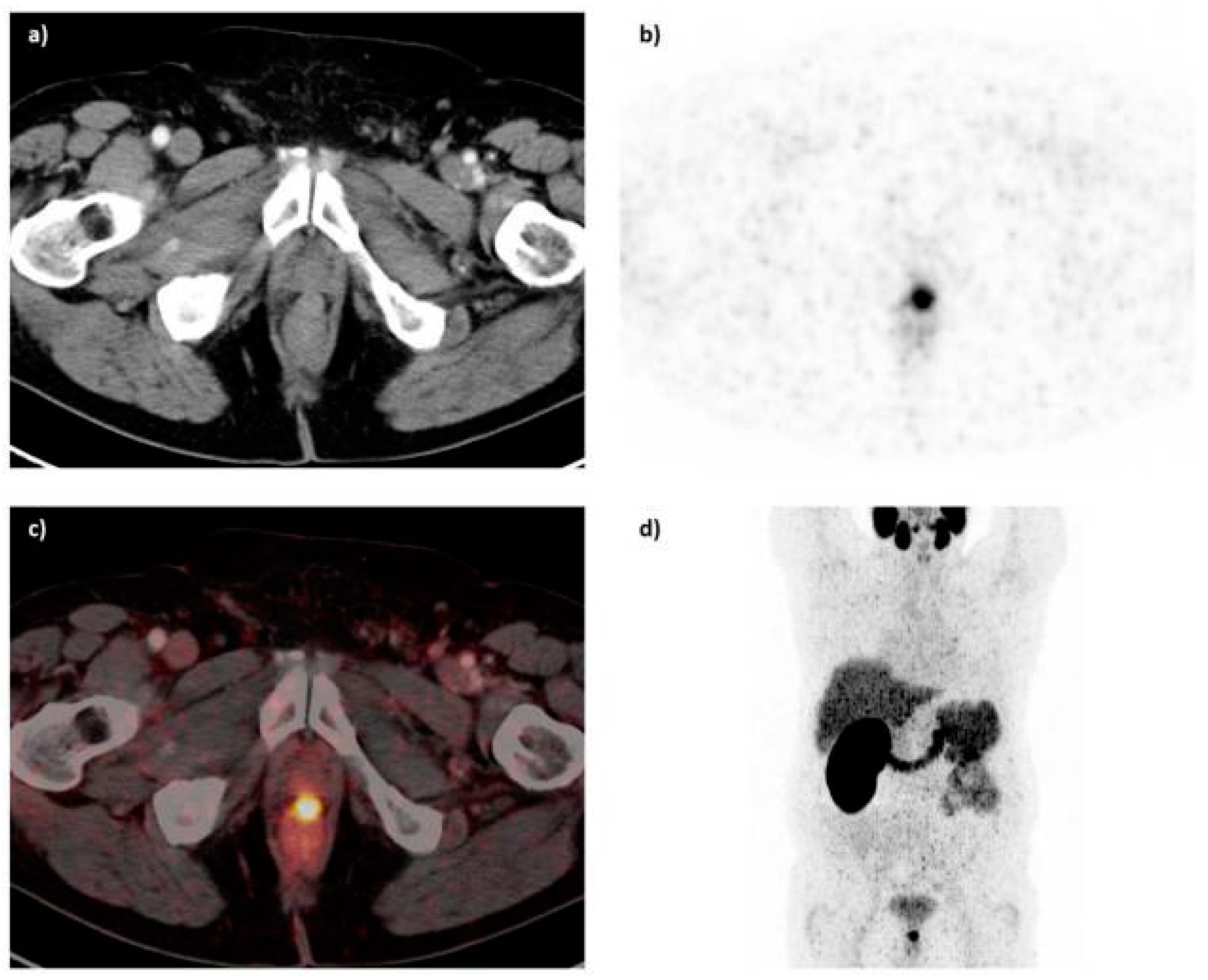{kind=link}
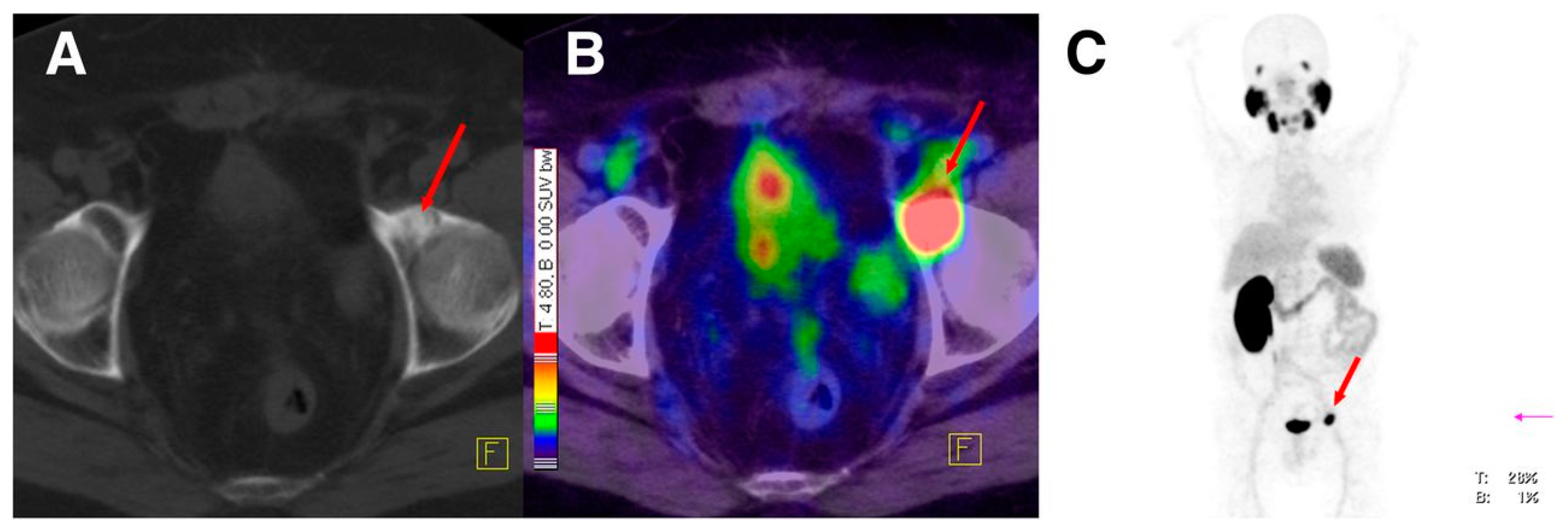{kind=link}
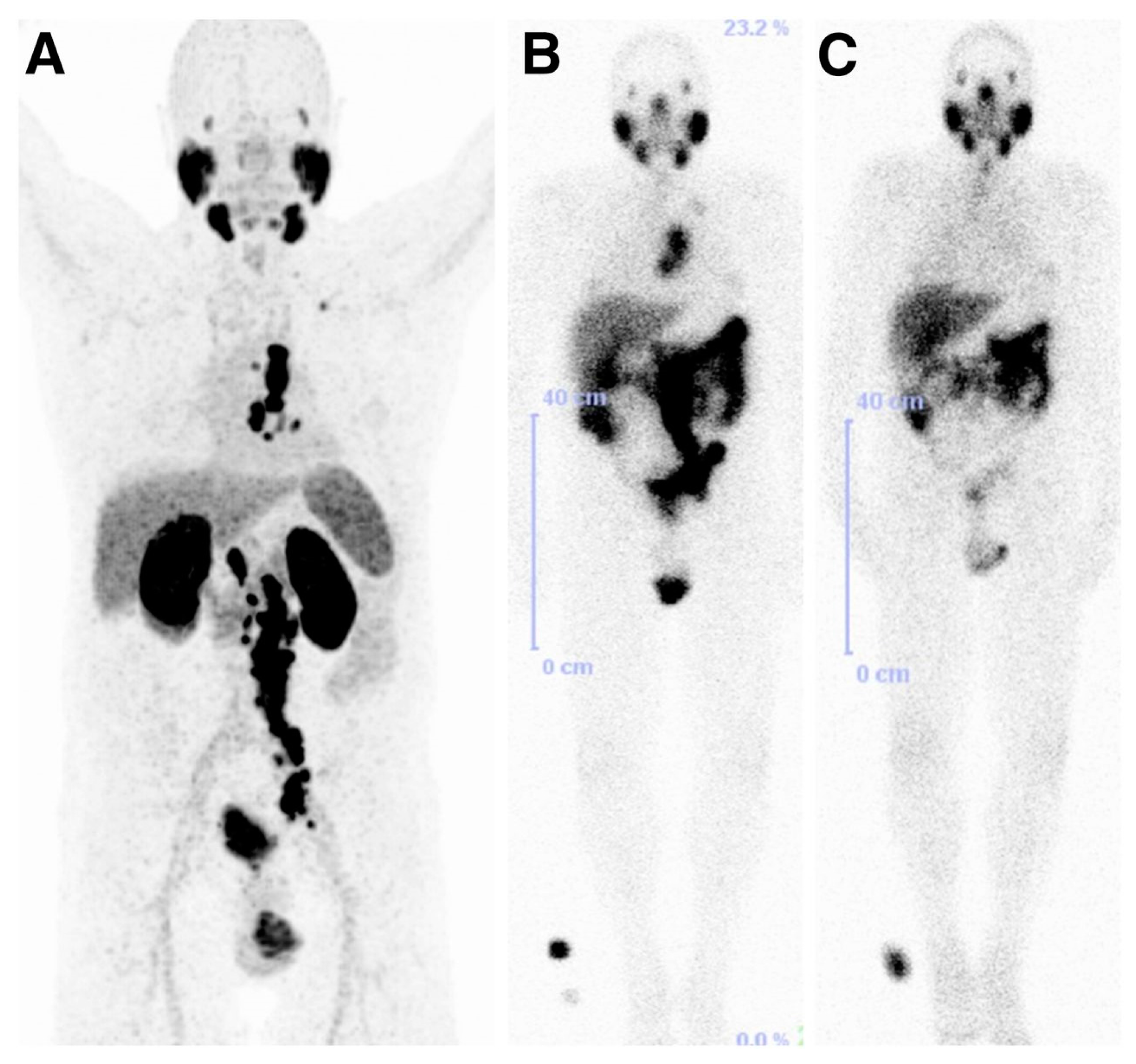{kind=link}
Table 1.
Affinity, labeling and lipophilicity data of the 99mTc-labeled PSMA inhibitors (13–25).
| Compound | Inhibitory Activity (Ki (nM)) | Labeling | LogPoct/water | Stage of Translation | Reference |
|---|---|---|---|---|---|
| 13–20 | 0.03 (13) | 0.25 M ammonium acetate/tartrate buffer, SnCl2 (4 mg/mL), 99mTcO4−, pH 8–8.5, 90–95 °C, 10–15 min, yield: >80%, purity: >99%, specific activity: >411 GBq/μmol | −2.38 (13) | no clinical translation | [99] |
| 0.13 (14) | −2.25 (14) | ||||
| 1.91 (15) | −2.14 (15) | ||||
| 7.99 (16) | −2.35 (16) | ||||
| 0.18 (17) | −3.04 (17) | ||||
| 0.048 (18) | N.D. (18) | ||||
| 0.17 (19) | −2.96 (19) | ||||
| 2.73 (20) | −2.94 (20) | ||||
| 21–24 | 15.25 (21) | [Tc(CO)3(H2O)3]+, pH 7.2, 95 °C, 30 min, yield: 70–95%, purity: >98%, specific activity: >411 GBq/μmol | −2.03 (21) | no clinical translation | [98,99] |
| 1.12 (22) | −2.37 (22) | ||||
| 12.60 (23) | −2.05 (23) | ||||
| 16.30 (24) | −2.07 (24) | ||||
| 25 | 1.74 (25) | HYNIC (2 mg/mL), 0.5 M NaHCO3, tricine (7 mg/mL), SnCl2 (1 mg/mL), 99mTcO4−, r.t., 30 min, yield: >80%, purity: >99% | −3.05 (25) | no clinical translation | [99] |
Table 2.
Affinity, labeling and lipophilicity data of the 99mTc-labeled PSMA inhibitors.
| Compound | KD (nM) | Labeling | LogPoct/water | Stage of Translation | Reference |
|---|---|---|---|---|---|
| 26–31 | 176 (26) | SnCl2, α-d-glucoheptonate, 99mTcO4−, pH 6.8, 90–100 °C, 18 min, yield: >98%, purity: >98%, Kit formulation was also performed | −3.94 (26) | no clinical translation | [100] |
| 60 (27) | −3.45 (27) | ||||
| 13.8 (28) | −0.27 (28) | ||||
| 31 (29) | −2.11 (29) | ||||
| 102 (30) | −4.64 (30) | ||||
| 338 (31) | −2.16 (31) |
Table 3.
Affinity, labeling and lipophilicity data of 99mTc-MAS3 (32)/mas3 (33)-y-nal-k-Sub-KuE.
| Compound | IC50 (nM) | Labeling | LogPoct/water | Stage of Translation | Reference |
|---|---|---|---|---|---|
| 32 (MES3 chelator) | 47.6 ± 2.5 | SnCl2,ascorbic acid, tartrate, ammonium acetate, 99mTcO4−, pH 7.5–8, 90 °C, 20 min. Cartridge purification to remove colloidal 99mTc species, specific activity: 25–37 GBq/μmol Kit formulation was also performed | −2.9 | [101] | |
| 33 (mes3 chelator) | 39.7 ± 1.2 | −3.0 | Initial stage of clinical assessment |
Table 4.
Affinity, labeling and lipophilicity data of 99mTc-TMCE.
| Compound | IC50 (nM) | Labeling | Stage of Translation | Reference |
|---|---|---|---|---|
| 99mTc-TMCE (34) | 3.4 | Tc(CO)3(H2O)3]+ (Isolink kit), pH 7.2, H2O, microwave, 110 °C, yield: 12–17%, purity: >98% (after HPLC purification) | no clinical translation | [102] |
Table 5.
Affinity, labeling and lipophilicity data of 99mTc-MIP-1428 (35), 99mTc-MIP-1405 (36), 99mTc-MIP-1404 (37), 99mTc-MIP-1427 (38).
Table 5.
Affinity, labeling and lipophilicity data of 99mTc-MIP-1428 (35), 99mTc-MIP-1405 (36), 99mTc-MIP-1404 (37), 99mTc-MIP-1427 (38).
| Compound | KD (nM) | Labeling | Stage of Translation | Reference |
|---|---|---|---|---|
| 99mTc-MIP-1428 (35) | 1.75 ± 0.32 | Tc(CO)3(H2O)3]+ (Isolink kit), pH 7.2, 100 °C, 30 min, purification via HPLC, yield: 70%, purity: >95%, specific activity: >37 TBq/mmol, Kit formulation was also performed | 99mTc-MIP-1404 is under clinical investigation | [103] |
| 99mTc-MIP-1405 (36) | 4.35 ± 0.35 | |||
| 99mTc-MIP-1404 (37) | 1.07 ± 0.89 | |||
| 99mTc-MIP-1427 (38) | 0.64 ± 0.46 |
Table 6.
Affinity, labeling and lipophilicity data of the 64Cu-labeled PSMA inhibitors 50–54.
| Compound | Ki (nM) (63/65Cu-…) | Labeling | Yields (%) | LogPoct/water | Stage of Translation | Reference |
|---|---|---|---|---|---|---|
| 64Cu-50 | 6.23 | Acetate buffer, pH 5.5–6, 65 °C, 30 min | 65–70 | −1.17 | no clinical translation | [139] |
| 64Cu-51 | 10.76 | Acetate buffer, pH 5.5–6, 65 °C, 30 min | 70–90 | −1.42 | ||
| 64Cu-52 | 5.47 | Acetate buffer, pH 5.5–6, 65 °C, 30 min | 75–85 | −1.56 | ||
| 64Cu-53A | 3.98 | Acetate buffer, pH 7.5–8, 95 °C, 1 h | 40–45 | −2.68 | ||
| 64Cu-53B | 4.65 | 30–35 | −2.31 | |||
| 64Cu-54 | 13.26 | Acetate buffer, pH 5.5–6, 65 °C, 30 min | 65–70 | ND |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gourni, E.; Henriksen, G. Metal-Based PSMA Radioligands. Molecules 2017, 22, 523. https://doi.org/10.3390/molecules22040523
AMA Style
Gourni E, Henriksen G. Metal-Based PSMA Radioligands. Molecules. 2017; 22(4):523. https://doi.org/10.3390/molecules22040523
Chicago/Turabian StyleGourni, Eleni, and Gjermund Henriksen. 2017. "Metal-Based PSMA Radioligands" Molecules 22, no. 4: 523. https://doi.org/10.3390/molecules22040523