
CDC25 Inhibition in Acute Myeloid Leukemia–A Study of Patient Heterogeneity and the Effects of Different Inhibitors


 ,
, 

Abstract
:1. Introduction
2. Results
2.1. CDC25 Inhibition has Antiproliferative Effects in Primary AML Cells for a Subset of Patients
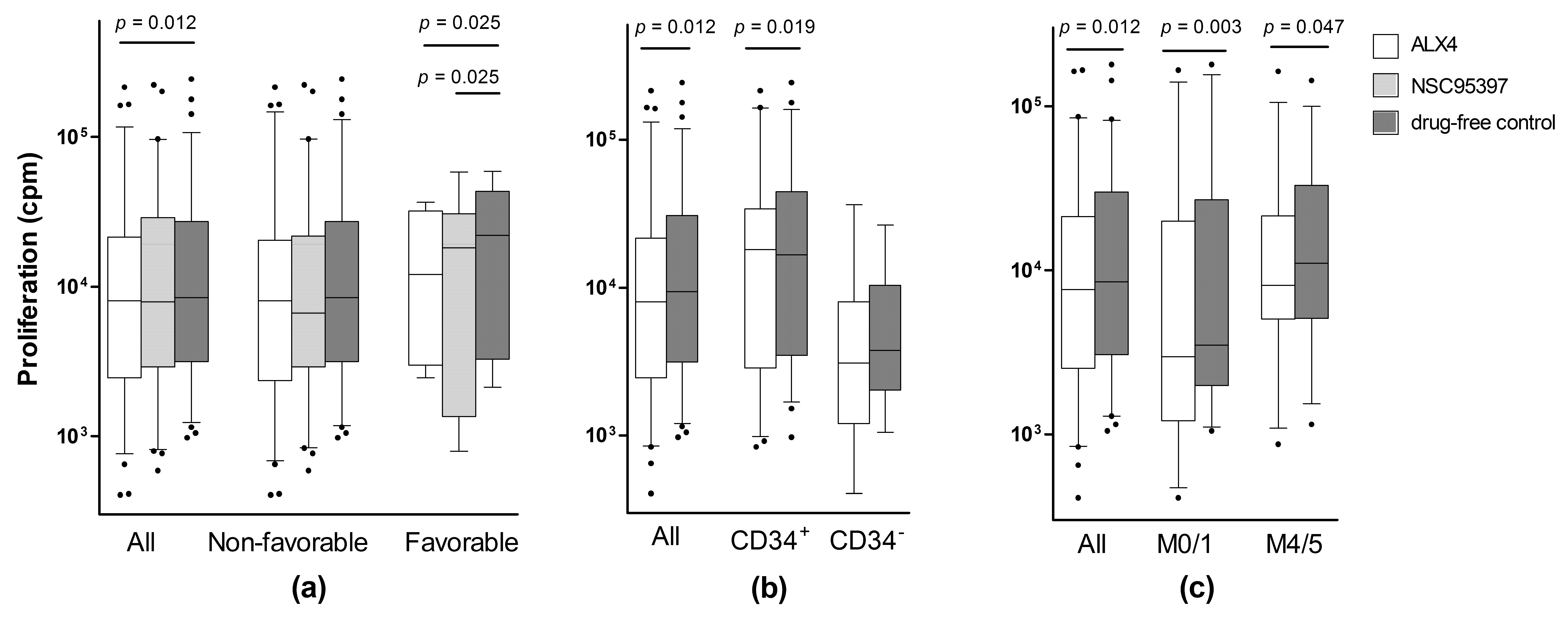
2.2. Identification of a Patient Subset Showing Growth Inhibition by Various CDC25 Inhibitors
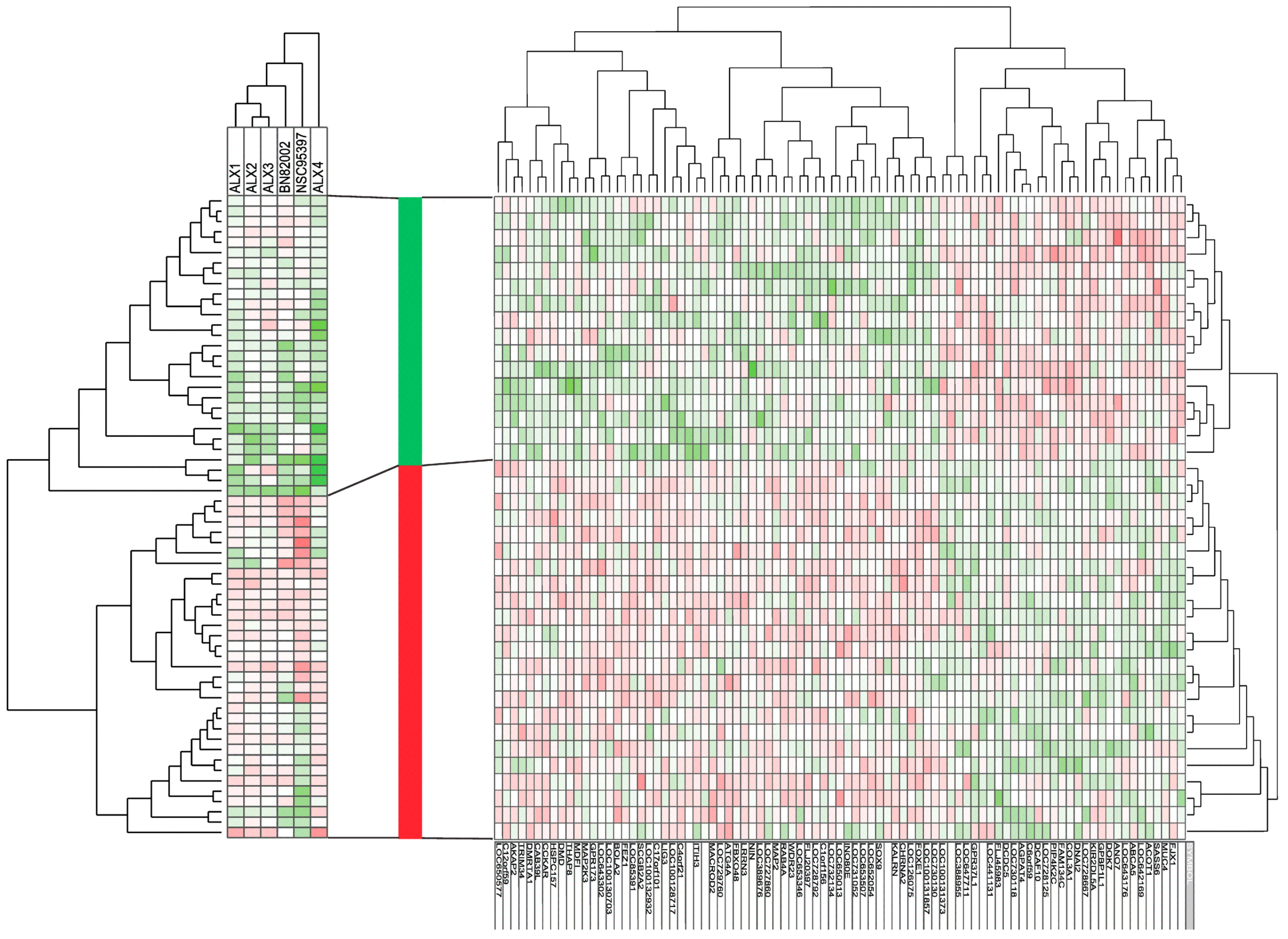
2.3. Responders to CDC25 Inhibition Can Be Identified by Analysis of Gene Expression Profiles
2.4. Combined CDC25 and PI3K/mTOR Inhibition Has Additive Antiproliferative Effects Only for Subsets of Patients
2.5. CDC25 Inhibition Has Only Minor Effects on the Constitutive Cytokine Release by AML Cells
2.6. CDC25 Inhibition Does Not Alter the Viability of In Vitro Cultured AML Cells
3. Discussion
4. Materials and Methods
4.1. AML Patient Population and Cell Isolation
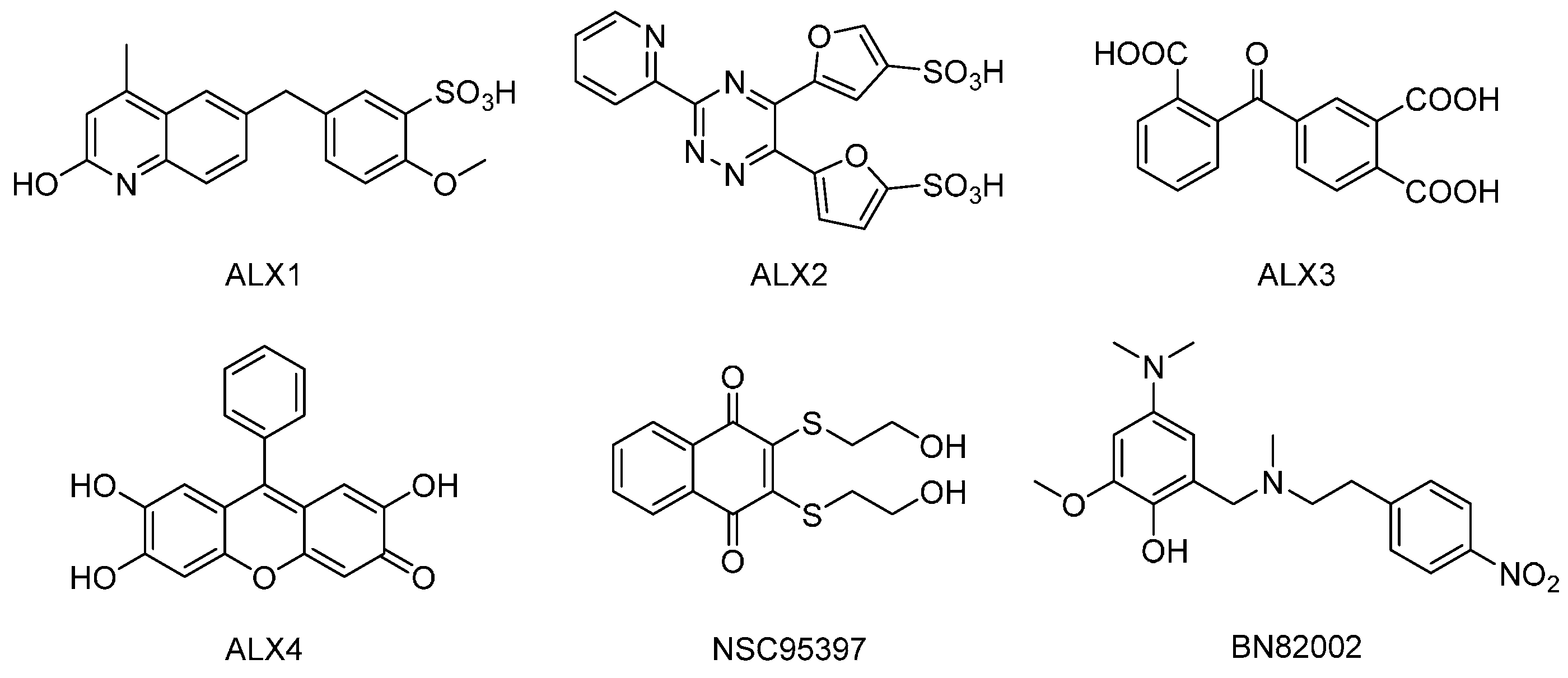
4.2. Reagents
4.3. Analysis of AML Cell Apoptosis and CD34 Expression by the Leukemic Cells
4.4. Analysis of Spontaneous and Cytokine-Dependent Proliferation
4.5. Analysis of Cytokine Levels in Culture Supernatants
4.6. RNA Preparation and Microarray Analysis
4.7. Bioinformatic and Statistical Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Hasserjian, R.P. Acute myeloid leukemia: Advances in diagnosis and classification. Int. J. Lab. Hematol. 2013, 35, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.; Wetzler, M.; Löwenberg, B. Therapeutic advances in acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Nicoletti, I.; Bolli, N.; Martelli, M.P.; Liso, A.; Gorello, P.; Mandelli, F.; Mecucci, C.; Martelli, M.F. Translocations and mutations involving the nucleophosmin (NPM1) gene in lymphomas and leukemias. Haematologica 2007, 92, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Döhner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. The phosphatidylinositol 3-kinase/AKT/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 2010, 1, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; O’Donnell, M.R.; Sekeres, M.A. Acute myeloid leukemia. Hematol. Am. Soc. Hematol. Educ. Progr. 2004, 98–117. [Google Scholar] [CrossRef] [PubMed]
- Perwitasari, O.; Torrecilhas, A.C.; Yan, X.; Johnson, S.; White, C.; Tompkins, S.M.; Tripp, R.A. Targeting cell division cycle 25 homolog B to regulate influenza virus replication. J. Virol. 2013, 87, 13775–13784. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C.; Novellino, E. CDC25 phosphatase inhibitors: An update. Mini Rev. Med. Chem. 2012, 12, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, M.; Draetta, G.F. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003, 4, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, B.G.; De Souza, C.P.; Tonks, I.D.; Clark, J.M.; Hayward, N.K.; Ellem, K.A. Cytoplasmic accumulation of cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J. Cell Sci. 1996, 109 Pt 5, 1081–1093. [Google Scholar] [PubMed]
- Karlsson, C.; Katich, S.; Hagting, A.; Hoffmann, I.; Pines, J. Cdc25B and Cdc25C differ markedly in their properties as initiators of mitosis. J. Cell Biol. 1999, 146, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, H.; Nishitani, H.; Seki, T.; Nishimoto, T. A dual-specificity phosphatase Cdc25B is an unstable protein and triggers p34cdc2/cyclin B activation in hamster BHK21 cells arrested with hydroxyurea. J. Cell Biol. 1997, 138, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, M.; Squatrito, M.; Ganoth, D.; Hershko, A.; Pagano, M.; Draetta, G.F. Dual mode of degradation of Cdc25 A phosphatase. EMBO J. 2002, 21, 4875–4884. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M.; Mercurio, C.; Dominguez, J.; Goubin, F.; Draetta, G.F. Human Cdc25 A inactivation in response to S phase inhibition and its role in preventing premature mitosis. EMBO Rep. 2000, 1, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; White, L.S.; Hurov, K.E.; Stappenbeck, T.S.; Piwnica-Worms, H. Response of small intestinal epithelial cells to acute disruption of cell division through CDC25 deletion. Proc. Natl. Acad. Sci. USA 2009, 106, 4701–4706. [Google Scholar] [CrossRef] [PubMed]
- Tamir, A.; Petrocelli, T.; Stetler, K.; Chu, W.; Howard, J.; Croix, B.S.; Slingerland, J.; Ben-David, Y. Stem cell factor inhibits erythroid differentiation by modulating the activity of G1-cyclin-dependent kinase complexes: A role for p27 in erythroid differentiation coupled G1 arrest. Cell Growth Differ. 2000, 11, 269–277. [Google Scholar] [PubMed]
- Nakamura, S.; Nagata, Y.; Tan, L.; Takemura, T.; Shibata, K.; Fujie, M.; Fujisawa, S.; Tanaka, Y.; Toda, M.; Makita, R.; et al. Transcriptional repression of Cdc25B by IER5 inhibits the proliferation of leukemic progenitor cells through NF-YB and p300 in acute myeloid leukemia. PLoS ONE 2011, 6, e28011. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Coluccia, A.; Di Giovanni, C.; Novellino, E. Cdc25B phosphatase inhibitors in cancer therapy: Latest developments, trends and medicinal chemistry perspective. Anticancer Agents Med. Chem. 2008, 8, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C.; Novellino, E. CDC25A and B dual-specificity phosphatase inhibitors: Potential agents for cancer therapy. Curr. Med. Chem. 2009, 16, 1831–1849. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C.; Novellino, E. Inhibitors of Cdc25 phosphatases as anticancer agents: A patent review. Expert Opin. Ther. Pat. 2010, 20, 405–425. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Agrawal, D.K. Phosphatases and kinases regulating CDC25 activity in the cell cycle: Clinical implications of CDC25 overexpression and potential treatment strategies. Mol. Cell. Biochem. 2016, 416, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.; Mondesert, O.; Goddard, M.L.; Jullien, D.; Villoutreix, B.O.; Ducommun, B.; Garbay, C.; Braud, E. Development of novel thiazolopyrimidines as CDC25B phosphatase inhibitors. ChemMedChem 2009, 4, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Cosconati, S.; Limongelli, V.; Novellino, E. Modeling of Cdc25B dual specifity protein phosphatase inhibitors: Docking of ligands and enzymatic inhibition mechanism. ChemMedChem 2006, 1, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Lazo, J.S.; Nemoto, K.; Pestell, K.E.; Cooley, K.; Southwick, E.C.; Mitchell, D.A.; Furey, W.; Gussio, R.; Zaharevitz, D.W.; Joo, B.; et al. Identification of a potent and selective pharmacophore for Cdc25 dual specificity phosphatase inhibitors. Mol. Pharmacol. 2002, 61, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, M.; Choi, J.; Cho, H.; Ham, S.W. Structure-based virtual screening approach to identify novel classes of Cdc25B phosphatase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4372–4375. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Lefterov, I.M.; Wang, M.; Lazo, J.S.; Scott, C.N.; Wilcox, C.S.; Carr, B.I. Binding and inhibition of Cdc25 phosphatases by vitamin K analogues. Biochemistry 2003, 42, 10490–10497. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Amoscato, A.A.; Bier, M.E.; Lazo, J.S. Dual G1 and G2 phase inhibition by a novel, selective Cdc25 inhibitor 6-chloro-7-(2-morpholin-4-ylethyl-amino)-quinoline-5,8-dione. J. Biol. Chem. 2002, 277, 46877–46885. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C.; Pesapane, A.; Montuori, N.; Ragno, P.; Martucci, N.M.; Masullo, M.; De Vendittis, E.; Novellino, E. Discovery of new inhibitors of Cdc25B dual specificity phosphatases by structure-based virtual screening. J. Med. Chem. 2012, 55, 4142–4158. [Google Scholar] [CrossRef] [PubMed]
- Capasso, A.; Cerchia, C.; Di Giovanni, C.; Granato, G.; Albano, F.; Romano, S.; De Vendittis, E.; Ruocco, M.R.; Lavecchia, A. Ligand-based chemoinformatic discovery of a novel small molecule inhibitor targeting CDC25 dual specificity phosphatases and displaying in vitro efficacy against melanoma cells. Oncotarget 2015, 6, 40202–40222. [Google Scholar] [PubMed]
- Lin, T.C.; Lin, P.L.; Cheng, Y.W.; Wu, T.C.; Chou, M.C.; Chen, C.Y.; Lee, H. MicroRNA-184 Deregulated by the microRNA-21 promotes tumor malignancy and poor outcomes in non-small cell lung cancer via targeting CDC25a and c-Myc. Ann. Surg. Oncol. 2015, 22 (Suppl. S3), S1532–S1539. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, S.; Boutzen, H.; David, L.; Larrue, C.; Vergez, F.; Fernandez-Vidal, A.; Yuan, L.; Hospital, M.A.; Tamburini, J.; Demur, C.; et al. CDC25A governs proliferation and differentiation of FLT3-ITD acute myeloid leukemia. Oncotarget 2015, 6, 38061–38078. [Google Scholar] [PubMed]
- Evain-Bana, E.; Schiavo, L.; Bour, C.; Lanfranchi, D.A.; Berardozzi, S.; Ghirga, F.; Bagrel, D.; Botta, B.; Hanquet, G.; Mori, M. Synthesis, biological evaluation and molecular modeling studies on novel quinonoid inhibitors of CDC25 phosphatases. J. Enzyme Inhib. Med. Chem. 2017, 32, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Huber-Villaume, S.; Revelant, G.; Sibille, E.; Philippot, S.; Morabito, A.; Dunand, S.; Chaimbault, P.; Bagrel, D.; Kirsch, G.; Hesse, S.; et al. 2-(Thienothiazolylimino)-1,3-thiazolidin-4-ones inhibit cell division cycle 25 A phosphatase. Bioorg. Med. Chem. 2016, 24, 2920–2928. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.W.; Zhang, Z.; Zhang, C.F.; Anderson, S.; Liu, Q.; Yuan, C.S.; Wang, C.Z. Anti-colon cancer effects of 6-shogaol through G2/M cell cycle arrest by p53/p21-cdc2/cdc25A crosstalk. Am. J. Chin. Med. 2015, 43, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Lin, X.; Kang, D.; Li, X.; Zhan, P.; Liu, X.; Zhang, Q. Discovery and characterization of novel imidazopyridine derivative CHEQ-2 as a potent CDC25 inhibitor and promising anticancer drug candidate. Eur. J. Med. Chem. 2014, 82, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Brezak, M.C.; Quaranta, M.; Contour-Galcera, M.O.; Lavergne, O.; Mondesert, O.; Auvray, P.; Kasprzyk, P.G.; Prevost, G.P.; Ducommun, B. Inhibition of human tumor cell growth in vivo by an orally bioavailable inhibitor of CDC25 phosphatases. Mol. Cancer Ther. 2005, 4, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Brezak, M.C.; Quaranta, M.; Mondesert, O.; Galcera, M.O.; Lavergne, O.; Alby, F.; Cazales, M.; Baldin, V.; Thurieau, C.; Harnett, J.; et al. A novel synthetic inhibitor of CDC25 phosphatases: BN82002. Cancer Res. 2004, 64, 3320–3325. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vidal, A.; Ysebaert, L.; Didier, C.; Betous, R.; De Toni, F.; Prade-Houdellier, N.; Demur, C.; Contour-Galcera, M.O.; Prevost, G.P.; Ducommun, B.; et al. Cell adhesion regulates CDC25A expression and proliferation in acute myeloid leukemia. Cancer Res. 2006, 66, 7128–7135. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Tamburini, J.; Skrede, S.; Holdhus, R.; Poulain, L.; Ersvær, E.; Hatfield, K.J.; Bruserud, Ø. Antileukaemic effect of PI3K-mTOR inhibitors in acute myeloid leukaemia-gene expression profiles reveal CDC25B expression as determinate of pharmacological effect. Br. J. Haematol. 2014, 164, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.-Q.; Yang, Q.-K.; Lu, B.-W.; Yi, W.; Cantin, G.; Chen, Y.-L.; Fearns, C.; Yates, J.R., III; Lee, J.-D. CDC25B Mediates Rapamycin-induced Oncogenic Responses in Cancer Cells. Cancer Res. 2009, 69, 2663–2668. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Gjertsen, B.T.; Foss, B.; Huang, T.S. New strategies in the treatment of acute myelogenous leukemia (AML): In vitro culture of aml cells—The present use in experimental studies and the possible importance for future therapeutic approaches. Stem Cells 2001, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Keefer, M.C.; Bonnez, W.; Roberts, N.J., Jr.; Dolin, R.; Reichman, R.C. Human immunodeficiency virus (HIV-1) gp160-specific lymphocyte proliferative responses of mononuclear leukocytes from HIV-1 recombinant gp160 vaccine recipients. J. Infect. Dis. 1991, 163, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.K.; Reikvam, H.; Lavecchia, A.; Bruserud, Ø. Therapeutic targeting the cell division cycle 25 (CDC25) phosphatases in human acute myeloid leukemia—The possibility to target several kinases through inhibition of the various CDC25 isoforms. Molecules 2014, 19, 18414–18447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryningen, A.; Ersvær, E.; Øyan, A.M.; Kalland, K.H.; Vintermyr, O.K.; Gjertsen, B.T.; Bruserud, Ø. Stress-induced in vitro apoptosis of native human acute myelogenous leukemia (AML) cells shows a wide variation between patients and is associated with low BCL-2:Bax ratio and low levels of heat shock protein 70 and 90. Leuk. Res. 2006, 30, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Gjertsen, B.T.; Øyan, A.M.; Marzolf, B.; Hovland, R.; Gausdal, G.; Døskeland, S.O.; Dimitrov, K.; Golden, A.; Kalland, K.H.; Hood, L.; et al. Analysis of acute myelogenous leukemia: Preparation of samples for genomic and proteomic analyses. J. Hematother. Stem Cell Res. 2002, 11, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Hovland, R.; Wergeland, L.; Huang, T.S.; Gjertsen, B.T. Flt3-mediated signaling in human acute myelogenous leukemia (AML) blasts: A functional characterization of Flt3-ligand effects in AML cell populations with and without genetic Flt3 abnormalities. Haematologica 2003, 88, 416–428. [Google Scholar] [PubMed]
- Damm, F.; Heuser, M.; Morgan, M.; Wagner, K.; Görlich, K.; Grosshennig, A.; Hamwi, I.; Thol, F.; Surdziel, E.; Fiedler, W.; et al. Integrative prognostic risk score in acute myeloid leukemia with normal karyotype. Blood 2011, 117, 4561–4568. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, H.J.; Valk, P.J.; de Bont, E.S.; Schuringa, J.J.; Ossenkoppele, G.; Vellenga, E.; Huls, G. Prognostic impact of white blood cell count in intermediate risk acute myeloid leukemia: Relevance of mutated NPM1 and FLT3-ITD. Haematologica 2011, 96, 1310–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheatley, K.; Burnett, A.K.; Goldstone, A.H.; Gray, R.G.; Hann, I.M.; Harrison, C.J.; Rees, J.K.; Stevens, R.F.; Walker, H. A simple, robust, validated and highly predictive index for the determination of risk-directed therapy in acute myeloid leukaemia derived from the MRC AML 10 trial. Br. J. Haematol. 1999, 107, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y.; Hamaki, T.; Yamamoto, R.; Chizuka, A.; Suguro, M.; Matsuyama, T.; Takezako, N.; Miwa, A.; Kami, M.; Hirai, H.; et al. The clinical significance of CD34 expression in response to therapy of patients with acute myeloid leukemia: An overview of 2483 patients from 22 studies. Cancer 2000, 88, 2529–2533. [Google Scholar] [CrossRef]
- Øyan, A.M.; Bø, T.H.; Jonassen, I.; Ulvestad, E.; Gjertsen, B.T.; Kalland, K.H.; Bruserud, Ø. CD34 expression in native human acute myelogenous leukemia blasts: Differences in CD34 membrane molecule expression are associated with different gene expression profiles. Cytom. B Clin. Cytom. 2005, 64, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Hajra, A.; Wijmenga, C.; Collins, F.S. Molecular pathogenesis of the chromosome-16 inversion in the M4Eo subtype of acute myeloid-leukemia. Blood 1995, 85, 2289–2302. [Google Scholar] [PubMed]
- Mrozek, K.; Heerema, N.A.; Bloomfield, C.D. Cytogenetics in acute leukemia. Blood Rev. 2004, 18, 115–136. [Google Scholar] [CrossRef]
- Didier, C.; Cavelier, C.; Quaranta, M.; Galcera, M.O.; Demur, C.; Laurent, G.; Manenti, S.; Ducommun, B. G2/M checkpoint stringency is a key parameter in the sensitivity of AML cells to genotoxic stress. Oncogene 2008, 27, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Wang, M.; Carr, B.I. 2-Methoxyestradiol inhibits hepatocellular carcinoma cell growth by inhibiting Cdc25 and inducing cell cycle arrest and apoptosis. Cancer Chemother. Pharmacol. 2008, 62, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Giessrigl, B.; Vonach, C.; Madlener, S.; Prinz, S.; Herbaceck, I.; Holzl, C.; Bauer, S.; Viola, K.; Mikulits, W.; et al. Berberine and a Berberis lycium extract inactivate Cdc25A and induce α-tubulin acetylation that correlate with HL-60 cell cycle inhibition and apoptosis. Mutat. Res. 2010, 683, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, W.S.; Yu, T.; Yi, Y.S.; Park, J.G.; Jeong, D.; Kim, J.H.; Oh, J.S.; Yoon, K.; Kim, J.H.; et al. Novel anti-inflammatory function of NSC95397 by the suppression of multiple kinases. Biochem. Pharmacol. 2014, 88, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Ryningen, A.; Olsnes, A.M.; Stordrange, L.; Øyan, A.M.; Kalland, K.H.; Gjertsen, B.T. Subclassification of patients with acute myelogenous leukemia based on chemokine responsiveness and constitutive chemokine release by their leukemic cells. Haematologica 2007, 92, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Ryningen, A.; Wergeland, L.; Glenjen, N.I.; Gjertsen, B.T. Osteoblasts increase proliferation and release of pro-angiogenic interleukin 8 by native human acute myelogenous leukemia blasts. Haematologica 2004, 89, 391–402. [Google Scholar] [PubMed]
- Bruserud, Ø. Effect of dipyridamole, theophyllamine and verapamil on spontaneous in vitro proliferation of myelogenous leukaemia cells. Acta Oncol. 1992, 31, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Vermes, I.; Haanen, C.; Steffensnakken, H.; Reutelingsperger, C. A novel assay for apoptosis—Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein-labeled annexin-V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef]
- Sample Availability: Not available.




{kind=link}
{kind=link}
{kind=link}
| Proliferative AML Cell Responses Corresponding to Activity >1000 cpm | ||||
|---|---|---|---|---|
| Drugs Added | Number a | Median (cpm) | Range (cpm) a | p-Value b |
| Drug-free control | 69 | 6929 | 1149–244,316 | – |
| NSC95397 | 64 | 6349 | 1045–223,371 | Ns |
| BN82002 | 69 | 5586 | 1094–260,005 | Ns |
| ALX1 | 67 | 5888 | 1051–220,146 | Ns |
| ALX2 | 68 | 6937 | 1151–213,939 | Ns |
| ALX3 | 68 | 6393 | 1062–180,671 | Ns |
| ALX4 | 64 | 6005 | 1051–215,655 | 0.012 |
| GDC0941 | 69 | 4580 | 1053–202,884 | <0.001 |
| NSC95397 + GDC0941 | 67 | 3892 | 1049–170,748 | <0.001 |
| Rapamycin | 61 | 3665 | 1030–105,449 | <0.001 |
| NSC95397 + Rapamycin | 60 | 3626 | 1032–142,996 | <0.001 |
| Functional Classification | Number | DOWN-Regulated mRNA Expression in Responders | UP-Regulated mRNA Expression in Responder |
|---|---|---|---|
| Cell cycle, mitosis DNA repair | 4 | SASS6 (centrosome), | NIN (centrosome) FEZ1 (centrosome) BOLA2 (cell cycle regulation) |
| Cytoskeleton, microtubule | 8 | DNAI2 (centrosome, spindle formation) DCDC5 (tubulin binding) | FLJ20397 (trafficking, organelle positioning, microtubule organization) WDR23 (actin cytoskeletal organization; ubiquitination) MAP2 (microtubule assembly) DMD (cytoskeletal) AKAP2 (anchoring protein) LIG3 (DNA repair) |
| Intracellular trafficking | 2 | KALRN (vesicle trafficking) RAB4A (GTPase, endosomes) | |
| Cell membrane molecules Extracellular molecules | 8 | MUC4 (glycoprotein) ABCA5 (transmembrane transport intra- and extracellular) KIR2DL5A (cell surface molecule) COL3A1 (collagen) | GPR173 (G protein coupled receptor) CCKAR (G protein coupled receptor) ITIH3 (matrix stabilization) CHRNA2 (ion channel) |
| Intracellular signaling | 10 | FAM134C PIP4K2C (PI3K signalling, endoplasmic reticulum) DCAF10 (ubiquitin) GPR37L1 (G protein coupled receptor) DOK7 (kinase phosphorylation) | CIorf156 (methyltransferase) WDR23 (ubiquitination) FBXO48 (ubiquitination) MAP2K3 (MAP kinase) CAB39L (LKB1 activation) TMEM52B (ubiquitination) |
| Transcription | 6 | GPBP1L1 | FOXE1, SOX9, INO8OE, MDFI, DMRTA1 (transcriptional regulators) |
| Metabolism, autophagy | 4 | ACOT1 (metabolism, acetyl-CoA) AGPAT4 (metabolism, phospholipid biosynthesis) | ATG4A (autophagy) MACROD2 (apoptosis) |
| Unknown | 40 | FJX1, LOC643176, ANO7, LOC728667, LOC728125, C6orf59, LOC730118, FLJ45983, LOC441131, LOC647711, LOC388955, LOC100131373 | LOC730130, LOC131857, LOC126075, LOC652054, LOC653507, LOC731052, LOC650013, LOC732134, LOC728792, LOC653346, LOC727860, LOC389676, LNNR3, LOC729260, ZGRF1, LOC100128717, OGFOD3, LOC642169, LOC100132932, SCGB2A2, LOC85391, LOC100130703, LOC643302, THAP8, HSPC157, TRIM34, LOC650577 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brenner, A.K.; Reikvam, H.; Rye, K.P.; Hagen, K.M.; Lavecchia, A.; Bruserud, Ø. CDC25 Inhibition in Acute Myeloid Leukemia–A Study of Patient Heterogeneity and the Effects of Different Inhibitors. Molecules 2017, 22, 446. https://doi.org/10.3390/molecules22030446
Brenner AK, Reikvam H, Rye KP, Hagen KM, Lavecchia A, Bruserud Ø. CDC25 Inhibition in Acute Myeloid Leukemia–A Study of Patient Heterogeneity and the Effects of Different Inhibitors. Molecules. 2017; 22(3):446. https://doi.org/10.3390/molecules22030446
Chicago/Turabian StyleBrenner, Annette K., Håkon Reikvam, Kristin Paulsen Rye, Karen Marie Hagen, Antonio Lavecchia, and Øystein Bruserud. 2017. "CDC25 Inhibition in Acute Myeloid Leukemia–A Study of Patient Heterogeneity and the Effects of Different Inhibitors" Molecules 22, no. 3: 446. https://doi.org/10.3390/molecules22030446
APA StyleBrenner, A. K., Reikvam, H., Rye, K. P., Hagen, K. M., Lavecchia, A., & Bruserud, Ø. (2017). CDC25 Inhibition in Acute Myeloid Leukemia–A Study of Patient Heterogeneity and the Effects of Different Inhibitors. Molecules, 22(3), 446. https://doi.org/10.3390/molecules22030446