
Intramolecular Hydrogen Bonding Involving Organic Fluorine: NMR Investigations Corroborated by DFT-Based Theoretical Calculations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
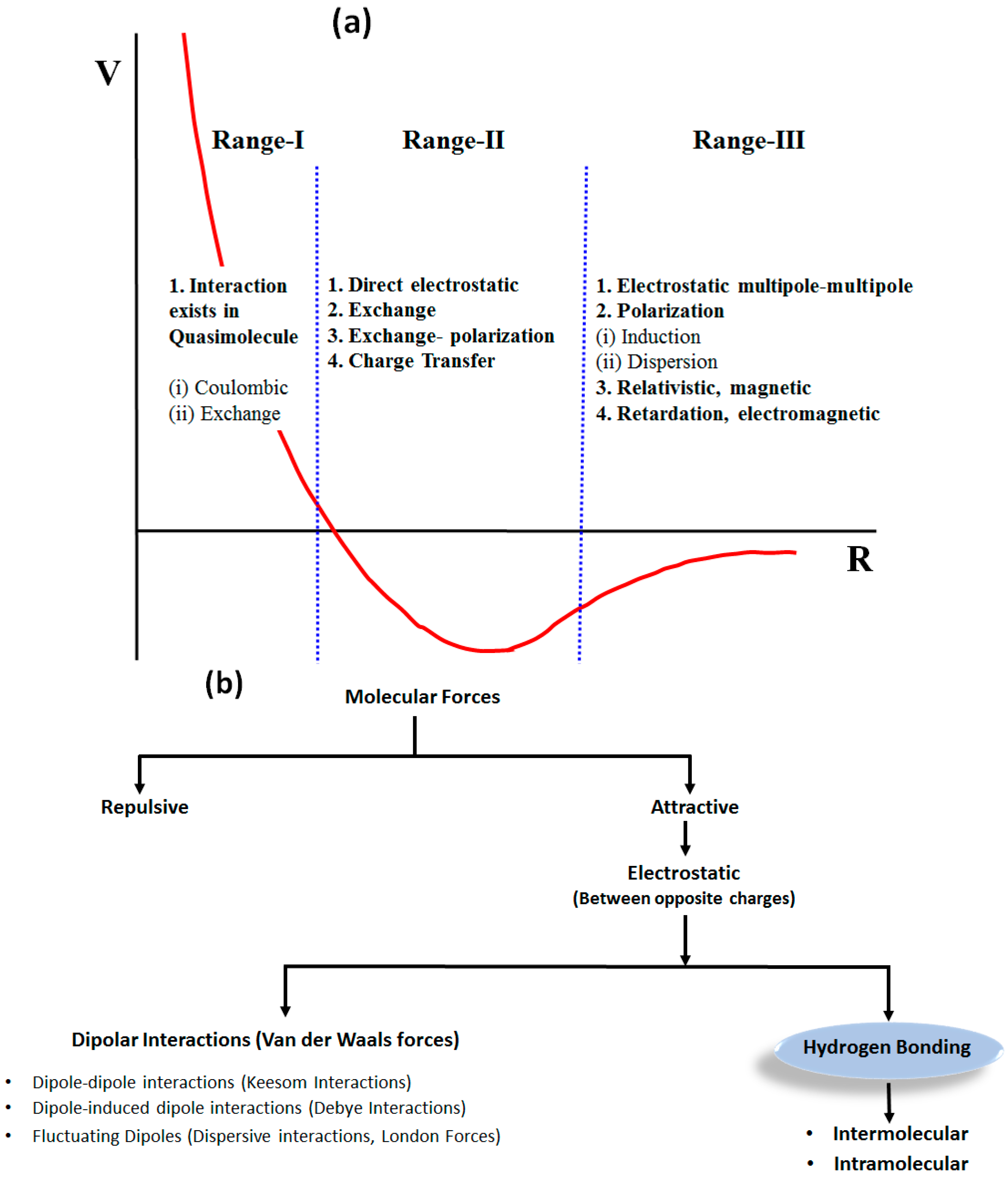
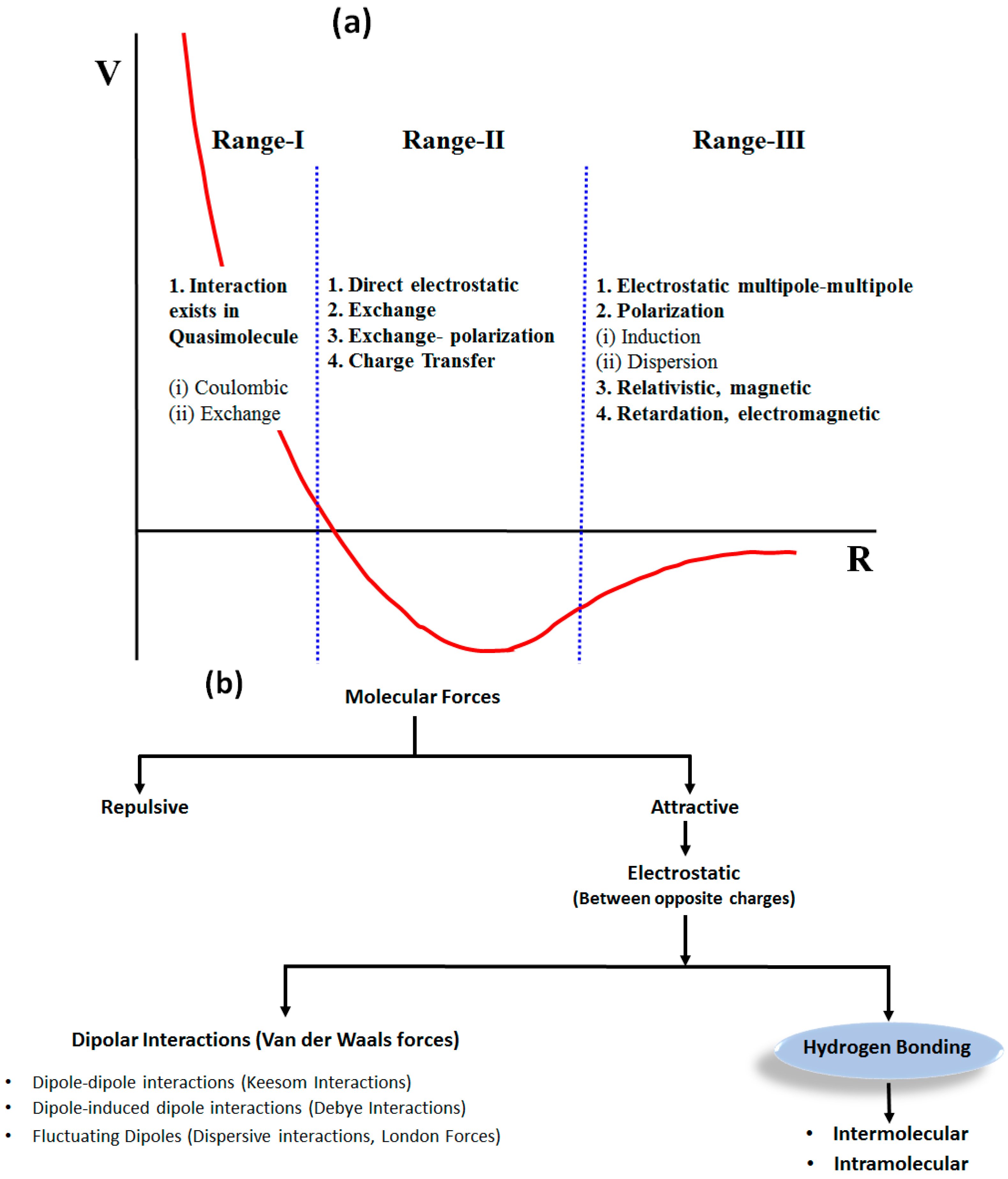
1.1. Weak Molecular Interactions
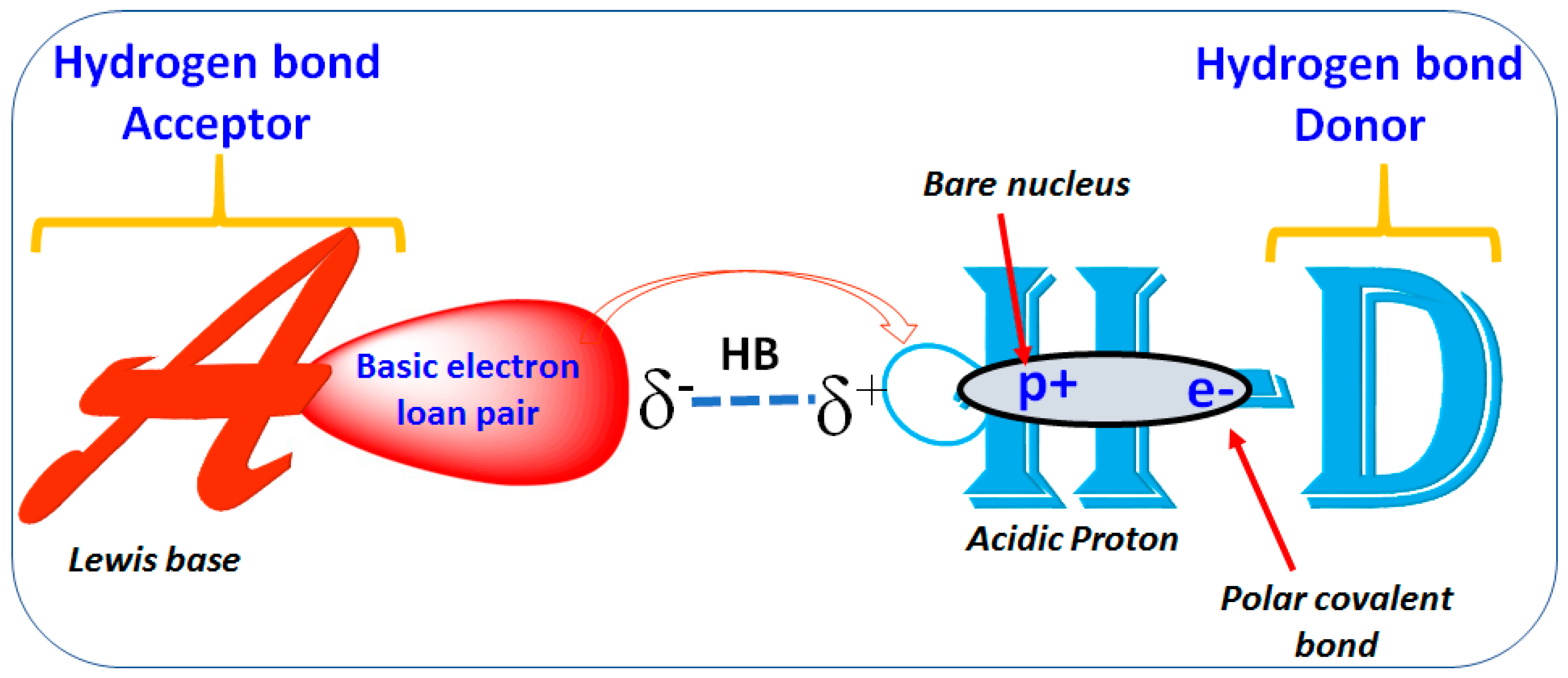
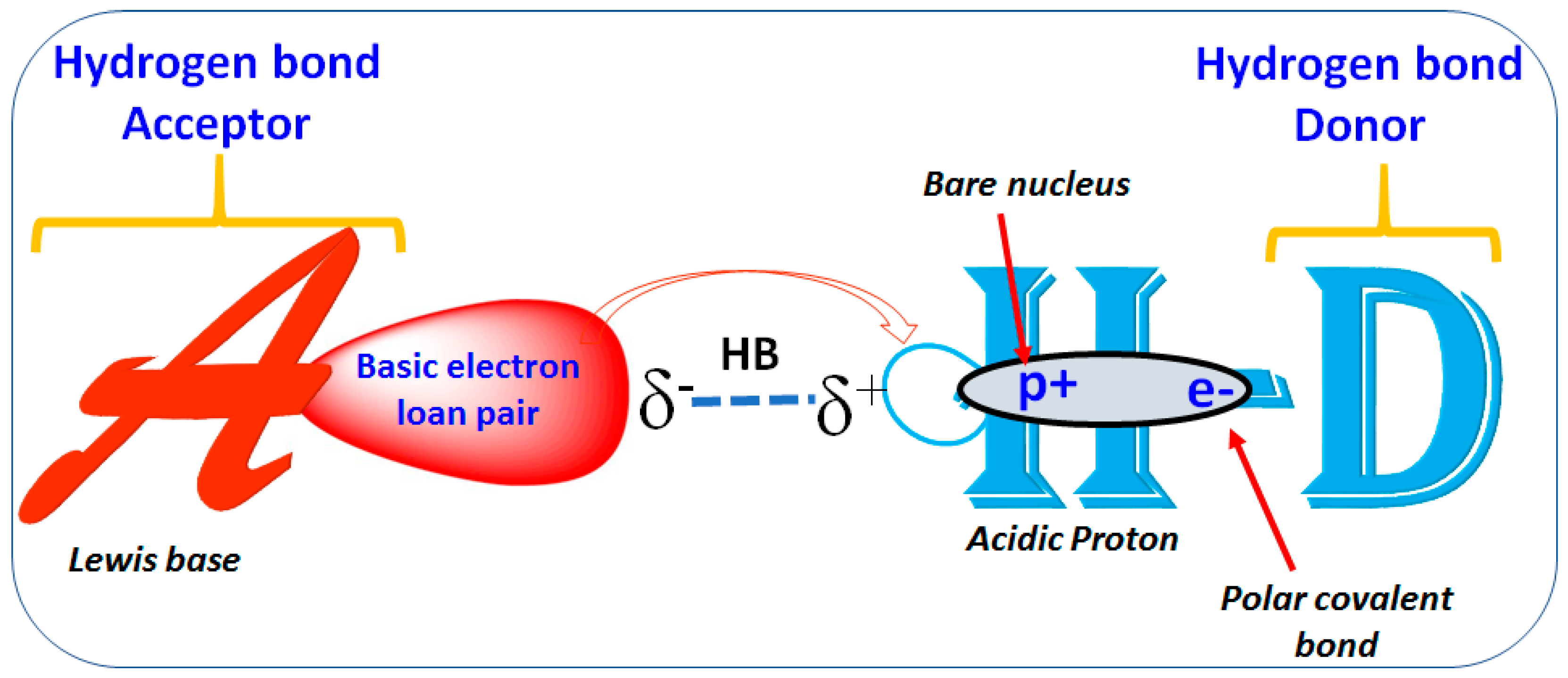
1.2. Hydrogen Bond
Importance of Hydrogen Bond
1.3. Organic Fluorine in the HB
1.4. NMR Spectroscopic Techniques for the Detection of Hydrogen Bond
1.5. Theoretical Calculations
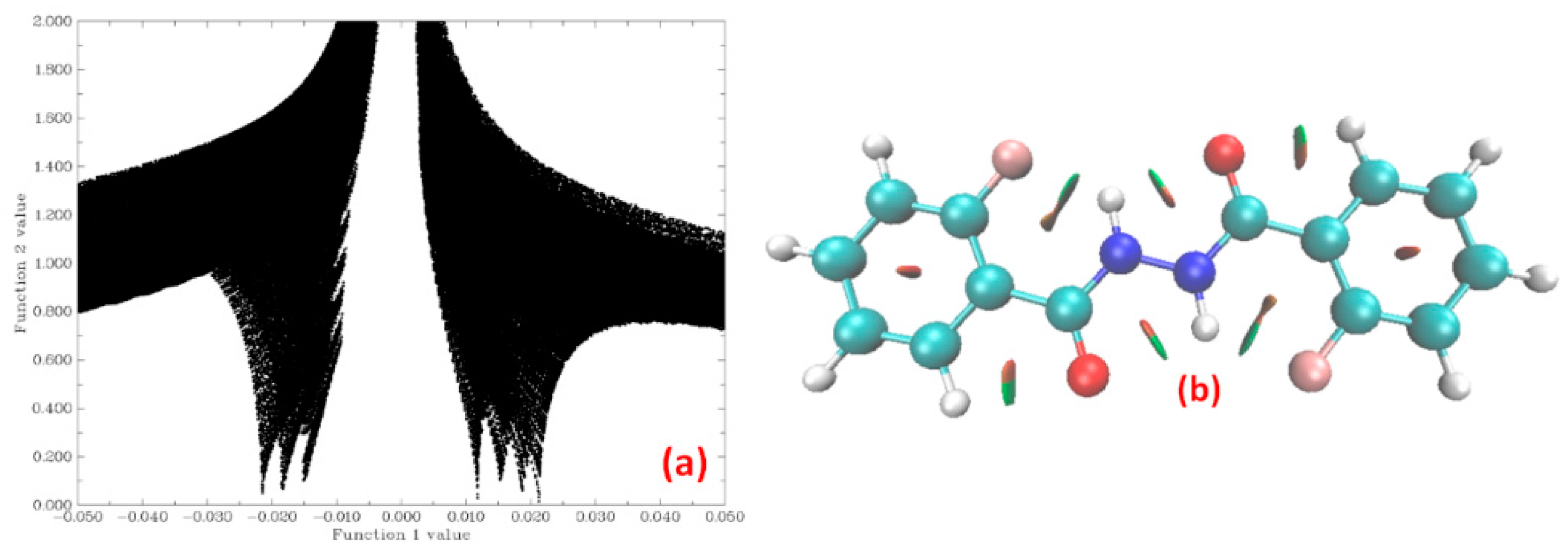
1.5.1. Non-Covalent Interaction (NCI) Calculations
1.5.2. Atoms in Molecules (AIM) Calculations
1.5.3. Natural Bond Orbitals (NBO)
1.5.4. Molecular Dynamics (MD)
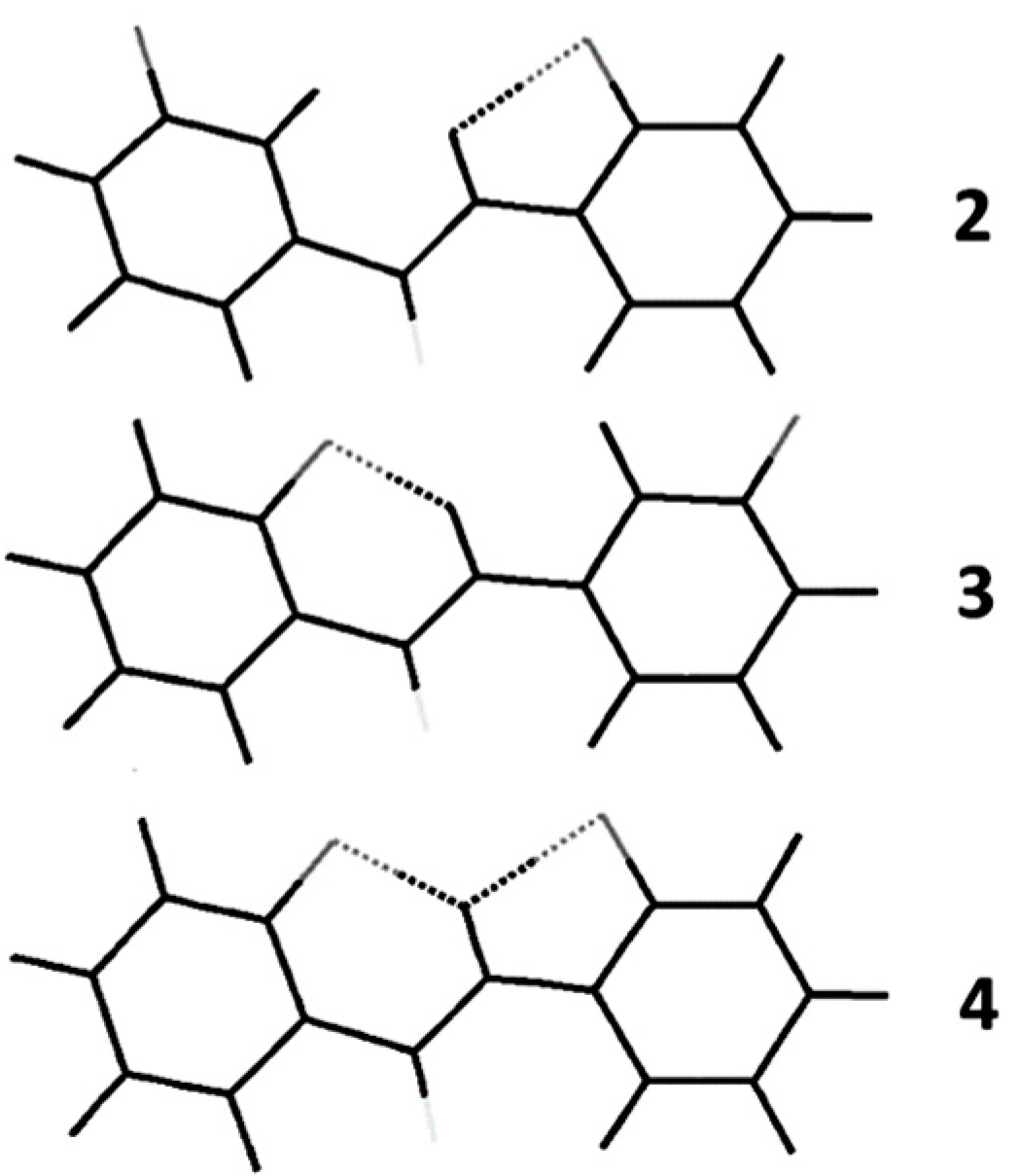
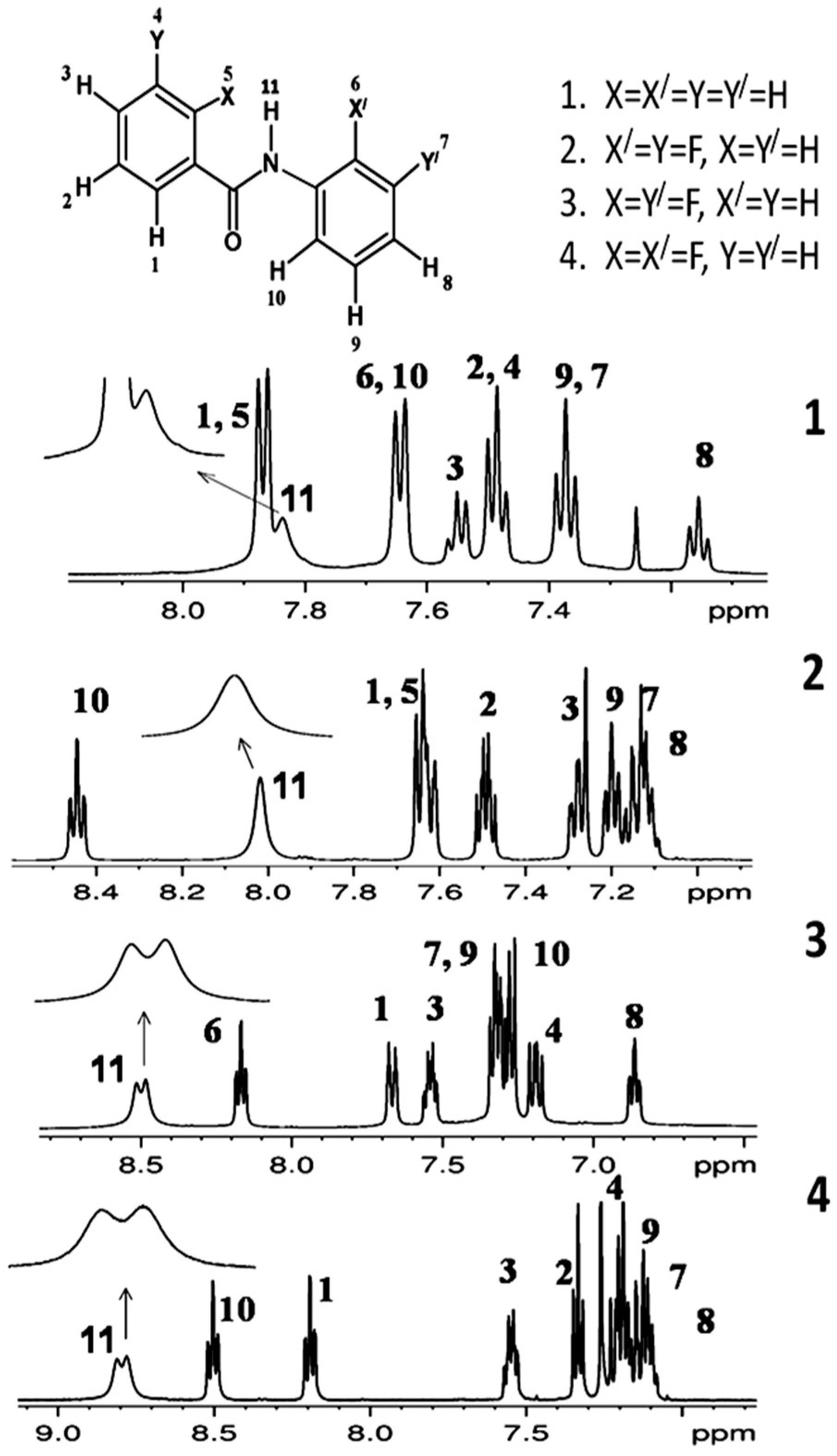
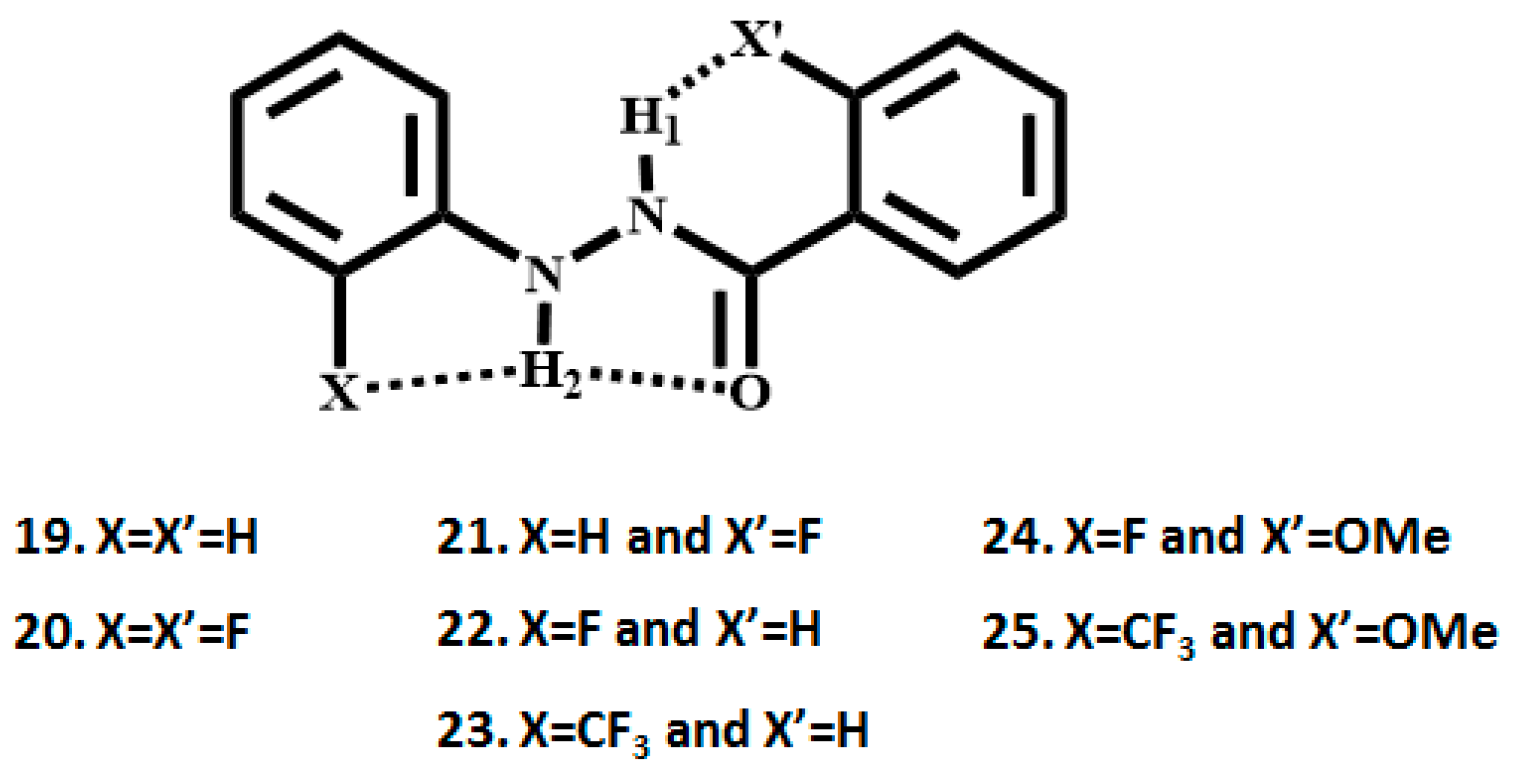
2. Studies on Benzanilides
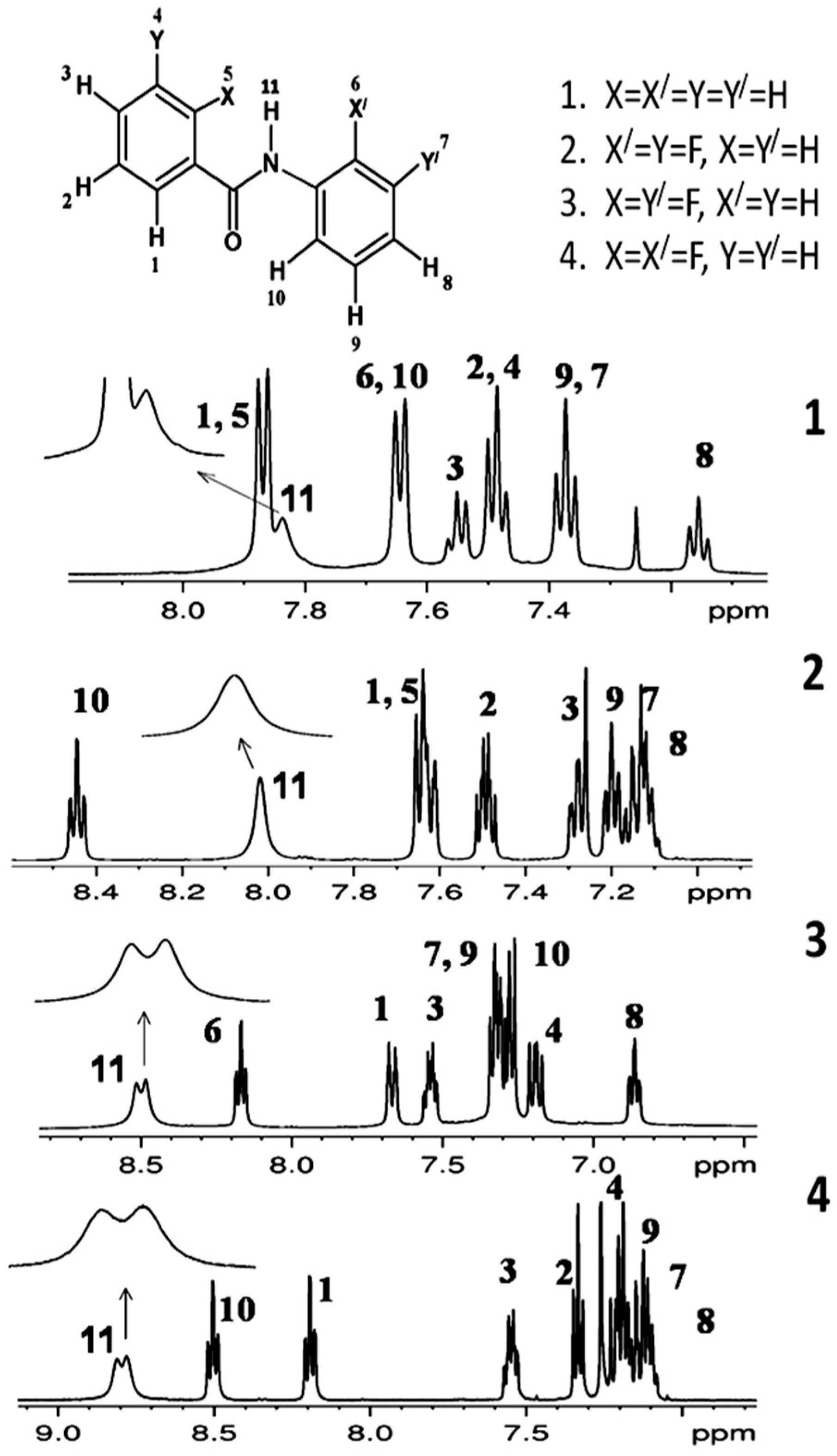
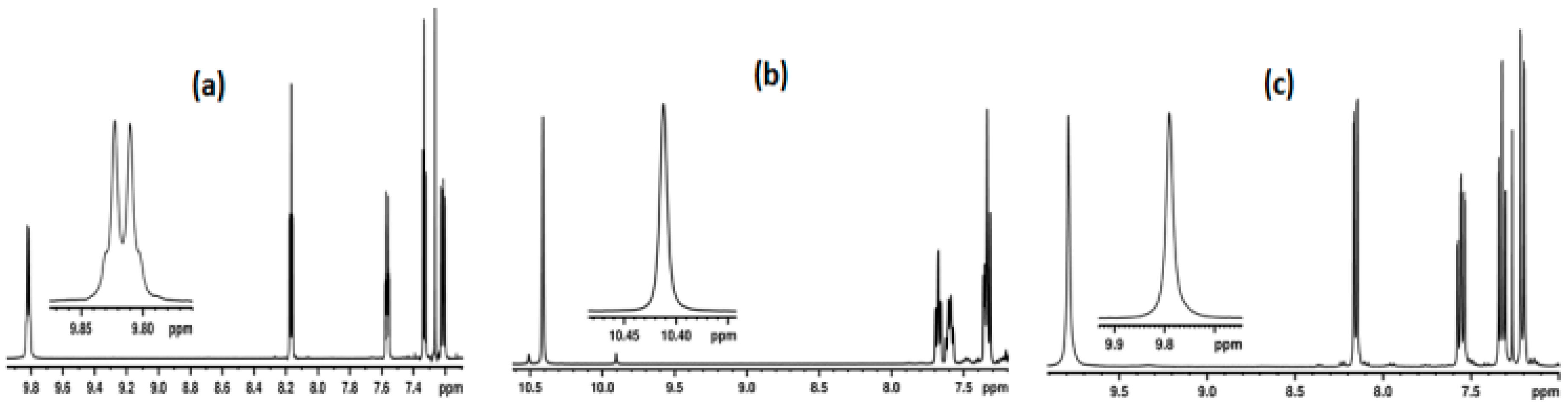
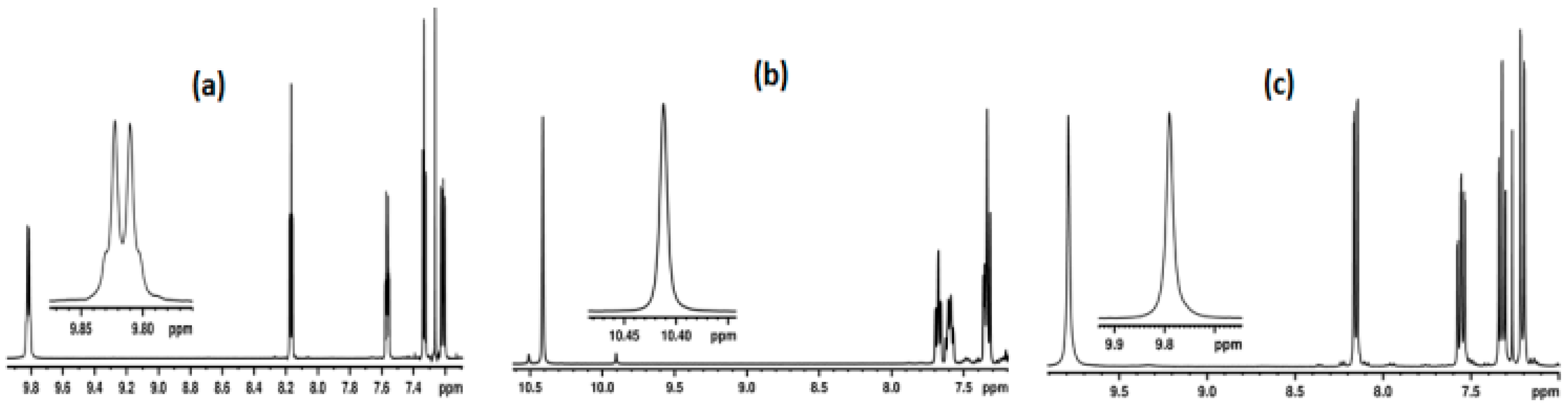
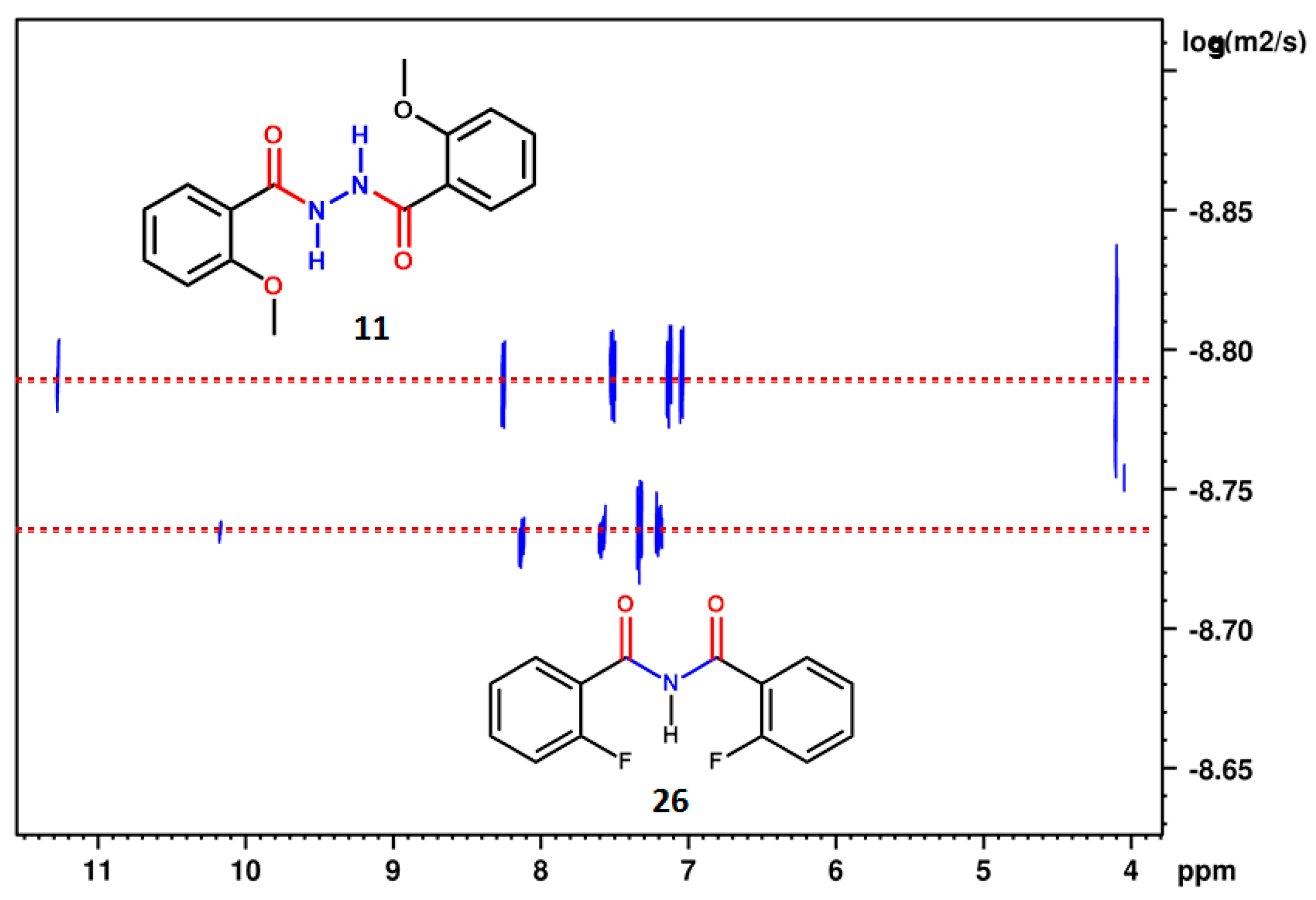
2.1. NMR Spectroscopic Experimental Findings
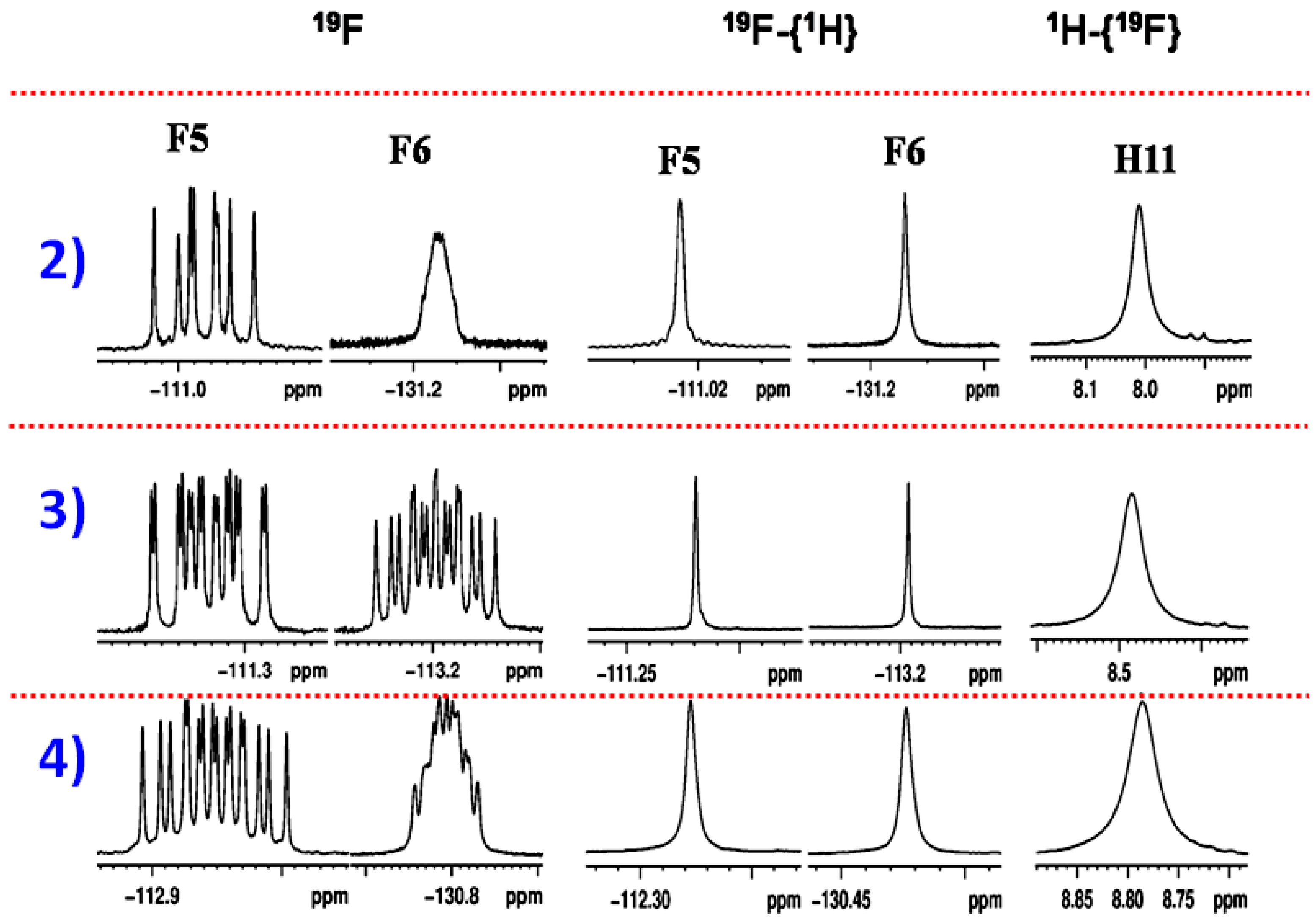
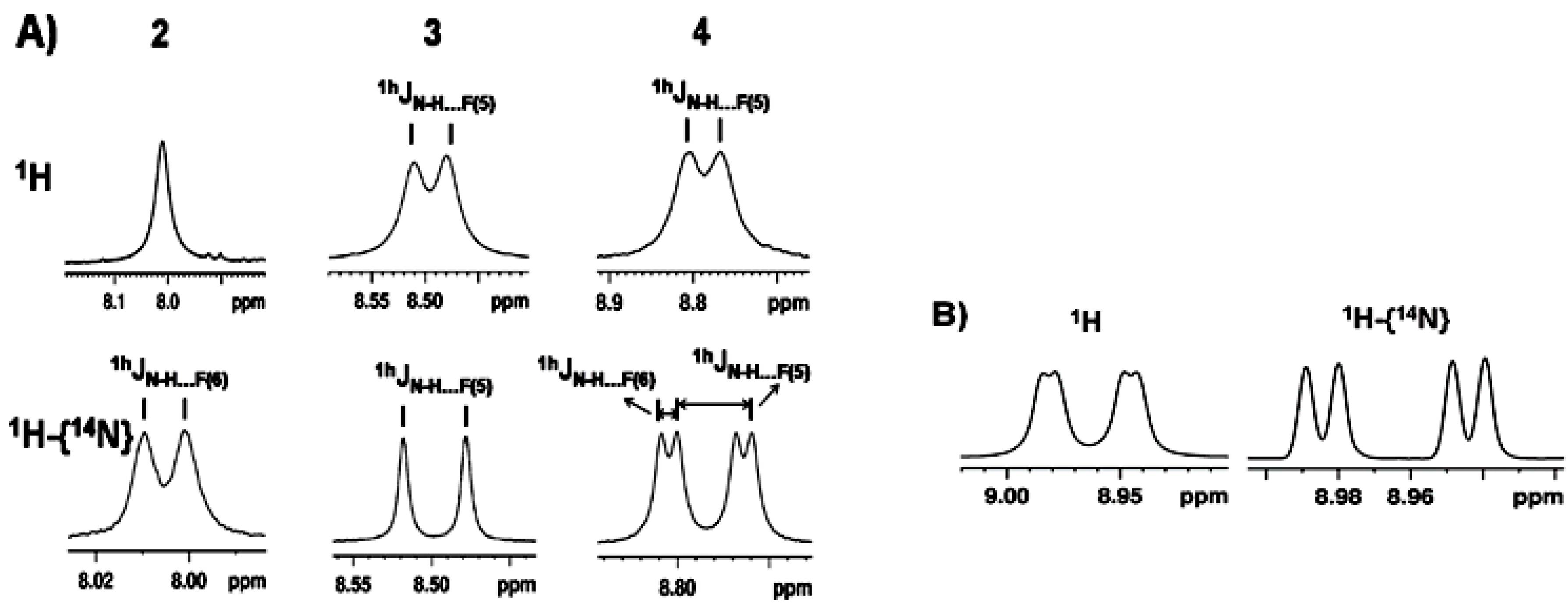
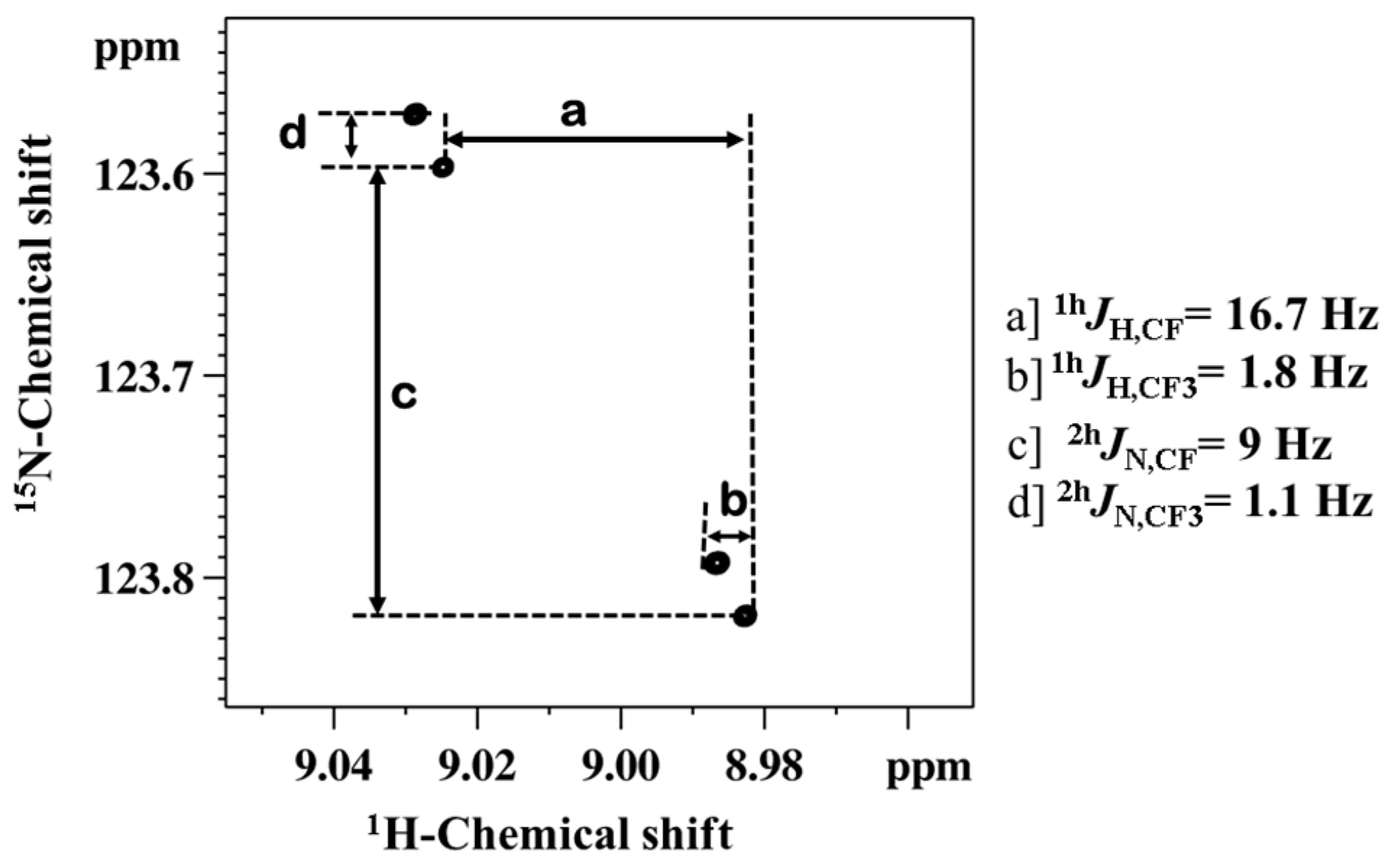
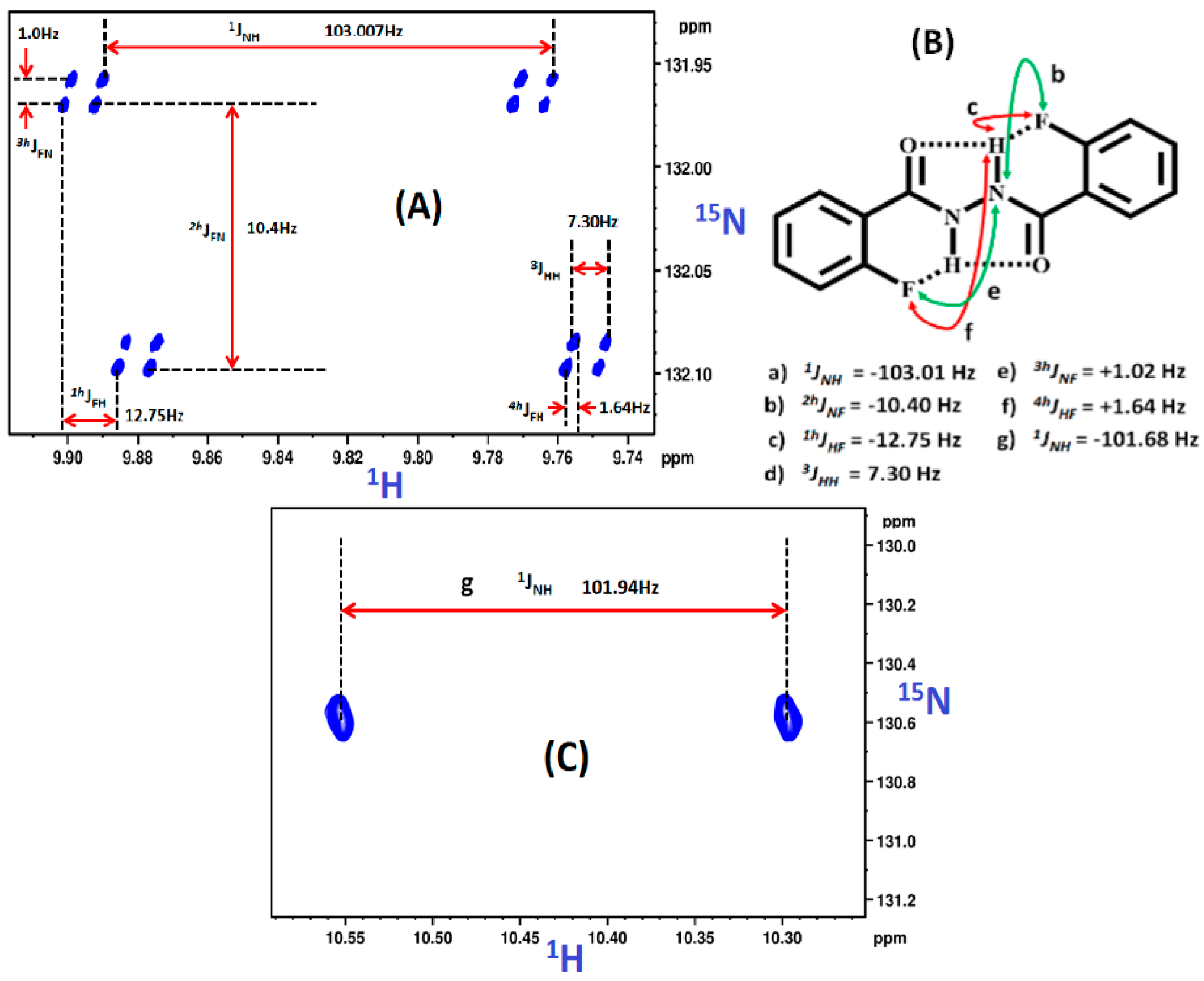
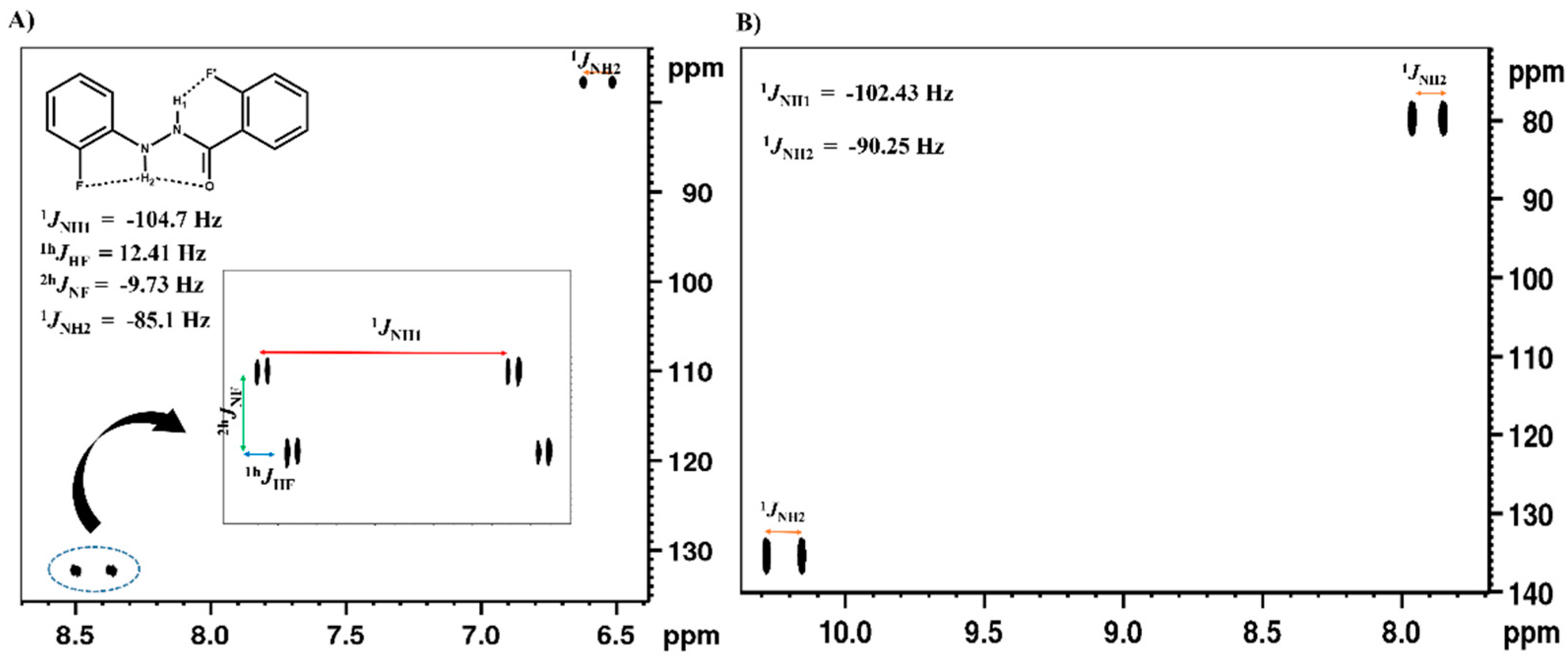
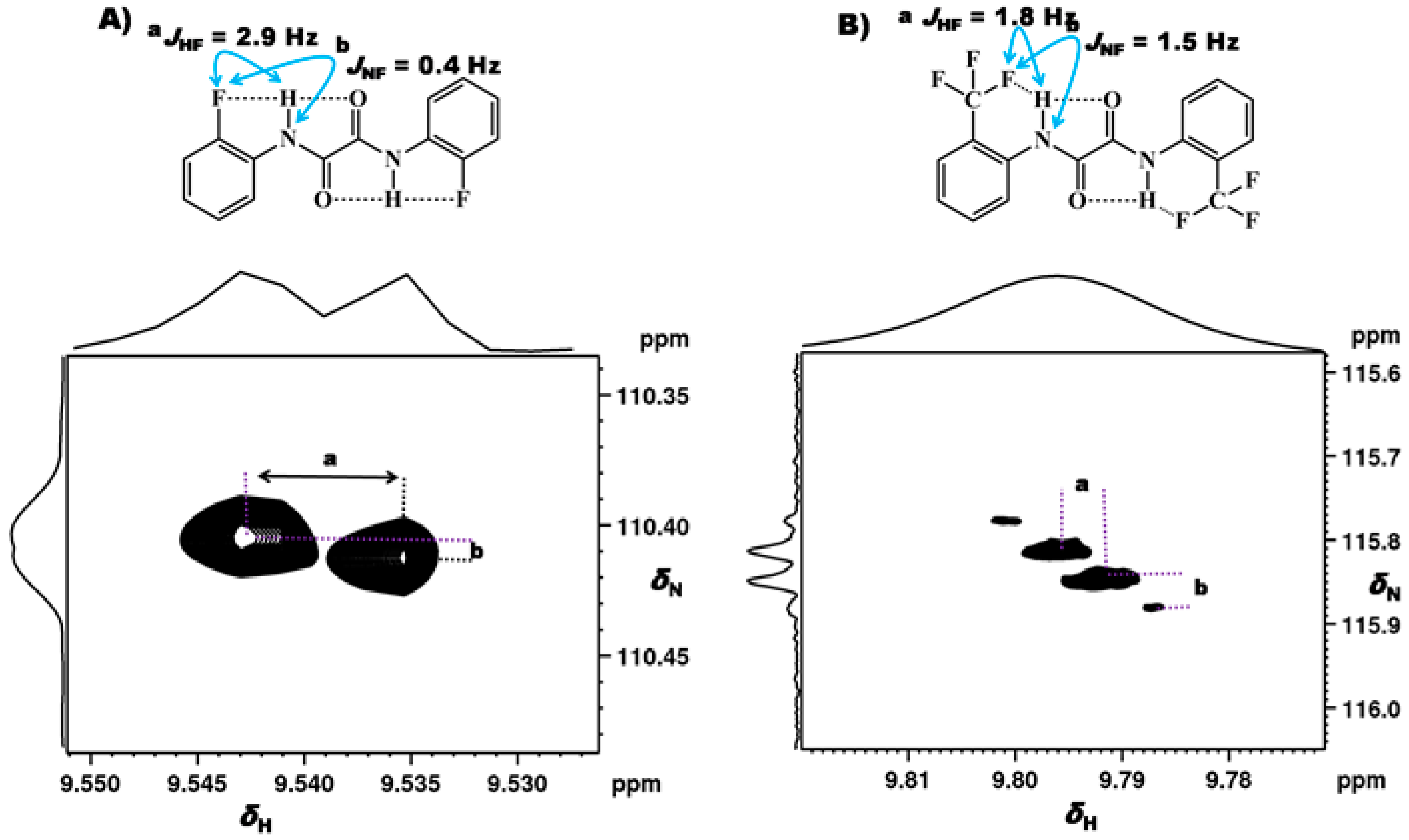
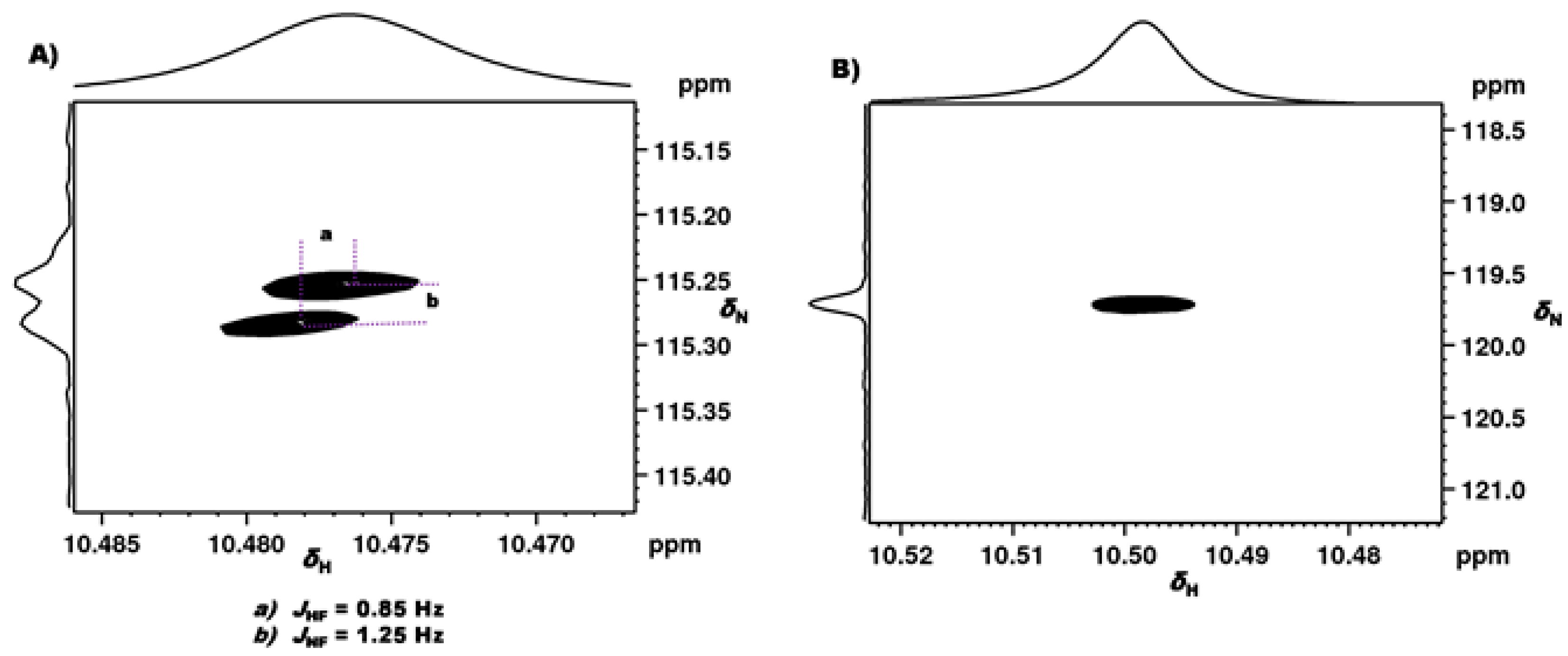
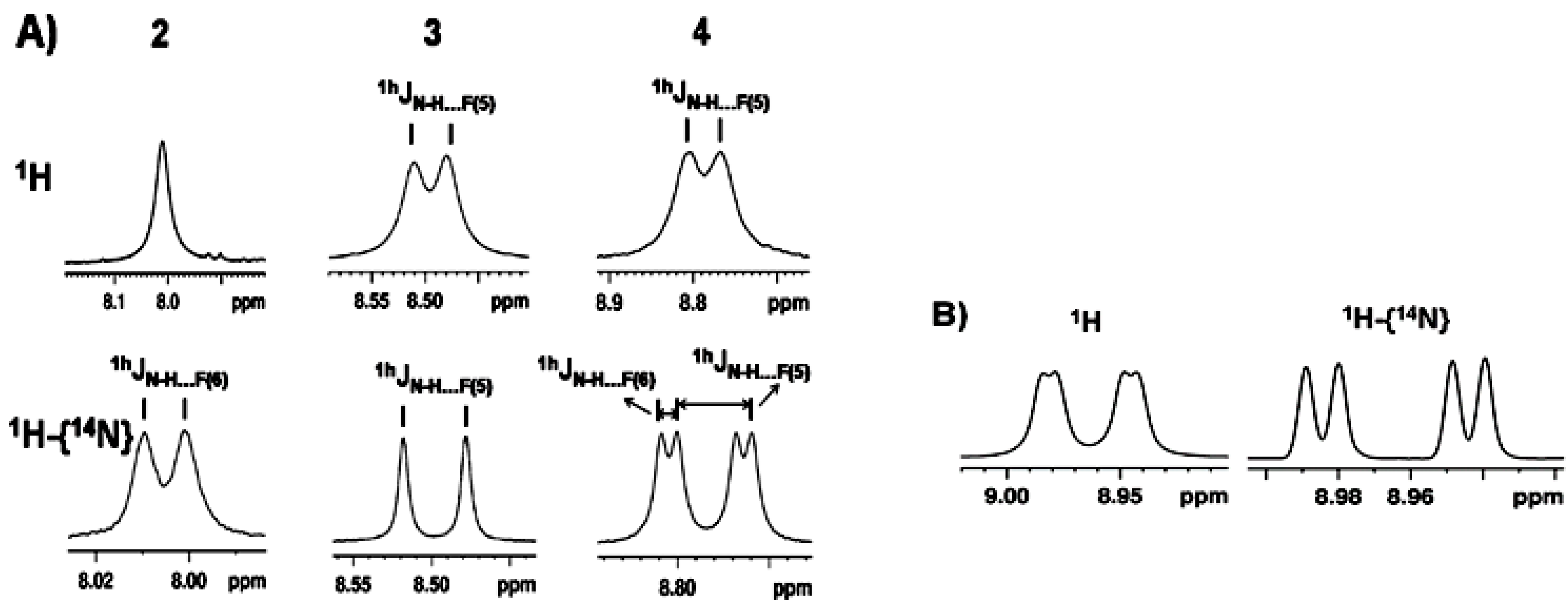
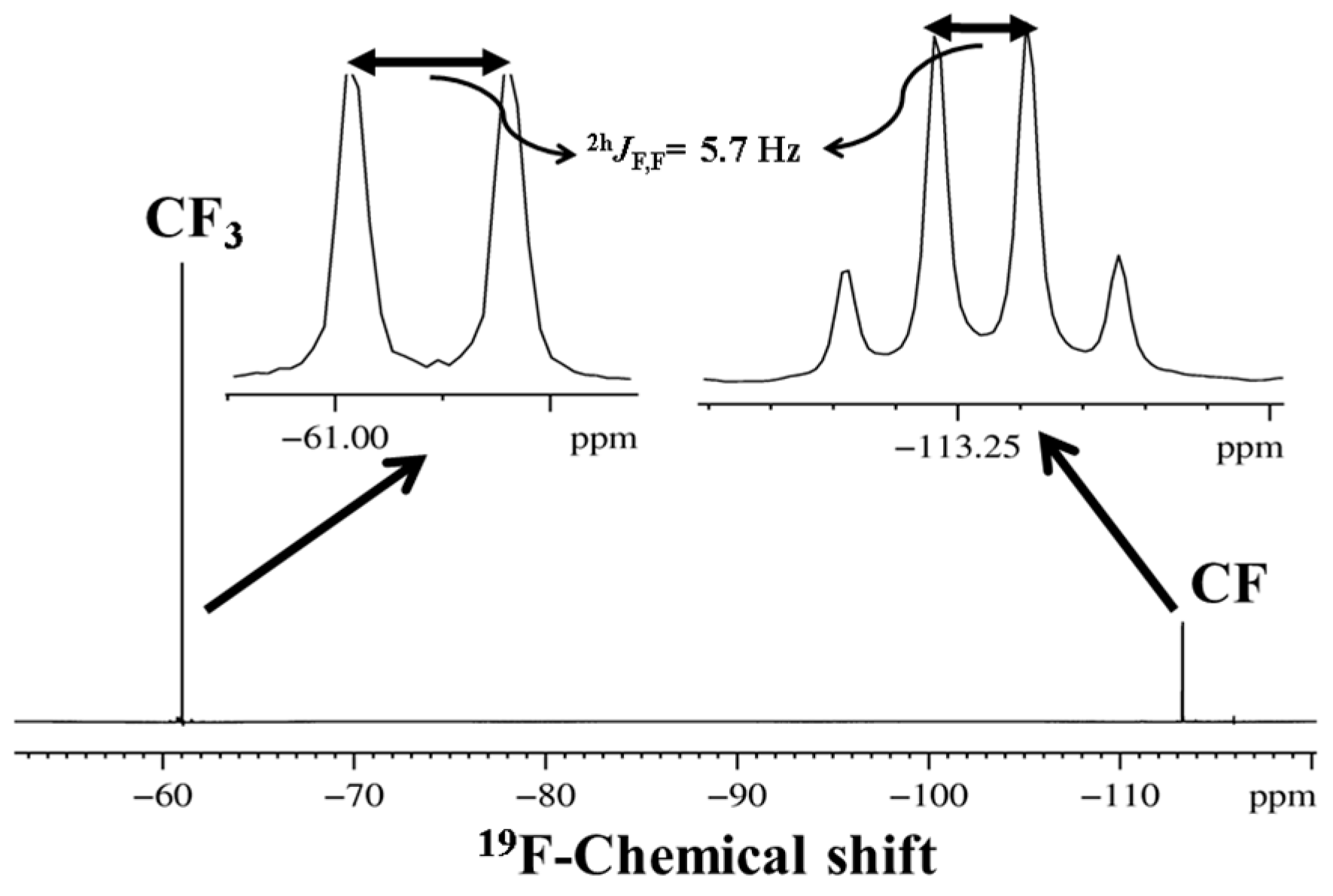
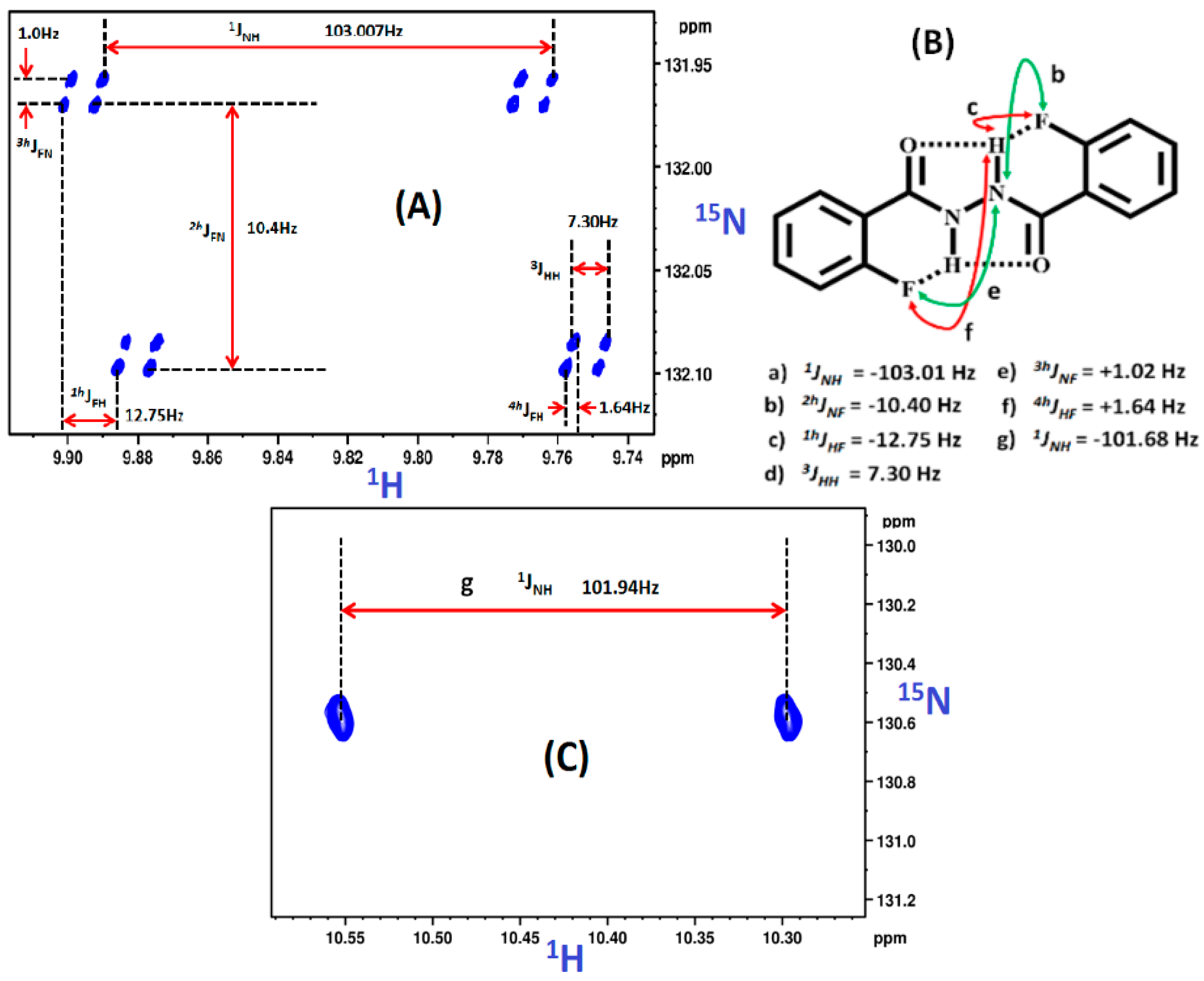
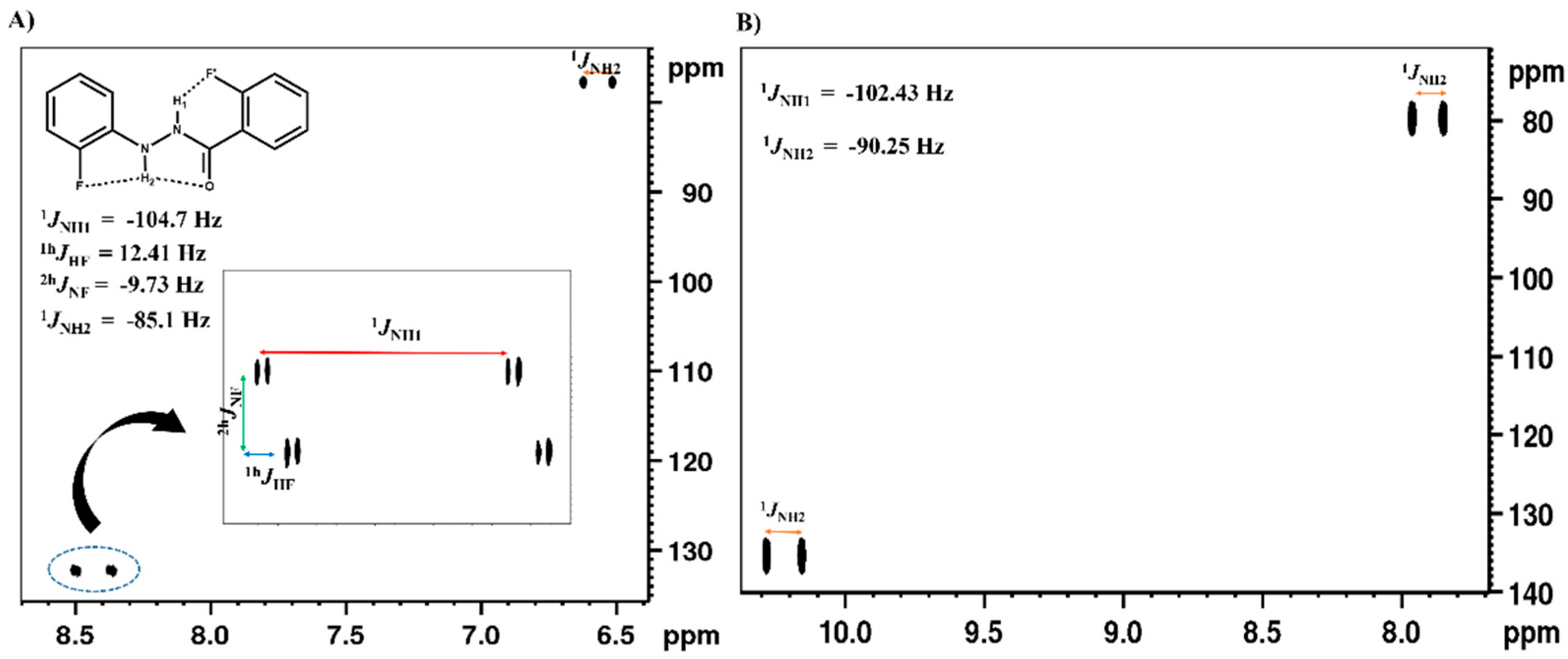
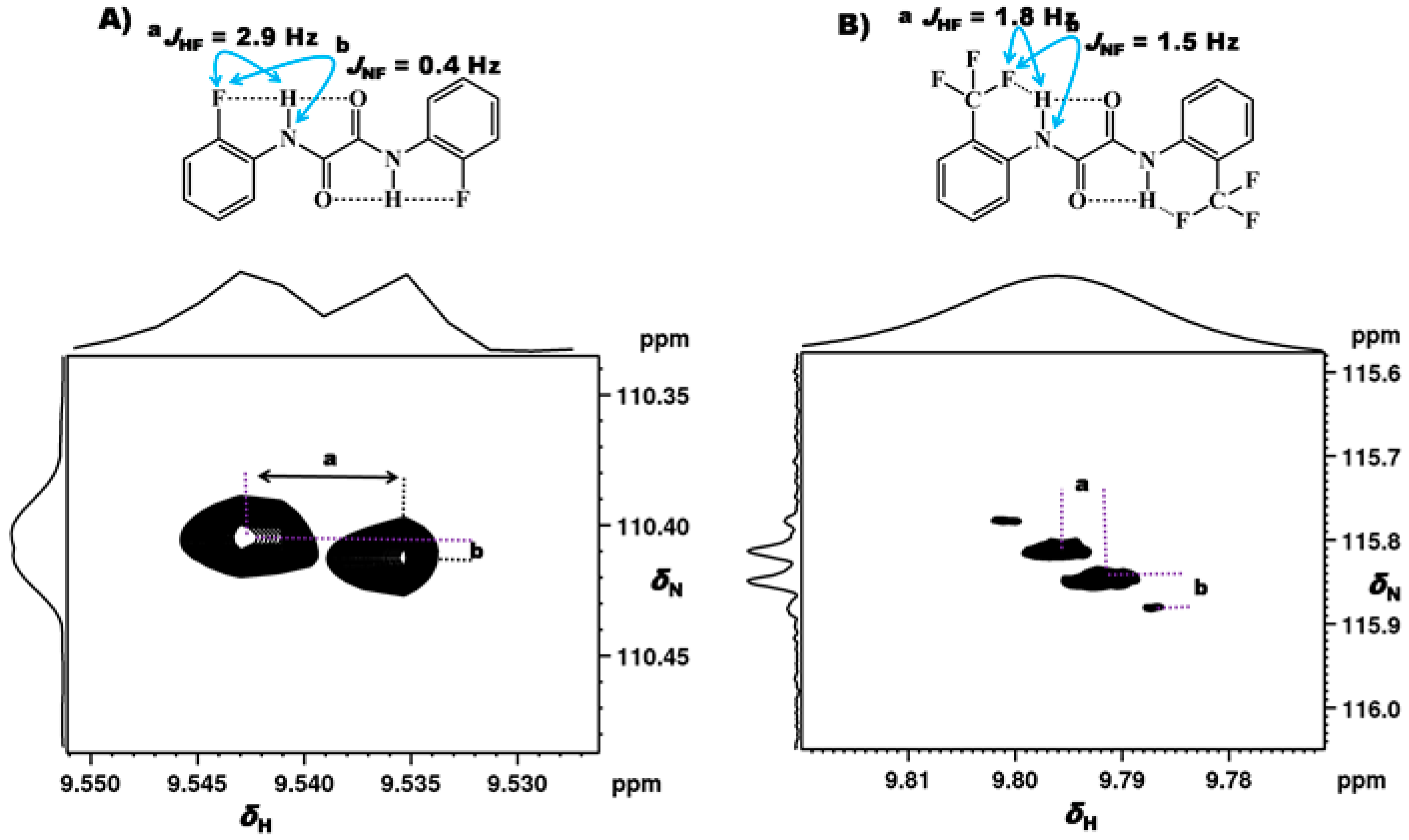
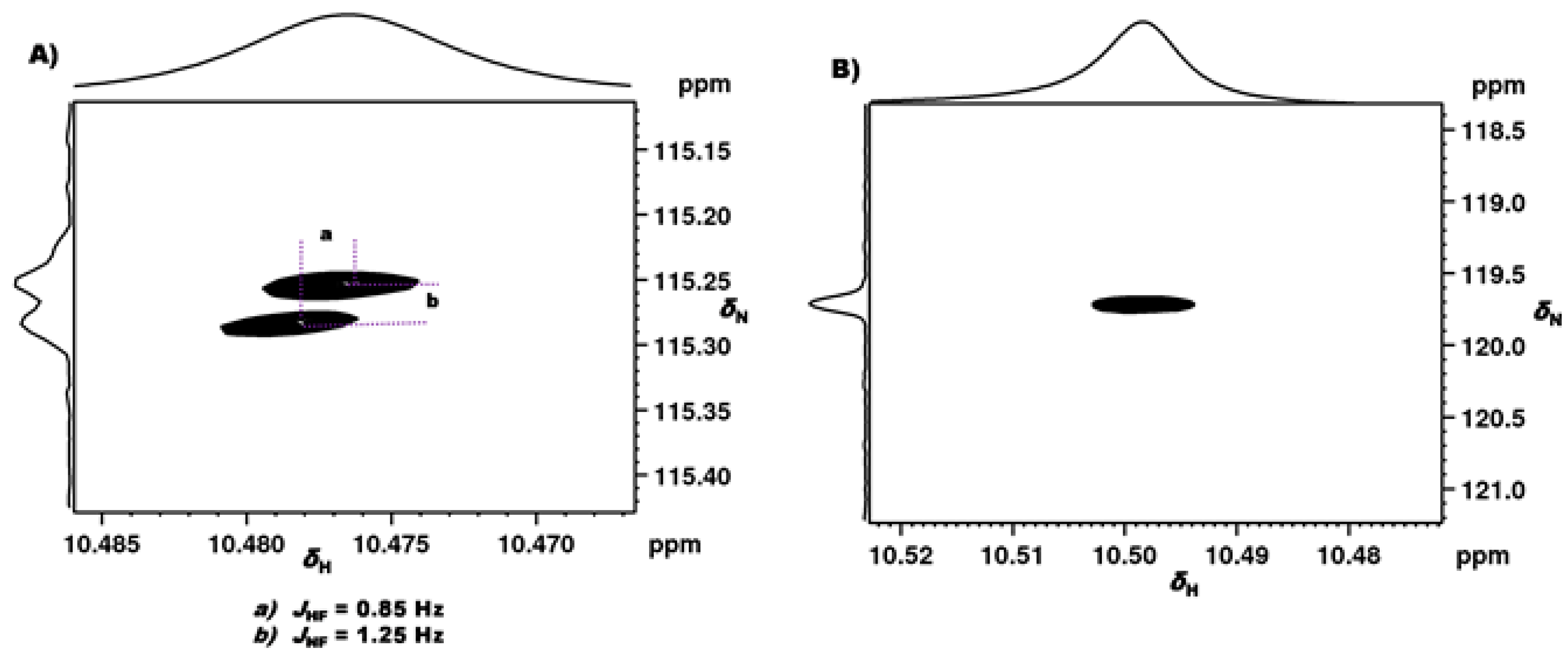
2.1.1. Detection of Spin-Spin Couplings Mediated by Hydrogen Bonds
2.1.2. Simultaneous Detection of Two through Space Couplings
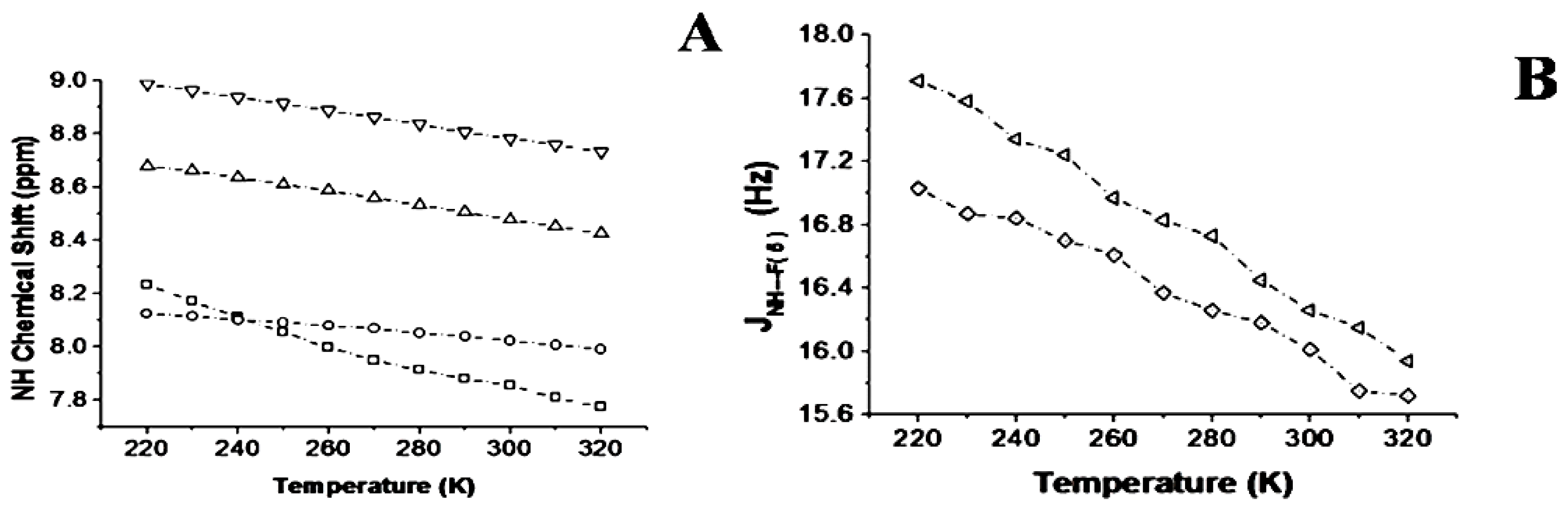
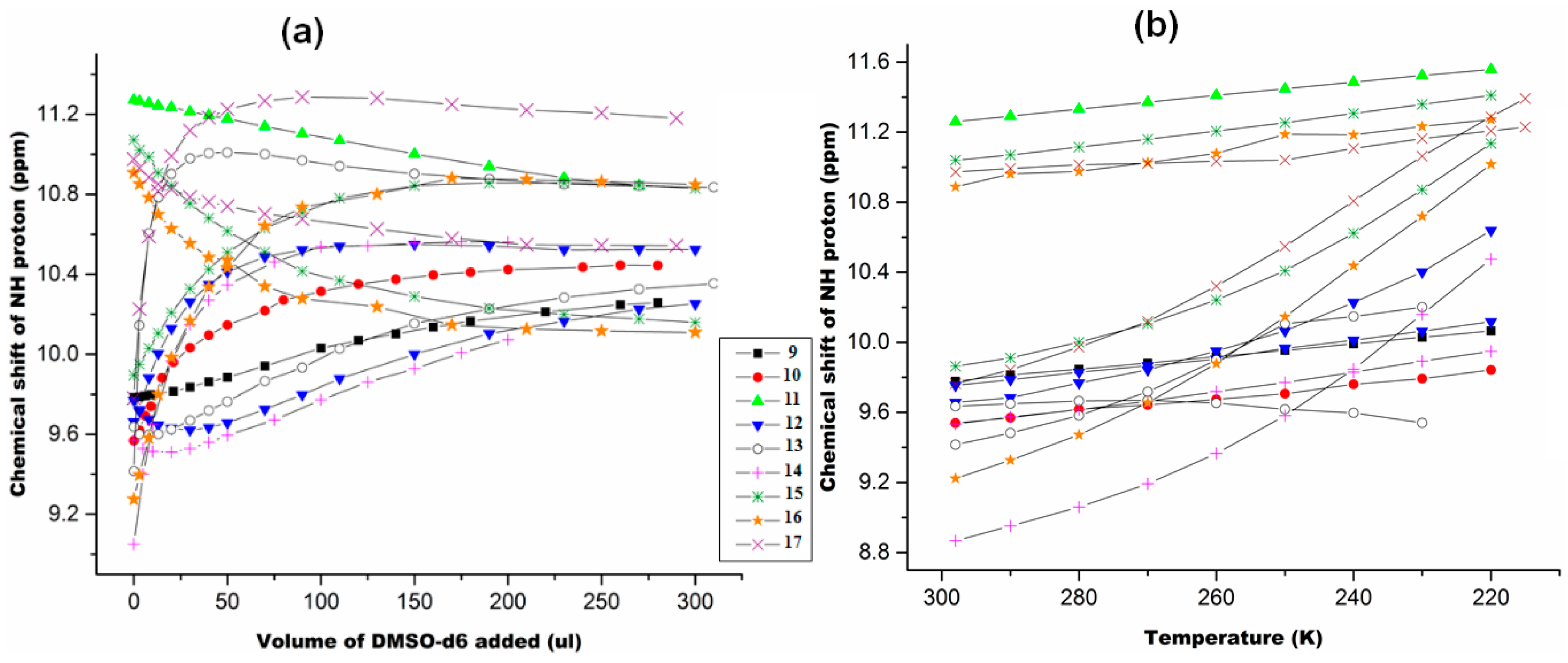
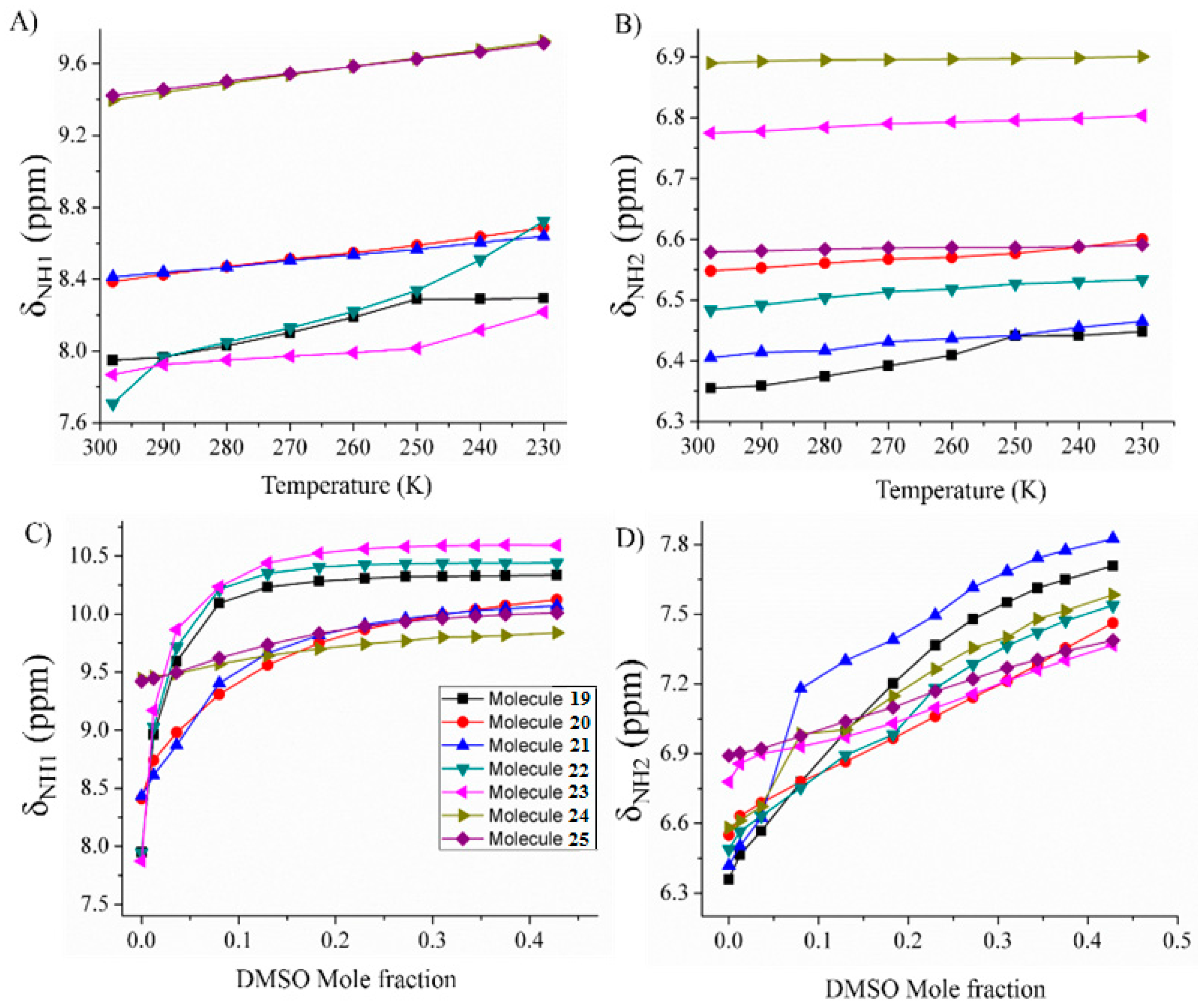
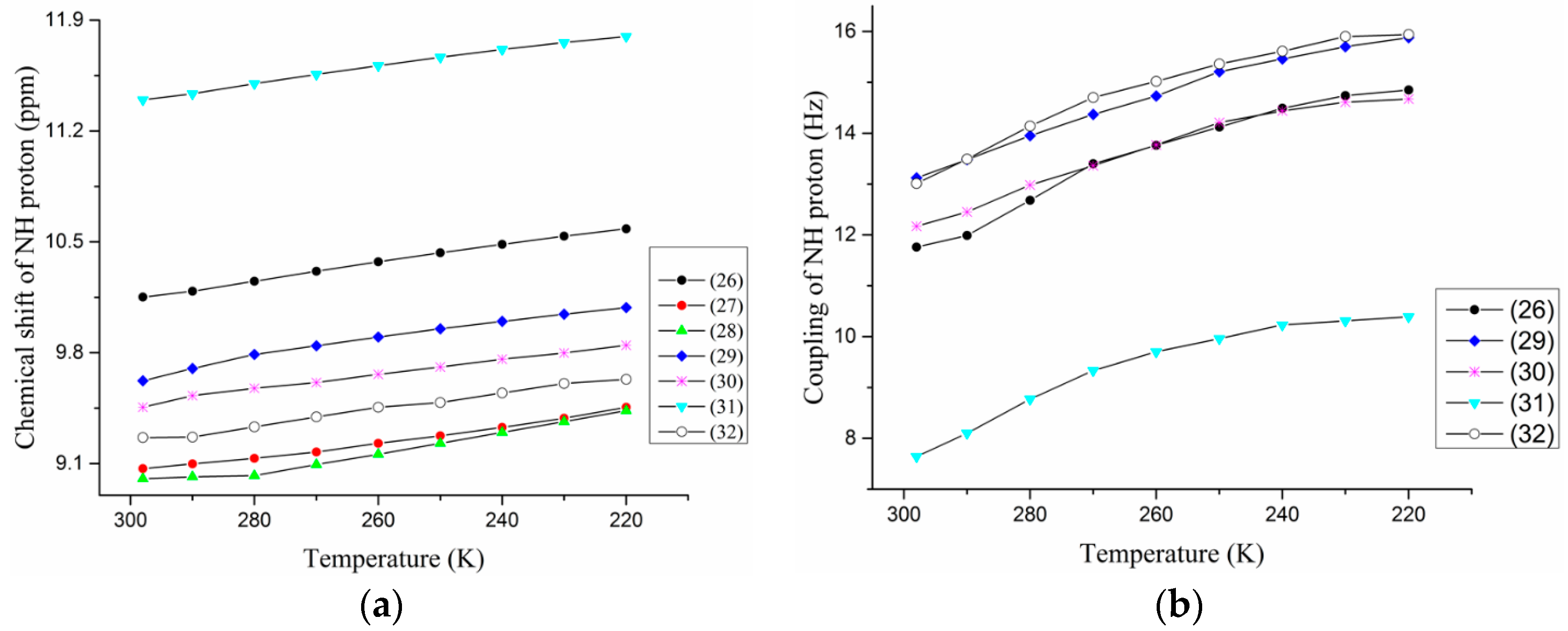
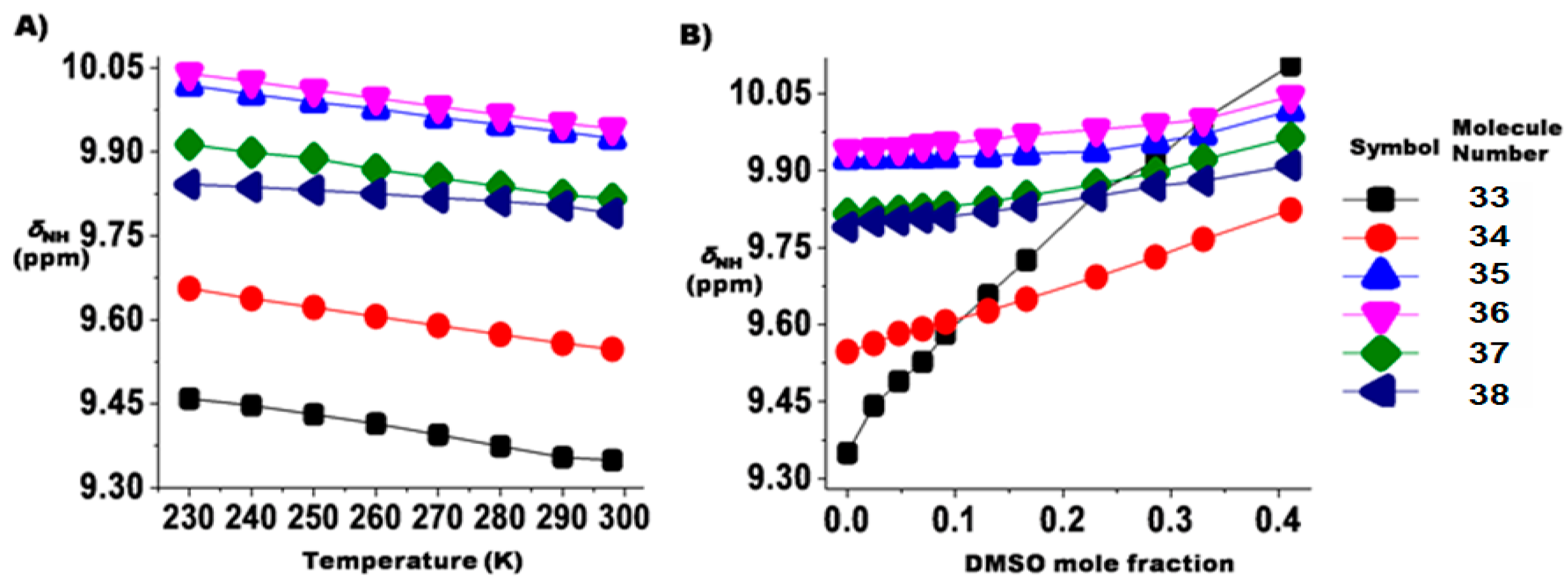
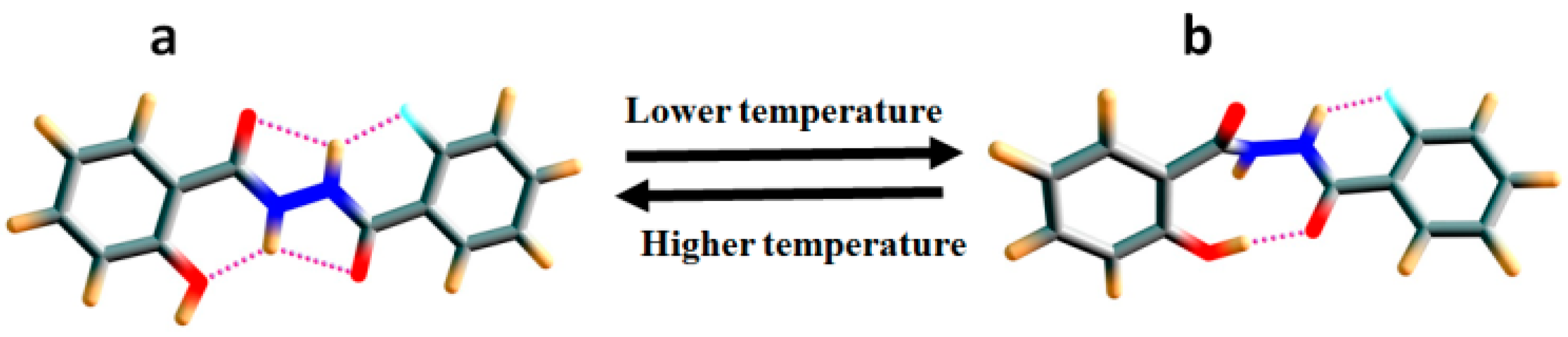
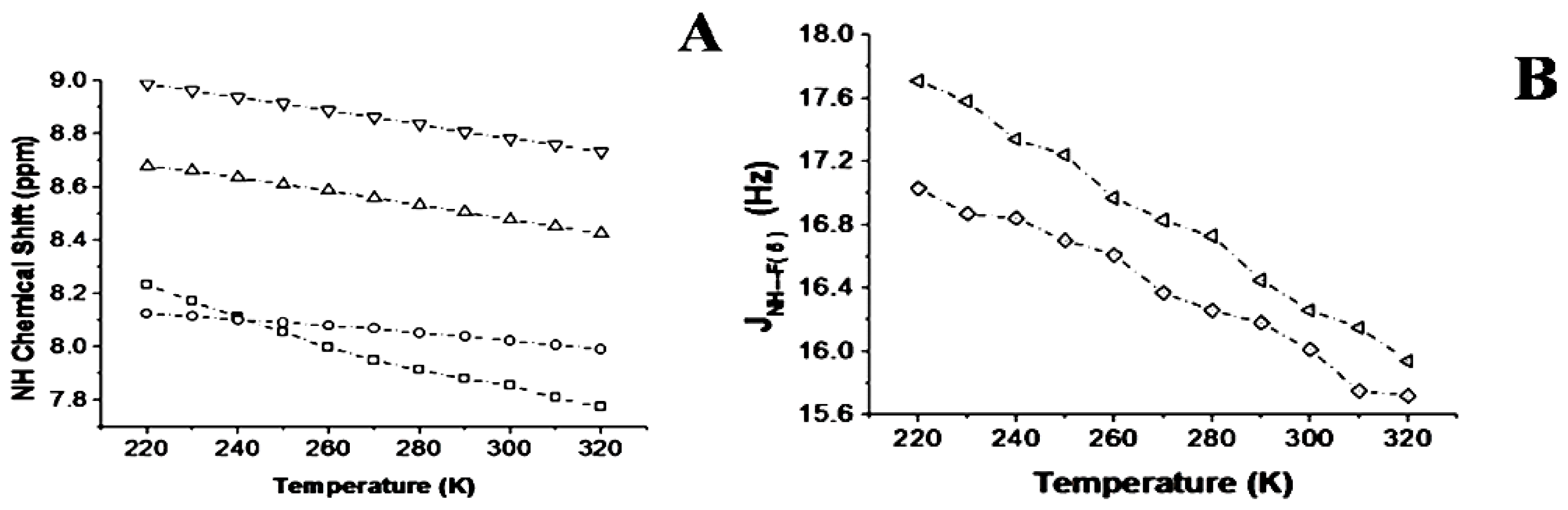
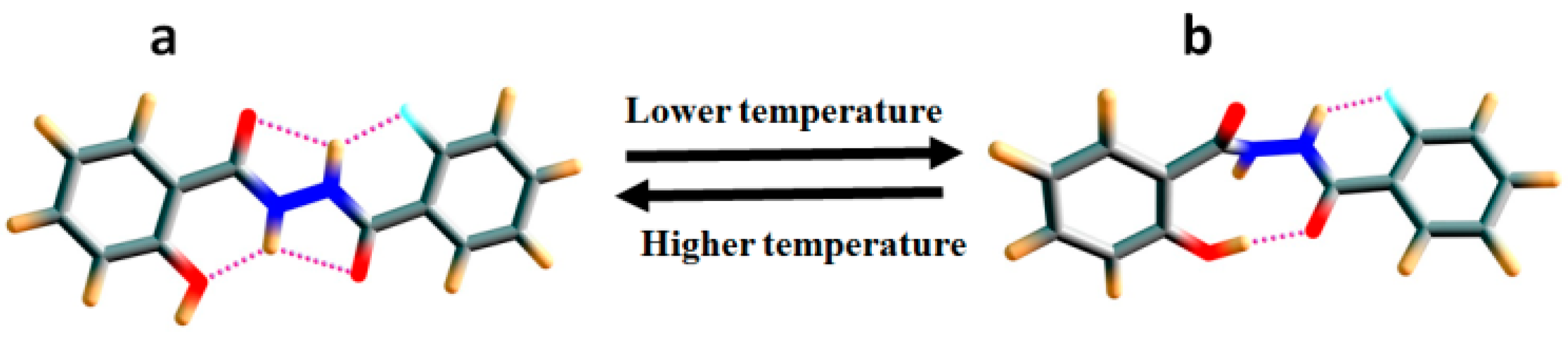
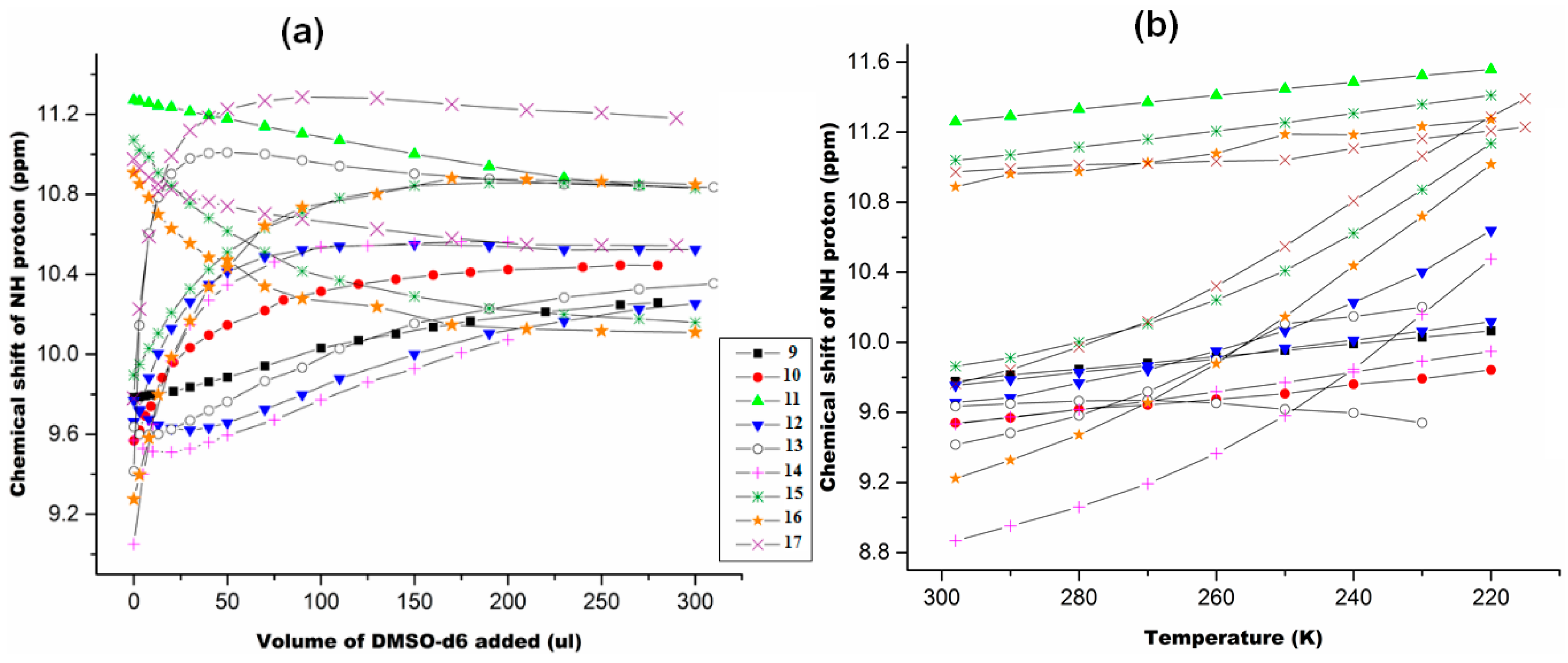
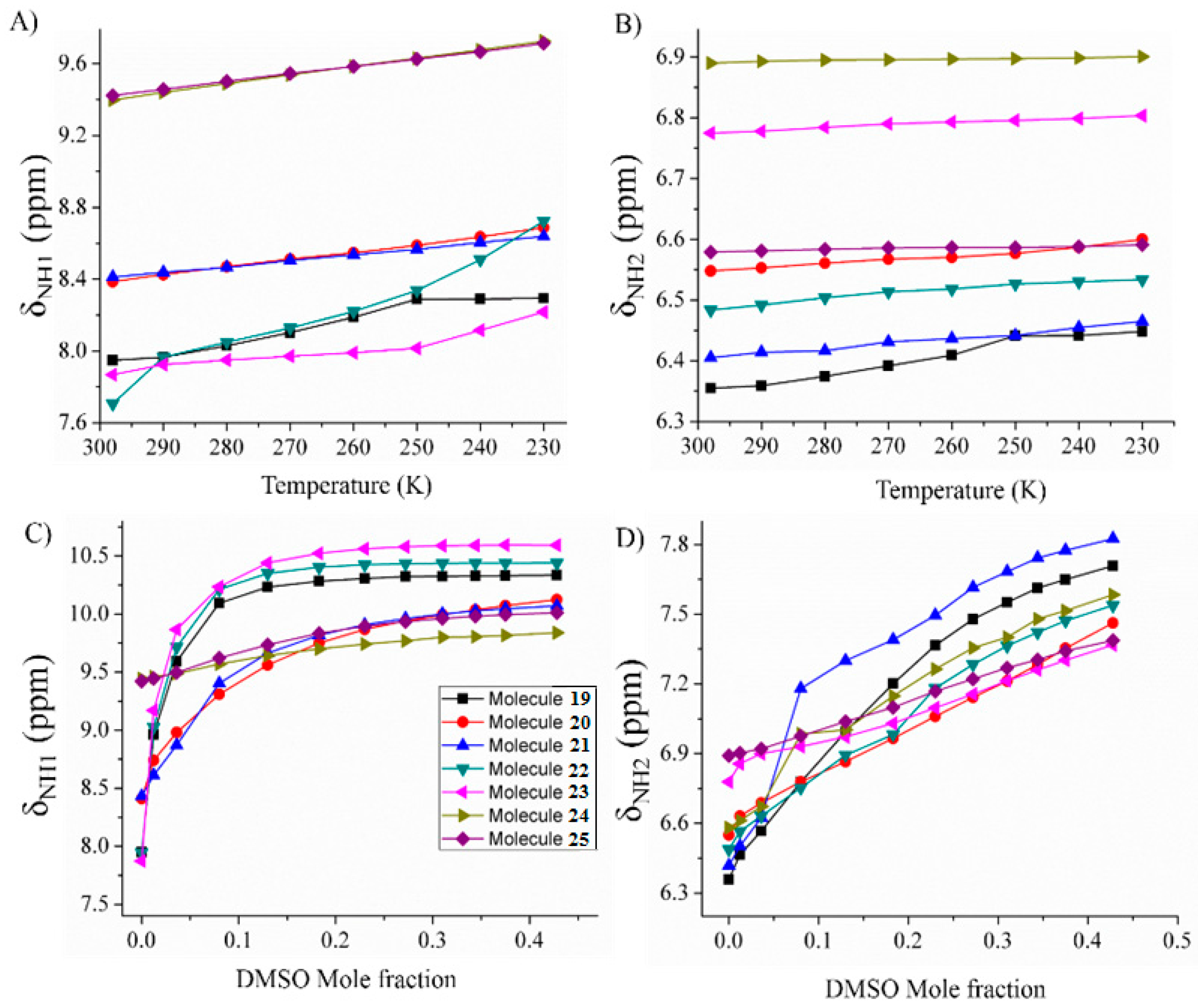
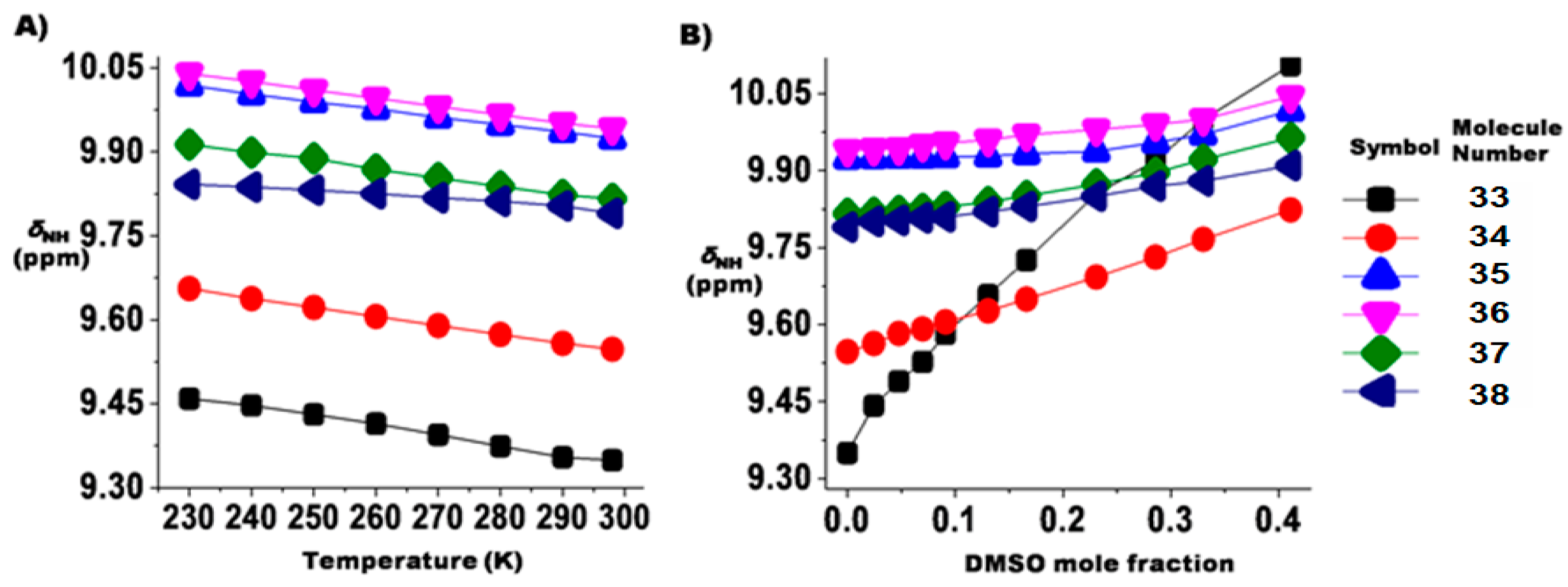
2.1.3. Temperature and Solvent Induced Perturbations
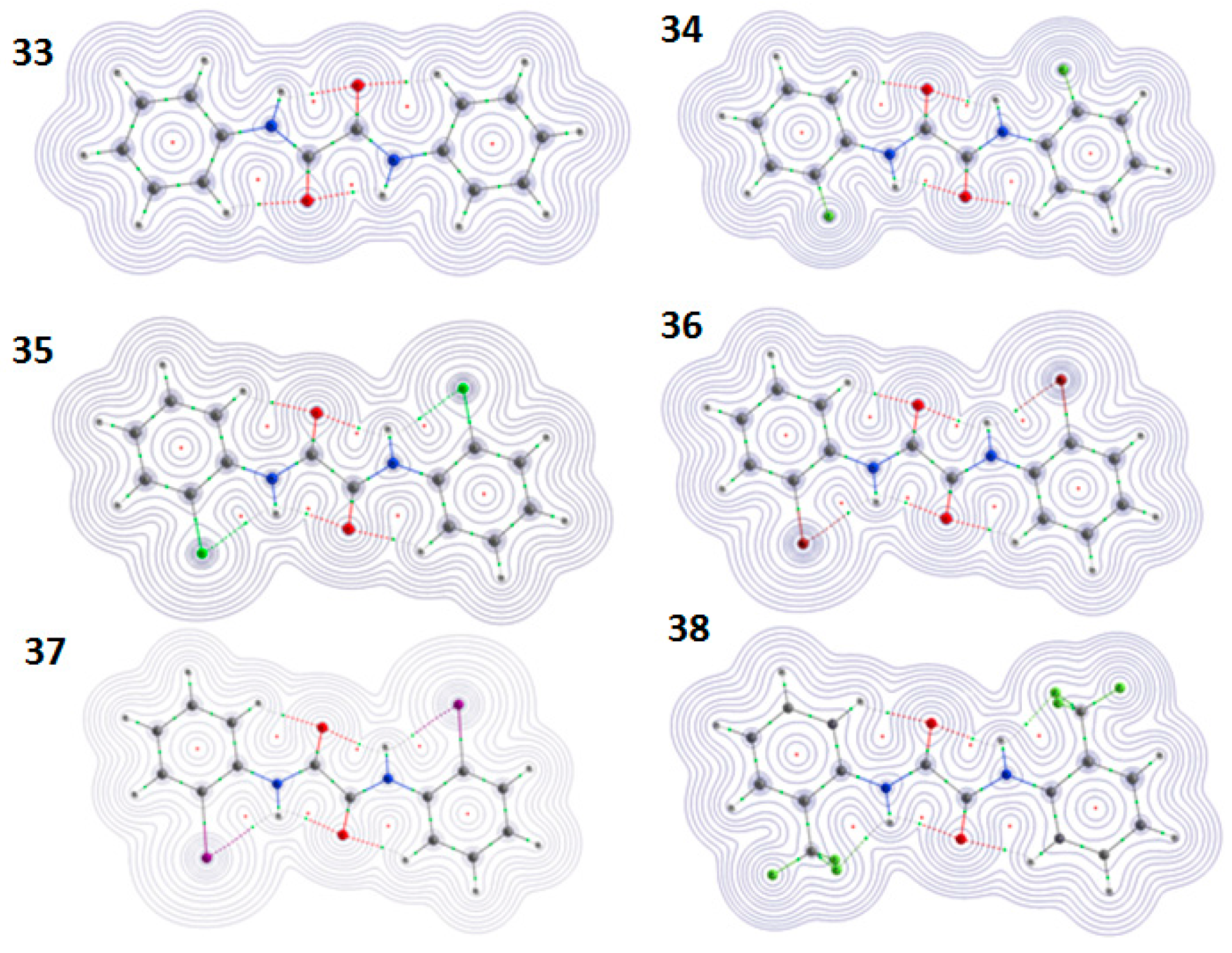
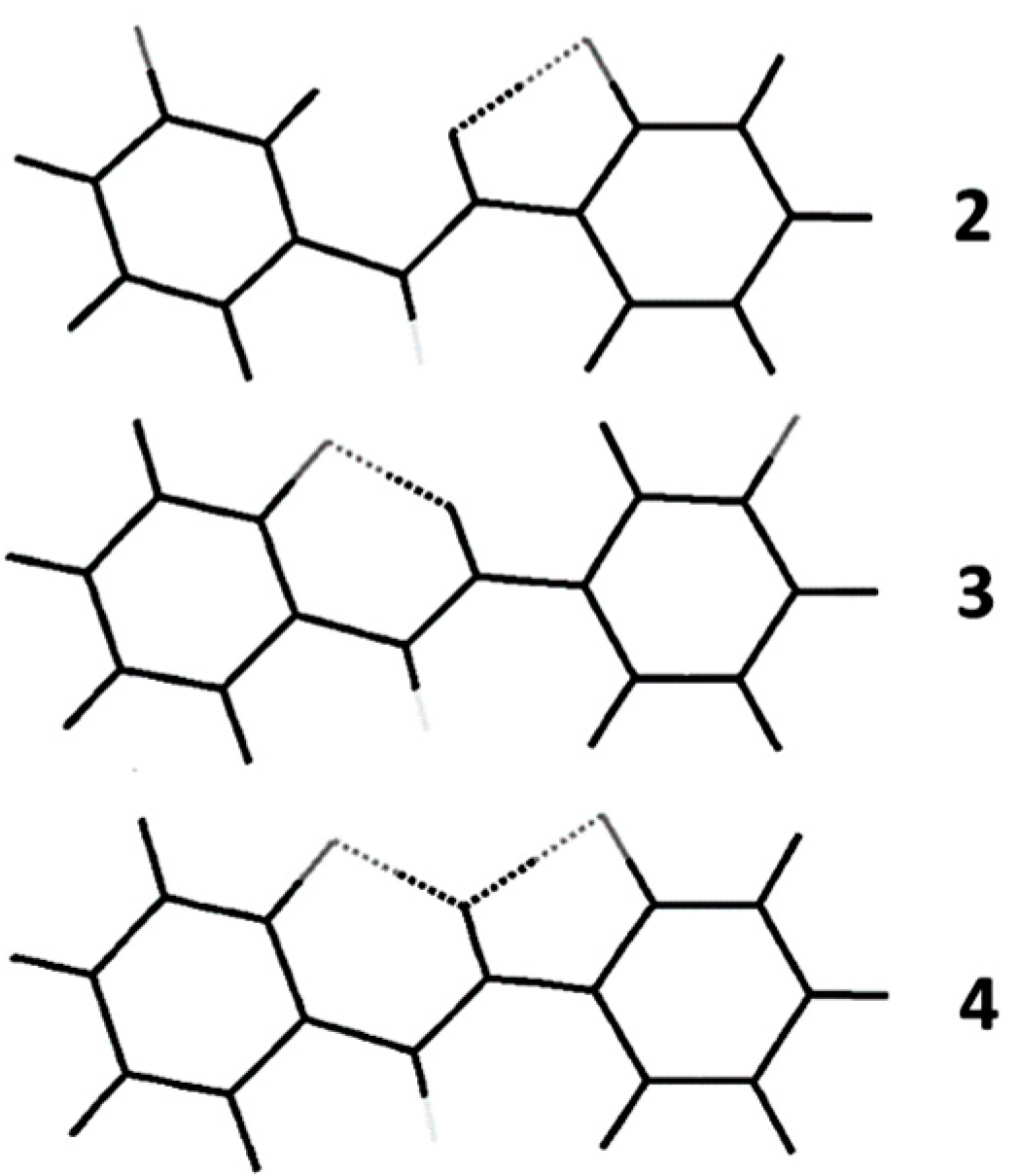
2.2. Theoretical Calculations
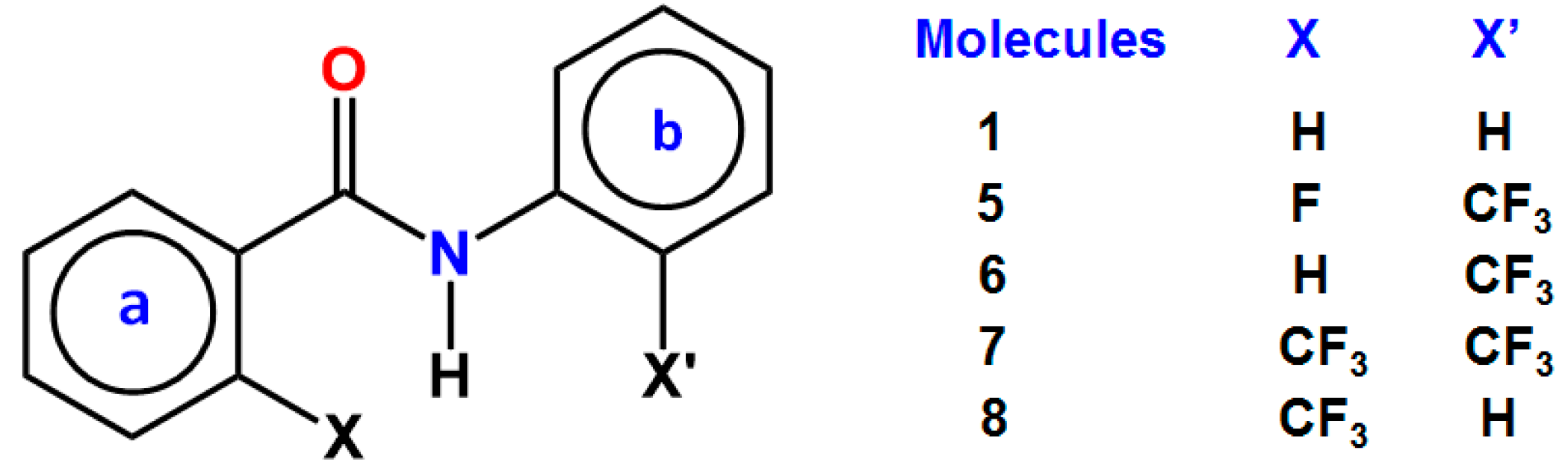
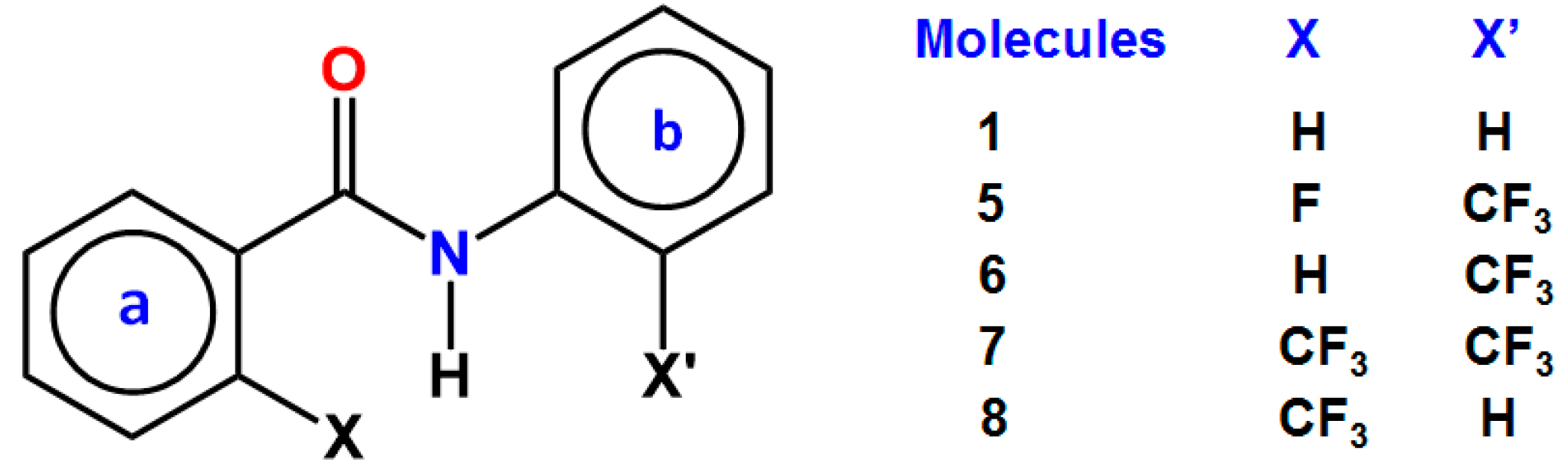
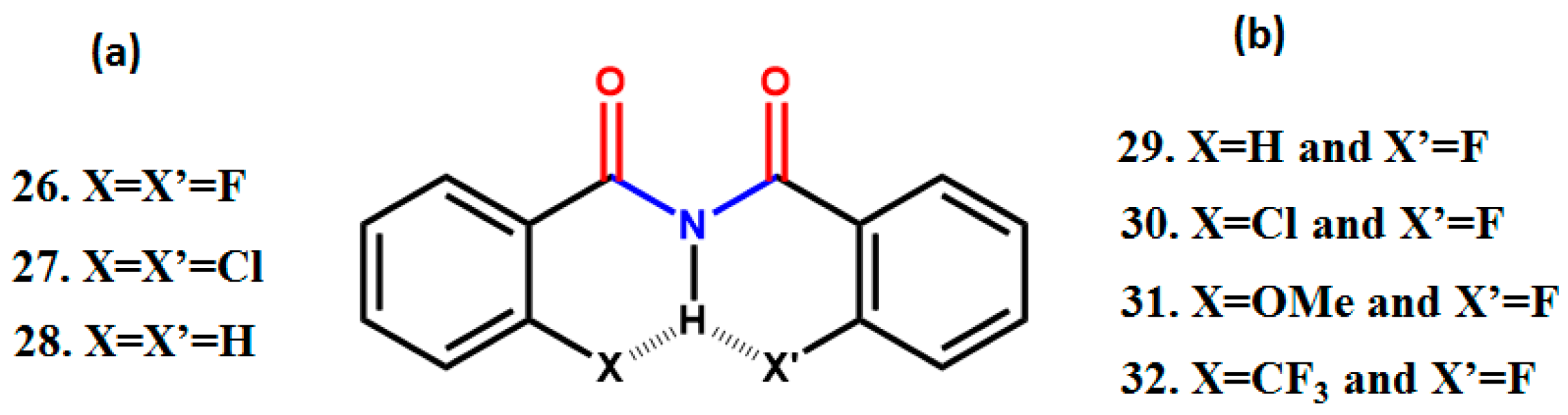
3. CF3 Substituted Benzanilides
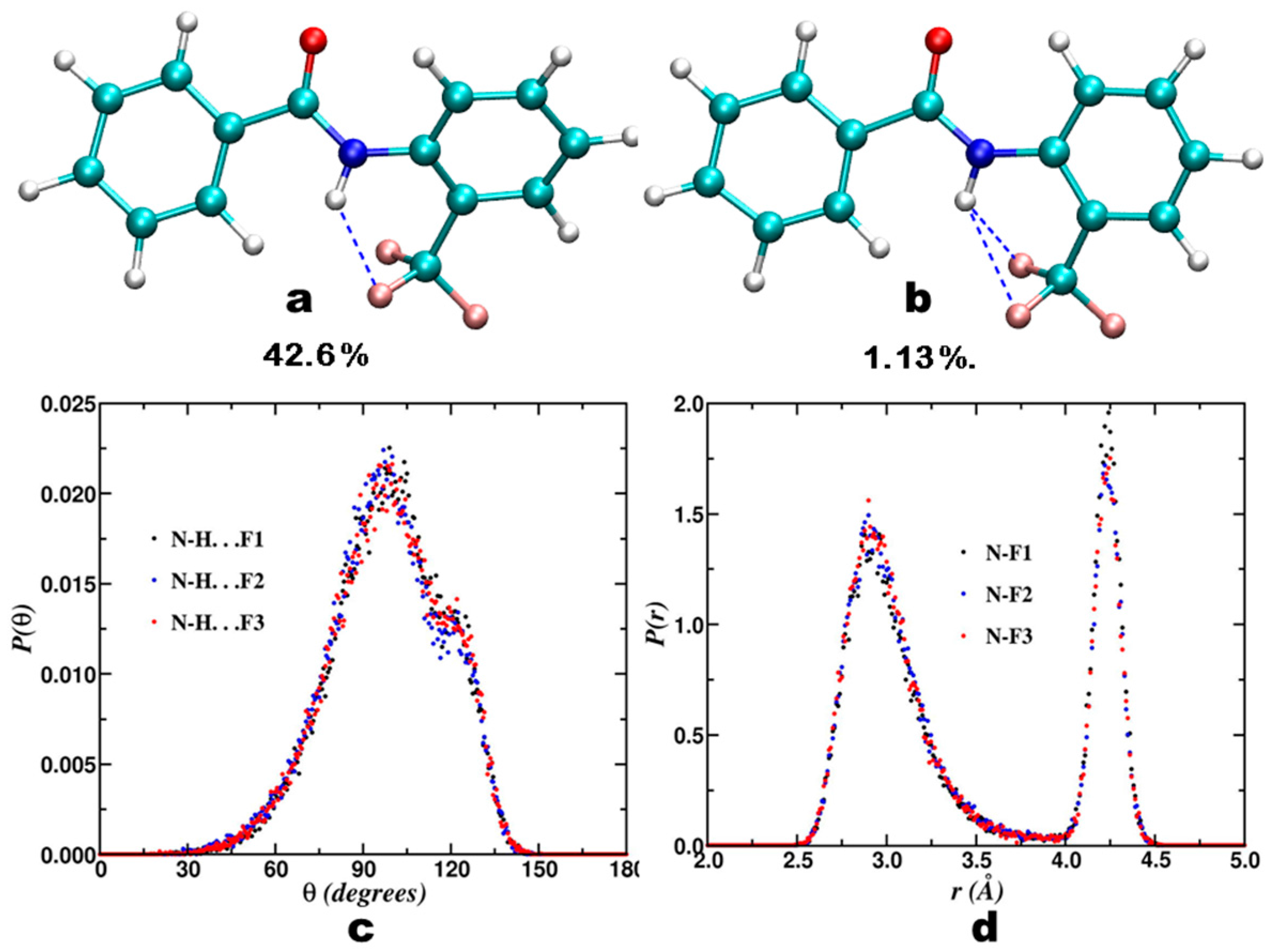
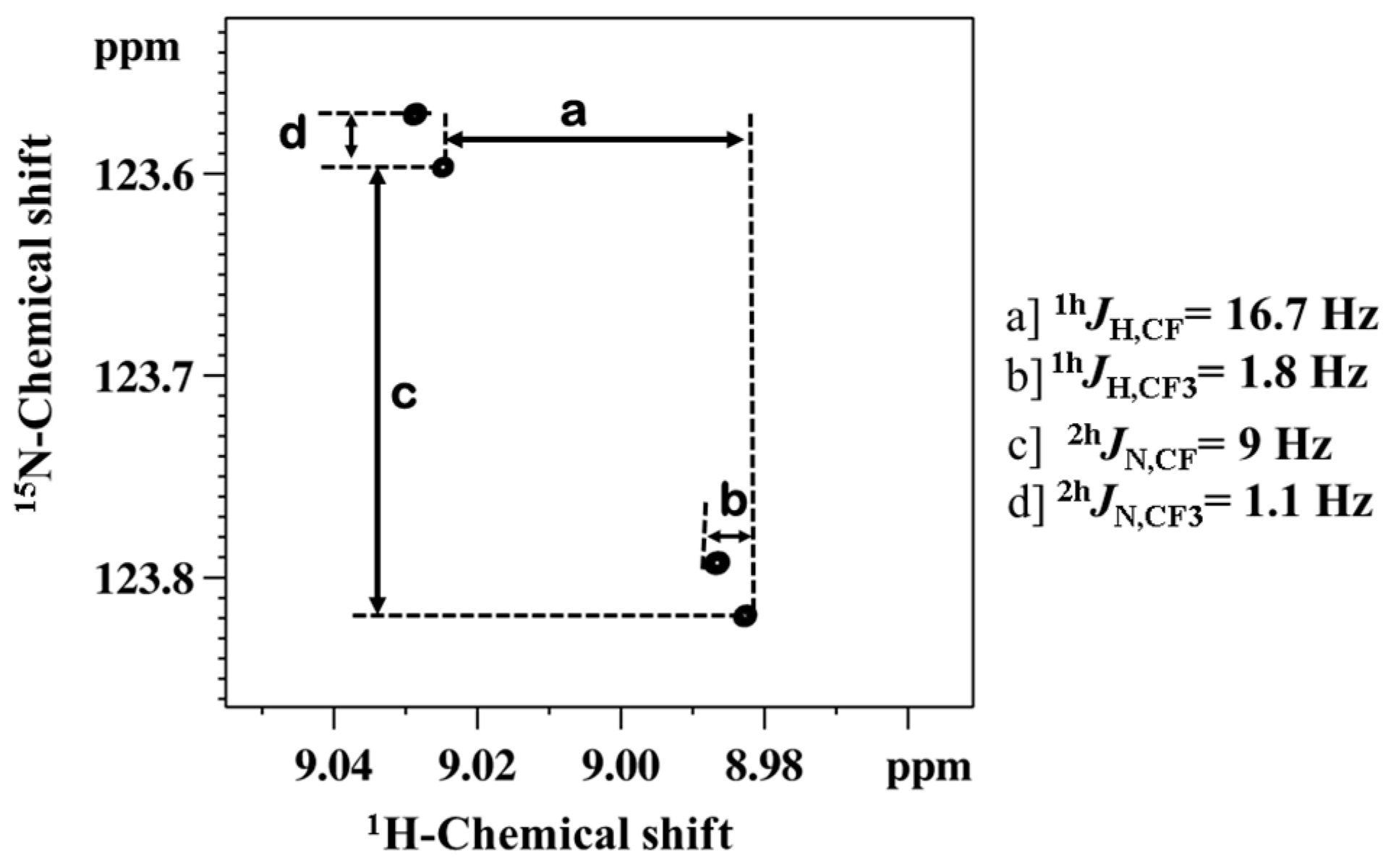
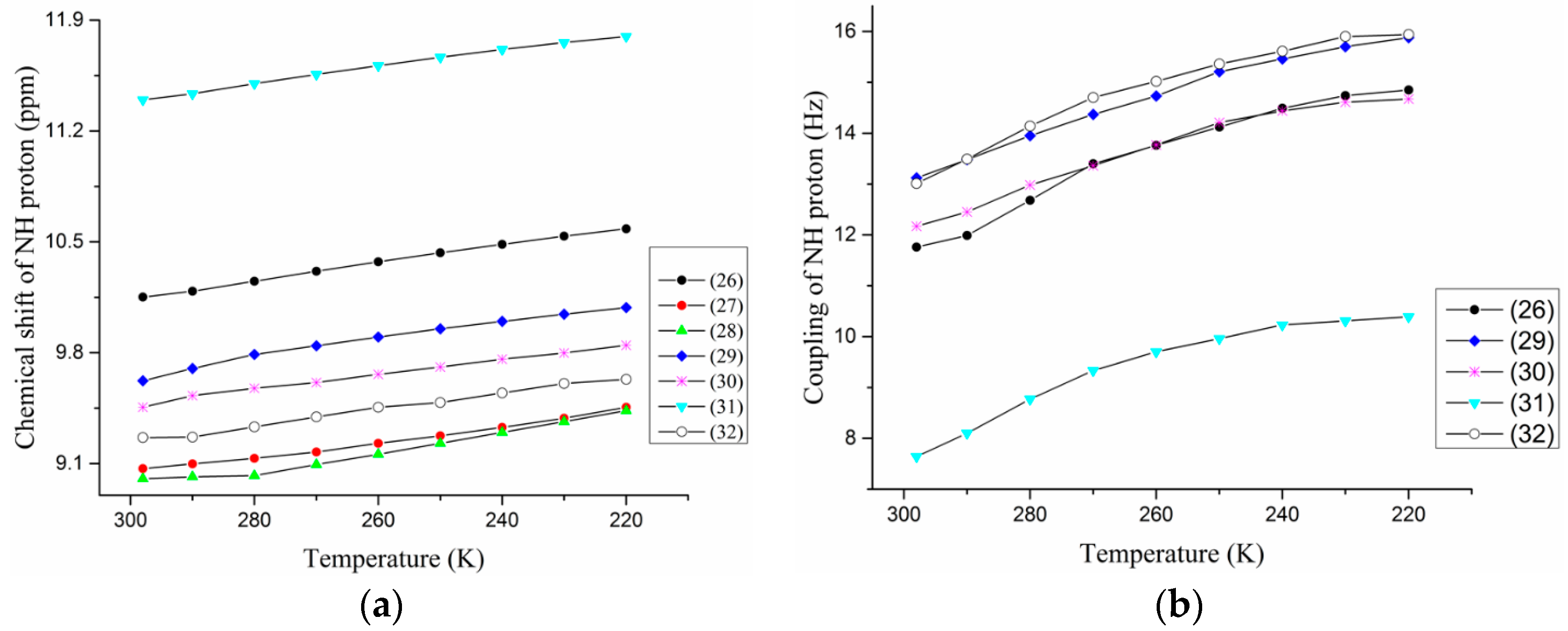
3.1. NMR Experimental Findings
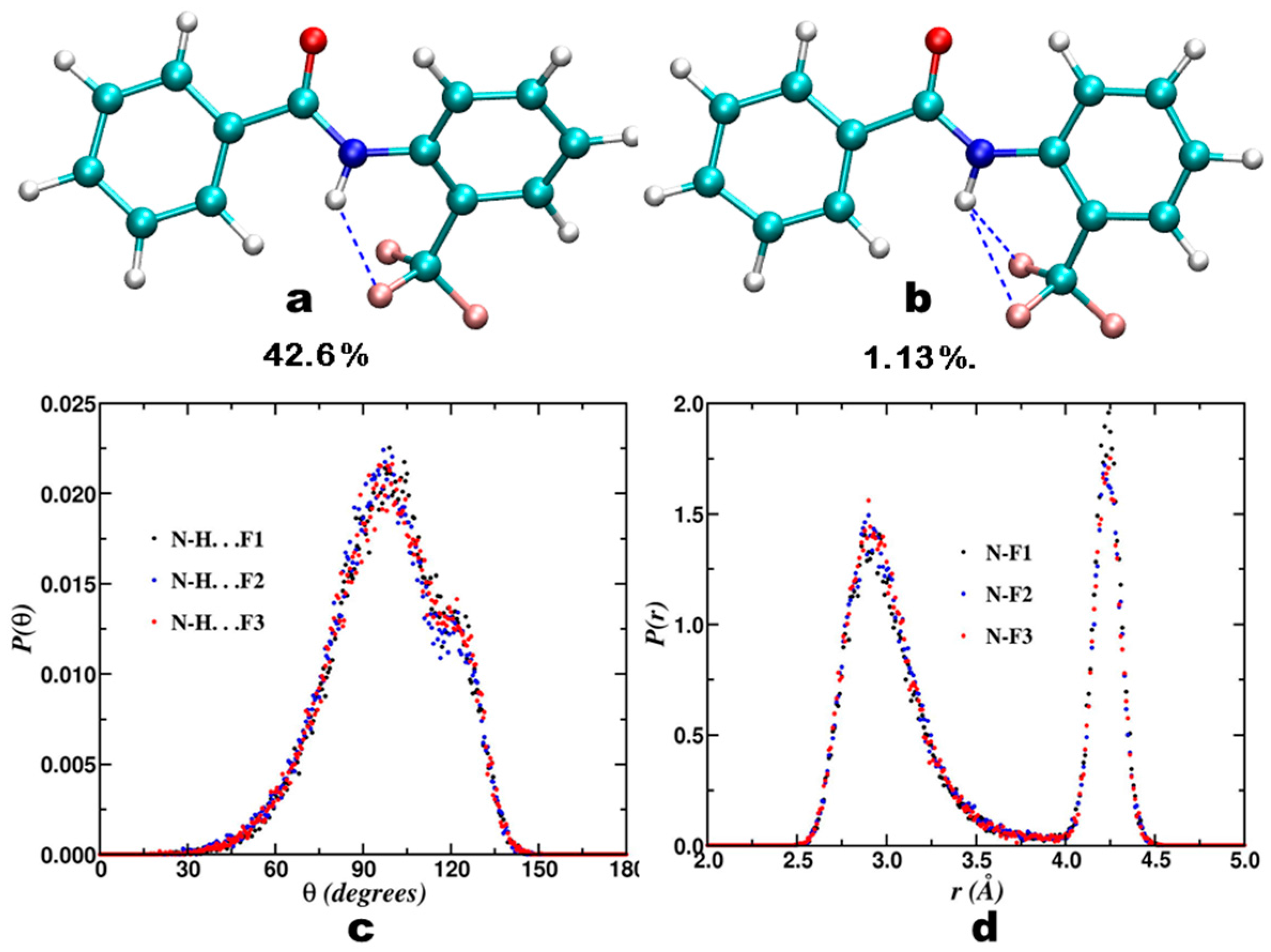
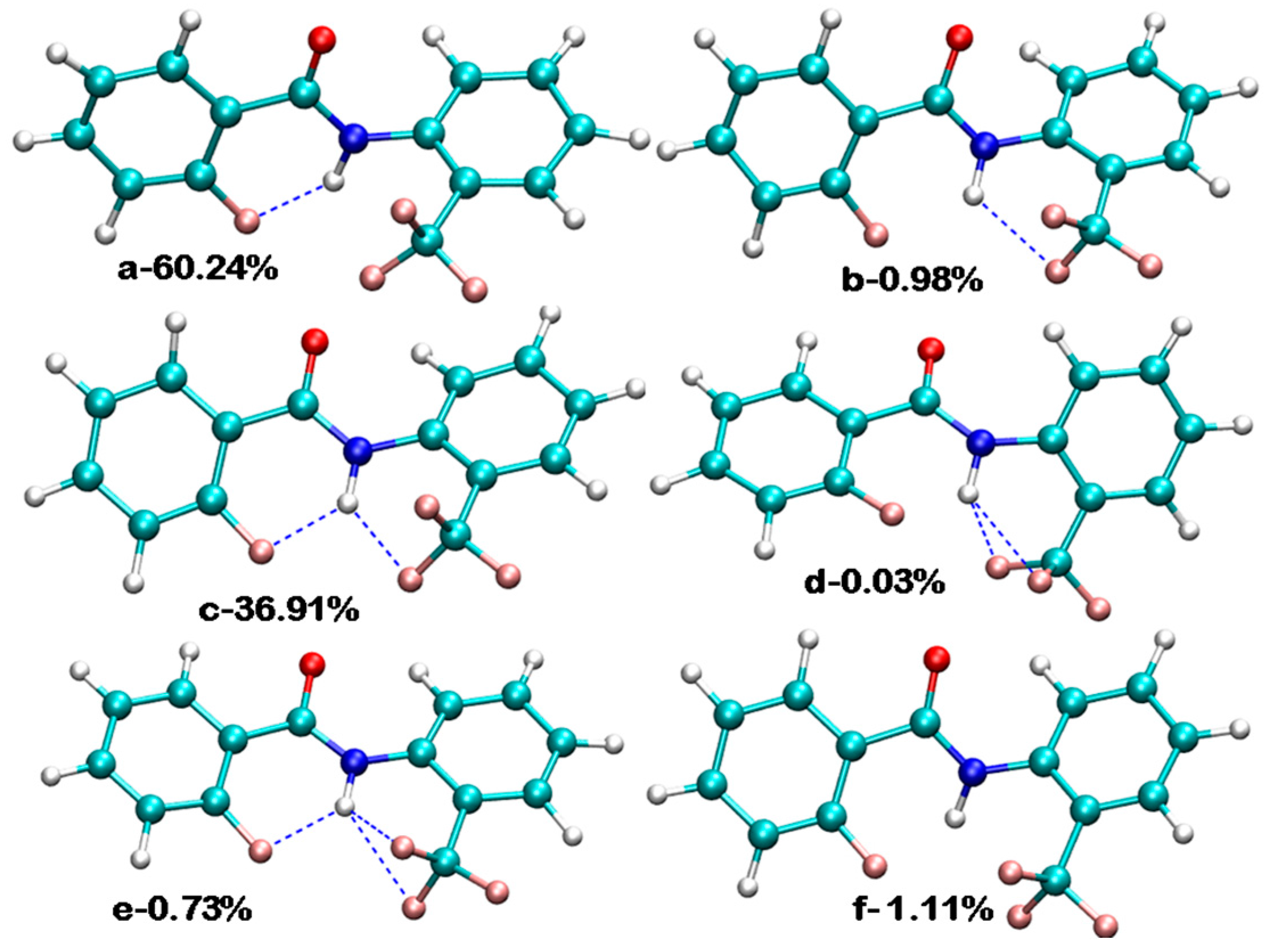
3.2. Theoretical Calculations
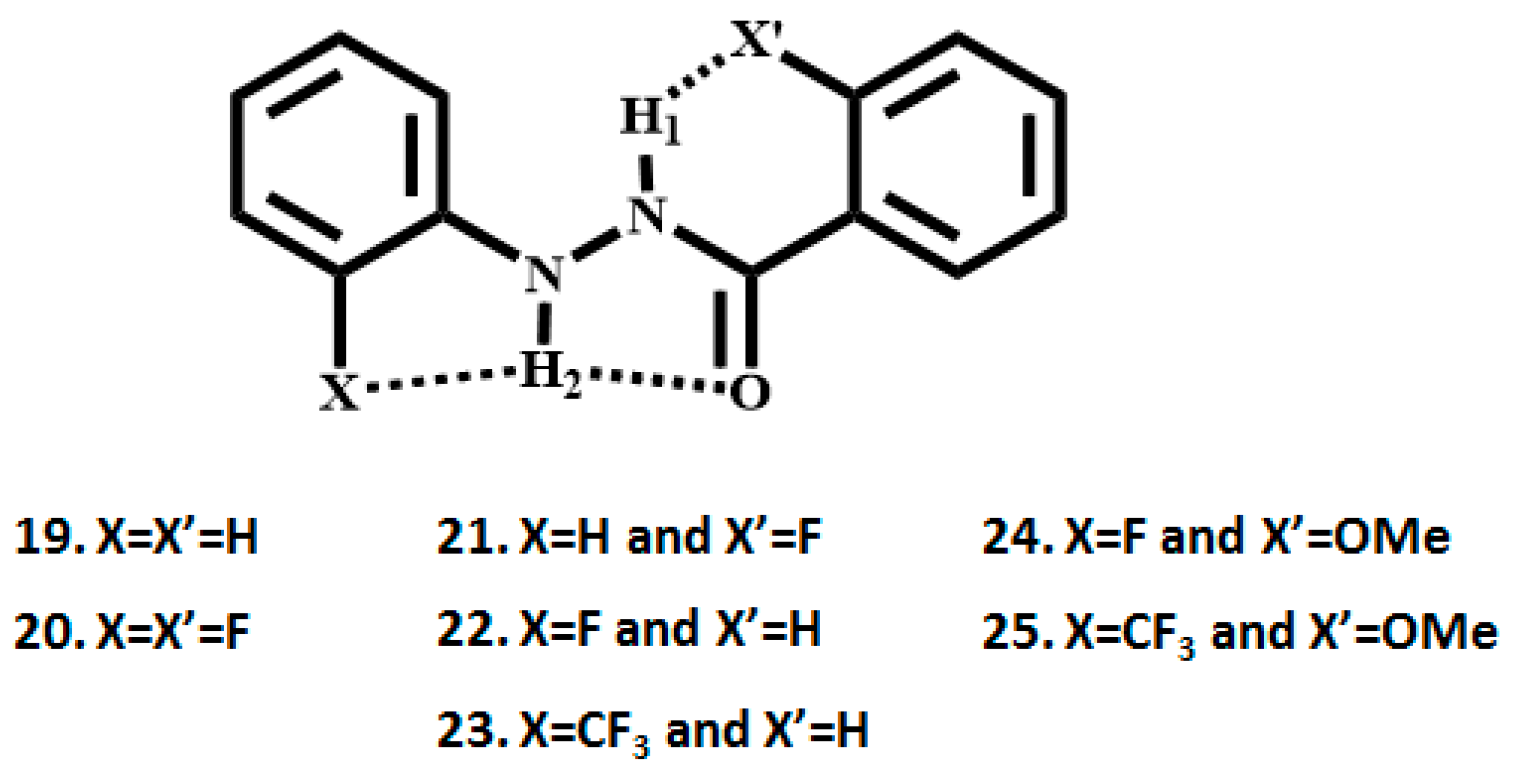
4. Studies on Hydrazides
4.1. N,N-Diacyl Substituted Derivatives of Hydrazides
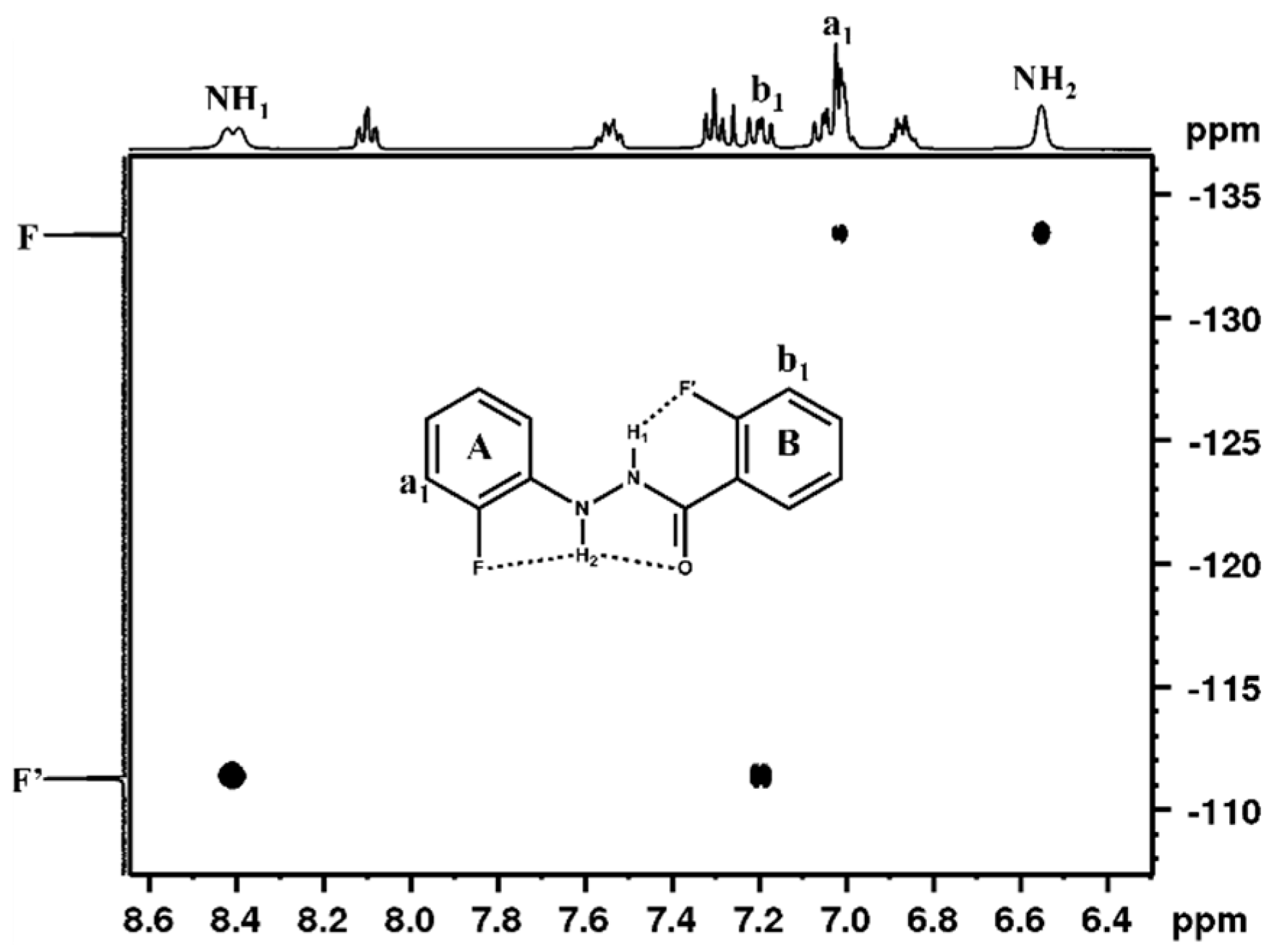
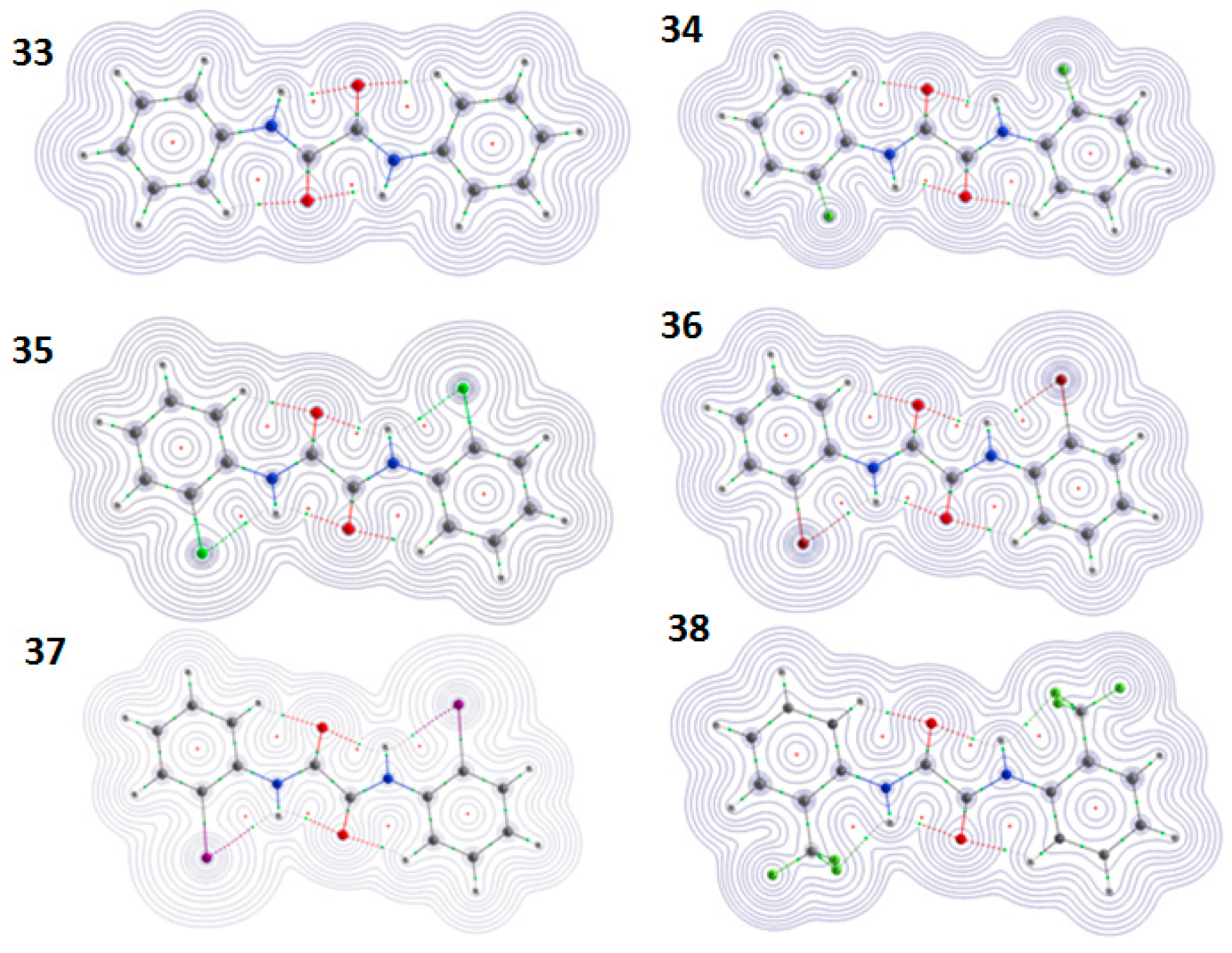
4.1.1. NMR Spectroscopic Detection
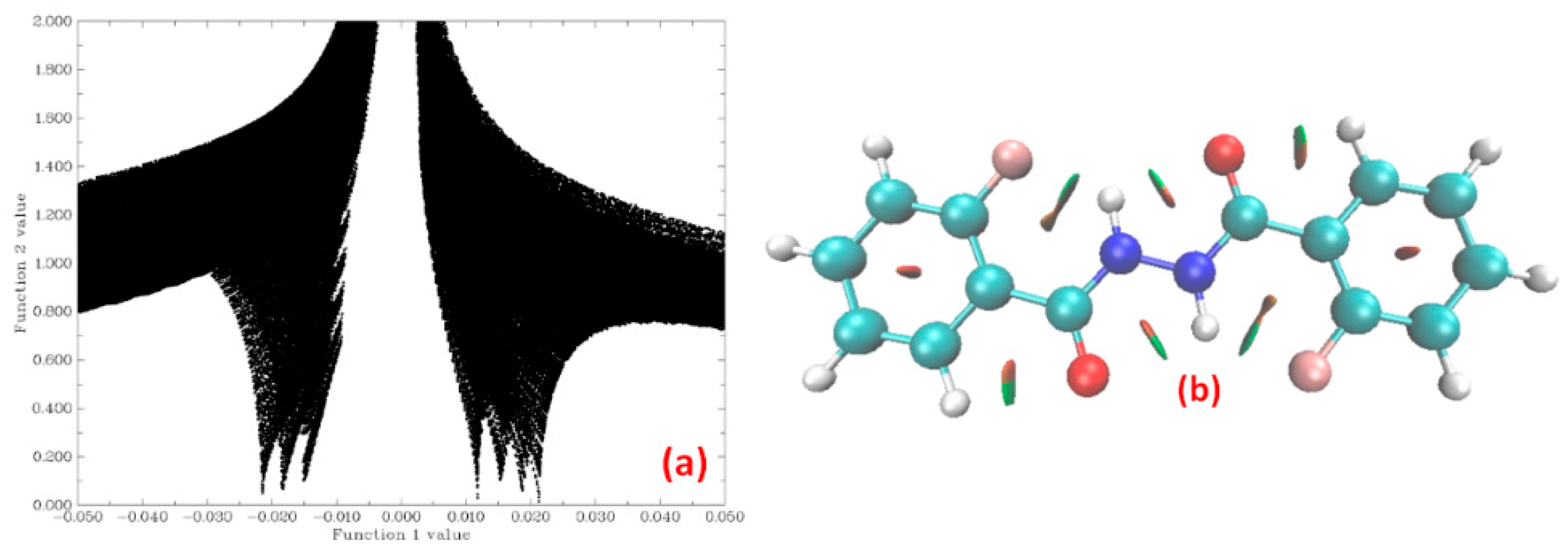
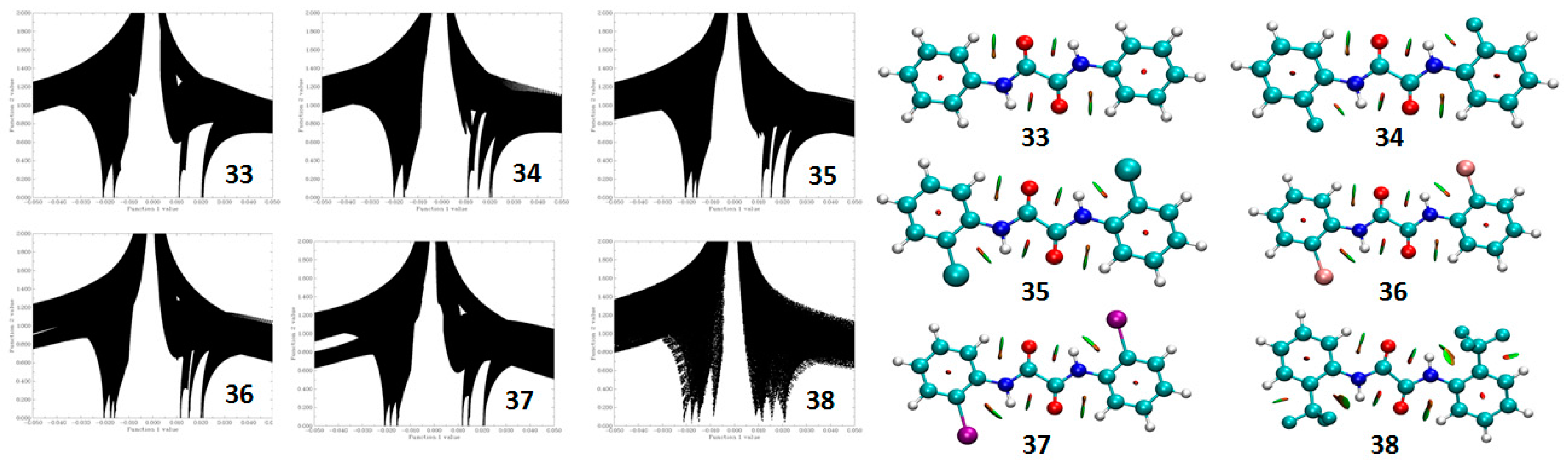
4.1.2. NCI Plot
4.1.3. Atoms in Molecules (AIM) Calculations
4.2. N, N Acyl-Phenyl Substituted Derivatives of Hydrazides
4.2.1. Spectroscopic Experimental Observations
4.2.2. Theoretical Calculations
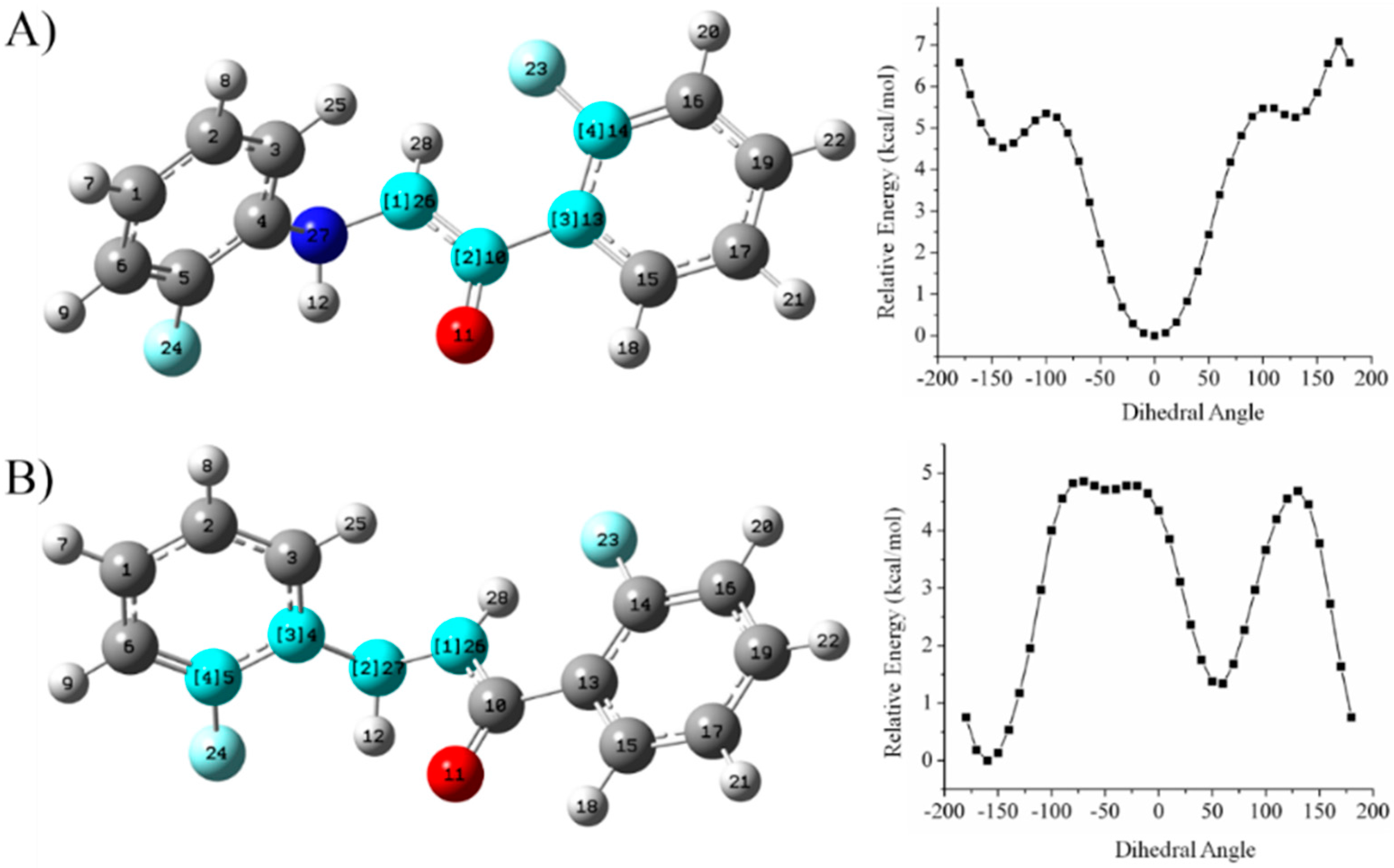
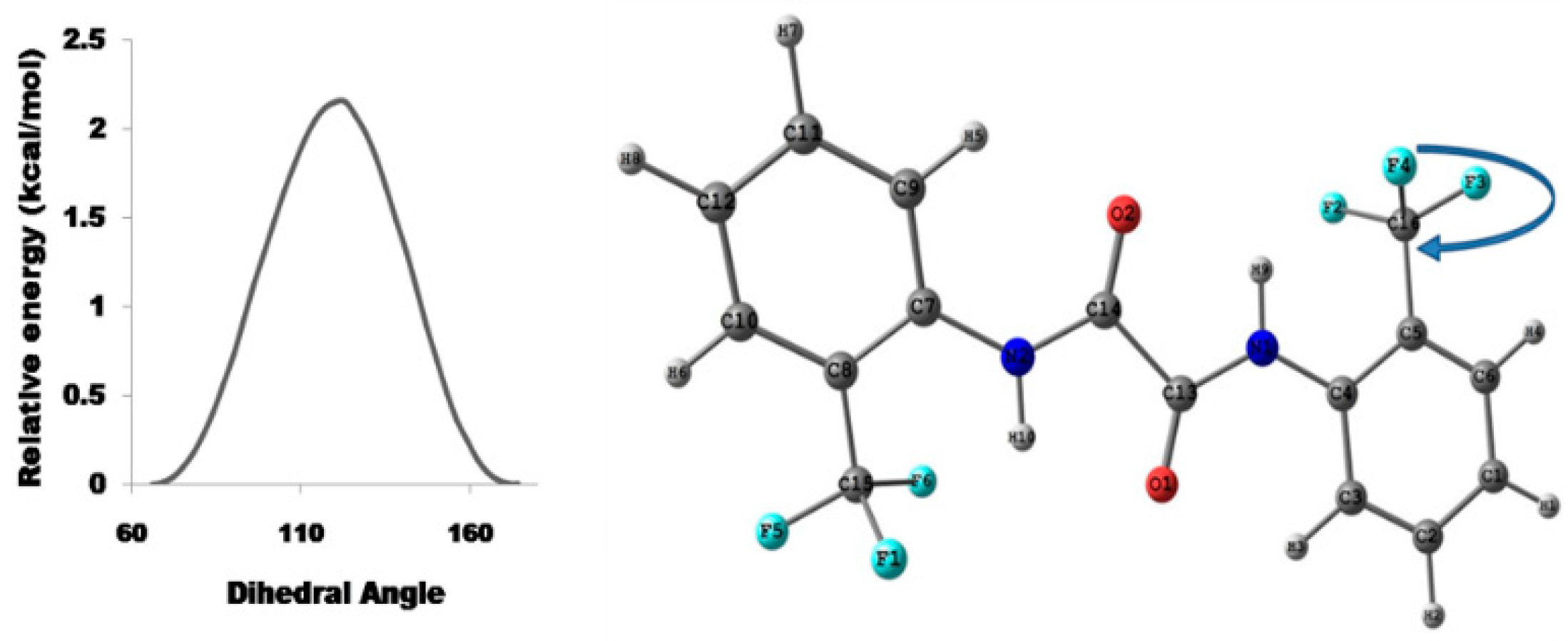
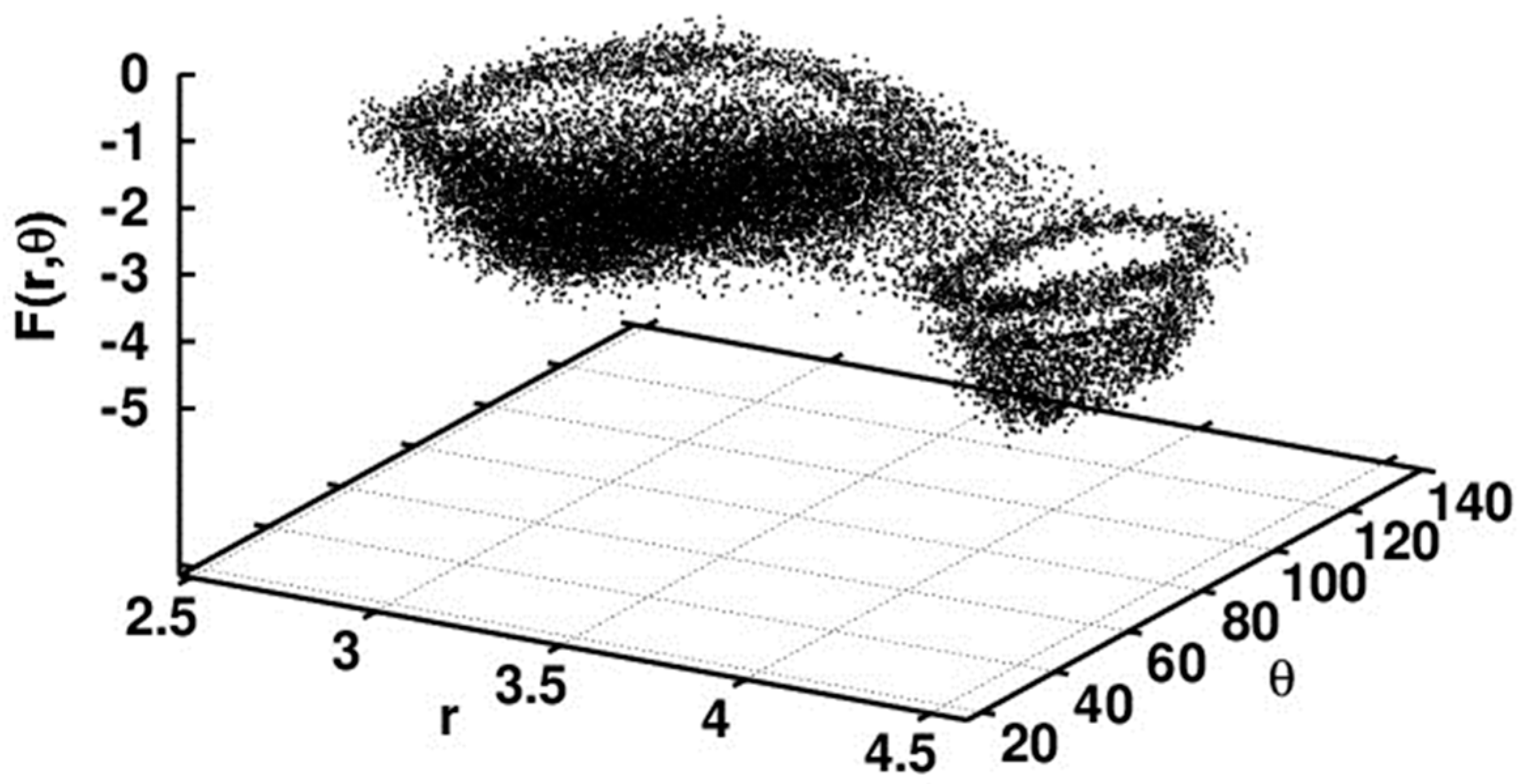
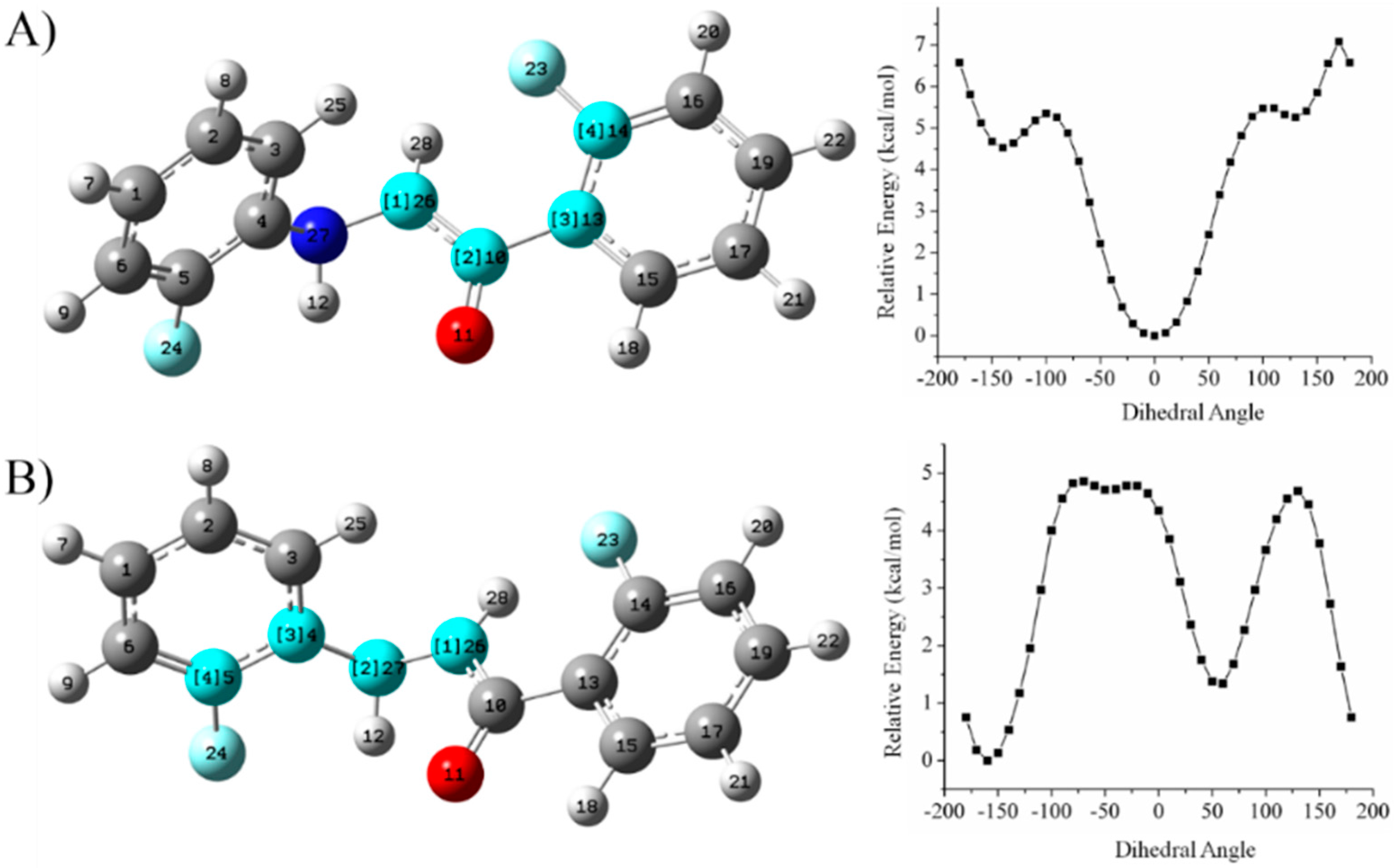
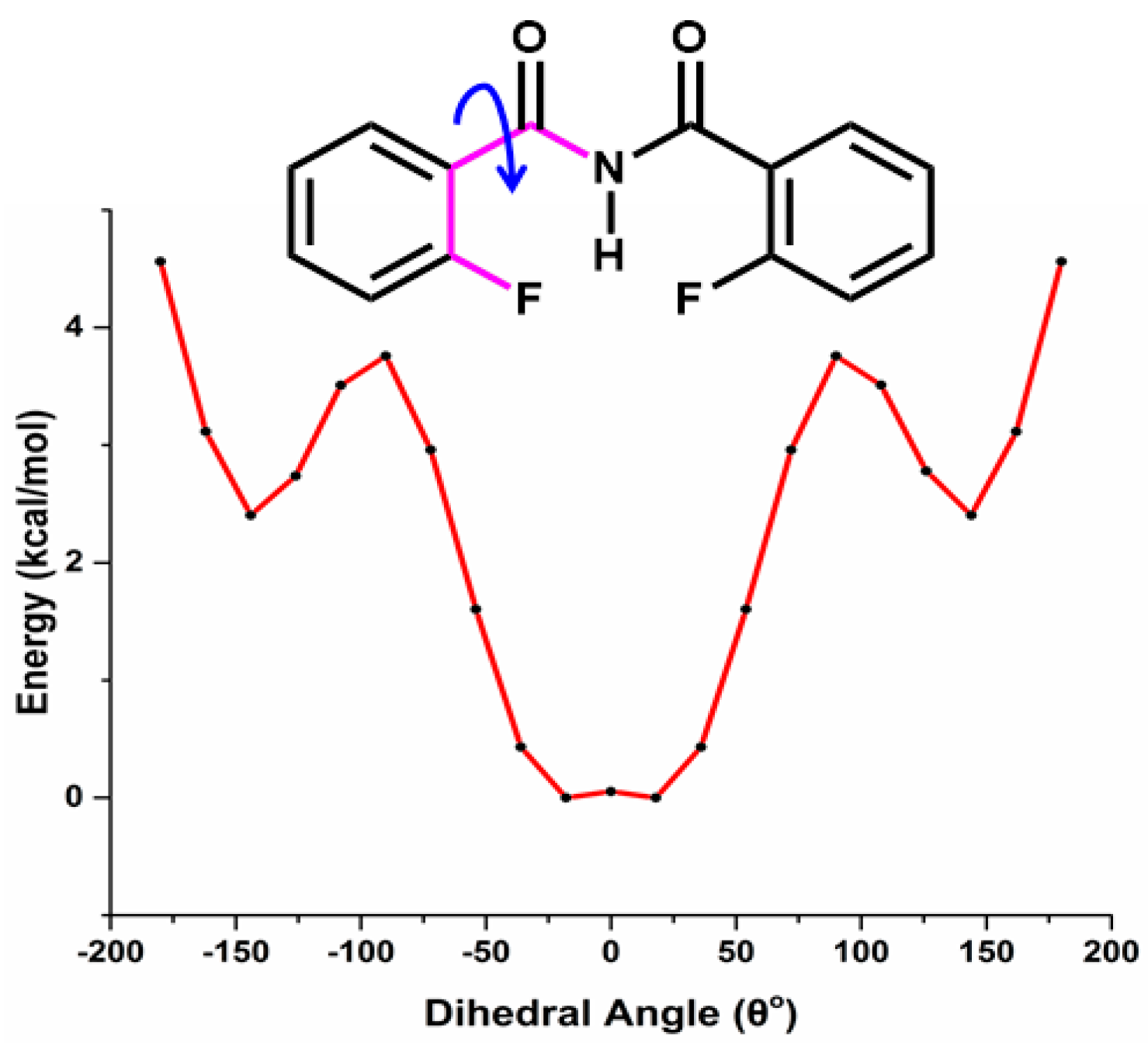
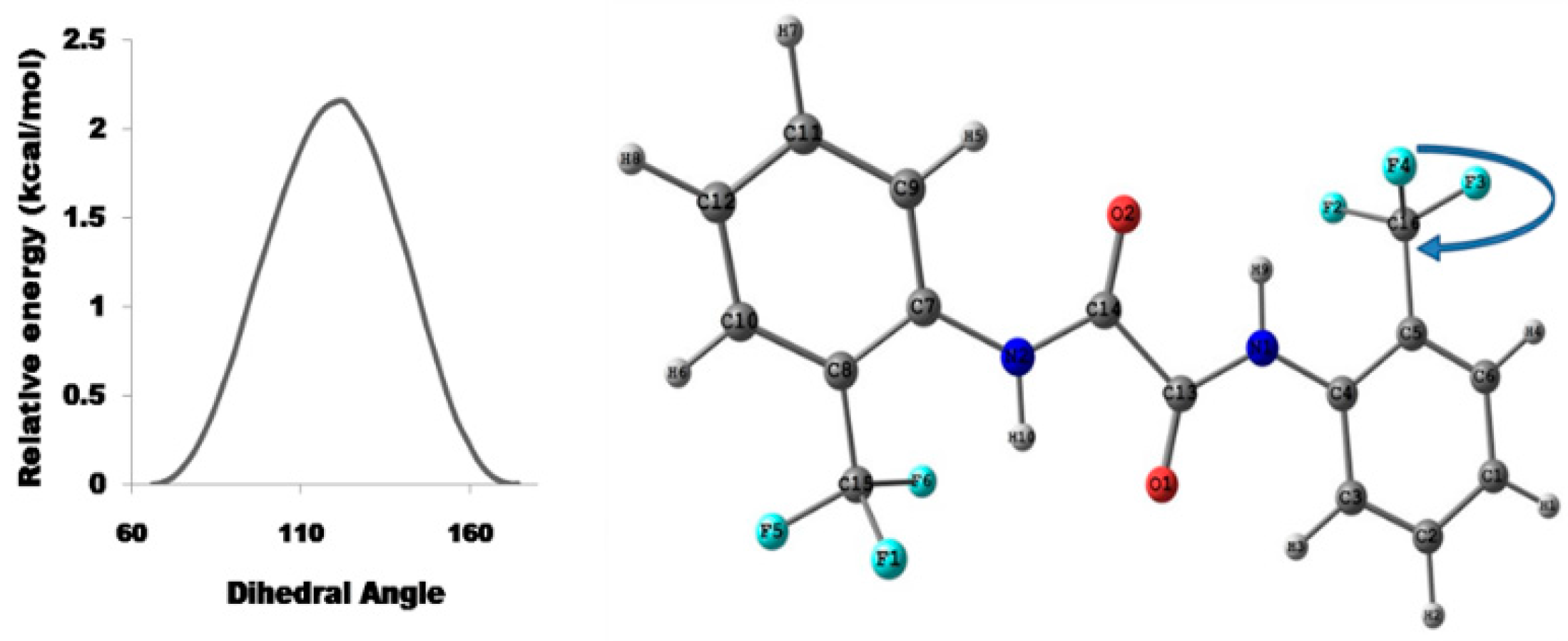
Relaxed Potential Energy Scan
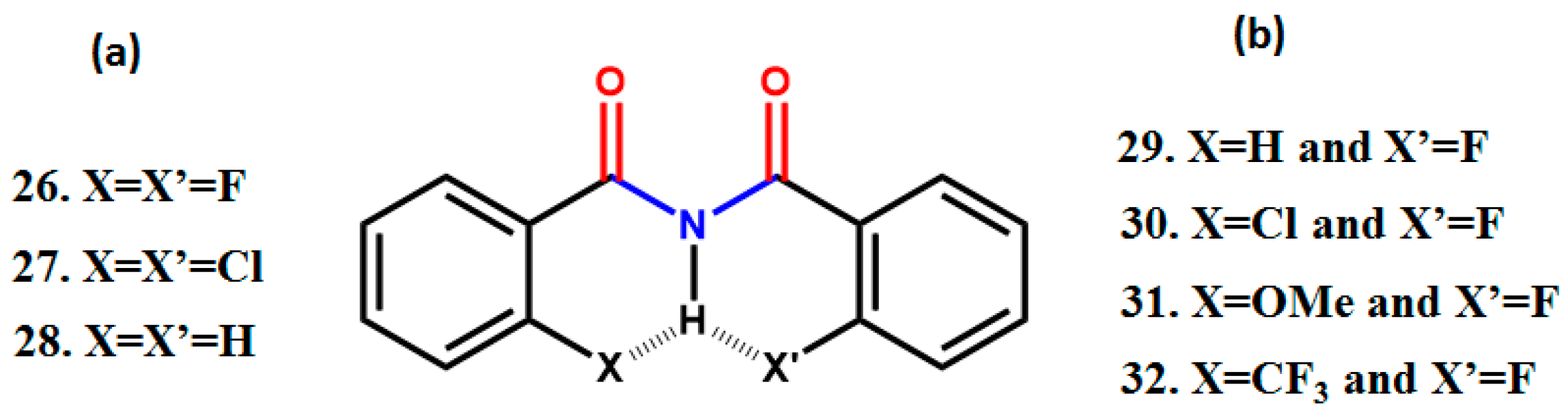
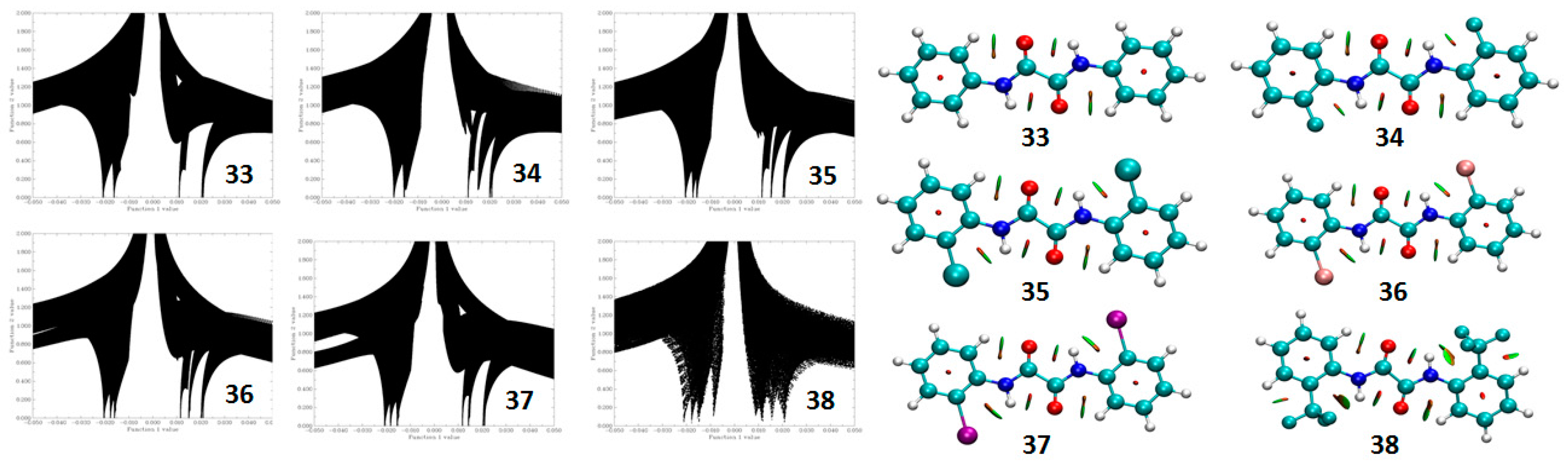
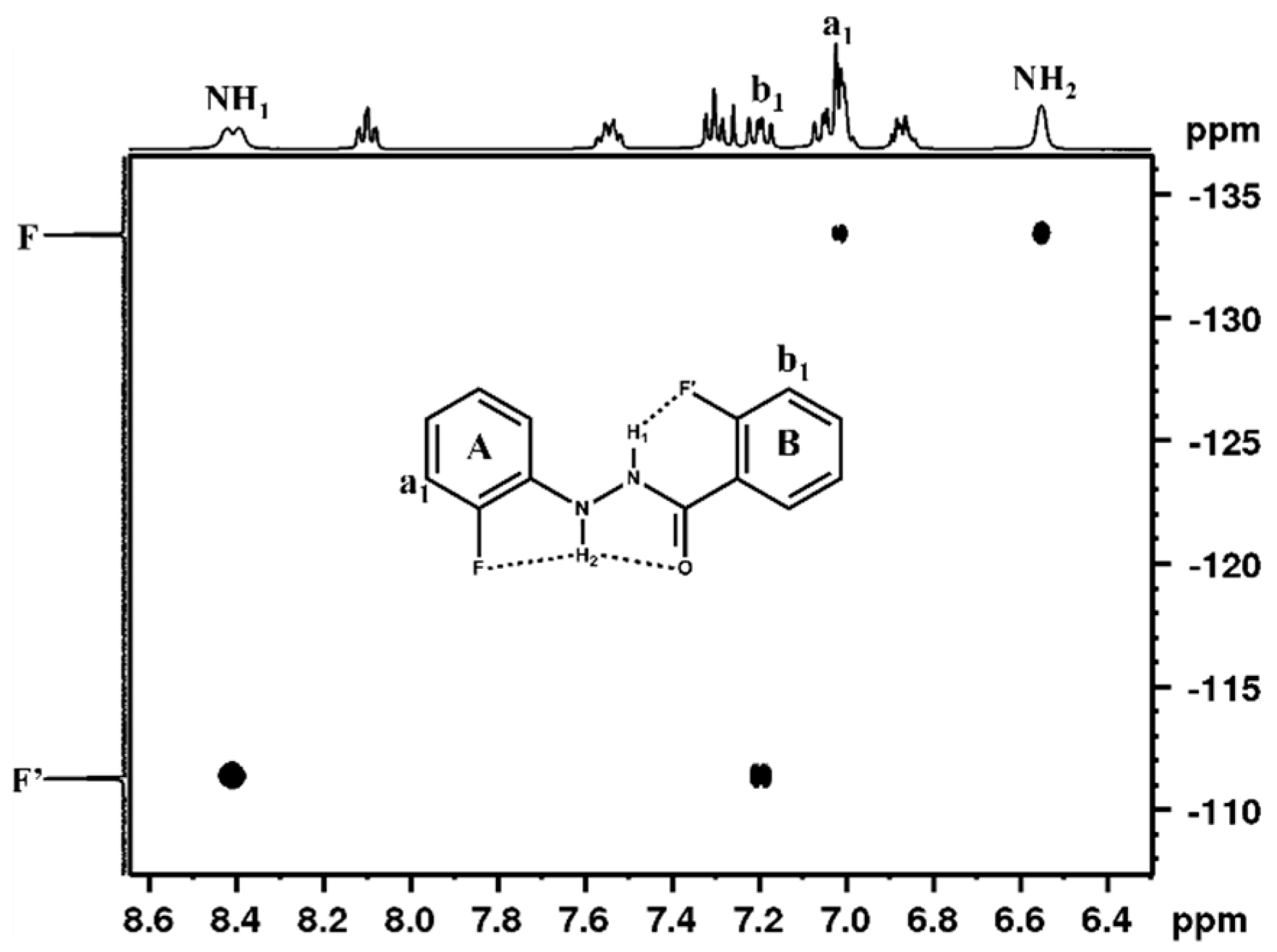
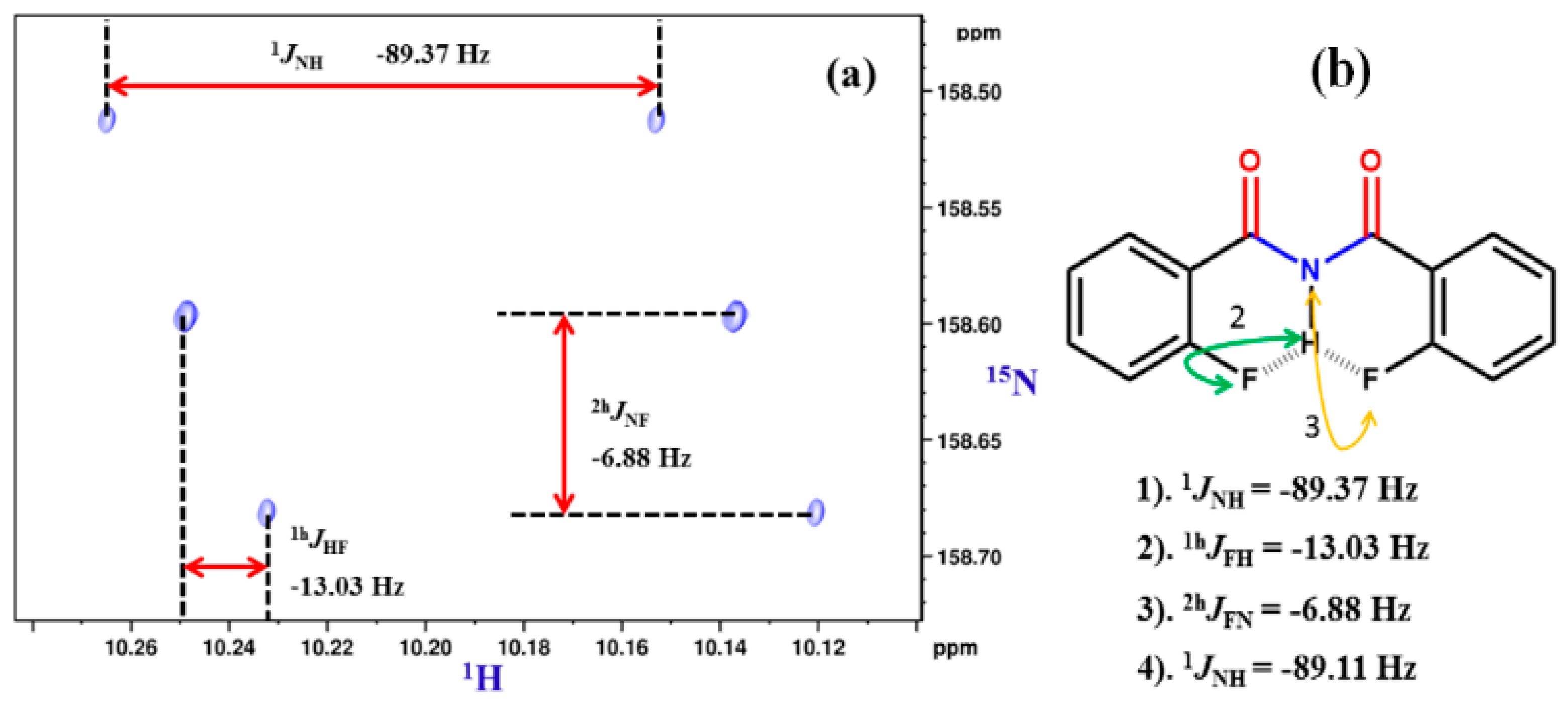
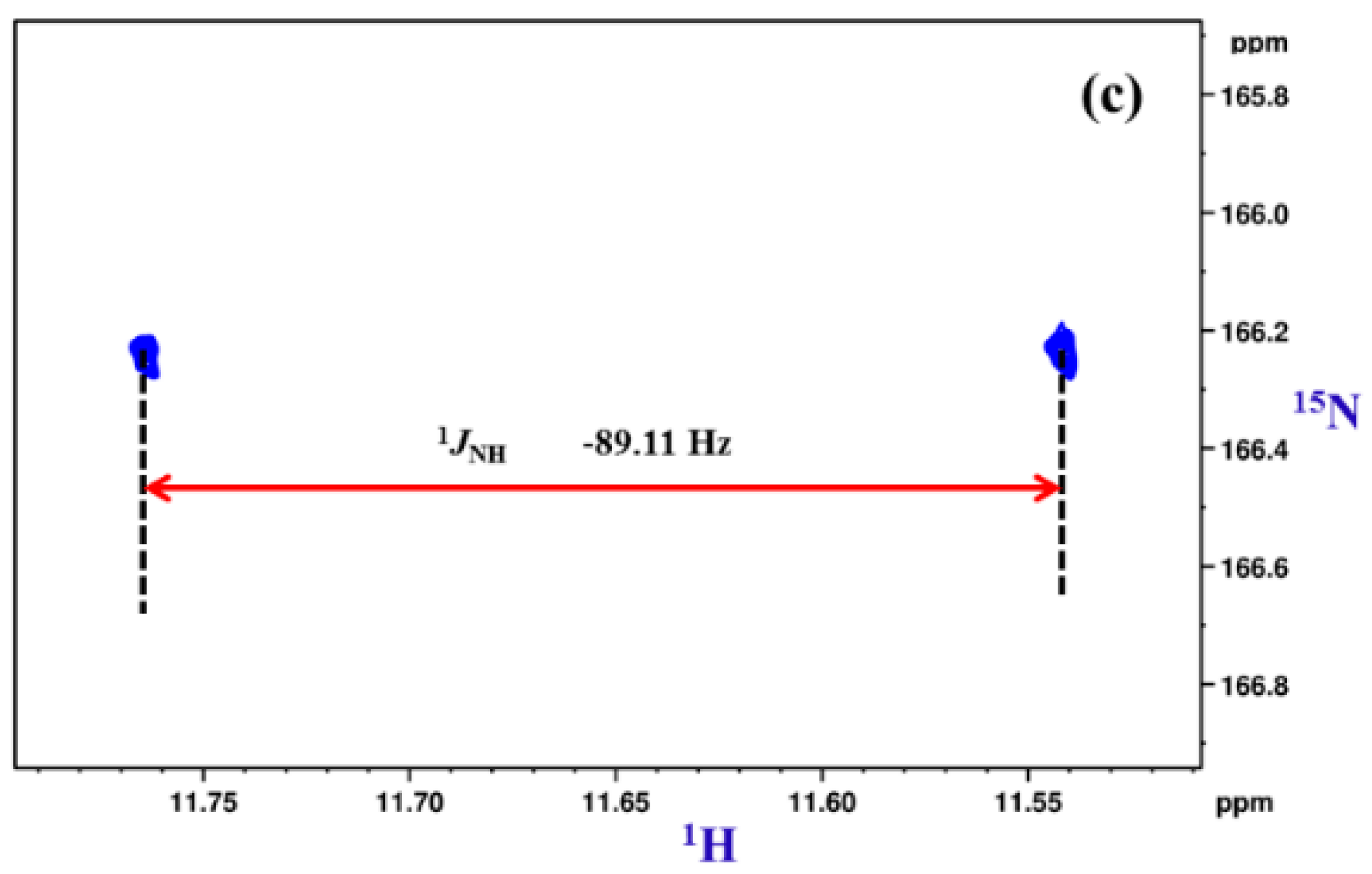
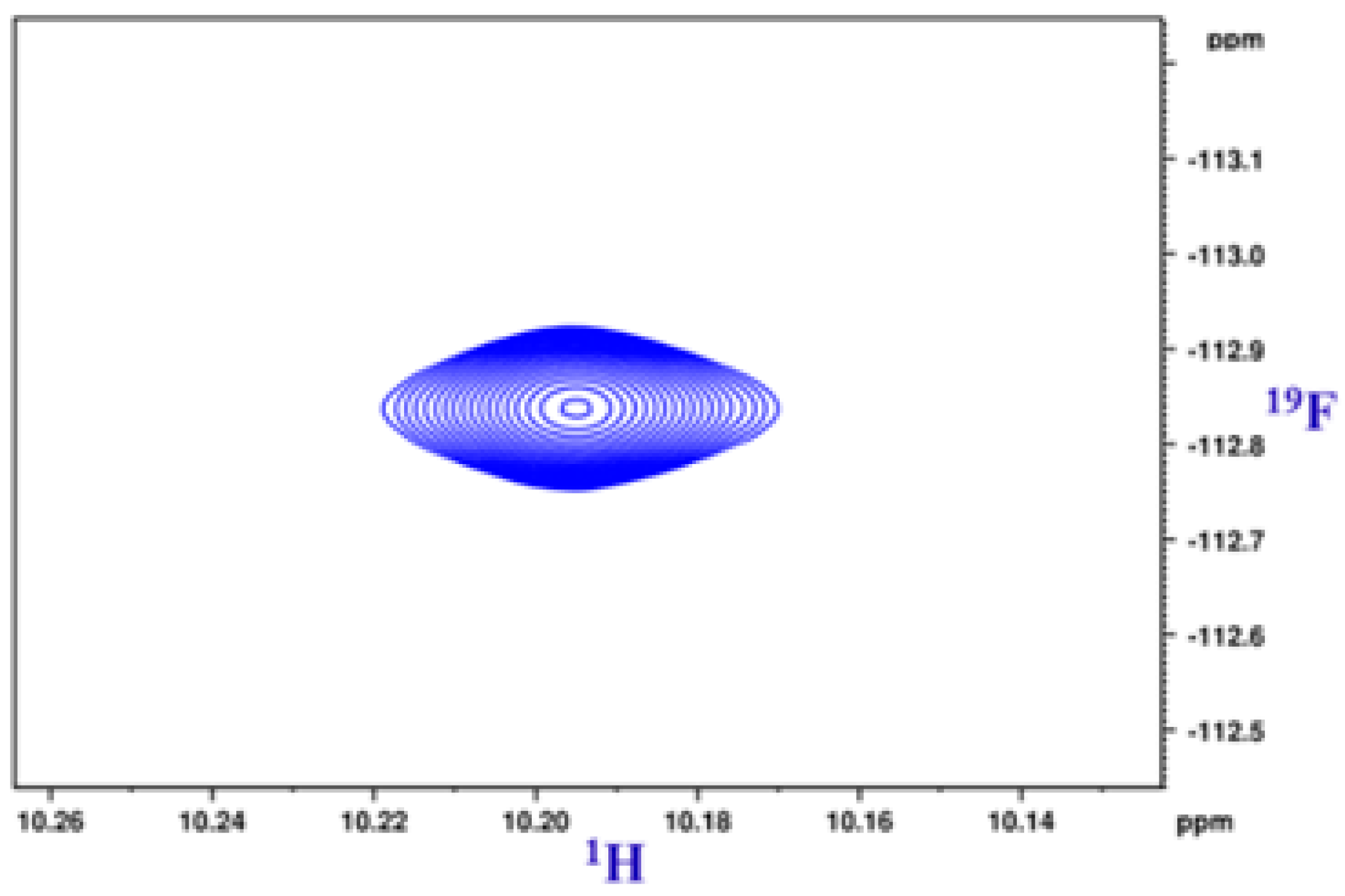
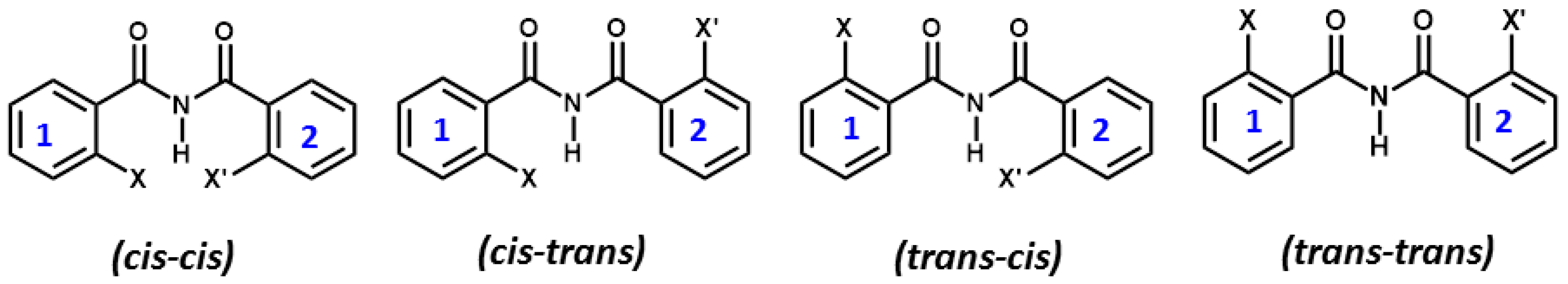
5. Studies on Derivatives of Imides
5.1. Information Derived by NMR Spectroscopy
5.2. Theoretical Calculations
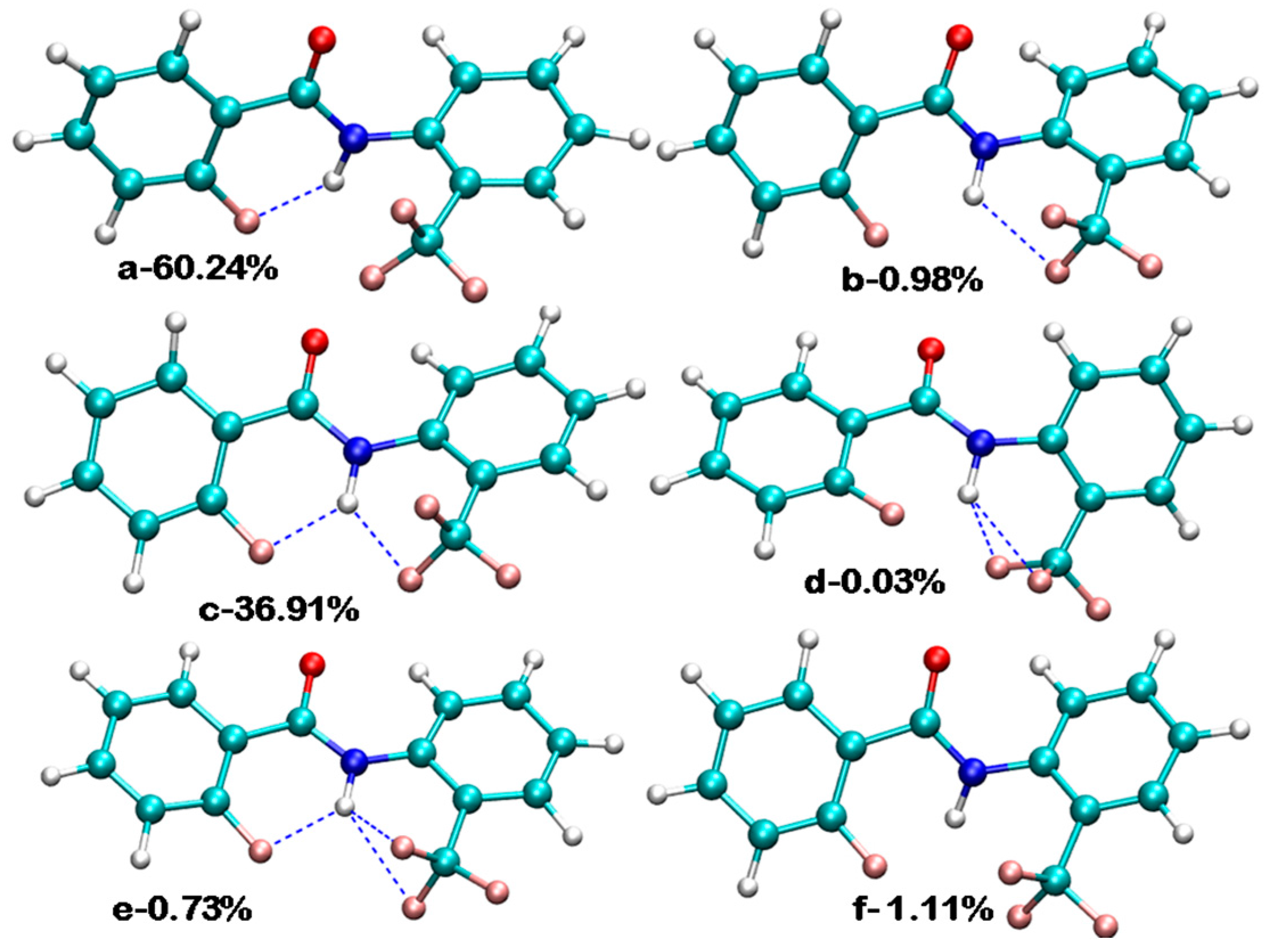
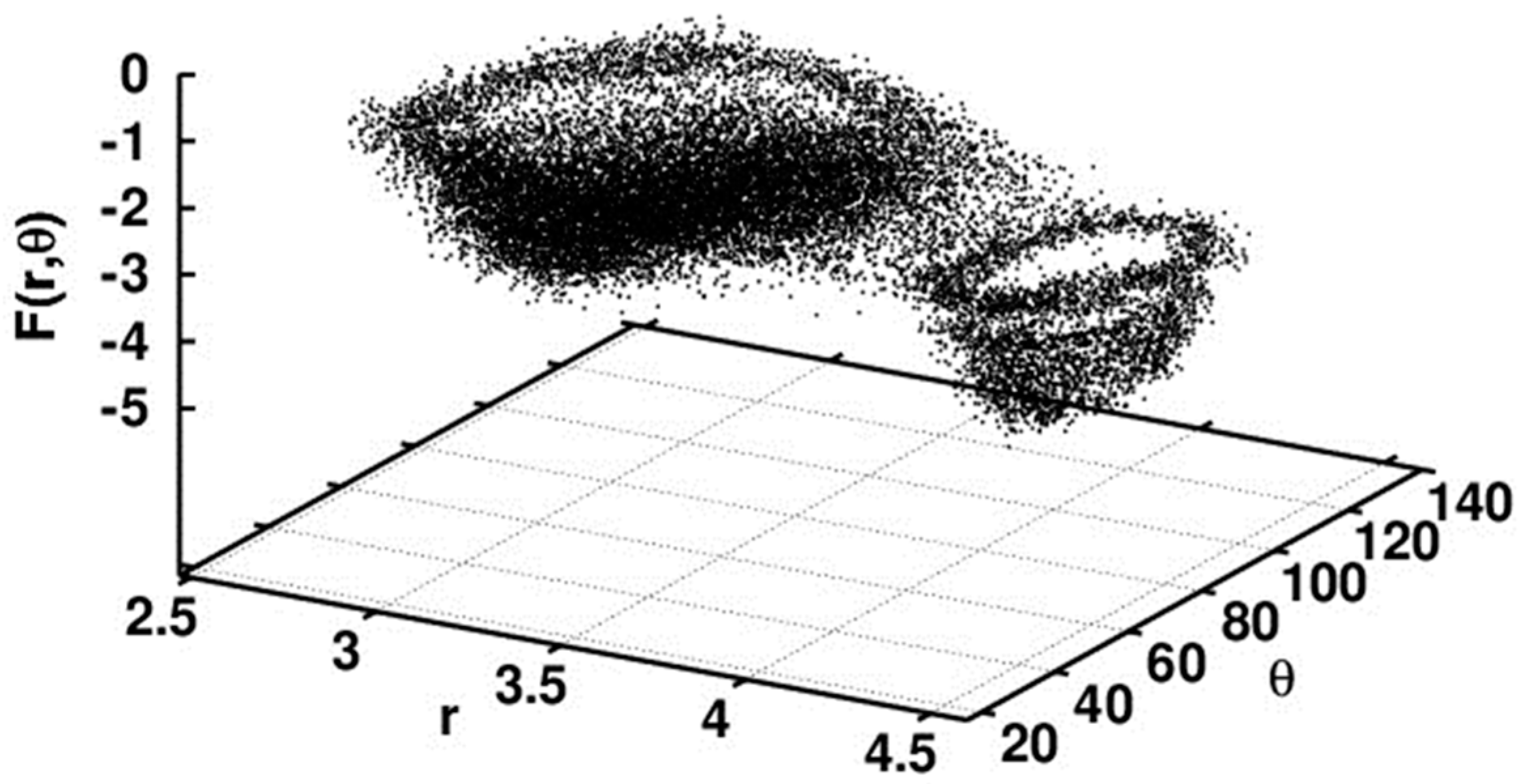
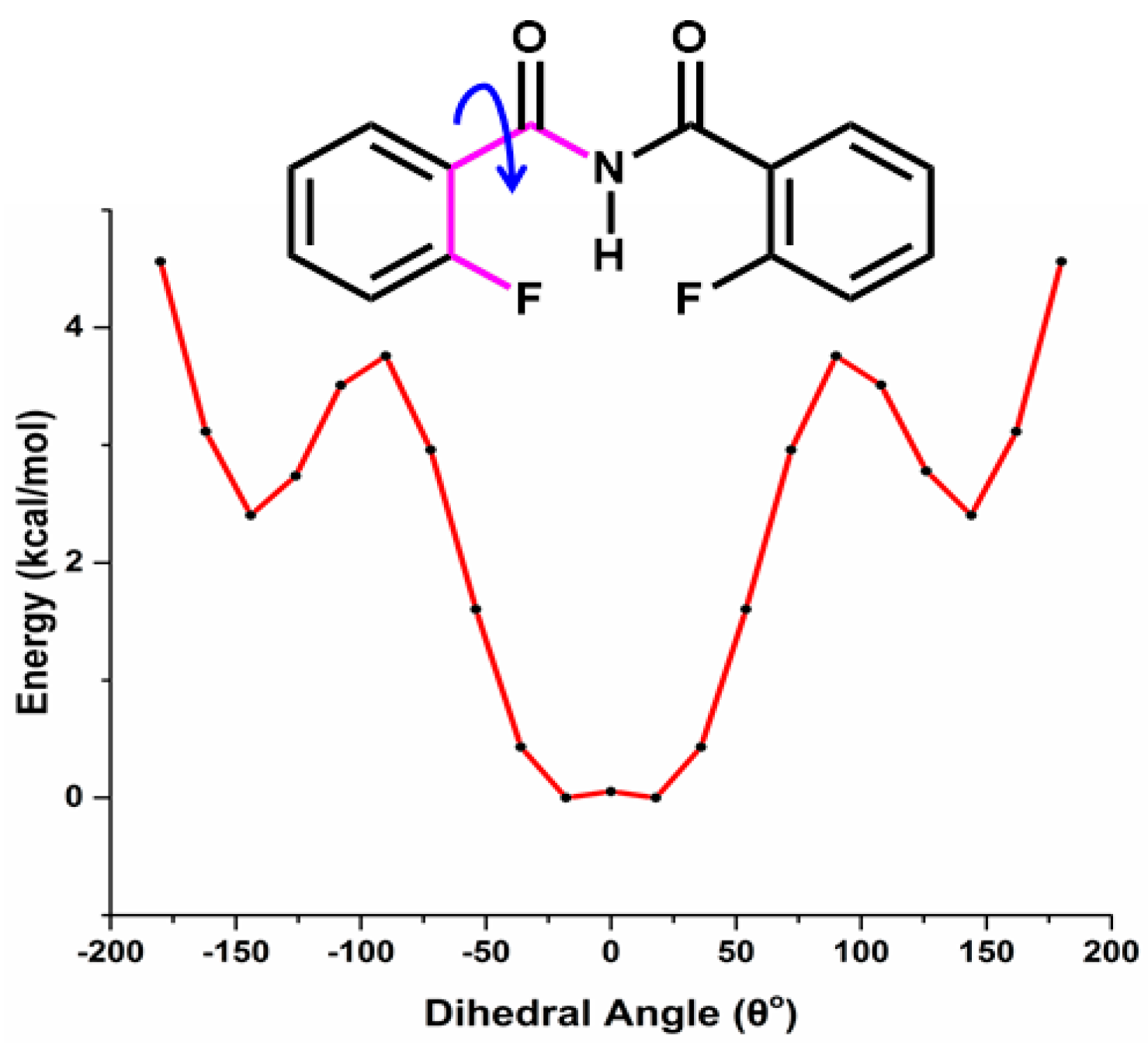
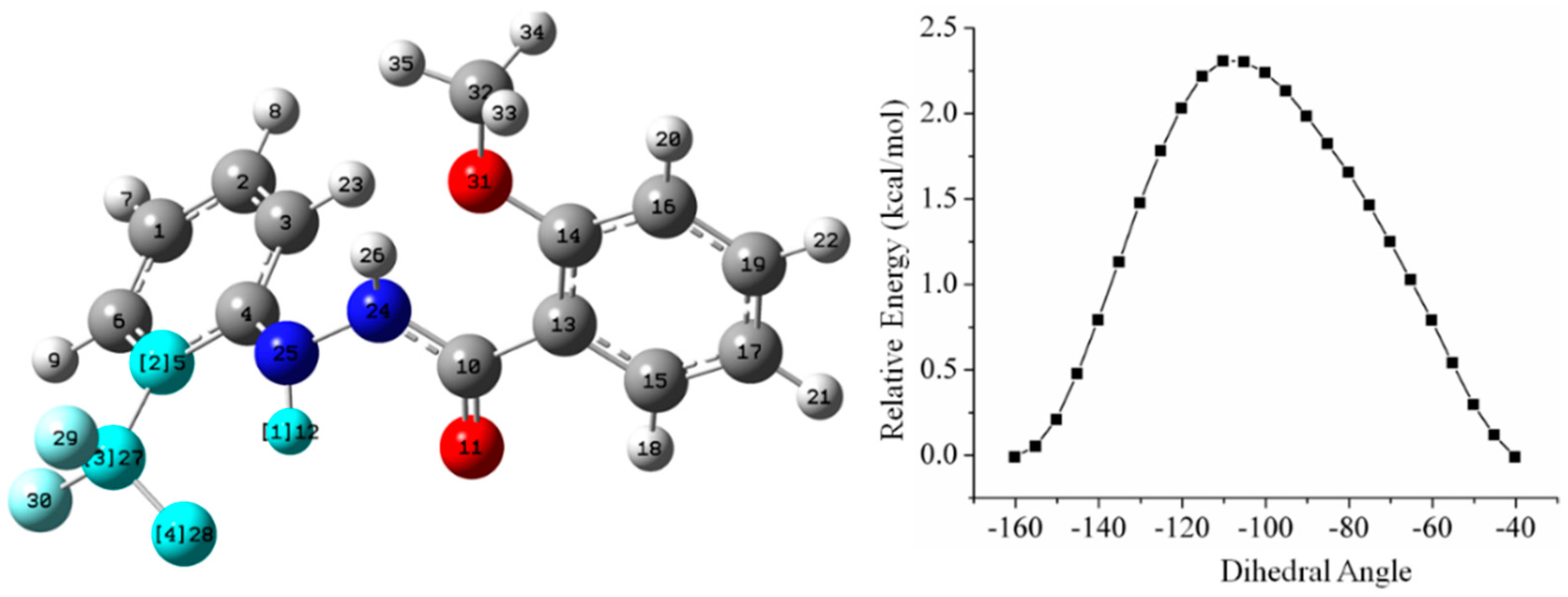
5.2.1. Conformational Study
Relaxed Potential Energy Scan
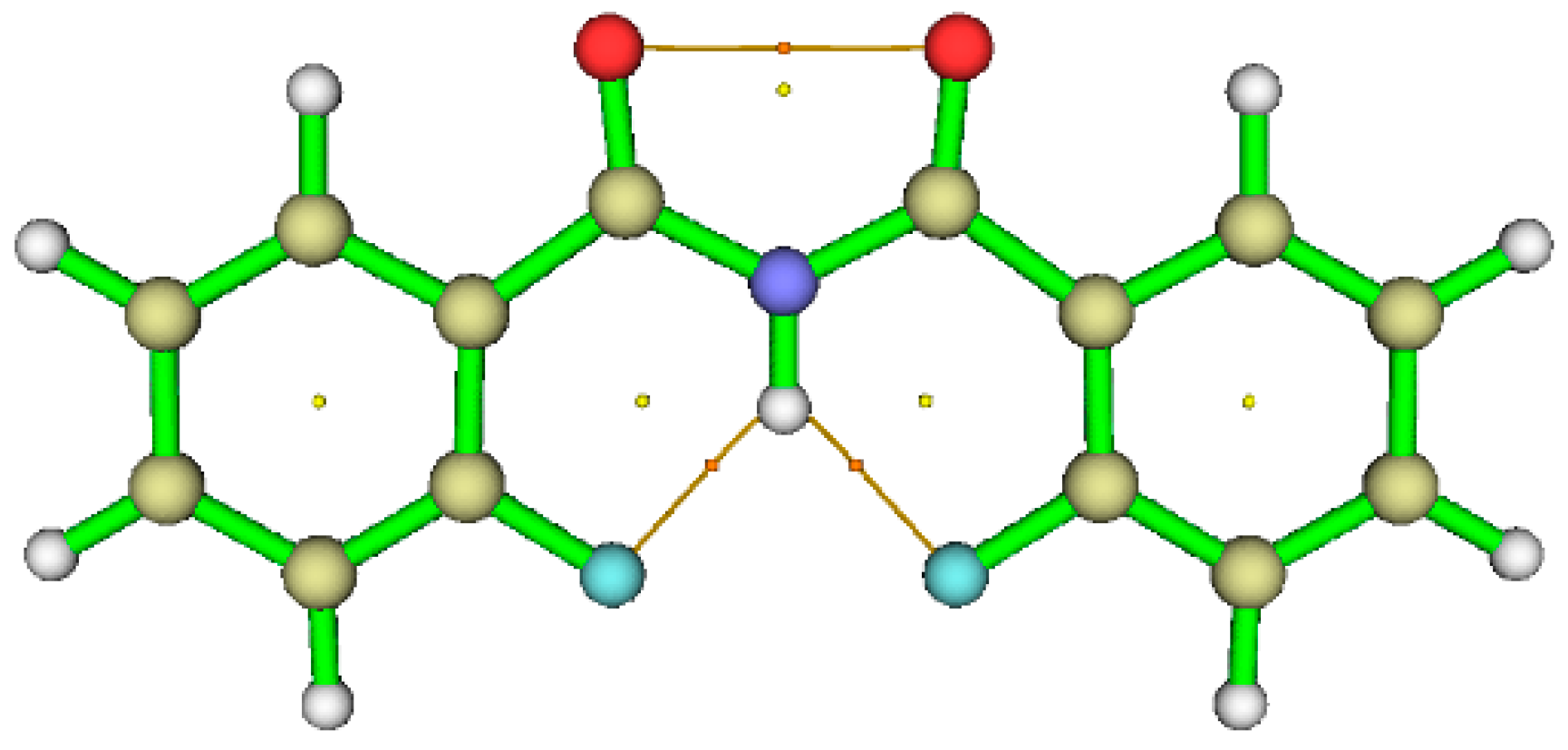
5.2.2. Non Covalent Interaction (NCI) Calculations
5.2.3. Atoms in Molecules (AIM) Calculations
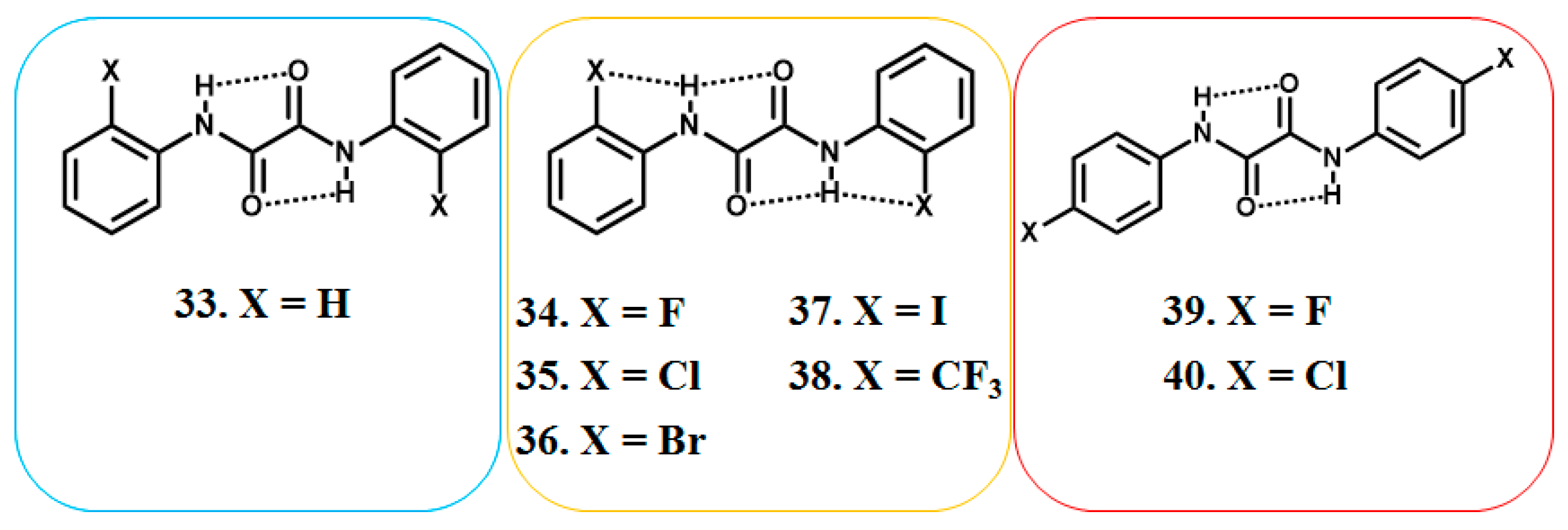
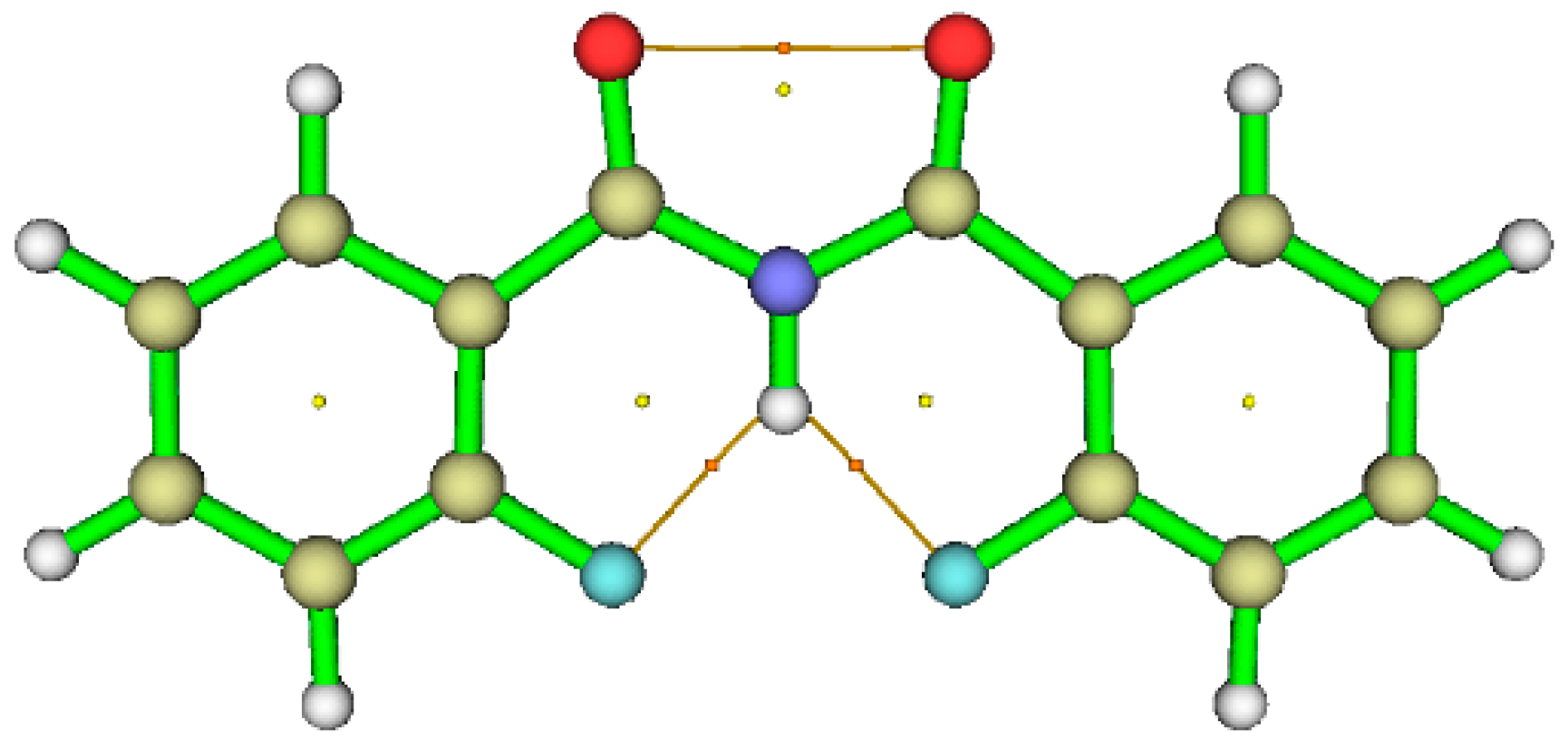
6. Studies on Diphenyloxamides
6.1. Experimental Observation by NMR Spectroscopy
6.2. Theoretical Studies
6.2.1. QTAIM Calculations
6.2.2. NBO Analysis
6.2.3. NCI Analysis
6.2.4. Relaxed Potential Energy Scan
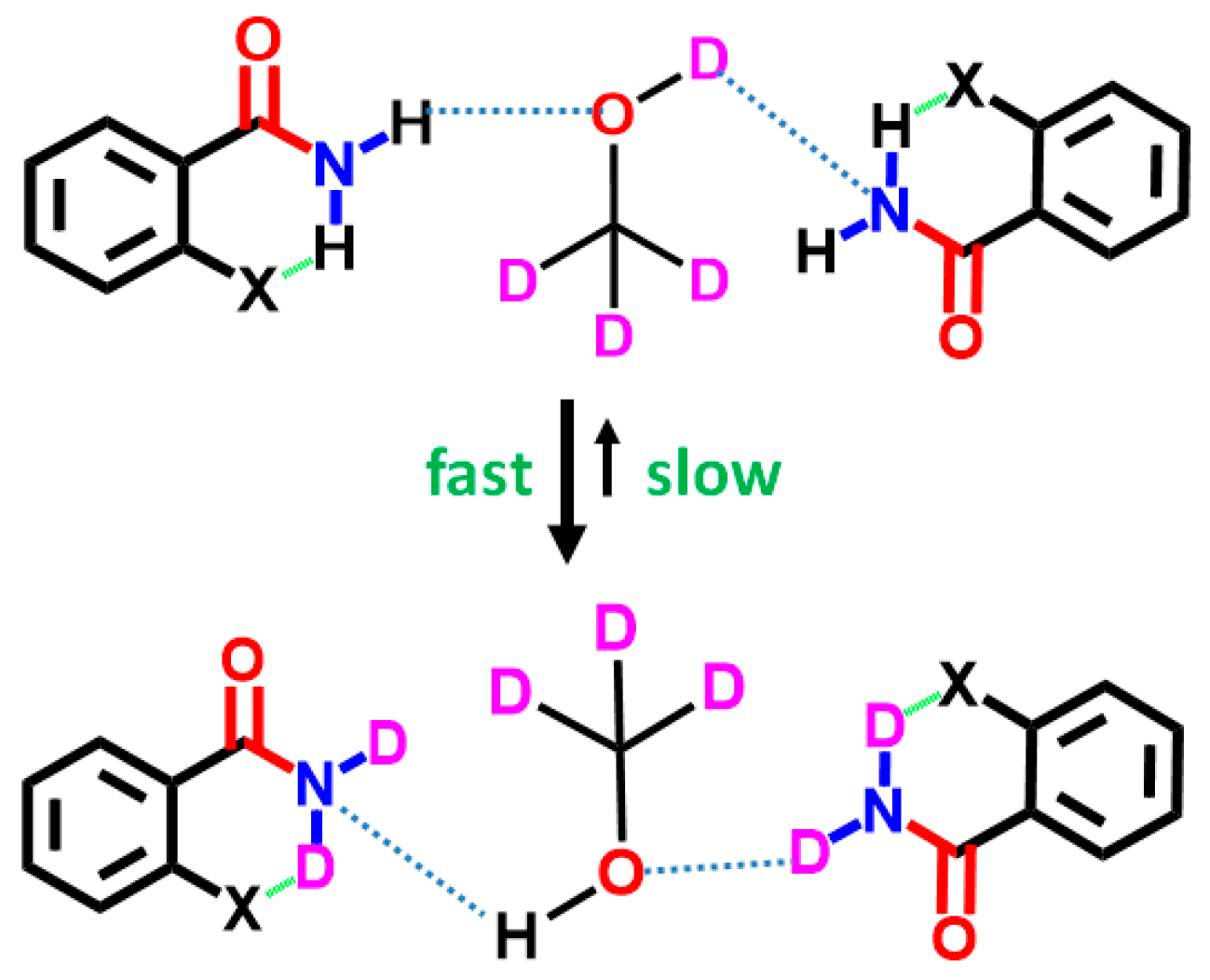
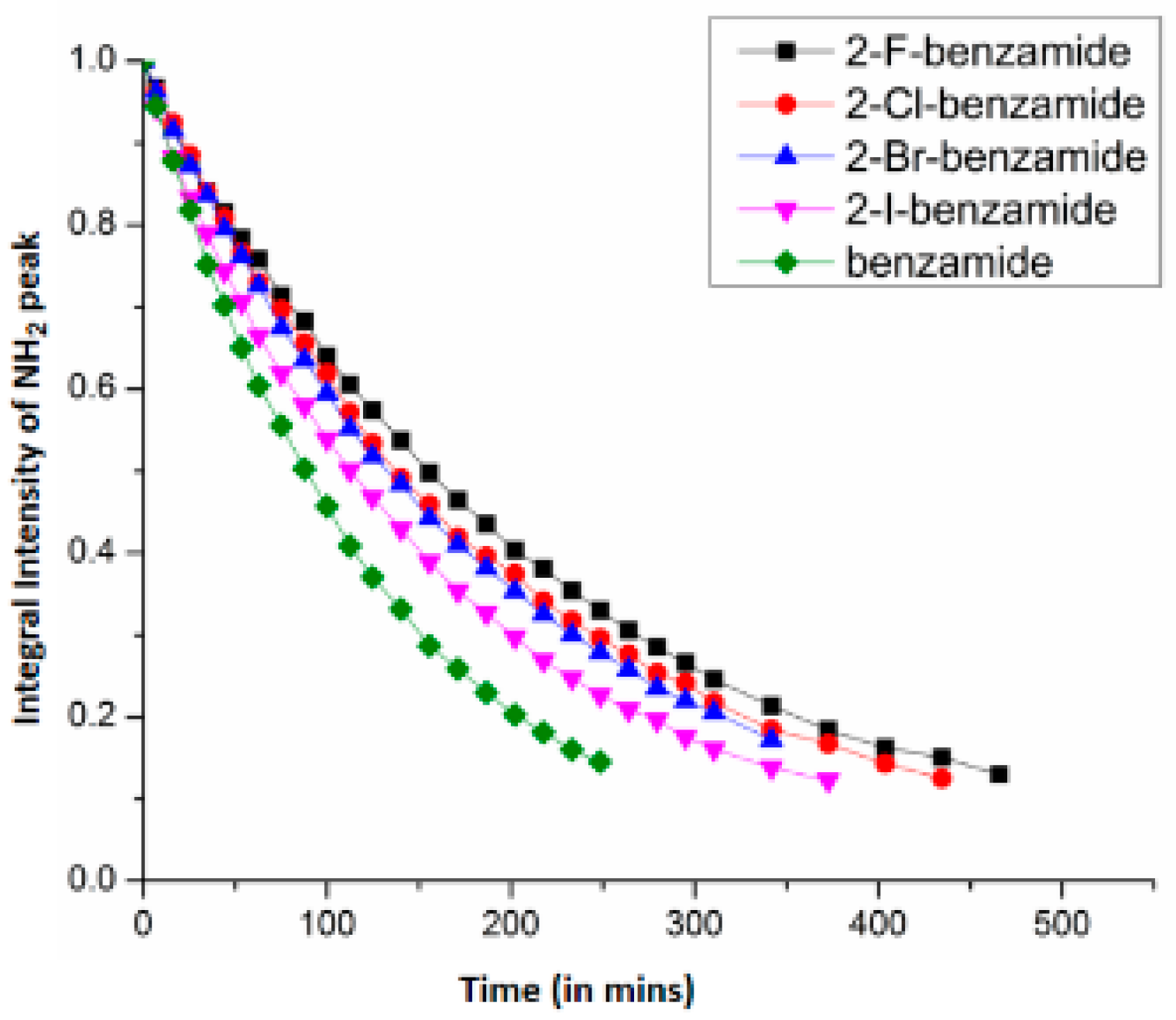
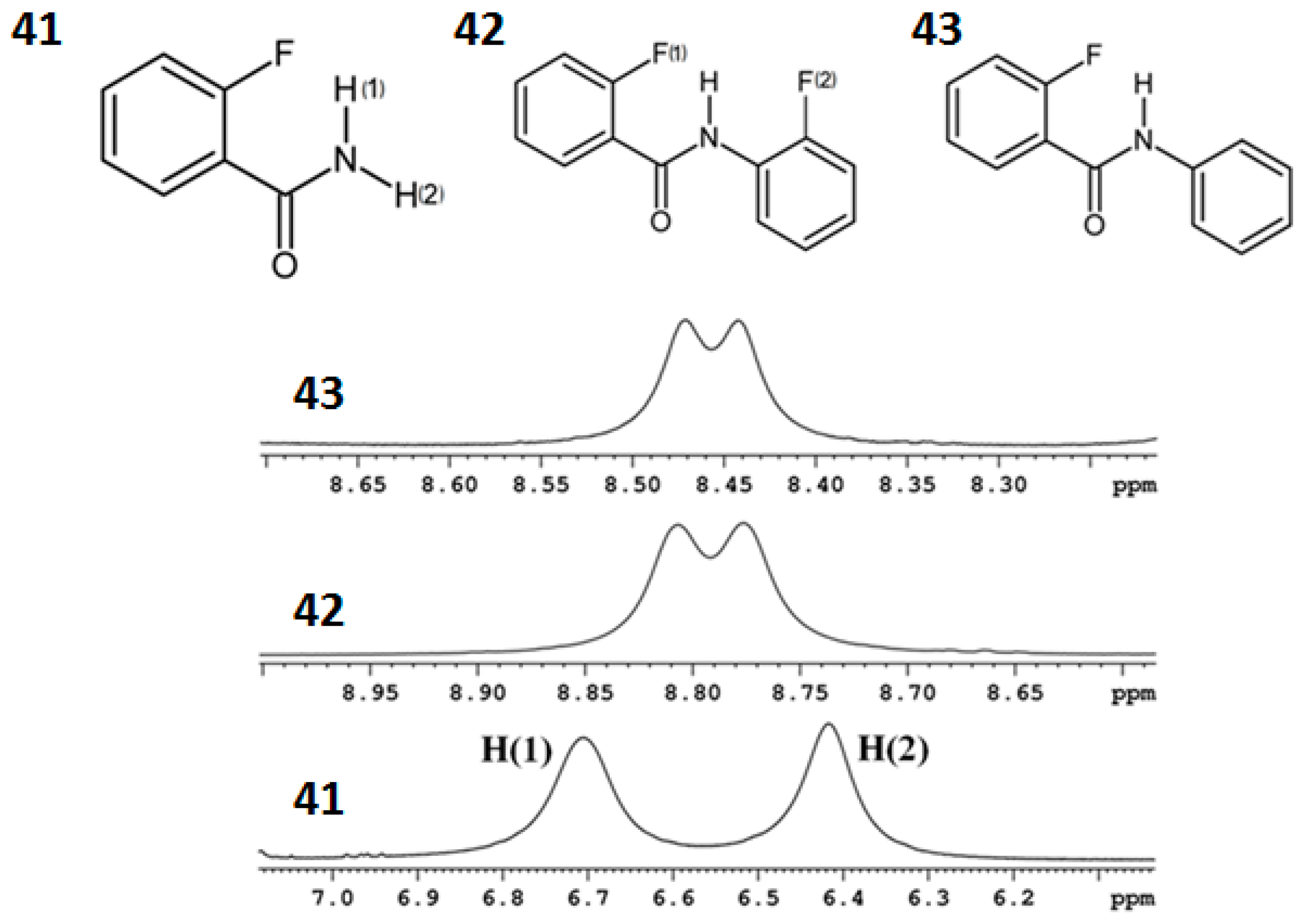
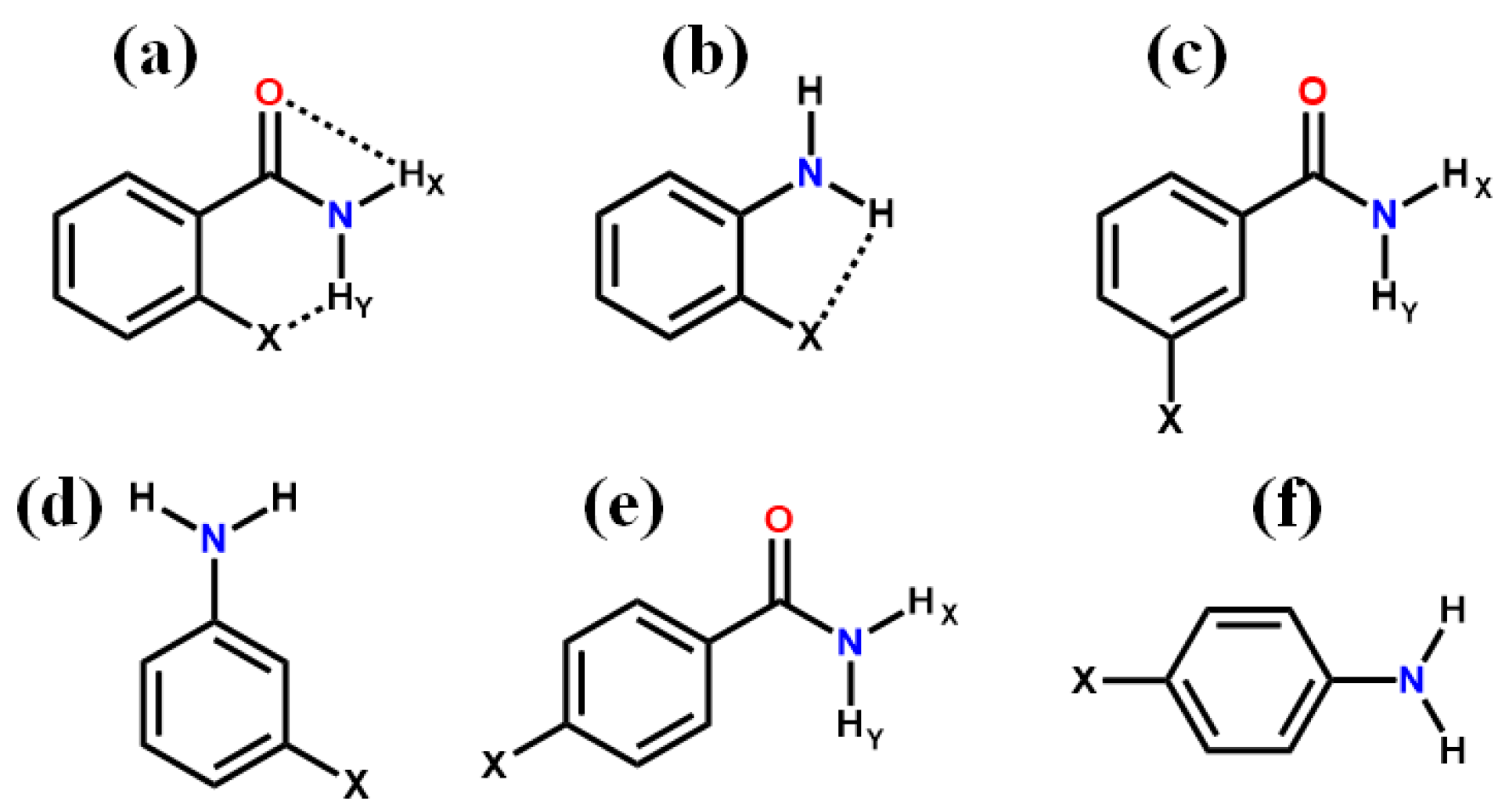
7. Utility of H/D Exchange for Study of HB
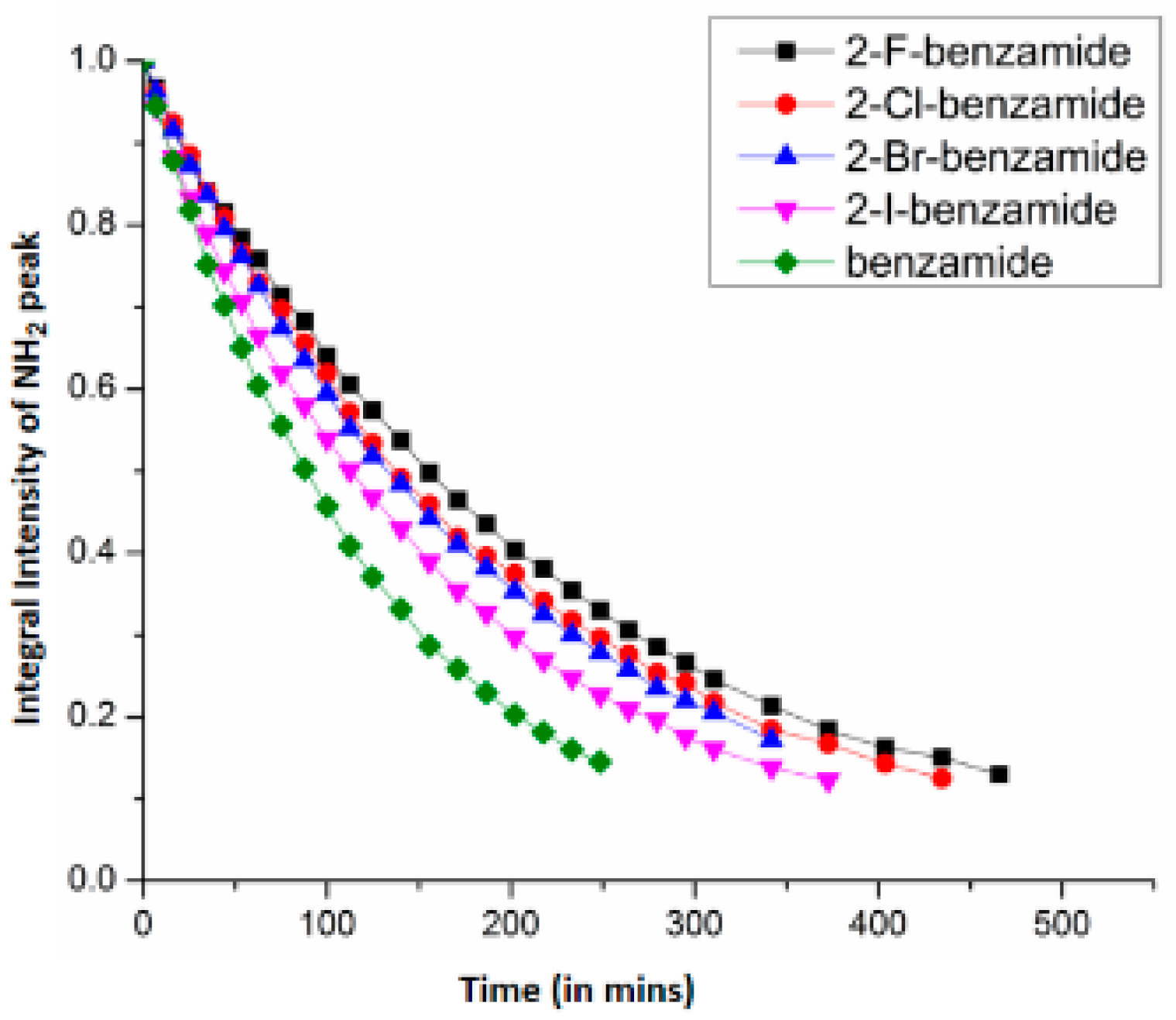
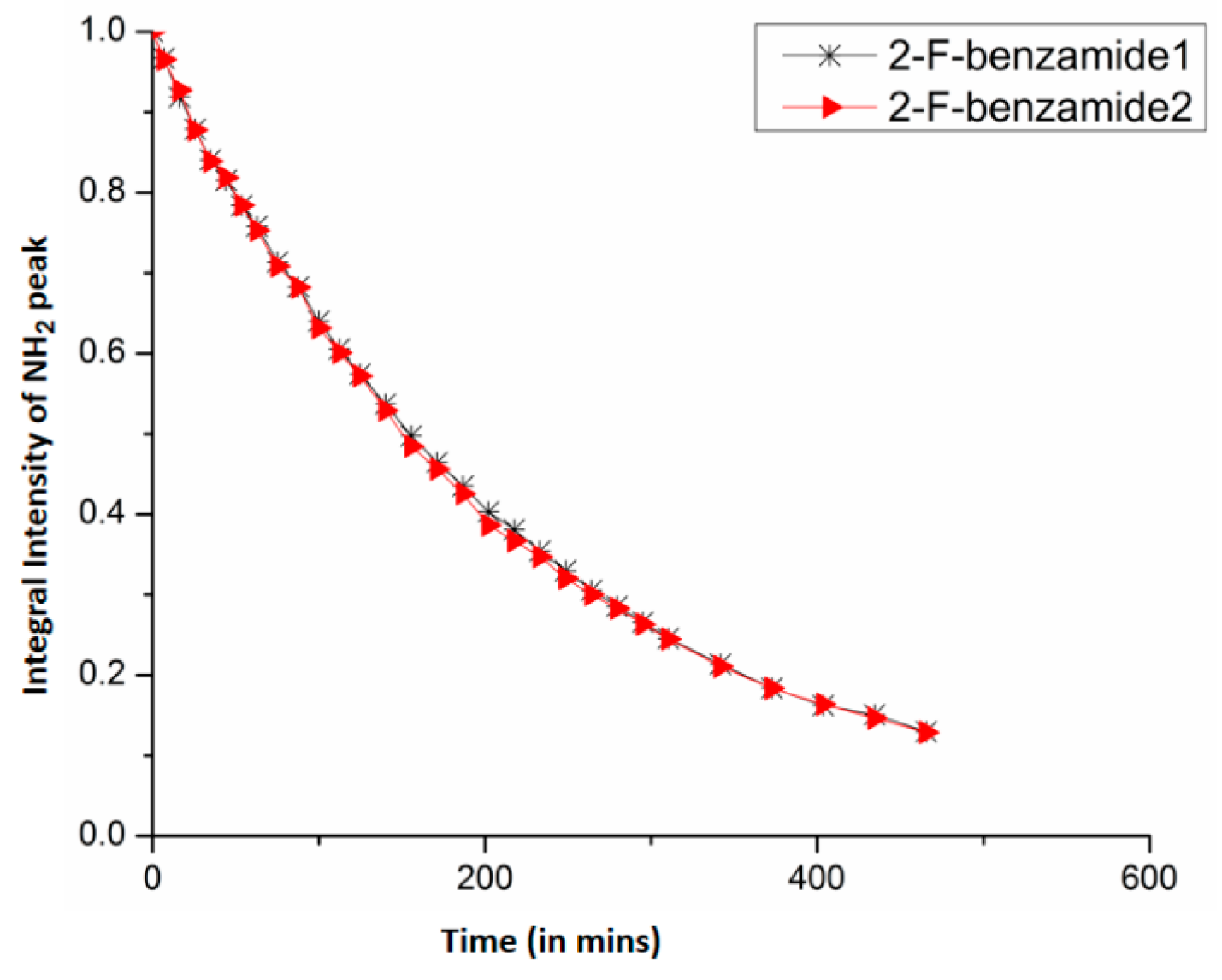
7.1. Factors Affecting the H/D Exchange
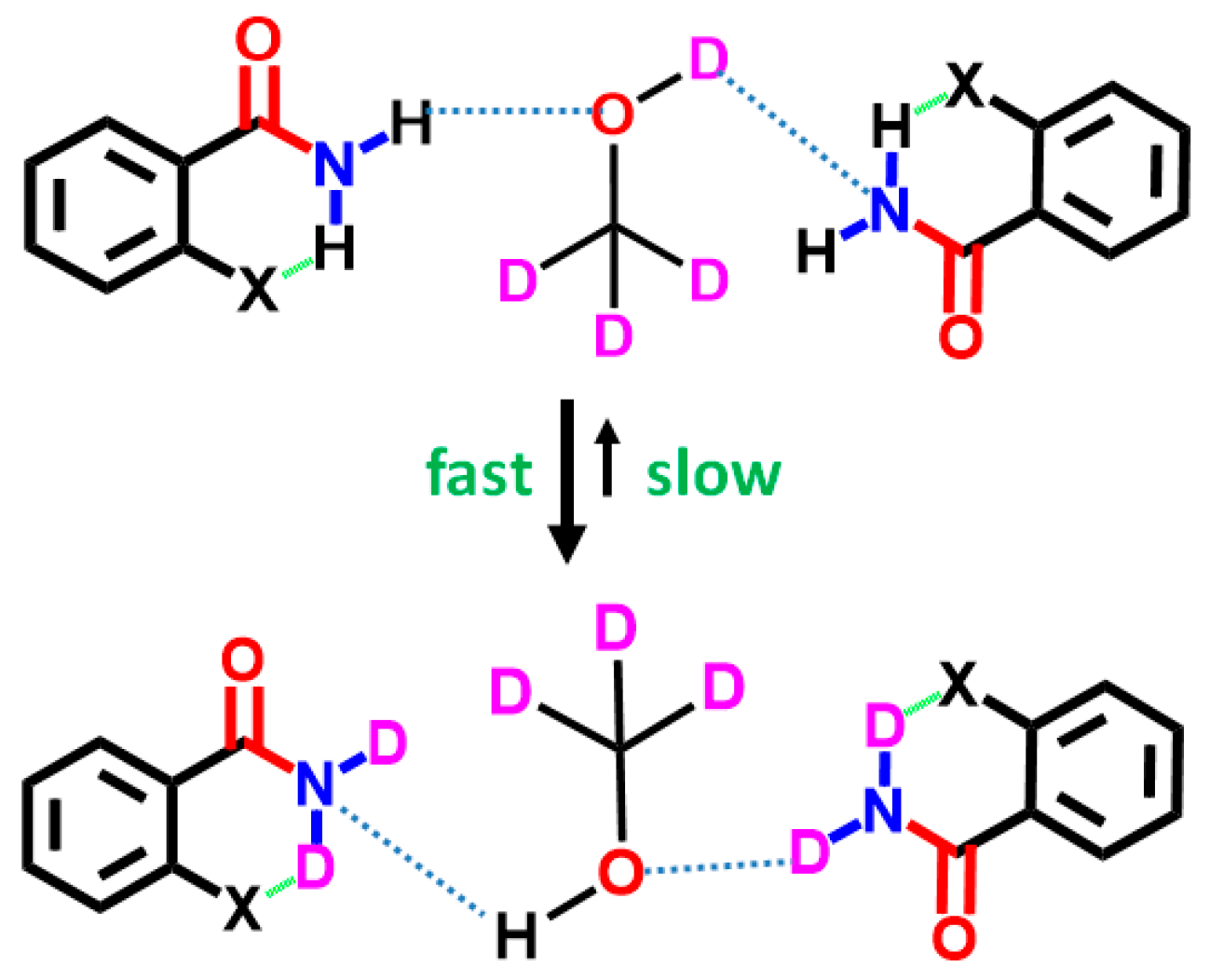
7.2. Exchange Mechanism
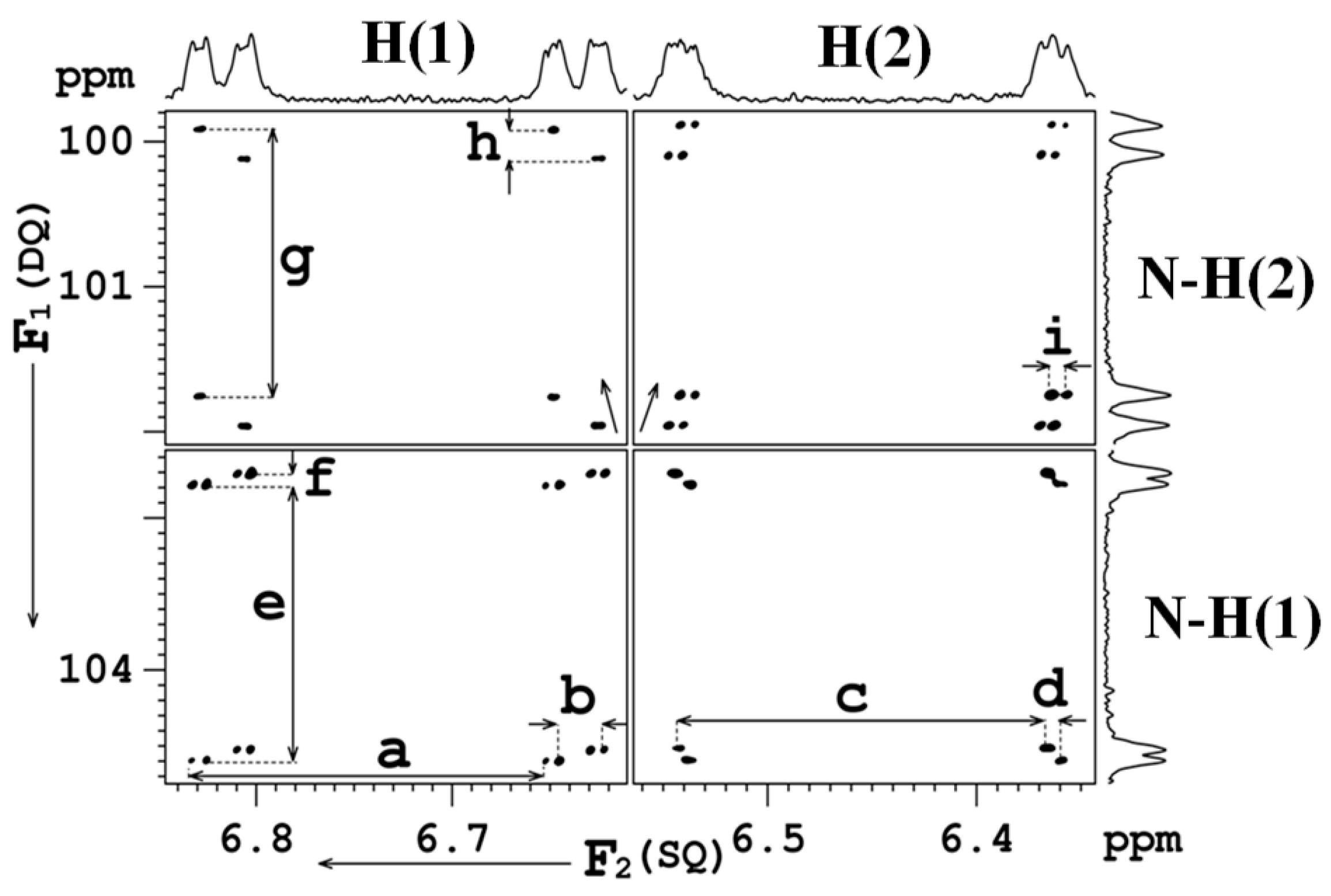
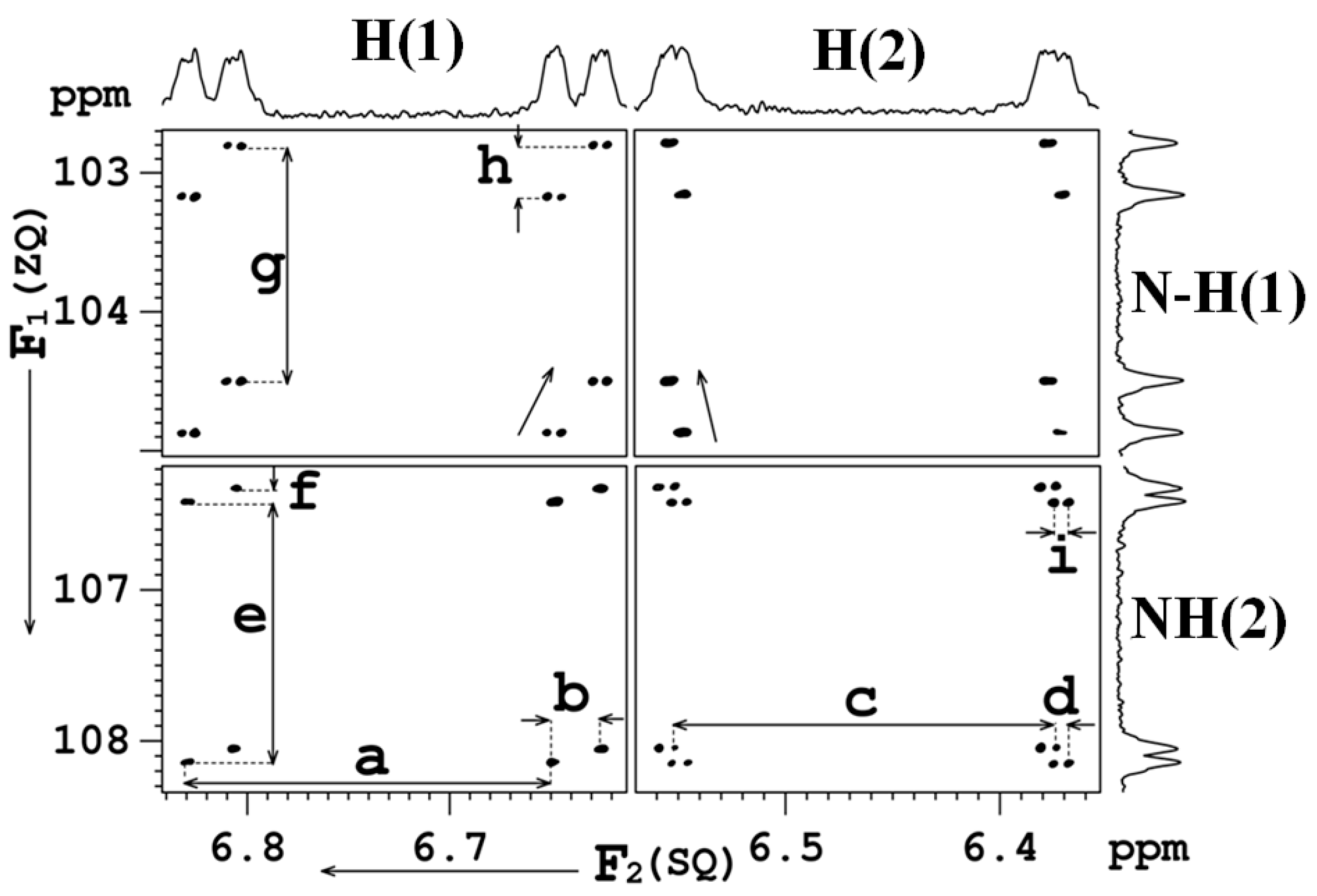
8. Multiple Quantum (MQ) NMR for the Detection of Intramolecular HBs
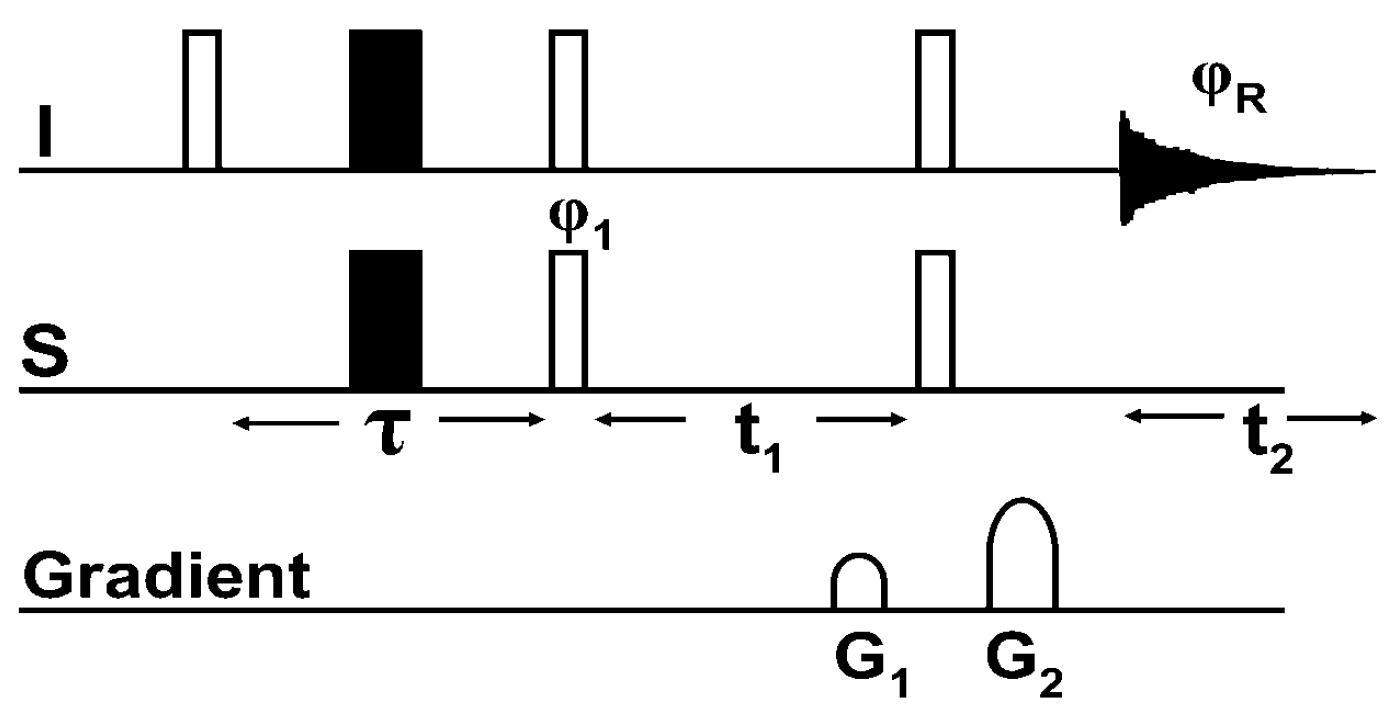
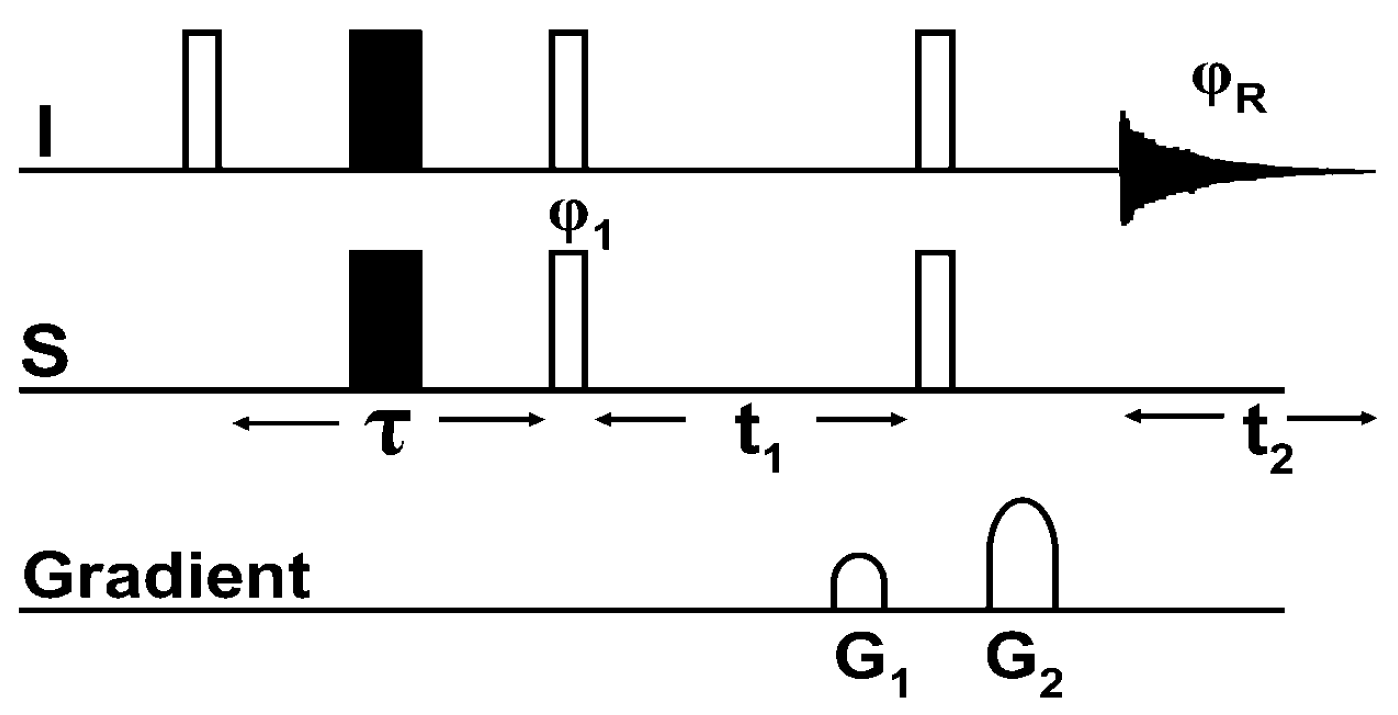
8.1. The Pulse Sequence
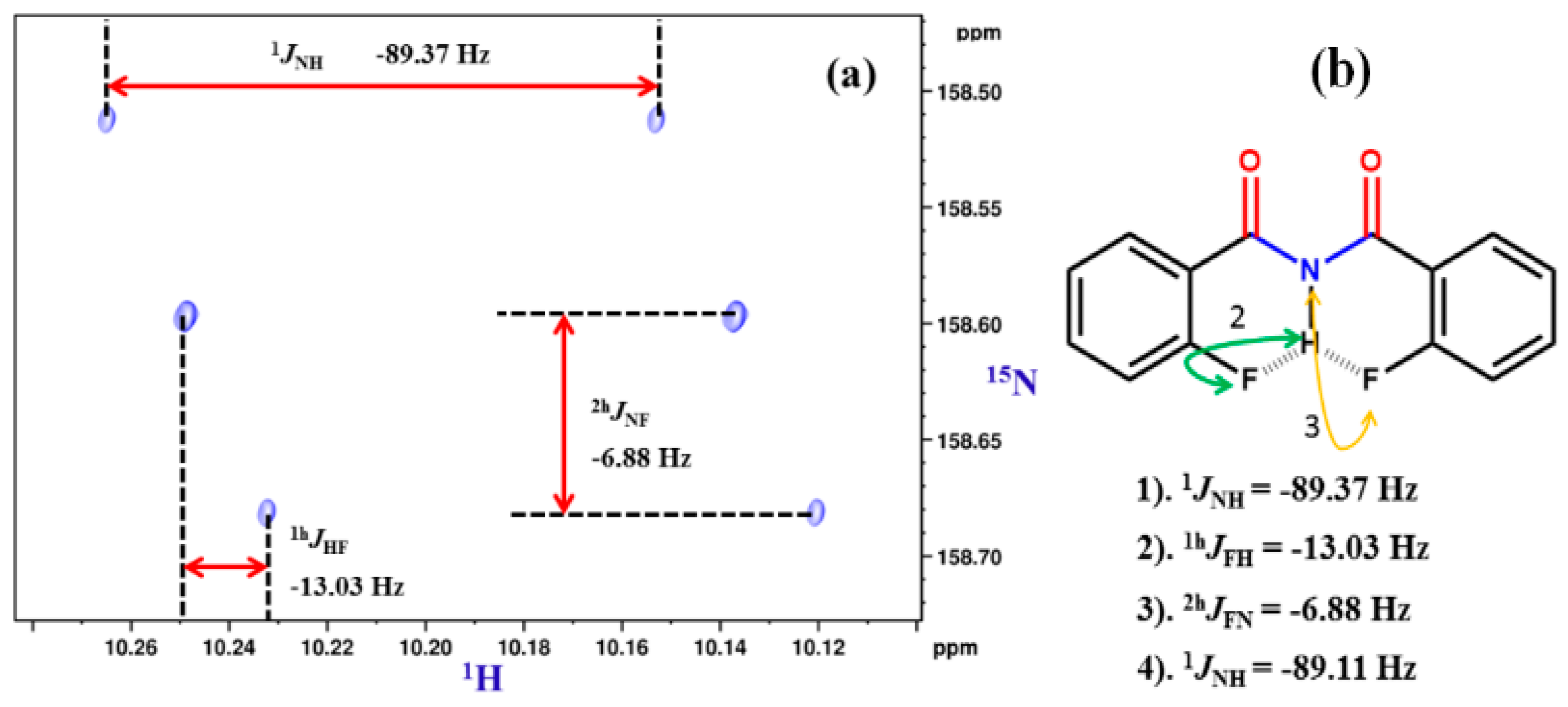
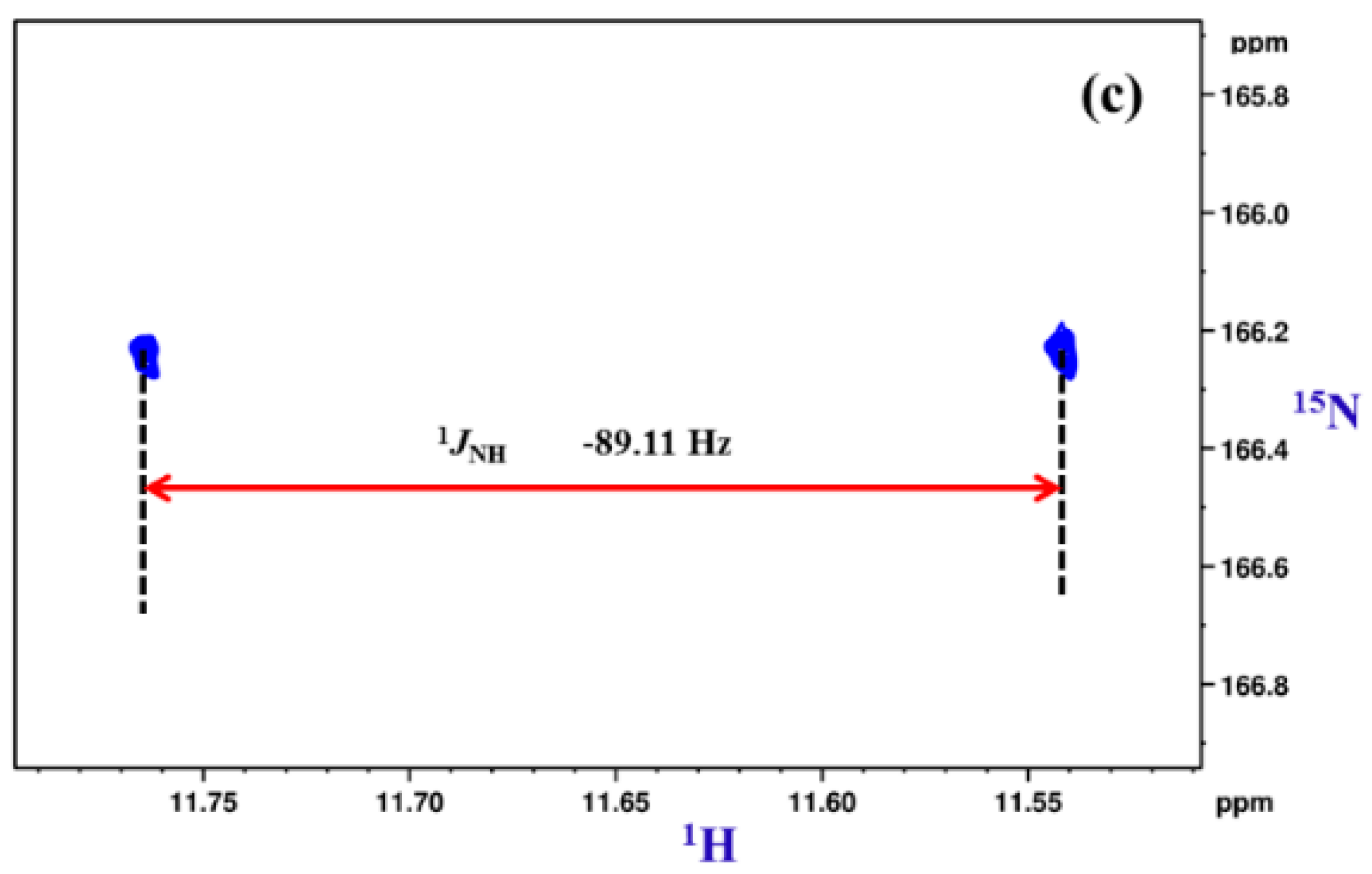
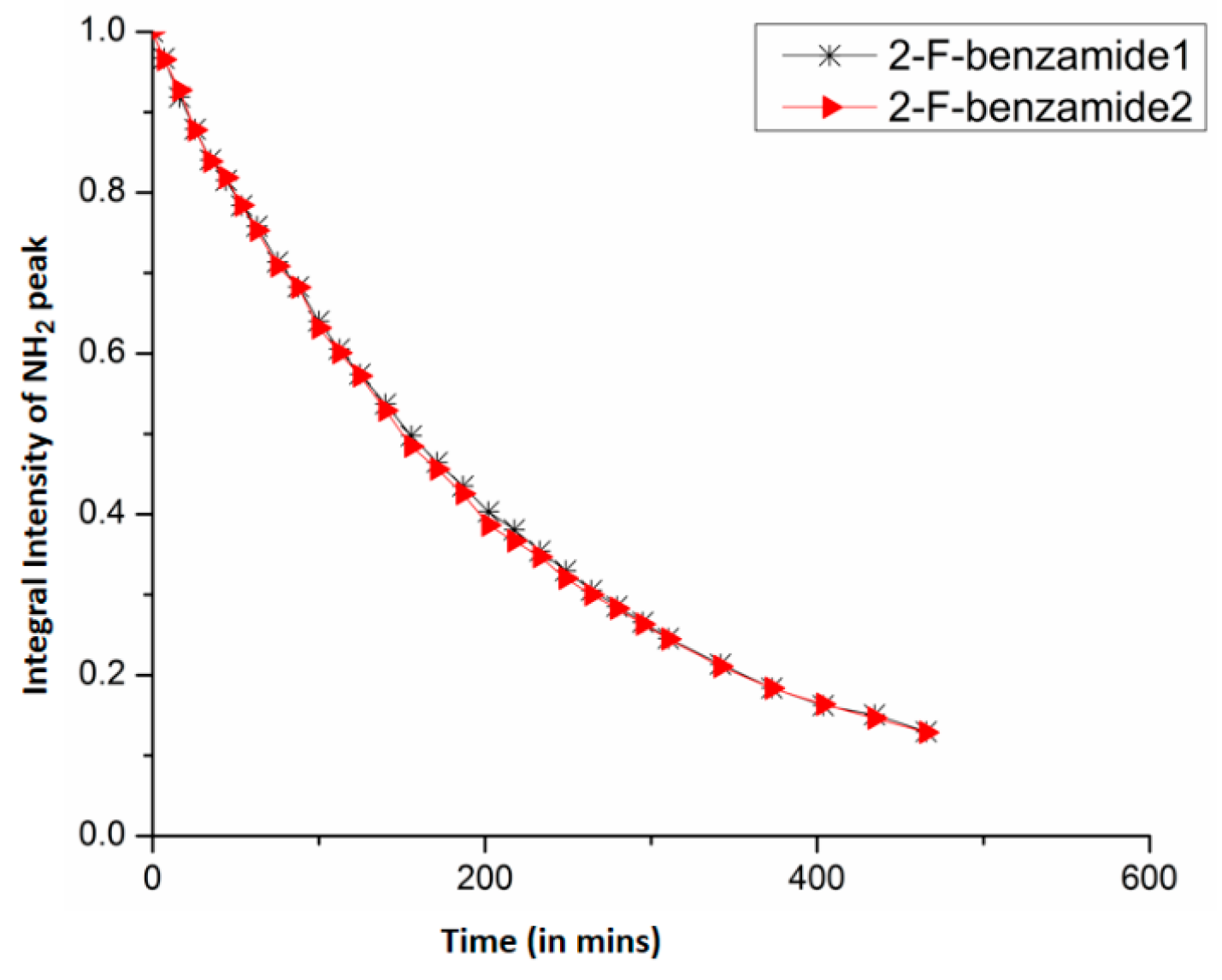
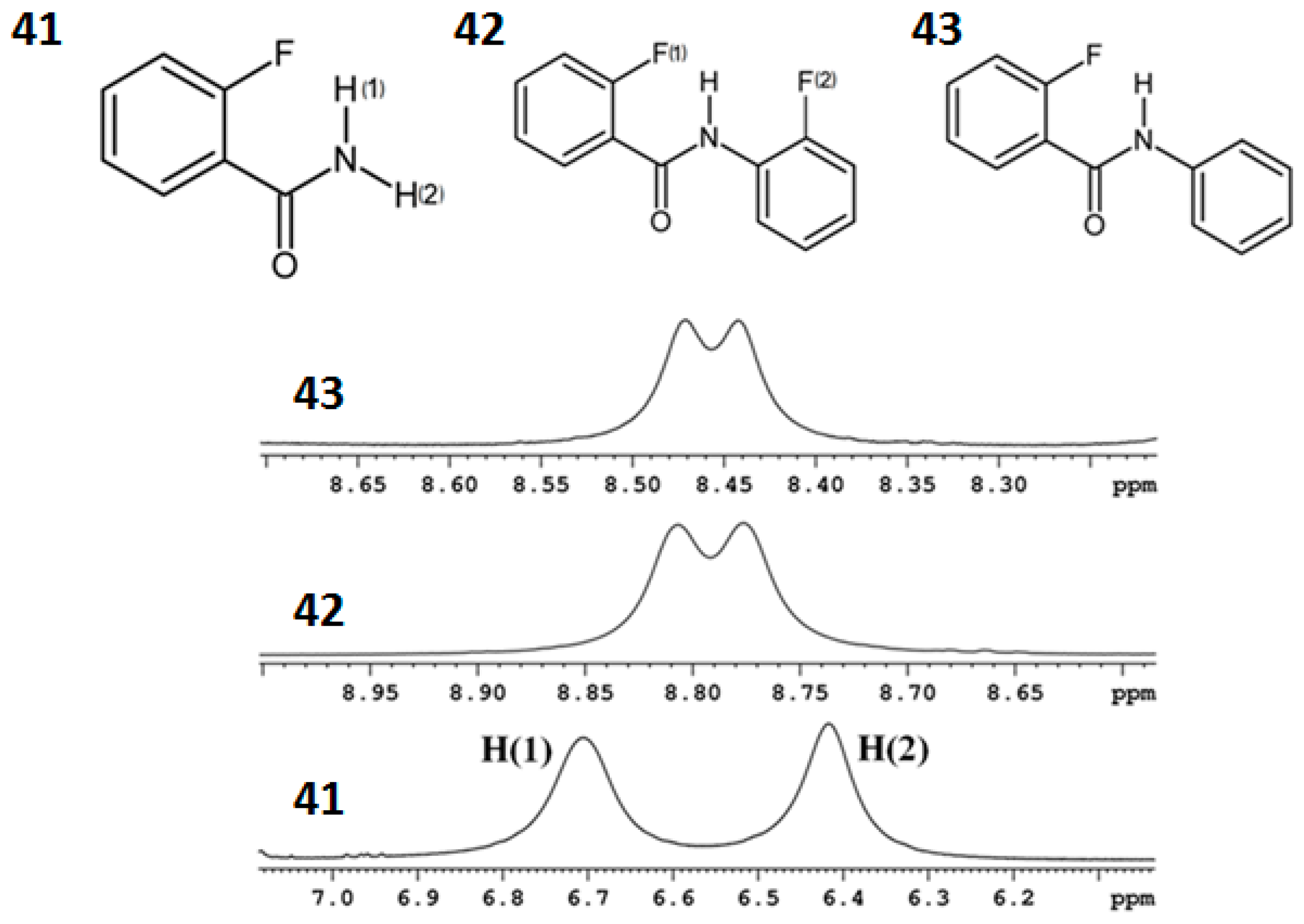
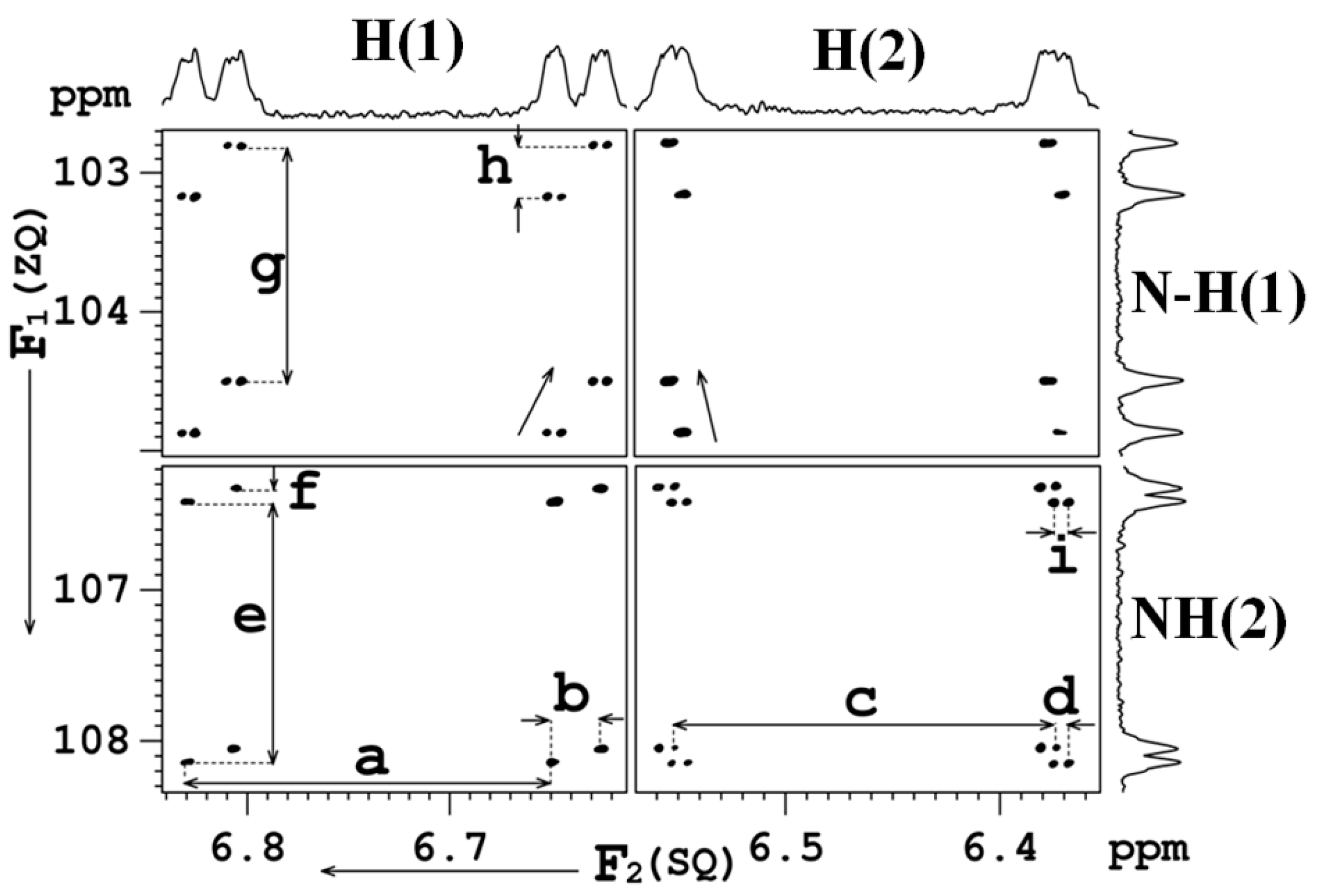
8.2. C-F…N-H Hydrogen Bond in 2-Fluorobenzamide
9. Conclusions
10. Computational Methods and the Employed Programs
Acknowledgments
Conflicts of Interest
References
- Müller-Dethlefs, K.; Hobza, P. Noncovalent Interactions: A Challenge for Experiment and Theory. Chem. Rev. 2000, 100, 143–167. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, I.G. Intermolecular Interactions: Physical Picture, Computational Methods and Model Potentials; John Wiley Sons, Ltd.: New York, NY, USA, 2006. [Google Scholar]
- Huggins, M.L. Quantum Mechanics of the Interaction of Gravity with Electrons: Theory of a Spin-two Field Coupled to Energy. PhD. Thesis, University of California, Oakland, CA, USA, 1919. [Google Scholar]
- Huggins, M.L. Atomic structure. Science 1922, 55, 459–460. [Google Scholar] [CrossRef]
- Huggins, M.L. Electronic Structures of Atoms. J. Phys. Chem. 1921, 26, 601–625. [Google Scholar] [CrossRef]
- Huggins, M.L. Atomic radii. Phys. Rev. 1922, 19, 346–353. [Google Scholar] [CrossRef]
- Latimerand, W.M.; Rodebush, W.H. Polarity and ionization from the standpoint of the Lewis theory of valence. J. Am. Chem. Soc. 1920, 42, 1419–1433. [Google Scholar] [CrossRef]
- Williams, L.D. Molecular Interactions (Noncovalent Interactions) and the Behaviors of Biological Macromolecules. Available online: http://ww2.chemistry.gatech.edu/~lw26/structure/molecular_interactions/mol_int.html (accessed on 26 February 2019).
- Hu, W.H.; Huang, K.-W.; Chiou, C.W.; Kuo, S.W. Complementary Multiple Hydrogen Bonding Interactions Induce the Self-Assembly of Supramolecular Structures from Heteronucleobase-Functionalized Benzoxazine and Polyhedral Oligomeric Silsesquioxane Nanoparticles. Macromolecules 2012, 45, 9020–9028. [Google Scholar] [CrossRef]
- Saccone, M.; Dichiarante, V.; Forni, A.; Goulet-Hanssens, A.; Cavallo, G.; Vapaavuori, J.; Terraneo, G.; Barrett, C.J.; Resnati, G.; Metrangolo, P.; Priimagi, A. Supramolecular hierarchy among halogen and hydrogen bond donors in light-induced surface patterning. J. Mater. Chem. C 2015, 3, 759–768. [Google Scholar] [CrossRef]
- Teunissen, A.J.P.; Nieuwenhuizen, M.M.L.; Rodríguez-Llansola, F.; Palmans, A.R.A.; Meijer, E.W. Mechanically Induced Gelation of a Kinetically Trapped Supramolecular Polymer. Macromolecules 2014, 47, 8429–8436. [Google Scholar] [CrossRef]
- Xiao, T.; Feng, X.; Ye, S.; Guan, Y.; Li, S.L.; Wang, Q.; Ji, Y.; Zhu, D.; Hu, X.; Lin, C.; Pan, Y.; Wang, L. Highly Controllable Ring−Chain Equilibrium in Quadruply Hydrogen Bonded Supramolecular Polymers. Macromolecules 2012, 45, 9585–9594. [Google Scholar] [CrossRef]
- Ajayaghosh, A.; George, S.J.; Schenning, A.P.H.J. Hydrogen-Bonded Assemblies of Dyes and Extended π-Conjugated Systems. In Topics in Current Chemistry; Würthner, F., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 258, pp. 83–118. [Google Scholar]
- Archer, E.A.; Gong, H.; Krische, M.J. Hydrogen bonding in noncovalent synthesis: selectivity and the directed organization of molecular strands. Tetrahedron 2001, 57, 1139–1159. [Google Scholar] [CrossRef]
- Brunsveld, L.; Folmer, B.J.B.; Meijer, E.W.; Sijbesma, R.P. Supramolecular Polymers. Chem. Rev. 2001, 101, 4071–4098. [Google Scholar] [CrossRef]
- Conn, M.M.; Rebek, J. Self-Assembling Capsules. Chem. Rev. 1997, 97, 1647–1668. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Gibson, H.W. Polypseudorotaxanes and polyrotaxanes. Prog. Polym. Sci. 2005, 30, 982–1018. [Google Scholar] [CrossRef]
- Lawrence, D.S.; Jiang, T.; Levett, M. Self-Assembling Supramolecular Complexes. Chem. Rev. 1995, 95, 2229–2260. [Google Scholar] [CrossRef]
- Lukin, O.; Vögtle, F. Knotting and Threading of Molecules: Chemistry and Chirality of Molecular Knots and Their Assemblies. Angew. Chem. Int. Ed. 2005, 44, 1456–1477. [Google Scholar] [CrossRef] [PubMed]
- Nowick, J.S. Chemical Models of Protein β-Sheets. Acc. Chem. Res. 1999, 32, 287–296. [Google Scholar] [CrossRef]
- Prins, L.J.; Reinhoudt, D.N.; Timmerman, P. Noncovalent Synthesis Using Hydrogen Bonding. Angew. Chem. Int. Ed. 2001, 40, 2382–2426. [Google Scholar] [CrossRef]
- Schmuck, C.; Wienand, W. Self-Complementary Quadruple Hydrogen-Bonding Motifs as a Functional Principle: From Dimeric Supramolecules to Supramolecular Polymers. Angew. Chem. Int. Ed. 2001, 40, 4363–4369. [Google Scholar] [CrossRef]
- Shenhar, R.; Rotello, V.M. Polymers Nanoparticles: Scaffolds and Building Blocks. Acc. Chem. Res. 2003, 36, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Sijbesma, R.P.; Meijer, E.W. Quadruple hydrogen bonded systems. Chem. Commun. 2003, 5–16. [Google Scholar] [CrossRef]
- Zeng, F.; Zimmerman, S.C. Dendrimers in Supramolecular Chemistry: From Molecular Recognition to Self-Assembly. Chem. Rev. 1997, 97, 1681–1712. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-H.; Wang, D.; Wan, L.J. Surface Tectonics of Nanoporous Networks of Melamine-Capped Molecular Building Blocks formed through Interface Schiff-Base Reactions. Chem. Asian J. 2013, 8, 2466–2470. [Google Scholar] [CrossRef] [PubMed]
- Rotondi, K.S.; Gierasch, L.M. Natural Polypeptide Scaffolds: β-Sheets, β-Turns, and β-Hairpins. Pept. Sci. 2006, 84, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Seebach, D.; Hook, D.F.; Glättli, A. Helices and Other Secondary Structures of β- and γ-Peptides. Pept. Sci. 2006, 84, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Užarević, K.; Ðilović, I.; Bregović, N.; Tomišić, V.; Matković-Čalogović, D.; Cindrić, M. Anion-Templated Supramolecular C3 Assembly for Efficient Inclusion of Charge-Dispersed Anions into Hydrogen-Bonded Networks. Chem. Eur. J. 2011, 17, 10889–10897. [Google Scholar] [CrossRef] [PubMed]
- Venugopalan, P.; Kishore, R. Unusual Folding Propensity of an Unsubstituted β,γ-Hybrid Model Peptide: Importance of the C_H···O Intramolecular Hydrogen Bond. Chem. Eur. J. 2013, 19, 9908–9915. [Google Scholar] [CrossRef] [PubMed]
- Cougnon, F.B.L.; Sanders, J.K.M. Evolution of Dynamic Combinatorial Chemistry. Acc. Chem. Res. 2011, 45, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.M.; Choi, S.; Shandler, S.; DeGrado, W.F. Hydrothermal contribution to the oceanic dissolved iron inventory. Nat. Chem. Biol. 2007, 3, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Nowick, J.S. What I have learned by using chemical model systems to study biomolecular structure and interactions. Org. Biomol. Chem. 2006, 4, 3869–3885. [Google Scholar] [CrossRef] [PubMed]
- Altintas, O.; Lejeune, E.; Gerstel, P.; Barner-Kowollik, C. Bioinspired dual self-folding of single polymer chains via reversible hydrogen bonding. Polym. Chem. 2012, 3, 640–651. [Google Scholar] [CrossRef]
- Moure, A.; Sanclimens, G.; Bujons, J.; Masip, I.; Alvarez-Larena, A.; Pérez-Payá, E.; Alfonso, I.; Messeguer, A. Chemical Modulation of Peptoids: Synthesis and Conformational Studies on Partially Constrained Derivatives. Chem. Eur. J. 2011, 17, 7927–7939. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, M.; Dal Corso, A.; Marchini, M.; Guzzetti, I.; Civera, M.; Piarulli, U.; Arosio, D.; Belvisi, L.; Potenza, D.; Pignataro, L.; Gennari, C. Cyclic isoDGR Peptidomimetics as Low-Nanomolar αvβ3 Integrin Ligands. Chem. Eur. J. 2013, 19, 3563–3567. [Google Scholar] [CrossRef] [PubMed]
- Rufo, C.M.; Moroz, Y.S.; Moroz, O.V.; Stöhr, J.; Smith, T.A.; Hu, X.; DeGrado, W.F.; Korendovych, I.V. Short peptides self-assemble to produce catalytic amyloids. Nat. Chem. 2014, 6, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Wennemers, H. Asymmetric catalysis with peptides. Chem. Comm. 2011, 47, 12036–12041. [Google Scholar] [CrossRef] [PubMed]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Ripka, A.S.; Rich, D.H. Peptidomimetic design. Curr. Opin. Chem. Biol. 1998, 2, 441–452. [Google Scholar] [CrossRef]
- Dingley, A.J.; Masse, J.E.; Peterson, R.D.; Barfield, M.; Feigon, J.; Grzesiek, S. Internucleotide Scalar Couplings Across Hydrogen Bonds in Watson-Crick and Hoogsteen Base Pairs of a DNA Triplex. J. Am. Chem. Soc. 1999, 121, 6019–6027. [Google Scholar] [CrossRef]
- Fonseca-Guerra, C.; Bickelhaupt, F.M.; Snijders, J.G.; Baerends, E.J. Hydrogen Bonding in DNA Base Pairs: Reconciliation of Theory and Experiment. J. Am. Chem. Soc. 2000, 122, 4117–4128. [Google Scholar] [CrossRef]
- Smith, J.D.; Cappa, C.D.; Wilson, K.R.; Cohen, R.C.; Geissler, P.L.; Saykally, R.J. Unified description of temperature-dependent hydrogen-bond rearrangements in liquid water. Proc. Natl. Acad. Sci. USA 2005, 102, 14171–14174. [Google Scholar] [CrossRef] [PubMed]
- Liskamp, R.M.J.; Rijkers, D.T.S.; Kruijtzer, J.A.; Kemmink, W.J. Peptides and Proteins as a Continuing Exciting Source of Inspiration for Peptidomimetics. Chembiochem 2011, 12, 1626–1653. [Google Scholar] [CrossRef] [PubMed]
- Panciera, M.; Amorín, M.; Castedo, L.; Granja, J.R. Design of Stable b-Sheet-Based Cyclic Peptide Assemblies Assisted by Metal Coordination: Selective Homo- and Heterodimer Formation. Chem. Eur. J. 2013, 19, 4826–4834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremey, E.; Bonnot, F.; Moreau, Y.; Berthomieu, C.; Desbois, A.; Favaudon, V.; Blondin, G.; Houée-Levin, C.; Nivière, V. Hydrogen bonding to the cysteine ligand of superoxide reductase: Acid–base control of the reaction intermediates. J. Biol. Inorg. Chem. 2013, 18, 815–830. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, K.; Kulp, J.L.; Arora, P.S. Evaluation of Biologically Relevant Short r-Helices Stabilized by a Main-Chain Hydrogen-Bond Surrogate. J. Am. Chem. Soc. 2006, 128, 9248–9256. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.; Liu, J.; Perez, L.M.; Lu, G.; Schaefer, A.; Burgess, K. Universal Peptidomimetics. J. Am. Chem. Soc. 2010, 133, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Meinhardt, N.; Wu, Y.; Kulkarni, S.; Hu, X.; Low, K.E.; Davies, P.L.; DeGrado, W.F.; Greenbaum, D.C. Development of α‑Helical Calpain Probes by Mimicking a Natural Protein−Protein Interaction. J. Am. Chem. Soc. 2012, 134, 17704–17713. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.; Liu, J.H.; Alayoglu, S.; Marcus, M.A.; Fakra, S.C.; Toste, F.D.; Somorjai, G.A. Asymmetric Catalysis at the Mesoscale: Gold Nanoclusters Embedded in Chiral Self-Assembled Monolayer as Heterogeneous Catalyst for Asymmetric Reactions. J. Am. Chem. Soc. 2013, 135, 3881–3886. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.C.; Bartlett, P.A. Synthesis of Amino Acid-Derived Cyclic Acyl Amidines for Use in β-Strand Peptidomimetics. J. Org. Chem. 2007, 72, 3104–3107. [Google Scholar] [CrossRef] [PubMed]
- Emenike, B.U.; Liu, A.T.; Naveo, E.P.; Roberts, J.D. Substituent Effects on Energetics of Peptide-Carboxylate Hydrogen Bonds as Studied by 1H NMR Spectroscopy: Implications for Enzyme Catalysis. J. Org. Chem. 2013, 78, 11765–11771. [Google Scholar] [CrossRef] [PubMed]
- Linton, B.R.; Reutershan, M.H.; Aderman, C.M.; Richardson, E.A.; Brownell, K.R.; Ashley, C.W.; Evans, C.A.; Miller, S.J. Asymmetric Michael addition of α-nitro-ketones using catalytic peptides. Tetrahedron Lett. 2007, 48, 1993–1997. [Google Scholar] [CrossRef]
- Ghorai, A.; Padmanaban, E.; Mukhopadhyay, C.; Achari, B.; Chattopadhyay, P. Design and synthesis of regioisomeric triazole based peptidomimetic macrocycles and their dipole moment controlled self-assembly. Chem. Commun. 2012, 48, 11975–11977. [Google Scholar] [CrossRef] [PubMed]
- Gong, B. Crescent Oligoamides: From Acyclic “Macrocycles” to Folding Nanotubes. Chem. Eur. J. 2001, 7, 4336–4342. [Google Scholar] [CrossRef]
- Huc, I. Aromatic Oligoamide Foldamers. Eur. J. Org. Chem. 2004, 1, 17–29. [Google Scholar] [CrossRef]
- Li, Z.T.; Hou, J.L.; Li, C.; Yi, H.P. Shape-Persistent Aromatic Amide Oligomers: New Tools for Supramolecular Chemistry. Chem. Asian J. 2006, 1, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Lockman, J.W.; Paul, N.M.; Parquette, J.R. The role of dynamically correlated conformational equilibria in the folding of macromolecular structures. A model for the design of folded dendrimers. Prog. Polym. Sci. 2005, 30, 423–452. [Google Scholar] [CrossRef]
- Sanford, A.R.; Yamato, K.; Yang, X.; Yuan, L.; Han, Y.; Gong, B. Well-defined secondary structures, Information-storing molecular duplexes and helical foldamers based on unnatural peptide backbones. Eur. J. Biochem. 2004, 271, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Chopra, D. Is Organic Fluorine Really “Not” Polarizable? Cryst. Growth Des. 2012, 12, 541–546. [Google Scholar] [CrossRef]
- Dunitz, J.D. Organic Fluorine: Odd Man Out. ChemBioChem 2004, 5, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D.; Taylor, R. Organic Fluorine Hardly Ever Accepts Hydrogen Bonds. Chem. Eur. J. 1997, 3, 89–98. [Google Scholar] [CrossRef]
- Howard, J.A.K.; Hoy, V.J.; O’Hagan, D.; Smith, G.T. How Good is Fluorine as a Hydrogen Bond Acceptor? Tetrahedron 1996, 52, 12613–12622. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Limbach, H.H.; Shenderovich, I.G.; Winkler, T. A DFT and AIM analysis of the spin–spin couplings across the hydrogen bond in the 2-fluorobenzamide and related compounds. Magn. Reson. Chem. 2009, 47, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, C.; Espinet, P.; Martin-Alvarez, J.M. Is there any bona fide example of O–H…F–C bond in solution? The cases of HOC(CF3)2(4-X-2,6-C6H2(CF3)2) (X = Si(i-Pr)3, CF3). Chem. Comm. 2007, 4384–4386. [Google Scholar] [CrossRef] [PubMed]
- Kareev, I.E.; Quiñones, G.S.; Kuvychko, I.V.; Khavrel, P.A.; Ioffe, I.N.; Goldt, I.V.; Lebedkin, S.F.; Seppelt, K.; Strauss, S.H.; Boltalina, O.V. Variable-Temperature 19F NMR and Theoretical Study of 1,9- and 1,7-C60F(CF3) and CS- and C1-C60F17(CF3): Hindered CF3 Rotation and Through-Space JFF Coupling. J. Am. Chem. Soc. 2005, 127, 11497–11504. [Google Scholar] [CrossRef] [PubMed]
- Prakash, G.K.S.; Wang, F.; Rahm, M.; Shen, J.; Ni, C.; Haiges, R.; Olah, G.A. On the Nature of C-H···F-C Interactions in Hindered CF-C(sp3) Bond Rotations. Angew. Chem. Int. Ed. 2011, 50, 11761–11764. [Google Scholar] [CrossRef] [PubMed]
- Ledbetter, M.P.; Saielli, G.; Bagno, A.; Tran, N.; Romalis, M.V. Observation of scalar nuclear spin–spin coupling in van der Waals complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 12393–12397. [Google Scholar] [CrossRef]
- Saielli, G.; Bini, R.; Bagno, A. Computational 19F-NMR. 2. Organic compounds. RSC. Adv. 2014, 4, 41605–41611. [Google Scholar] [CrossRef]
- Rae, I.D.; Weigold, J.A.; Contreras, R.H.; Biekofsky, R.R. Analysis of Long-Range Through-Space Couplings via an Intramolecular Hydrogen Bond. Magn. Reson. Chem. 1993, 31, 836–840. [Google Scholar] [CrossRef]
- Benedict, H.; Shenderovich, I.G.; Malkina, O.L.; Malkin, V.G.; Denisov, G.S.; Golubev, N.S.; Limbach, H.H. Nuclear Scalar Spin-Spin Couplings and Geometries of Hydrogen Bonds. J. Am. Chem. Soc. 2000, 122, 1979–1988. [Google Scholar] [CrossRef]
- Golubev, N.S.; Shenderovich, I.G.; Smirnov, S.N.; Denisov, G.S.; Limbach, H.-H. Nuclear Scalar Spin ± Spin Coupling Reveals Novel Properties of Low-Barrier Hydrogen Bonds in a Polar Environment. Chem. Eur. J. 1999, 5, 492–497. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Smirnov, S.N.; Denisov, G.S.; Gindin, V.A.; Golubev, N.S.; Dunger, A.; Reibke, R.; Kirpekar, S.; Malkina, O.L.; Limbach, H.H. Nuclear Magnetic Resonance of Hydrogen Bonded Clusters between F- and (HF)n: Experiment and Theory. Ber. Bunsenges. Phys. Chem. 1998, 102, 422–428. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Tolstoy, P.M.; Golubev, N.S.; Smirnov, S.N.; Denisov, G.S.; Limbach, H.H. Low-Temperature NMR Studies of the Structure and Dynamics of a Novel Series of Acid-Base Complexes of HF with Collidine Exhibiting Scalar Couplings Across Hydrogen Bonds. J. Am. Chem. Soc. 2003, 125, 11710–11720. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluor. Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Chopra, D.; Row, T.N.G. Role of organic fluorine in crystal engineering. CrystEngComm 2011, 13, 2175–2186. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef] [PubMed]
- Reichenbacher, K.; Suss, H.I.; Hulliger, J. Fluorine in crystal engineering—“the little atom that could”. Chem. Soc. Rev. 2005, 34, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Organic fluorine compounds: a great opportunity for enhanced materials properties. Chem. Soc. Rev. 2011, 40, 3496–3508. [Google Scholar] [CrossRef] [PubMed]
- Abeles, R.H.; Alston, T.A. A Rationale for the Design of an Inhibitor of Tyrosyl Kinase*. J. Biol. Chem. 1990, 265, 16705–16708. [Google Scholar] [PubMed]
- Chapeau, M.C.; Frey, P.A. Synthesis of UDP-4-deoxy-4-fluoroglucose and UDP-4-deoxy-4-fluorogalactose and Their Interactions with Enzymes of Nucleotide Sugar Metabolism. J. Org. Chem. 1994, 59, 6994–6998. [Google Scholar] [CrossRef]
- Kovacs, T.; Pabuccuoglu, A.; Lesiak, K.; Torrence, P.F. Fluorodeoxy Sugar Analogues of 2′,5′-Oligoadenylates as Probes of Hydrogen Bonding in Enzymes of the 2-5A System. Bioorg. Chem. 1993, 21, 192–208. [Google Scholar] [CrossRef]
- O’Hagan, D.; Rzepa, H.S. Some influences of fluorine in bioorganic chemistry. Chem. Commun. 1997, 645–652. [Google Scholar] [CrossRef]
- Takahashi, L.H.; Radhakrishnan, R.; Rosenfield, R.E.; Meyer, E.F.; Trainor, D.A. Crystal Structure of the Covalent Complex Formed by a Peptidyl α,α-Difluoro-β-keto Amide with Porcine Pancreatic Elastase at 1.78-Å Resolution. J. Am. Chem. Soc. 1989, 111, 3368–3374. [Google Scholar] [CrossRef]
- Banerjee, R.; Desiraju, G.R.; Mondal, R.; Howard, J.A.K. Organic Chlorine as a Hydrogen-Bridge Acceptor: Evidence for the Existence of Intramolecular O-H…Cl-C Interactions in Some gem-Alkynols. Chem. Eur. J. 2004, 10, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ren, S.F.; Hou, J.L.; Yi, H.P.; Zhu, S.Z.; Jiang, X.K.; Li, Z.T. F···H-N Hydrogen Bonding Driven Foldamers: Efficient Receptors for Dialkylammonium Ions**. Angew. Chem. Int. Ed. 2005, 44, 5725–5729. [Google Scholar] [CrossRef] [PubMed]
- Mido, Y.; Okuno, T. The Intramolecular NH…Cl Hydrogen Bond in Urea Derivatives Containing the O-Chlorophenyl Group. J. Mol. Struct. 1982, 82, 29–34. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W.; Anderson, S.G.; Duke, A.J. Quantum Topology of Molecular Charge Distributions, 1. J. Am. Chem. Soc. 1979, 101, 1389–1395. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essén, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Nguyen-Dang, T.T.; Tal, Y. A topological theory of molecular structure. Rep. Prog. Phys. 1981, 44, 893–948. [Google Scholar] [CrossRef]
- Runtz, G.R.; Bader, R.F.W.; Messer, R.R. Definition of bond paths and bond directions in terms of the molecular charge distribution. Can. J. Chem. 1977, 55, 3040–3045. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Fermi, E.; Pasta, J.; Ulam, S. Studies of Nonlinear Problems. I, Los Alamos Report LA-1940. Los Alamos Scientific Laboratory of the University of California: Oakland, CA, USA, 1955. [Google Scholar]
- Chopra, D.; Guru Row, T.N. Evaluation of the interchangeability of C–H and C–F groups: insights from crystal packing in a series of isomeric fluorinated benzanilides. Cryst. Eng. Comm. 2008, 10, 54–67. [Google Scholar] [CrossRef]
- Böhm, H.J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in Medicinal Chemistry. Chem. Biochem. 2004, 5, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Wickenden, O.; Gross, M.F.; Smith, G.A.M. Benzanilides as Potassium Channel Openers. U.S. Patent 6,989,398, 24 January 2006. [Google Scholar]
- Calderone, V.; Fiamingo, F.L.; Giorgi, I.; Leonardi, M.; Livi, O.; Martelli, A.; Martinotti, E. Heterocyclic analogs of benzanilide derivatives as potassium channel activators, IX. Eur. J. Med. Chem. 2006, 41, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Biagi, G.; Giorgi, I.; Livi, O.; Nardi, A.; Calderone, V.; Martelli, A.; Martinotti, E.; Salerni, O.L. Synthesis and biological activity of novel substituted benzanilides as potassium channel activators, V. Eur. J. Med. Chem. 2004, 39, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Bondiwell, W.E.; Chan, J.A. Substituted Benzanilides as CCR5 Receptors Ligands, Anti-inflammatory Agents and Antiviral Agents. U.S. Patent 6,515,027, 4 February 2003. [Google Scholar]
- Reddy, G.N.M.; Vasantha Kumar, M.V.; Guru Row, T.N.; Suryaprakash, N. N–H…F hydrogen bonds in fluorinated benzanilides: NMR and DFT study. Phys. Chem. Chem. Phys. 2010, 12, 13232–13237. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, S.N.; Golubev, N.S.; Denisov, G.S.; Benedict, H.; Schah-Mohammedi, P.; Limbach, H.H. Hydrogen/Deuterium Isotope Effects on the NMR Chemical Shifts and Geometries of Intermolecular Low-Barrier Hydrogen-Bonded Complexes. J. Am. Chem. Soc. 1996, 118, 4094–4101. [Google Scholar] [CrossRef]
- Golubev, N.S.; Smirnov, S.N.; Gindin, V.A.; Denisov, G.S.; Benedict, H.; Limbach, H.H. Formation of Charge Relay Chains between Acetic Acid and Pyridine Observed by Low-Temperature Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1994, 116, 12055–12056. [Google Scholar] [CrossRef]
- Golubev, N.S.; Denisov, G.S.; Smirnov, S.N.; Shchepkin, D.N.; Limbach, H.H. Evidence by NMR of Temperature-dependent Solvent Electric Field Effects on Proton Transfer and Hydrogen Bond Geometries. Z. Phys. Chem. 1996, 196, 73–84. [Google Scholar] [CrossRef]
- Borman, S. Speedy Route to Folded DNA. Chem. Eng. News 1999, 77, 36–38. [Google Scholar] [CrossRef]
- Dingley, A.J.; Grzesiek, S. Direct Observation of Hydrogen Bonds in Nucleic Acid Base Pairs by Internucleotide 2JNN Couplings. J. Am. Chem. Soc. 1998, 120, 8293–8297. [Google Scholar] [CrossRef]
- Pervushin, K.; Ono, A.; Fernández, C.; Szyperski, T.; Kainosho, M.; Wüthrich, K. NMR scalar couplings across Watson–Crick base pair hydrogen bonds in DNA observed by transverse relaxation-optimized spectroscopy. Proc. Natl. Acad. Sci. USA 1998, 95, 14147–14151. [Google Scholar] [CrossRef] [PubMed]
- Fritz, H.; Winkler, T.; Küng, W. Weitreichende Kernspin-Kopplungen in 2-Fluorbenzamiden II. [15N]-2-Fluorbenzamid. Helv. Chim. Acta 1975, 58, 1822–1824. [Google Scholar] [CrossRef]
- Hennig, L.; Ayala-Leon, K.; Angulo-Cornejo, J.; Richter, R.; Beyer, L. Fluorine hydrogen short contacts and hydrogen bonds in substituted benzamides. J. Fluor. Chem. 2009, 130, 453–460. [Google Scholar] [CrossRef]
- Pople, J.A.; Schneider, W.G.; Bernstein, H.J. High Resolution Nuclear Magnetic Resonance; McGraw Hill: New York, NY, USA, 1959. [Google Scholar]
- Günther, H. NMR Spectroscopy: Basic Principles, Concepts and Applications in Chemistry; John Wiley Sons: New York, NY, USA, 1995. [Google Scholar]
- Reddy, G.N.M.; Row, T.N.G.; Suryaprakash, N. Discerning the degenerate transitions of scalar coupled 1H NMR spectra: Correlation and resolved techniques at higher quantum. J. Magn. Reson. 2009, 196, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Bikash, B.; Reddy, G.N.M.; Uday, R.P.; Row, T.N.G.; Suryaprakash, N. Simplifying the Complex 1H-NMR Spectra of Fluorine-Substituted Benzamides by Spin System Filtering and Spin-State Selection: Multiple-Quantum-Single-Quantum Correlation. J. Phys. Chem. A 2008, 112, 10526–10532. [Google Scholar]
- Reddy, G.N.M.; Caldarelli, S. Demixing of Severely Overlapping NMR Spectra through Multiple-Quantum NMR. Anal. Chem. 2010, 82, 3266–3269. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Foresman, J.B.; Keith, T.A.; Wiberg, K.B.; Snoonian, J.; Frisch, M.J. Solvent Effects. 5. Influence of Cavity Shape, Truncation of Electrostatics, and Electron Correlation on ab Initio Reaction Field Calculations. J. Phys. Chem. 1996, 100, 16098–16104. [Google Scholar] [CrossRef]
- Chaudhari, S.R.; Mogurampelly, S.; Suryaprakash, N. Engagement of CF3 Group in N−H···F−C Hydrogen Bond in the Solution State: NMR Spectroscopy and MD Simulation Studies. J. Phys. Chem. B 2013, 117, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.J. Hydrogen bonds with fluorine. Studies in solution, in gas phase and by computations, conflicting conclusions from crystallographic analyses. Chem. Sci. 2012, 3, 1381–1394. [Google Scholar] [CrossRef]
- Jiang, J.C.; Tsa, M.H. Ab Initio Study of the Hydrogen Bonding between Pyrrole and Hydrogen Fluoride: A Comparison of NH…F and FH…π Interactions. J. Phys. Chem. A 1997, 101, 1982–1988. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S. Folding Studies of a Linear Pentamer Peptide Adopting a Reverse Turn Conformation in Aqueous Solution through Molecular Dynamics Simulation. J. Phys. Chem. B 2000, 104, 8023–8034. [Google Scholar] [CrossRef]
- Pophristic, V.; Vemparala, S.; Ivanov, I.; Liu, Z.; Klein, M.L.; DeGrado, W.F. Controlling the Shape and Flexibility of Arylamides: A Combined ab Initio, ab Initio Molecular Dynamics, and Classical Molecular Dynamics Study. J. Phys. Chem. B 2006, 110, 3517–3526. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, T.; Srivastava, A.; Vrabec, J.; Hasse, H. Hydrogen Bonding of Methanol in Supercritical CO2: Comparison between 1H NMR Spectroscopic Data and Molecular Simulation Results. J. Phys. Chem. B 2007, 111, 9871–9878. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Remsing, R.C.; Liu, D.; Moyna, G.; Pophristic, V. Hydrogen Bonding in ortho-Substituted Arylamides: The Influence of Protic Solvents. J. Phys. Chem. B 2009, 113, 7041–7044. [Google Scholar] [CrossRef] [PubMed]
- Galan, J.F.; Brown, J.; Wildin, J.L.; Liu, Z.; Liu, D.; Moyna, G.; Pophristic, V. Intramolecular Hydrogen Bonding in ortho-Substituted Arylamide Oligomers: A Computational and Experimental Study of ortho-Fluoro- and ortho-Chloro-N-methylbenzamides. J. Phys. Chem. B. 2009, 113, 12809–12815. [Google Scholar] [CrossRef] [PubMed]
- International Union of Pure and Applied Chemistry (IUPAC). Compendium of Chemical Terminology, 2nd ed.; The “Gold Book”; McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Friedman, L.; Litle, R.L.; Reichle, W.R. p-Toluenesulphonylhydrazide [p-Toluenesulphonic acid, hydrazide]. Org. Synth. 1960, 40, 93–96. [Google Scholar]
- Mohareb, R.M.; Fleita, D.H.; Sakka, O.K. Novel Synthesis of Hydrazide-Hydrazone Derivatives and Their Utilization in the Synthesis of Coumarin, Pyridine, Thiazole and Thiophene Derivatives with Antitumor Activity. Molecules 2011, 16, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Wadrdakhan, W.W.; El-saeed, N.N.E.; Mohereb, R.M. Development of coated beads for oral controlled delivery of cefaclor: In vitro evaluation. Acta Pharm. 2013, 63, 45–57. [Google Scholar]
- Swietlińska, Z.; Zuk, J. Cytotoxic Effects of Maleic Hydrazide. Mutat. Res. Rev. Gen. Toxicol. 1978, 55, 15–30. [Google Scholar] [CrossRef]
- Kaplancikli, Z.A.; Turan-Zitouni, G.; Ozdemir, A.; Teulade, J.C. Synthesis and Antituberculosis Activity of New Hydrazide Derivatives. Arch. Pharm. 2008, 341, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Suryaprakash, N. Intramolecular hydrogen bonds involving organic fluorine in the derivatives of hydrazides: an NMR investigation substantiated by DFT based theoretical calculations. Phys. Chem. Chem. Phys. 2015, 17, 15226–15235. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Morris, K.F.; Johnson, C.S. Diffusion-Ordered Two-Dimensional Nuclear Magnetic Resonance Spectroscopy. J. Am. Chem. Soc. 1992, 114, 3139–3141. [Google Scholar] [CrossRef]
- Morris, K.F.; Johnson, C.S. Resolution of Discrete and Continuous Molecular Size Distributions by Means of Diffusion-Ordered 2D NMR Spectroscopy. J. Am. Chem. Soc. 1993, 115, 4291–4299. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Axenrod, T.; Pregosin, P.S.; Wieder, M.J.; Becker, E.D.; Bradley, R.B.; Milne, G.W.A. Nitrogen- 15 Nuclear Magnetic Resonance Spectroscopy. Substituent Effects on 15N-H Coupling Constants and Nitrogen Chemical Shifts in Aniline Derivatives. J. Am. Chem. Soc. 1971, 93, 6536–6541. [Google Scholar] [CrossRef]
- Dunger, A.; Limbach, H.-H.; Weisz, K. Geometry and Strength of Hydrogen Bonds in Complexes of 2’-Deoxyadenosine with 2’-Deoxyuridine. J. Am. Chem. Soc. 2000, 122, 10109–10114. [Google Scholar] [CrossRef]
- Grzesiek, S.; Cordier, F.; Jaravine, V.; Barfield, M. Insights into biomolecular hydrogen bonds from hydrogen bond scalar couplings. Prog. Nucl. Magn. Reson. Spectrosc. 2004, 45, 275–300. [Google Scholar] [CrossRef]
- Afonin, A.V.; Ushakov, I.A.; Sobenina, L.N.; Stepanova, Z.V.; Petrova, O.G.V.; Trofimov, B.A. Different types of hydrogen bonds in 2-substituted pyrroles and 1-vinyl pyrroles as monitored by 1H, 13C and 15N NMR spectroscopy and ab initio calculations. Magn. Reson. Chem. 2006, 44, 59–65. [Google Scholar] [CrossRef] [PubMed]
- King, M.M.; Yeh, H.J.C.; Dudek, G.O. Nitrogen NMR Spectroscopy: Application to some Substituted Pyrroles. Org. Magn. Reson. 1976, 8, 208–212. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Nakahara, M.; Wakai, C. Monomeric and Cluster States of Water Molecules in Organic Solvent. Chem. Lett. 1992, 21, 809–812. [Google Scholar] [CrossRef]
- Vizioli, C.; de Azua, M.C.R.; Giribet, C.G.; Contreras, R.H.; Turi, L.; Dannenberg, J.J.; Rae, I.D.; Weigold, J.A.; Malagoli, M. Proximity Effects on Nuclear Spin-Spin Coupling Constants. 1. 1J(CH) Couplings in the Vicinity of an Atom Bearing Lone Pairs. J. Phys. Chem. 1994, 98, 8858–8861. [Google Scholar] [CrossRef]
- Lakshmipriya, A.; Suryaprakash, N. Two- and Three-Centered Hydrogen Bonds Involving Organic Fluorine Stabilize Conformations of Hydrazide Halo Derivatives: NMR, IR, QTAIM, NCI, and Theoretical Evidence. J. Phys. Chem. A 2016, 120, 7810–7816. [Google Scholar] [CrossRef] [PubMed]
- Walter, W.W.; Michael, H.A. “Polyimides” in Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Prinos, J.; Tselios, C.; Bikiaris, D.; Panayiotou, C. Properties of miscible blends of polyglutarimide with poly(styrene-co-maleic anhydride). Polymer 1997, 38, 5921–5930. [Google Scholar] [CrossRef]
- Luo, L.; Zheng, Y.; Huang, J.; Li, K.; Wang, H.; Feng, Y.; Wang, X.; Liu, X. High-performance copoly(benzimidazole-benzoxazole-imide) fibers: Fabrication, structure, and properties. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Kanaoka, Y. Photoreactions of Cyclic Imides. Examples of Synthetic Organic Photochemistry. Acc. Chem. Res. 1978, 11, 407–413. [Google Scholar] [CrossRef]
- Stec, J.; Huang, Q.; Pieroni, M.; Kaiser, M.; Fomovska, A.; Mui, E.; Witola, W.H.; Bettis, S.; McLeod, R.; Brun, R.; Kozikowski, A.P. Synthesis, Biological Evaluation, and Structure−Activity Relationships of N-Benzoyl-2-hydroxybenzamides as Agents Active against P. falciparum (K1 strain), Trypanosomes, and Leishmania. J. Med. Chem. 2012, 55, 3088–3100. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kelley, S.P.; Brantley, J.W.; Chatel, G.; Shamshina, J.; Pereira, J.F.B.; Debbeti, V.; Myerson, A.S.; Rogers, R.D. Ionic Fluids Containing Both Strongly and Weakly Interacting Ions of the Same Charge Have Unique Ionic and Chemical Environments as a Function of Ion Concentration. ChemPhysChem 2015, 16, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, D.; Maxwell, M.; Goodman, J.; White, B.; Tang, C.M.; Boullay, V.; de Gouville, A.C. Discovery and characterization of GSK256073, a non-flushing hydroxy-carboxylic acid receptor 2 (HCA2) agonist. Eur. J. Pharmacol., 2015, 756, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mhidia, R.; Boll, E.; Fécourt, F.; Ermolenko, M.; Ollivier, N.; Sasaki, K.; Crich, D.; Delpech, B.; Melnyk, O. Exploration of an imide capture/N,N-acyl shift sequence for asparagine native peptide bond formation. Bioorg. Med. Chem. 2013, 21, 3479–3485. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Wang, J. Microwave-Assisted Synthesis of Amides from Various Amines and Benzoyl Chloride under Solvent-Free Conditions: A Rapid and Efficient Method for Selective Protection of Diverse Amines. Russ. J. Organ. Chem. 2008, 44, 358–361. [Google Scholar] [CrossRef]
- Mishra, S.K.; Suryaprakash, N. Organic fluorine involved intramolecular hydrogen bonds in the derivatives of imides: NMR evidence corroborated by DFT based theoretical calculations. RSC Adv. 2015, 5, 86013–86022. [Google Scholar] [CrossRef]
- Contreras-García, J.; Yang, W.; Johnson, E.R. Analysis of Hydrogen-Bond Interaction Potentials from the Electron Density: Integration of Noncovalent Interaction Regions. J. Phys. Chem. A 2011, 115, 12983–12990. [Google Scholar] [CrossRef] [PubMed]
- Gellman, S.H.; Adams, B.R.; Dado, G.P. Temperature-Dependent Changes in the Folding Pattern of a Simple Triamide. J. Am. Chem. Soc. 1990, 112, 460–461. [Google Scholar] [CrossRef]
- Gellman, S.H.; Dado, G.P.; Liang, G.B.; Adams, B.R. Conformation-Directing Effects of a Single Intramolecular Amide-Amide Hydrogen Bond: Variable-Temperature NMR and IR Studies on a Homologous Diamide Series. J. Am. Chem. Soc. 1991, 113, 1164–1173. [Google Scholar] [CrossRef]
- Martinez-Martinez, F.J.; Ariza-Castolo, A.; Tlahuext, H.; Tlahuextl, M.; Contreras, R. 1H, 13C, 15N, 2D and Variable Temperature NMR Study of the Role of Hydrogen Bonding in the Structure and Conformation of Oxamide Derivatives. J. Chem. Soc. Perkin Trans. 2 1993, 1481–1485. [Google Scholar] [CrossRef]
- Rinaldi, P.L. Heteronuclear 2D-NOE Spectroscopy. J. Am. Chem. Soc. 1983, 105, 5167–5168. [Google Scholar] [CrossRef]
- Yu, C.; Levy, G.C. Solvent and Intramolecular Proton Dipolar Relaxation of the Three Phosphates of ATP: A Heteronuclear 2D NOE Study. J. Am. Chem. Soc. 1983, 105, 6994–6996. [Google Scholar] [CrossRef]
- Yu, C.; Levy, G.C. Two-Dimensional Heteronuclear NOE (HOESY) Experiments: Investigation of Dipolar Interactions between Heteronuclei and Nearby Protons. J. Am. Chem. Soc. 1984, 106, 6533–6537. [Google Scholar] [CrossRef]
- Lakshmipriya, A.; Chaudhari, S.R.; Shahi, A.; Arunan, E.; Suryaprakash, N. Three centered hydrogen bonds of the type C-O…H(N)…X–C in diphenyloxamide derivatives involving halogens and a rotating CF3 group: NMR, QTAIM, NCI and NBO studies. Phys. Chem. Chem. Phys. 2015, 17, 7528–7536. [Google Scholar] [CrossRef] [PubMed]
- Shahi, A.; Arunan, E. Hydrogen bonding, halogen bonding and lithium bonding: an atoms in molecules and natural bond orbital perspective towards conservation of total bond order, inter- and intra-molecular bonding. Phys. Chem. Chem. Phys. 2014, 16, 22935–22952. [Google Scholar] [CrossRef] [PubMed]
- Brotherhood, P.R.; Luck, I.J.; Blake, I.M.; Jensen, P.; Turner, P.; Crossley, M.J. Regioselective Reactivity of an Asymmetric Tetravalent Di[dihydroxotin(IV)] Bis-Porphyrin Host Driven by Hydrogen-Bond Templation. Chem. Eur. J. 2008, 14, 10967–10977. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Suryaprakash, N. Study of H/D exchange rates to derive the strength of intramolecular hydrogen bonds in halo substituted organic building blocks: An NMR spectroscopic investigation. Chem. Phys. Letts. 2015, 639, 254–260. [Google Scholar] [CrossRef]
- Pietrzak, M.; Limbach, H.H.; Torralba, M.P.; Sanz, D.; Claramunt, R.M.; Elguero, J. Scalar Coupling Constants Across the Intramolecular NHN-Hydrogen Bond of Symmetrically and Non-symmetrically substituted 6-Aminofulvene-1-aldimines. Magn. Reson. Chem. 2001, 39, S100–S108. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Burtsev, A.P.; Denisov, G.S.; Golubev, N.S.; Limbach, H.H. Influence of the temperature-dependent dielectric constant on the H/D isotope effects on the NMR chemical shifts and the hydrogen bond geometry of the collidine–HF complex in CDF3/CDClF2 solution. Magn. Reson. Chem. 2001, 39, S91–S99. [Google Scholar] [CrossRef]
- Wang, Y.X.; Marquardt, J.L.; Wingfield, P.; Stahl, S.J.; Huang, S.L.; Torchia, D.; Bax, A. Simultaneous Measurement of 1H-15N, 1H-13C’, and 15N-13C’ Dipolar Couplings in a Perdeuterated 30 kDa Protein Dissolved in a Dilute Liquid Crystalline Phase. J. Am. Chem. Soc. 1998, 120, 7385–7386. [Google Scholar] [CrossRef]
- Cornilescu, G.; Hu, J.S.; Bax, A. Identification of the Hydrogen Bonding Network in a Protein by Scalar Couplings. J. Am. Chem. Soc. 1999, 121, 2949–2950. [Google Scholar] [CrossRef]
- Norwood, T.J. Multiple-Quantum NMR Methods. Prog. NMR Spect. 1992, 24, 295–375. [Google Scholar] [CrossRef] [Green Version]
- Bodenhausen, G. Multiple-Quantum NMR. Prog. NMR Spect. 1981, 14, 137–173. [Google Scholar] [CrossRef]
- Divya, K.; Hebbar, S.; Suryaprakash, N. Intra-molecular hydrogen bonding with organic fluorine in the solution state: Deriving evidence by a two dimensional NMR experiment. Chem. Phys. Lett. 2012, 525–526, 129–133. [Google Scholar] [CrossRef]
- Hebbar, S.; Suryaprakash, N. Spin-selective multiple quantum excitation: Relative signs of the couplings and ambiguous situations. J. Magn. Reson. 2008, 194, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Munowitz, M.; Pines, A. Homogeneous NMR spectra in inhomogeneous fields. Science 1986, 233, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Bikash, B.; Suryaprakash, N. Spin selective multiple quantum NMR for spectral simplification, determination of relative signs, and magnitudes of scalar couplings by spin state selection. J. Chem. Phys. 2007, 127, 214510–214521. [Google Scholar]
- Griffey, R.H.; Poulter, C.D.; Bax, A.; Hawkins, B.L.; Yamaizumi, Z.; Nishimura, S. Multiple quantum two-dimensional 1H- 15N nuclear magnetic resonance spectroscopy: Chemical shift correlation maps for exchangeable imino protons of Escherichia coli tRNAfMet in water. Proc. Nat. Acad. Sci. USA 1983, 80, 5895–5897. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian G09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N∙log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (Version 13.11.04); TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Takemura, H.; Kon, N.; Yasutake, M.; Nakashima, S.; Shinmyozu, T.; Inazu, T. The C-F…Cation Interaction: An Ammonium Complex of a Hexafluoro Macrocyclic Cage Compound. Chem. Eur. J. 2000, 6, 2334–2337. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, X.Z.; Jiang, X.K.; Chen, Y.Q.; Li, Z.T.; Chen, G.J. Hydrazide-Based Quadruply Hydrogen-Bonded Heterodimers. Structure, Assembling Selectivity, and Supramolecular Substitution. J. Am. Chem. Soc. 2003, 125, 15128–15139. [Google Scholar] [CrossRef] [PubMed]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, S.K.; Suryaprakash, N. Intramolecular Hydrogen Bonding Involving Organic Fluorine: NMR Investigations Corroborated by DFT-Based Theoretical Calculations. Molecules 2017, 22, 423. https://doi.org/10.3390/molecules22030423
Mishra SK, Suryaprakash N. Intramolecular Hydrogen Bonding Involving Organic Fluorine: NMR Investigations Corroborated by DFT-Based Theoretical Calculations. Molecules. 2017; 22(3):423. https://doi.org/10.3390/molecules22030423
Chicago/Turabian StyleMishra, Sandeep Kumar, and N. Suryaprakash. 2017. "Intramolecular Hydrogen Bonding Involving Organic Fluorine: NMR Investigations Corroborated by DFT-Based Theoretical Calculations" Molecules 22, no. 3: 423. https://doi.org/10.3390/molecules22030423