
Biologic Stress, Oxidative Stress, and Resistance to Drugs: What Is Hidden Behind

Abstract
:
1. Introduction
2. Stress
3. Xenobiotic Metabolism-Resistance to Drugs
4. Stress and the Resistance of the Body to Drugs
5. Biologic Stress and Oxidative Stress
6. Effect of Stress on Hepatic Drug Metabolic Activity
7. Structure-Activity Relationships
8. Conclusions
Conflicts of Interest
References
- Selye, H. Stress in Health and Disease; Butterworths: Boston, MA, USA, 1976. [Google Scholar]
- Kobler, J. The Reluctant Surgeon; a Biography of John Hunter, 1st ed.; Doubleday: Garden City, NY, USA, 1960. [Google Scholar]
- Selye, H. The Stress of Life, Rev. ed.; McGraw-Hill: New York, NY, USA, 1978. [Google Scholar]
- Bernard, C. Lectures on the Phenomena of Life Common to Animals and Plants; Thomas: Springfield, IL, USA, 1974. [Google Scholar]
- Cannon, W.B. The Wisdom of the Body; W.W. Norton & Company: New York, NY, USA, 1939. [Google Scholar]
- Selye, H. Hormones and Resistance; Springer-Verlag: Berlin, Germany; New York, NY, USA, 1971. [Google Scholar]
- Kourounakis, P.N. Steroids, Drug Response and Metabolism: A Pharmaceutical Approach to Defensive Steroids; Ellis Horwwod: Chichester, UK, 1993. [Google Scholar]
- Kasahara, E.; Sekiyama, A.; Hori, M.; Kuratsune, D.; Fujisawa, N.; Chida, D.; Hiramoto, K.; Li, J.; Okamura, H.; Inoue, M.; et al. Stress-Induced Glucocorticoid Release Upregulates Uncoupling Protein-2 Expression and Enhances Resistance to Endotoxin-Induced Lethality. Neuroimmunomodulation 2015, 22, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Kourounakis, P.N.; Rekka, E. Induction of drug metabolism can be a homeostatic response. Arch. Pharm. 1991, 324, 161–164. [Google Scholar] [CrossRef]
- Toates, F.M. Stress: Conceptual and Biological Aspects; Wiley: Chichester, NY, USA, 1995. [Google Scholar]
- Kourounakis, P.N.; Rekka, E. The Pharmacochemistry of stress: effect of chlordiazepoxide and diazepam in some manifestations of stress. Sci. Pharm. 1990, 58, 389–393. [Google Scholar]
- Levy, A.; Grauer, E.; Ben-Nathan, D.; De Kloet, E.R. New Frontiers in Stress Research: Modulation of Brain Function; Harwood Academic Publishers: Amsterdam, The Netherlands, 1998. [Google Scholar]
- Shirazi, S.N.; Friedman, A.R.; Kaufer, D.; Sakhai, S.A. Glucocorticoids and the Brain: Neural Mechanisms Regulating the Stress Response. Adv. Exp. Med. Biol. 2015, 872, 235–252. [Google Scholar] [PubMed]
- Szabo, S.; Tache, Y.; Somogyi, A. The legacy of Hans Selye and the origins of stress research: A retrospective 75 years after his landmark brief “letter” to the editor# of nature. Stress 2012, 15, 472–478. [Google Scholar] [PubMed]
- Salas, M.; Tuchweber, B.; Kourounakis, P.; Selye, H. Temperature-dependence of stress-induced hepatic autophagy. Experientia 1977, 33, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Salas, M.; Tuchweber, B.; Kourounakis, P. Liver ultrastructure during acute stress. Pathol. Res. Pract. 1980, 167, 217–233. [Google Scholar] [CrossRef]
- Tsiakitzis, K.; Kourounakis, A.P.; Tani, E.; Rekka, E.A.; Kourounakis, P.N. Stress and active oxygen species--effect of alpha-tocopherol on stress response. Arch. Pharm. 2005, 338, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Tsiakitzis, K.C.; Rekka, E.A.; Kourounakis, A.P.; Kourounakis, P.N. Novel compounds designed as antistress agents. J. Med. Chem. 2009, 52, 7315–7318. [Google Scholar] [CrossRef] [PubMed]
- Keverling Buisman, J.A.; International Union of Pure and Applied Chemistry; Commission on Medicinal Chemistry; International Union of Pharmacology. Strategy in drug research. In Proceedings of the Second IUPAC-IUPHAR Symposium, Noordwijkerhout, The Netherlands, 25–28 August 1981; Elsevier Science Pub. Co.: Amsterdam, The Netherlands, 1982. [Google Scholar]
- Albert, A. Selective Toxicity: The Physico-Chemical Basis of Therapy, 7th ed.; Chapman and Hall: London, UK, 1985. [Google Scholar]
- Kourounakis, P.; Selye, H. Influence of steroids and stress on toxicity and disposition of tetraethylammonium bromide. J. Pharm. Sci. 1976, 65, 1838–1840. [Google Scholar] [CrossRef] [PubMed]
- Kourounakis, P.; Szabo, S.; Selye, H. Effect of fluorosteroids on drug response and metabolism. Biochem. Pharmacol. 1976, 25, 477–481. [Google Scholar] [CrossRef]
- Kourounakis, P.; Szabo, S.; Werringloer, J.; Selye, H. Effect of various steroids and ACTH on plasma levels of zoxazolamine and dicumarol. J. Pharm. Sci. 1973, 62, 690–692. [Google Scholar] [CrossRef] [PubMed]
- Rapp, U.; Kourounakis, P.; Selye, H. Effect of steroids and diethylstilbestrol on cocaine toxicity, plasma concentrations and urinary excretion. Arzneimittel-Forschung 1979, 29, 48–50. [Google Scholar] [PubMed]
- Kourounakis, P.N.; Rekka, E. Effect of spironolactone on dimethyl mercury toxicity. A possible molecular mechanism. Arzneimittel-Forschung 1994, 44, 1150–1153. [Google Scholar] [PubMed]
- Kourounakis, P.N.; Pouskoulelis, G.P.; Rekka, E. Interaction of spironolactone with mercury. A possible molecular mechanism. Arzneimittel-Forschung 1992, 42, 1025–1028. [Google Scholar] [PubMed]
- Rigalli, J.P.; Ruiz, M.L.; Perdomo, V.G.; Villanueva, S.S.; Mottino, A.D.; Catania, V.A. Pregnane X receptor mediates the induction of P-glycoprotein by spironolactone in HepG2 cells. Toxicology 2011, 285, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Kourounakis, P.; Szabo, S.; Selye, H. Letter: Effect of various steroids and ACTH on methyprylon plasma levels and sleeping time in rats. J. Pharm. Pharmacol. 1973, 25, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Selye, H. Homeostasis and heterostasis. Perspect. Biol. Med. 1973, 16, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Kourounakis, P.; Szabo, S.; Selye, H. Effect of various steroids and ACTH on distribution of zoxazolamine in rats. J. Pharm. Sci. 1973, 62, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.; Kourounakis, P.; Selye, H. Effect of ACTH and pregnenolone-16a-carbonitrile on free plasma zoxazolamine level. Stimulation of urinary and biliary excretion of zoxazolamine by ACTH. Fed. Proc. 1974, 33, 532. [Google Scholar]
- Szabo, S.; Kourounakis, P.; Selye, H. Influence of adrenocorticotropic hormone, somatotrophic hormone and pregnenolone-16alpha-carbonitrile on drug response and metabolism. Biochem. Pharmacol. 1975, 24, 1549–1551. [Google Scholar] [CrossRef]
- Pincus, G.; Thimann, K.V.; Astwood, E.B. The Hormones: Physiology, chemistry, and Applications; Academic Press: New York, NY, USA, 1948. [Google Scholar]
- Guseinov Sh, G.; Aliev, M.G.; Kurbanov, T.G. Changes in the function of the hypothalamo-hypophyseo-adrenal system and the thymus as affected by corticotropin in guinea pigs. Probl. Endokrinol. 1982, 28, 51–55. [Google Scholar]
- Martocchia, A.; Stefanelli, M.; Falaschi, G.M.; Toussan, L.; Rocchietti March, M.; Raja, S.; Romano, G.; Falaschi, P. Targets of anti-glucocorticoid therapy for stress-related diseases. Recent Pat. CNS Drug Discovery 2013, 8, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Natelson, B.H.; Ottenweller, J.E.; Pitman, D.; Tapp, W.N. An assessment of prolactin's value as an index of stress. Life Sci. 1988, 42, 1597–1602. [Google Scholar] [CrossRef]
- Matoulkova, P.; Pavek, P.; Maly, J.; Vlcek, J. Cytochrome P450 enzyme regulation by glucocorticoids and consequences in terms of drug interaction. Expert Opin. Drug Metab. Toxicol. 2014, 10, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, C.Y.; Kong, A.N. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 2005, 28, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yeo, H.C.; Overvik-Douki, E.; Hagen, T.; Doniger, S.J.; Chyu, D.W.; Brooks, G.A.; Ames, B.N. Chronically and acutely exercised rats: Biomarkers of oxidative stress and endogenous antioxidants. J. Appl. Physiol. 2000, 89, 21–28. [Google Scholar] [PubMed]
- Di Simplicio, P.; Rossi, R.; Falcinelli, S.; Ceserani, R.; Formento, M.L. Antioxidant status in various tissues of the mouse after fasting and swimming stress. Eur. J. Appl. Physiol. Occup. Physiol. 1997, 76, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Orellana, M.; Fuentes, O.; Valdes, E. Starvation effect on rat kidney peroxisomal and microsomal fatty acid oxidation. A comparative study between liver and kidney. FEBS Lett. 1993, 322, 61–64. [Google Scholar] [CrossRef]
- Longo, V.; Ingelman-Sundberg, M.; Amato, G.; Salvetti, A.; Gervasi, P.G. Effect of starvation and chlormethiazole on cytochrome P450s of rat nasal mucosa. Biochem. Pharmacol. 2000, 59, 1425–1432. [Google Scholar] [CrossRef]
- Bondoc, F.Y.; Bao, Z.; Hu, W.Y.; Gonzalez, F.J.; Wang, Y.; Yang, C.S.; Hong, J.Y. Acetone catabolism by cytochrome P450 2E1: Studies with CYP2E1-null mice. Biochem. Pharmacol. 1999, 58, 461–463. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Parke, D.V.; Ioannides, C.; Lewis, D.F. The 1990 Pharmaceutical Manufacturers Association of Canada keynote lecture. The role of the cytochromes P450 in the detoxication and activation of drugs and other chemicals. Can. J. Physiol. Pharmacol. 1991, 69, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; Stojdl, D.F.; Bell, J.C.; Hettmann, T.; Leiden, J.M.; Ron, D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Kelly, M. J. The pharmacology of vitamin E. Prog. Med. Chem. 1988, 25, 249–290. [Google Scholar] [PubMed]
- al-Shabanah, O.A.; Mostafa, Y.H.; Hassan, M.T.; Khairaldin, A.A.; al-Sawaf, H.A. Vitamin E protects against bacterial endotoxin-induced increase of plasma corticosterone and brain glutamate in the rat. Res. Commun. Mol. Pathol. Pharmacol. 1996, 92, 95–105. [Google Scholar] [PubMed]
- Azzi, A.; Stocker, A. Vitamin E: Non-antioxidant roles. Prog. Lipid Res. 2000, 39, 231–255. [Google Scholar] [CrossRef]
- Cerezo, M.; Laorden, M.L.; Milanes, M.V. Inhibition of protein kinase C but not protein kinase A attenuates morphine withdrawal excitation of rat hypothalamus-pituitary-adrenal axis. Eur. J. Pharmacol. 2002, 452, 57–66. [Google Scholar] [CrossRef]
- Ohta, Y.; Yashiro, K.; Ohashi, K.; Horikoshi, Y.; Kusumoto, C.; Matsura, T.; Fukuzawa, K. Effect of Dietary Vitamin E Supplementation on Liver Oxidative Damage in Rats with Water-Immersion Restraint Stress. J. Nutr. Sci. Vitaminol. 2015, 61, 113–122. [Google Scholar] [CrossRef] [PubMed]
- de Diego-Otero, Y.; Romero-Zerbo, Y.; el Bekay, R.; Decara, J.; Sanchez, L.; Rodriguez-de Fonseca, F.; del Arco-Herrera, I. Alpha-tocopherol protects against oxidative stress in the fragile X knockout mouse: An experimental therapeutic approach for the Fmr1 deficiency. Neuropsychopharmacology 2009, 34, 1011–1026. [Google Scholar] [CrossRef] [PubMed]
- Nur Azlina, M.F.; Nafeeza, M.I. Tocotrienol and alpha-tocopherol reduce corticosterone and noradrenalin levels in rats exposed to restraint stress. Pharmazie 2008, 63, 890–892. [Google Scholar] [PubMed]
- Bousquet, W.F.; Rupe, B.D.; Miya, T.S. Endocrine Modification of Drug Responses in the Rat. J. Pharmacol. Exp. Ther. 1965, 147, 376–379. [Google Scholar] [PubMed]
- Stitzel, R.E.; Furner, R.L. Stress-induced alterations in microsomal drug metabolism in the rat. Biochem. Pharmacol. 1967, 16, 1489–1494. [Google Scholar] [CrossRef]
- Alexidis, A.N.; Commandeur, J.N.; Rekka, E.A.; Groot, E.; Kourounakis, P.N.; Vermeulen, N.P. Novel piperidine derivatives: Inhibitory properties towards cytochrome P450 isoforms, and cytoprotective and cytotoxic characteristics. Environ. Toxicol. Pharmacol. 1996, 1, 81–88. [Google Scholar] [CrossRef]
- Rekka, E.; Ayalogu, E.O.; Lewis, D.F.; Gibson, G.G.; Ioannides, C. Induction of hepatic microsomal CYP4A activity and of peroxisomal beta-oxidation by two non-steroidal anti-inflammatory drugs. Arch. Toxicol. 1994, 68, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Daskalopoulos, E.P.; Malliou, F.; Rentesi, G.; Marselos, M.; Lang, M.A.; Konstand, M. Stress is a critical player in CYP3A, CYP2C, and CYP2D regulation: Role of adrenergic receptor signaling pathways. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E40–54. [Google Scholar] [CrossRef] [PubMed]
- Kourounakis, A.P.; Rekka, E.A.; Kourounakis, P.N. Effect of guaiazulene on some cytochrome P450 activities. Implication in the metabolic activation and hepatotoxicity of paracetamol. Arch. Pharm. 1997, 330, 7–11. [Google Scholar] [CrossRef]
- Avery, M.A.; Woolfrey, J.R. Anti-inflammatory steroids. In Burger’s Medicinal Chemistry and Drug Discovery; John Wiley & Sons, Inc.: New York, NY, USA, 2003; pp. 747–854. [Google Scholar]
- Dvorak, Z.; Pavek, P. Regulation of drug-metabolizing cytochrome P450 enzymes by glucocorticoids. Drug Metab. Rev. 2010, 42, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Kourounakis, P.N.; Rekka, E. Structural considerations of the 16-cyano and related pregnenolones on their drug metabolic inducing activity. Eur. J. Med. Chem. 1990, 25, 701–704. [Google Scholar] [CrossRef]
- Kourounakis, P.N.; Rekka, E.; Demopoulos, V.J.; Retsas, S. Effect of the position of the cyano-group of cyanopregnenolones on their drug metabolic inducing activity. Eur. J. Drug Metab. Pharmacokinet. 1991, 16, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Rekka, E.A.; Kourounakis, P.N. An approach to QSAR of 16-substituted pregnenolones as microsomal enzyme inducers. Eur. J. Drug Metab. Pharmacokinet. 1996, 21, 7–11. [Google Scholar] [CrossRef] [PubMed]
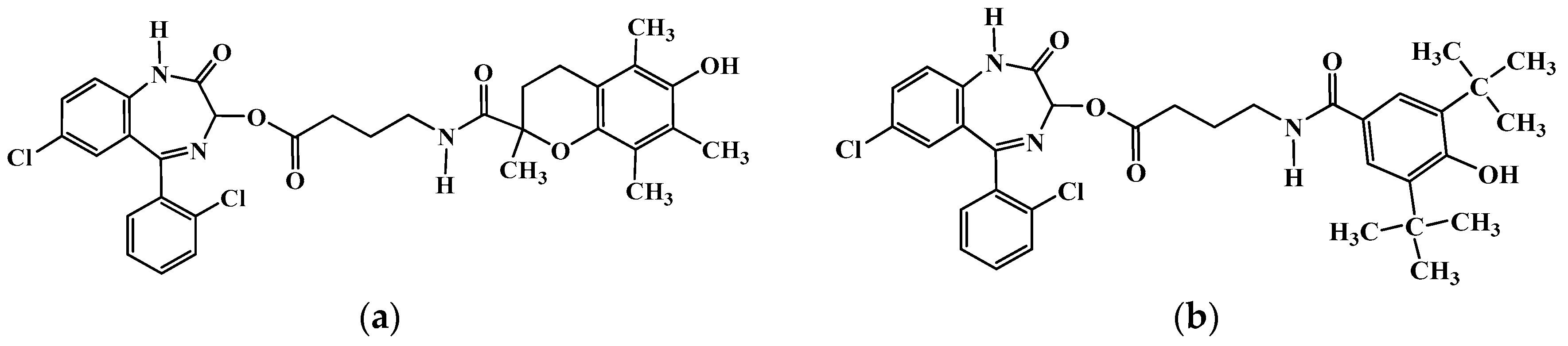

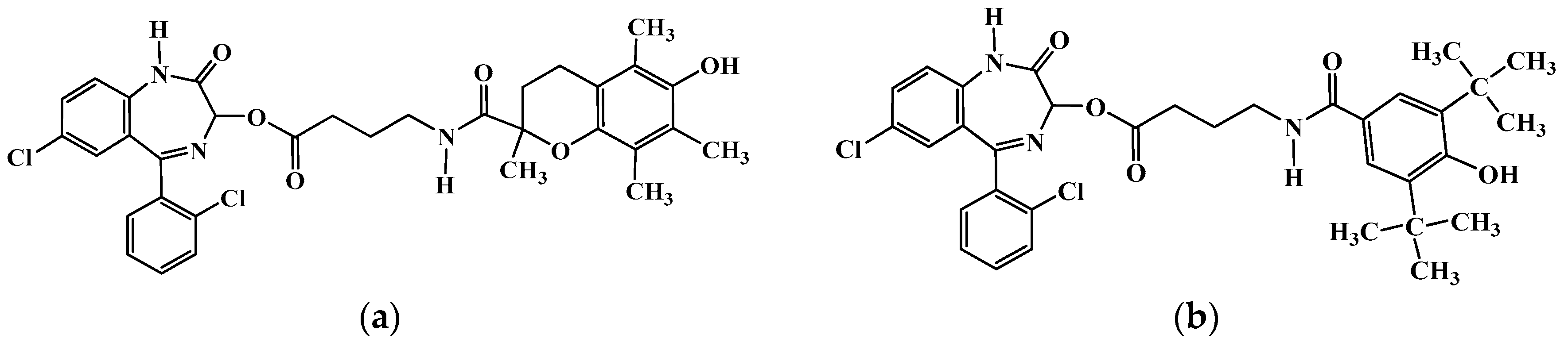
{kind=link}
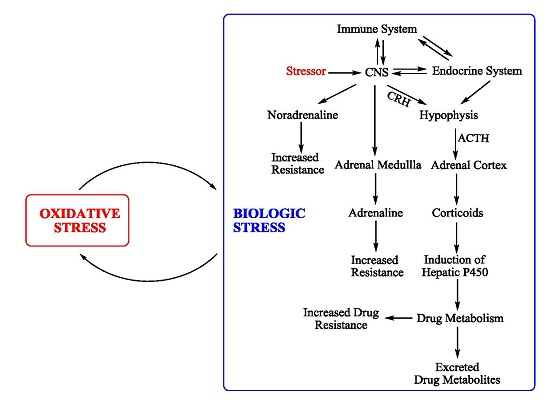
{kind=link}
| Treatment | Thymus (mg/100 g) | Adrenals (mg/100 g) | Spleen (mg/100 g) | Liver (mg/100 g) | Body Weight Change (%) | Uropepsinogen (KU/h) | Corticosterone (μg/100 mL) |
|---|---|---|---|---|---|---|---|
| control | 214 ± 9 | 33 ± 1 | 251 ± 10 | 4.70 ± 0.10 | 4.2 ± 0.1 | 15 ± 1 | 20 ± 1 |
| Control + α-Toc | 240 ± 10 ** ++ | 39 ± 2 ** ns | 281 ± 10 ** +++ | 4.90 ± 0.10 * ++ | 3.8 ± 0.3 ns +++ | 15 ± 1 ns ++ | 25 ± 2 ns ++ |
| stress | 92 ± 7 ** | 40 ± 1 *** | 131 ± 3 *** | 3.10 ± 0.02 *** | −28.1 ± 1.0 *** | 55 ± 3 ** | 58 ± 2 ** |
| Stress + α-Τοc | 114 ± 5 *** +++ xxx | 42 ± 3 ** ns ns | 155 ± 5 *** +++ xxx | 3.20 ± 0.04 *** + xxx | −21.0 ± 0.7 *** +++ xxx | 21 ± 2 ** ++ | 41 ± 2 ** ++ xx |
| Stress + 48 h rest | 142 ± 18 ** ++ | 35 ± 1 ns + | 229 ± 11 * +++ | 3.60 ± 0.20 ** ++ | −5.2 ± 2.5 *** +++ | 12 ± 1 ++ | ns ns |
| Treatment | Thymus (mg/100 g) | Adrenals (mg/100 g) | Spleen (mg/100 g) | Liver (mg/100 g) | Body Weight Change (%) | Uropepsinogen (KU/h) |
|---|---|---|---|---|---|---|
| control | 214 ± 9 | 32 ± 0.6 | 251 ± 10 | 4.6 ± 0.15 | +4.1 ± 0.08 | 15 ± 0.9 |
| stress | 87 ± 9 *** | 46 ± 1 *** | 127 ± 11 *** | 3.4 ± 0.009 *** | −16.8 ± 1.4 *** | 127 ± 18 *** |
| Stress + comp. I | 117 ± 22 *** + | 34 ± 2 ns ++ | 123 ± 3 *** ns | 4.2 ± 0.007 * +++ | −10 ± 1.1 *** ++ | 34 ± 8.4 *** +++ |
| Stress + comp. II | 151 ± 7 *** +++ | 40 ± 3 *** ++ | 124 ± 12 *** ns | 4.5 ± 0.31 ns + | −3.8 ± 1.6 *** +++ | 30 ± 13 ** +++ |
| Steroid (Treatment) a | Zoxazolamine (10 mg/100g b.w., i.p.) | |||
|---|---|---|---|---|
| Plasma Conc. (μg/mL) | Paralysis Time (min) | |||
| Treated | Control | Treated | Control | |
| PCN | 25.5 ± 0.7 *** | 41.4 ± 0.3 | 19 ± 1 *** | 115 ± 17 |
| ESTR | 25.3 ± 0.7 *** | 30.5 ± 1.1 | 166 ± 10 *** | 203 ± 23 |
| PROG | 22.0 ± 1.0 ** | 27.9 ± 0.7 | 103 ± 15 ns | 100 ± 3 |
| SNL | 23.0 ± 1.0 ** | 26.4 ± 0.9 | 78 ± 3 ** | 107 ± 8 |
| DEX | 18.9 ± 1.1 *** | 28.03 ± 1.7 | 31 ± 2 *** | 128 ± 7 |
| TRIAM | 22.0 ± 1.0 ** | 27.2 ± 1.2 | 115 ± 9 * | 160 ± 15 |
| Steroid (Treatment) a | Zoxazolamine (10mg/100 g b.w., i.p.) | |||
|---|---|---|---|---|
| Plasma Conc. (μg/mL) | Sleeping Time (min) | |||
| Treated | Control | Treated | Control | |
| PCN | 97.3 ± 7 *** | 144.5 ± 5.2 | 14 ± 6 *** | 100 ± 15 |
| SNL | 80.2 ± 2.3 *** | 136.4 ± 13 | 82 ± 6 ** | 220 ± 20 |
| TRIAM | 131.2 ± 11 ns | 119.5 ± 9 | 68 ± 4 * | 101 ± 12 |
| Steroid (Treatment) a | Tetraethylammonium Bromide (10mg/100 g b.w., i.p.) | |||
|---|---|---|---|---|
| Plasma Conc. (μg/mL) | Dyskinesia (Positive/Total) | Mortality (Positive/Total) | ||
| Treated | Control | Treated | Control | |
| PCN | 39.8 ± 2.8 * | 45.5 ± 2.3 | 13/18 * | 1/18 |
| SNL | 19.4 ± 0.7 ** | 45.3 ± 2.3 | 9/18 *** | 0/18 |
| TRIAM | 24.7 ± 1.6 ** | 45.1 ± 2.3 | 3/18 *** | 0/18 |
| Steroid | Zoxazolamine Metabolism | Ethylmorphine Metabolism | Protein a | |||
|---|---|---|---|---|---|---|
| μmol/g/h | % Increase | HCHO μmol/g/h | % Increase | mg/g | % Increase | |
| PCN | 42.8 ± 16 *** (16.0 ± 1.4) | 166 | 388.4 ± 22.7 *** (72.4 ± 2.3) | 437 | 104.2 ± 9.1 * (97.4 ± 2.0) | 7 |
| SNL | 45.7 ± 0.3 *** (19.8 ± 0.6) | 130 | 203.7 ± 13.0 *** (48.3 ± 20.6) | 132 | 99.7 ± 1.2 (97.4 ± 2.0) | 2 |
| DEX | 65.3 ± 3.0 *** (20.9 ± 2.0) | 213 | 483.0 ± 20.6 *** (68.6 ± 2.3) | 603 | 82.8 ± 1.3 (84.3 ± 1.3) | −2 |
| BET | 73.0 ± 1.5 *** (28.3 ± 2.1) | 198 | 422.5 ± 28.0 *** (116.8 ± 13.2) | 262 | 79.4 ± 2.4 (84.3 ± 1.3) | −6 |
| FLUDR | 24.8 ± 1.1 *** (16.0 ± 1.4) | 54 | 180.7 ± 17.0 *** (78.0 ± 0.6) | 131 | 77.7 ± 1.6 (84.3 ± 1.4) | −8 |
| TRIAM | 28.3 ± 2.1 (24.5 ± 2.2) | 15 | 151.2 ± 13.3 (116.8 ± 13.1) | 30 | 84.3 ± 1.7 (84.1 ± 1.4) | 1 |
| ACTH | 30.9 ± 1.4 (25.8 ± 1.7) | 19 | 132.3 ± 9.1 (166.0 ± 10.9) | -20 | 82.7 ± 1.7 (86.0 ± 2.0) | −4 |
| Pretreatment | Group | Zoxazolamine Concentration (μg/mL) | Cbr/Cbl | ||
|---|---|---|---|---|---|
| Plasma | Brain | Adipose Tissue | |||
| PCN | control pretreated | 35.4 ± 1.1 26.1 ± 1.4 *** | 98.2 ± 6.9 63.6 ± 4.8 *** | 92.9 ± 3.1 59.1 ± 2.9 *** | 2.8 2.4 |
| TRIAM | control pretreated | 28.8 ± 1.3 32.9 ± 1.7 * | 91.2 ± 4.1 85.4 ± 4.2 ns | 81.6 ± 4.1 79.1 ± 2.4 ns | 3.2 2.6 |
| FLUDR | control pretreated | 26.6 ± 1.4 23.1 ± 1.3 *** | 150.6 ± 3.6 101.3 ± 2.3 *** | 146.5 ± 7.5 87.8 ± 4.4 *** | 5.7 4.4 |
| ACTH | control pretreated | 36.9 ± 2.3 37.3 ± 2.1 ns | 111.9 ± 7.5 106.7 ± 4.3 ns | 106.0 ± 6.5 94.7 ± 3.9 ns | 3.0 2.9 |
| Group | Pretreatment | Paralysis Time min | Zoxazolamine Metabolism μmol/g Liver/h (%) | Protein mg/g Liver | Liver Weight g/100 g b.w. |
|---|---|---|---|---|---|
| 1 | control (none) | 188 ± 10 | 2.18 ± 0.12 (100) | 93.42 ± 0.66 | 4.7 ± 0.16 |
| 2 | ACTH | 89 ± 111 *** | 2.47 ± 0.18 ns (113) | 90.76 ± 1.76 ns | 5.41 ± 0.08 ** |
| 3 | PCN (1 mg) | 70 ± 9 *** | 4.77 ± 0.17 *** (218) | 93.36 ± 1.41 ns | 5.21 ± 0.08 * |
| 4 | PCN (0.1 mg) | 155 ± 12 * | 2.90 ± 0.22 * (133) | 92.16 ± 1.89 ns | 4.94 ± 0.06 ns |
| 5 | STH (twice) | 184 ± 12 ns | 1.79 ± 0.16 ns (82) | 105.52 ± 1.56 * | 4.58 ± 0.08 ns |
| 6 | PCN + ACTH | 45 ± 6 *** (***2) (*3) | 5.19 ± 0.16 *** (237) | 90.46 ± 0.97ns | 6.22 ± 0.08 *** (***3) |
| 7 | PCN + STH | 238 ± 16 ***4 | 2.56 ± 1.12 ** (117) | 94.92 ± 2.61ns | 4.84 ± 0.19 ns |
| Treatment | Liver | Brain | |||
|---|---|---|---|---|---|
| MDA (nmol/mg Protein) | GSH (nmol/mg Protein) | MDA (nmol/mg Protein) | GSH (nmol/mg Protein) | Protein Carbonyl Content (nmol/mg Protein) | |
| Control | 15.9 ± 4.1 | 2.0 ± 0.4 | 0.63 ± 0.06 | 0.27 ± 0.04 | 0.18 ± 0.01 |
| Control + α-Τοc | 2.7 ± 1.1 * ++ | 2.3 ± 0.3 ns ++ | 0.68 ± 0.10 ns ++ | 0.38 ± 0.05 ns ++ | 0.15 ± 0.01 ** ++ |
| Stress | 49.8 ± 5.8 ** | 0.4 ± 0.1 ** | 1.80 ± 0.19 ** | 0.14 ± 0.02 * | 0.28 ± 0.01 ** |
| Stress + α-Τοc | 18.8 ± 2.4 ns ++ x | 1.4 ± 0.1 ns ++ ns | 0.82 ± 0.13 ns ++ ns | 0.30 ± 0.02 ns ++ ns | 0.20 ± 0.02 ns ++ xx |
| Liver Microsomal Parameter | Control Group | Stressed Group | ||
|---|---|---|---|---|
| Absolute | α-Tocopherol Treated | Untreated | α-Tocopherol Treated | |
| Total P450 nmol/mg prot | 0.22 | 0.30 ** ++ | 0.41 ** | 0.40 ** ++ |
| Erythromycin N-demethylation HCHO nmol/min/nmol P450 | 0.95 | 2.20 ** + | 1.85 * | 2.70 ** ++ |
| Nitrocatechol hydroxylation nmol/min/nmol P450 | 18.5 | 28.0 ++ | 52.0 ** | 59.0 ** ++ |
| Liver Microsomal Parameter | Control Group | Stressed Group | |
|---|---|---|---|
| Untreated | Compound I Treated | ||
| Total P450 nmol/mg prot | 0.23 | 0.53 ** | 0.32 ** ++ |
| Erythromycin N-demethylation HCHO nmol/min/nmol P450 | 1.0 | 2.4 ** | 1.6 + |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pantelidou, M.; Tsiakitzis, K.; Rekka, E.A.; Kourounakis, P.N. Biologic Stress, Oxidative Stress, and Resistance to Drugs: What Is Hidden Behind. Molecules 2017, 22, 307. https://doi.org/10.3390/molecules22020307
Pantelidou M, Tsiakitzis K, Rekka EA, Kourounakis PN. Biologic Stress, Oxidative Stress, and Resistance to Drugs: What Is Hidden Behind. Molecules. 2017; 22(2):307. https://doi.org/10.3390/molecules22020307
Chicago/Turabian StylePantelidou, Maria, Karyofyllis Tsiakitzis, Eleni A. Rekka, and Panos N. Kourounakis. 2017. "Biologic Stress, Oxidative Stress, and Resistance to Drugs: What Is Hidden Behind" Molecules 22, no. 2: 307. https://doi.org/10.3390/molecules22020307