
Reaction of 3-Amino-1,2,4-Triazole with Diethyl Phosphite and Triethyl Orthoformate: Acid-Base Properties and Antiosteoporotic Activities of the Products

 and
and 
Abstract
:1. Introduction
2. Results and Discussion
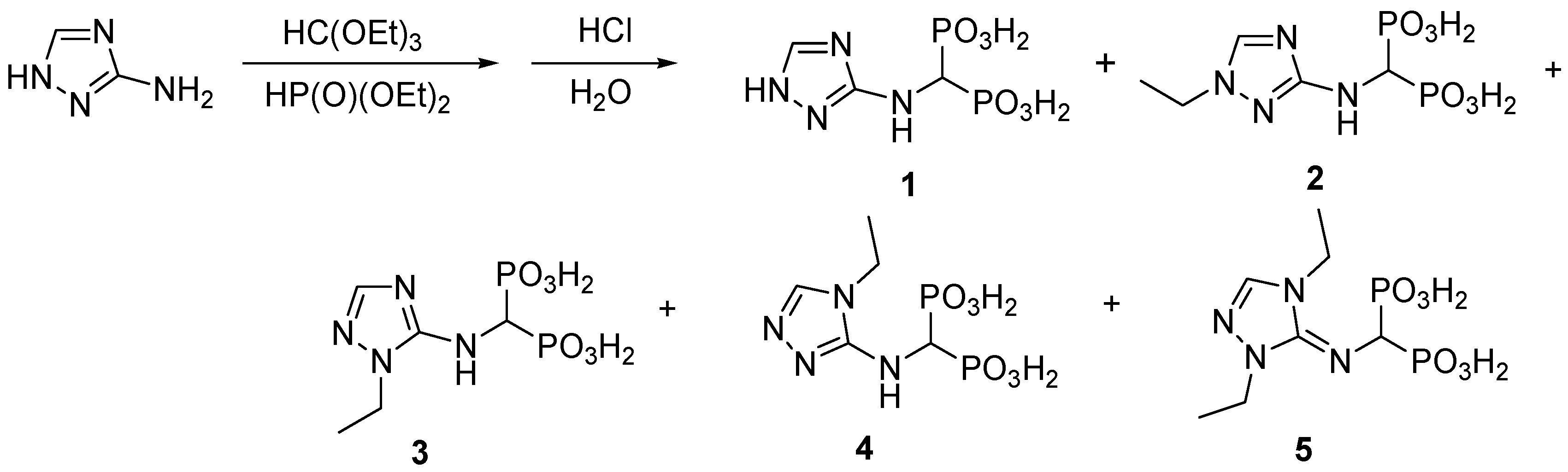
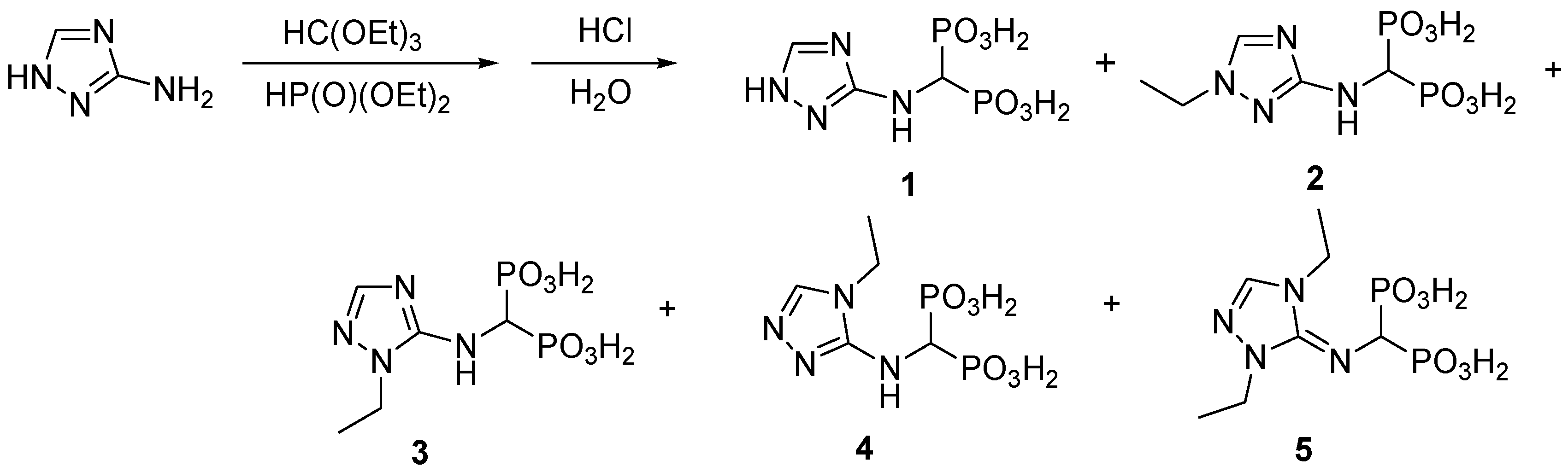
2.1. Chemistry
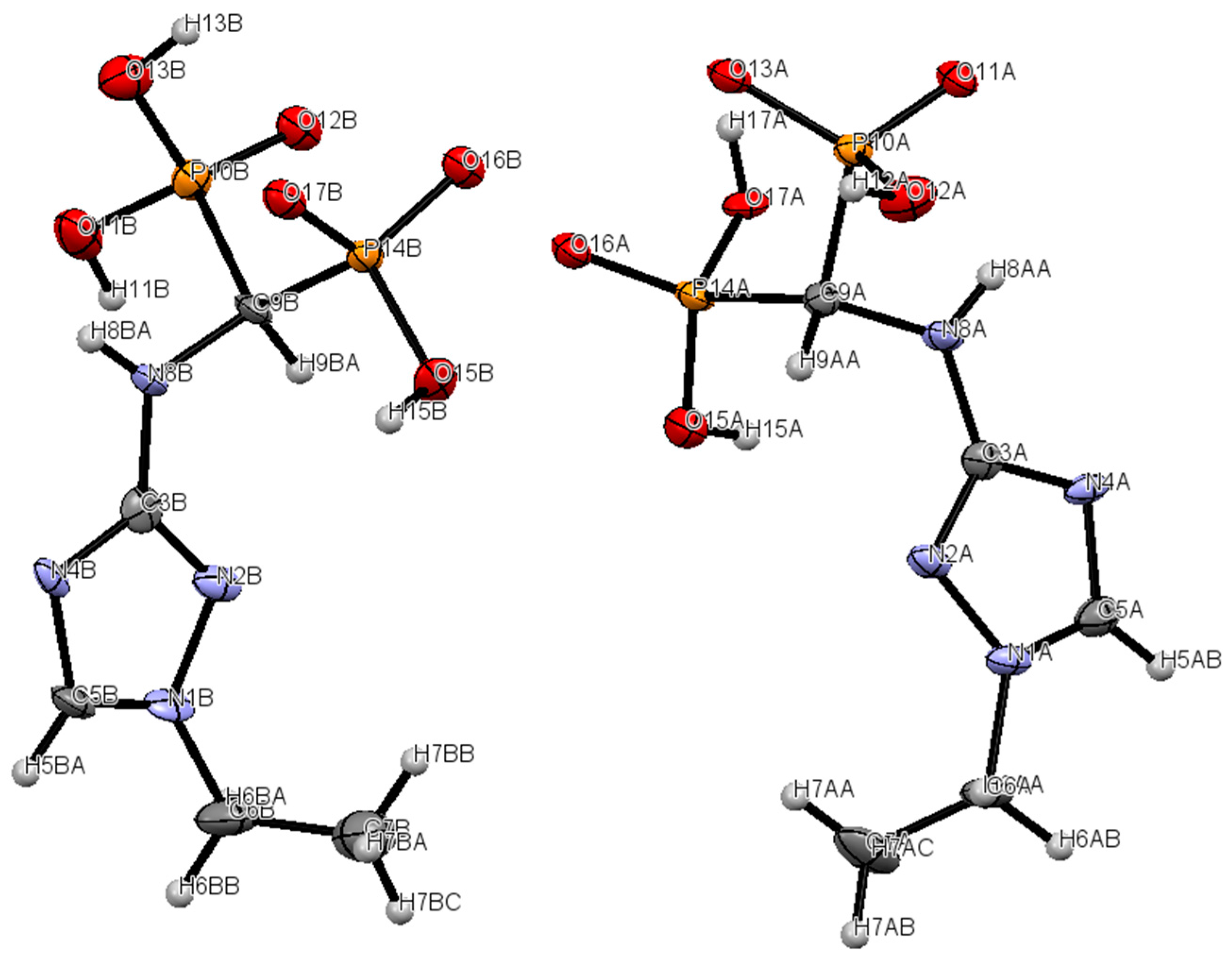
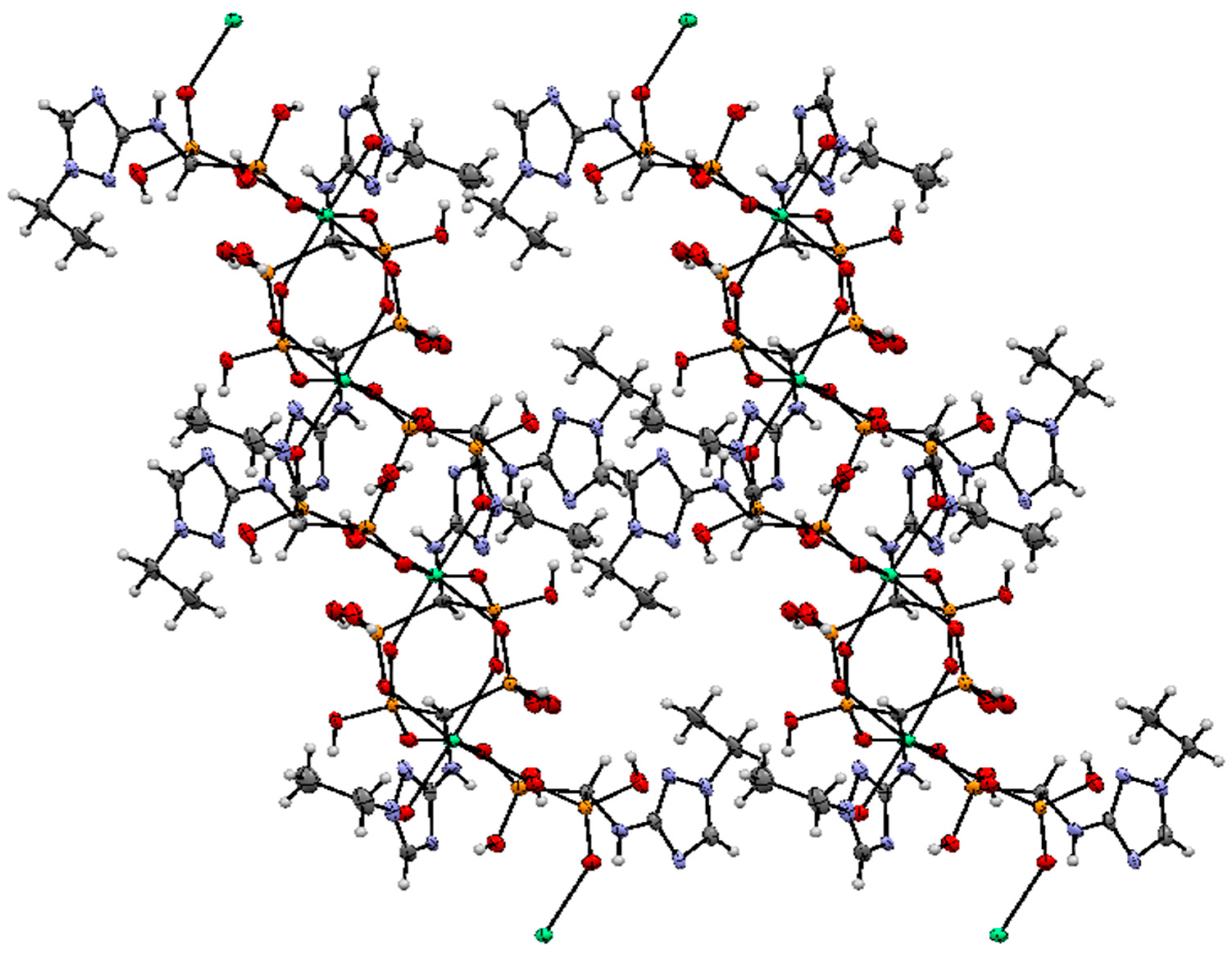
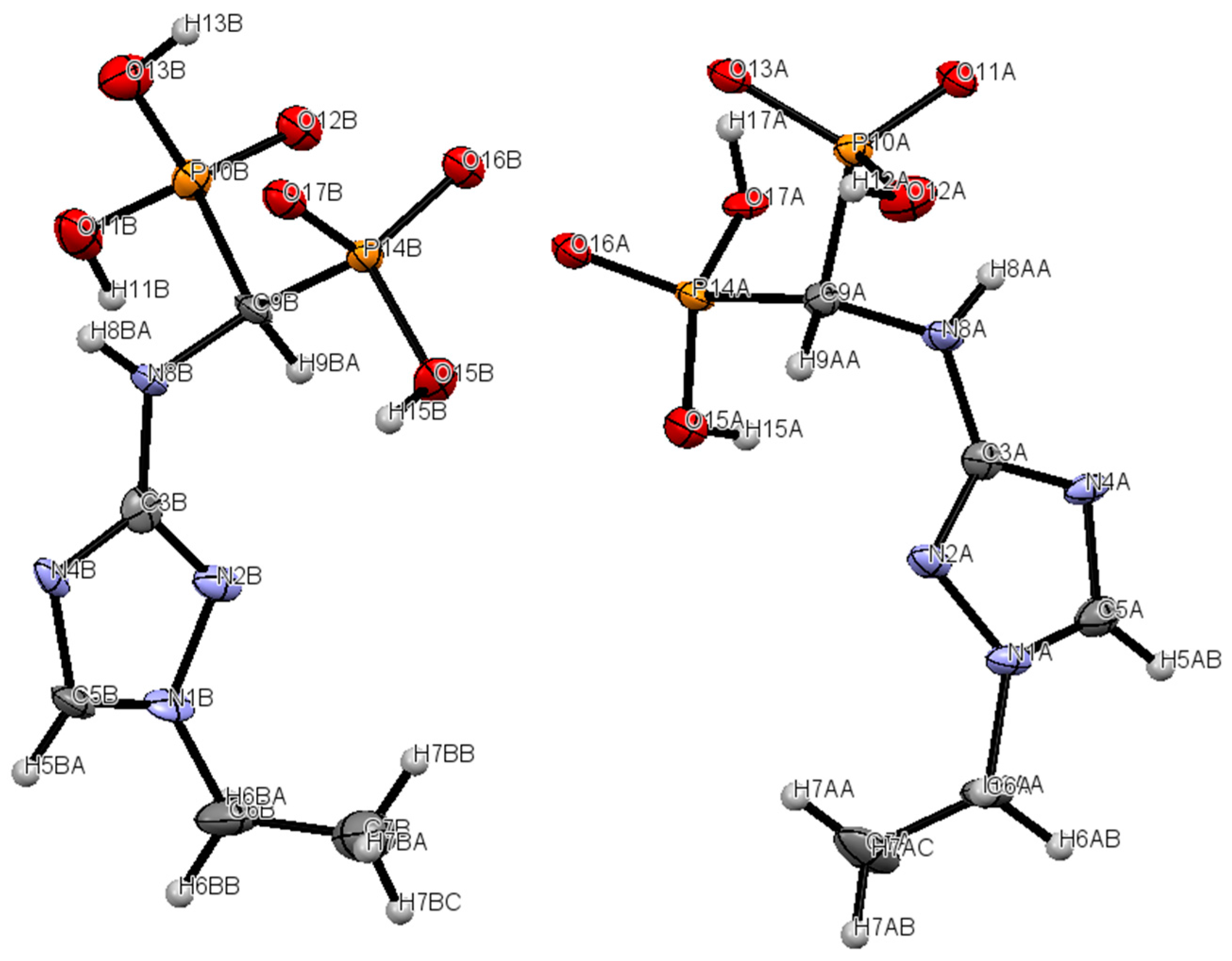
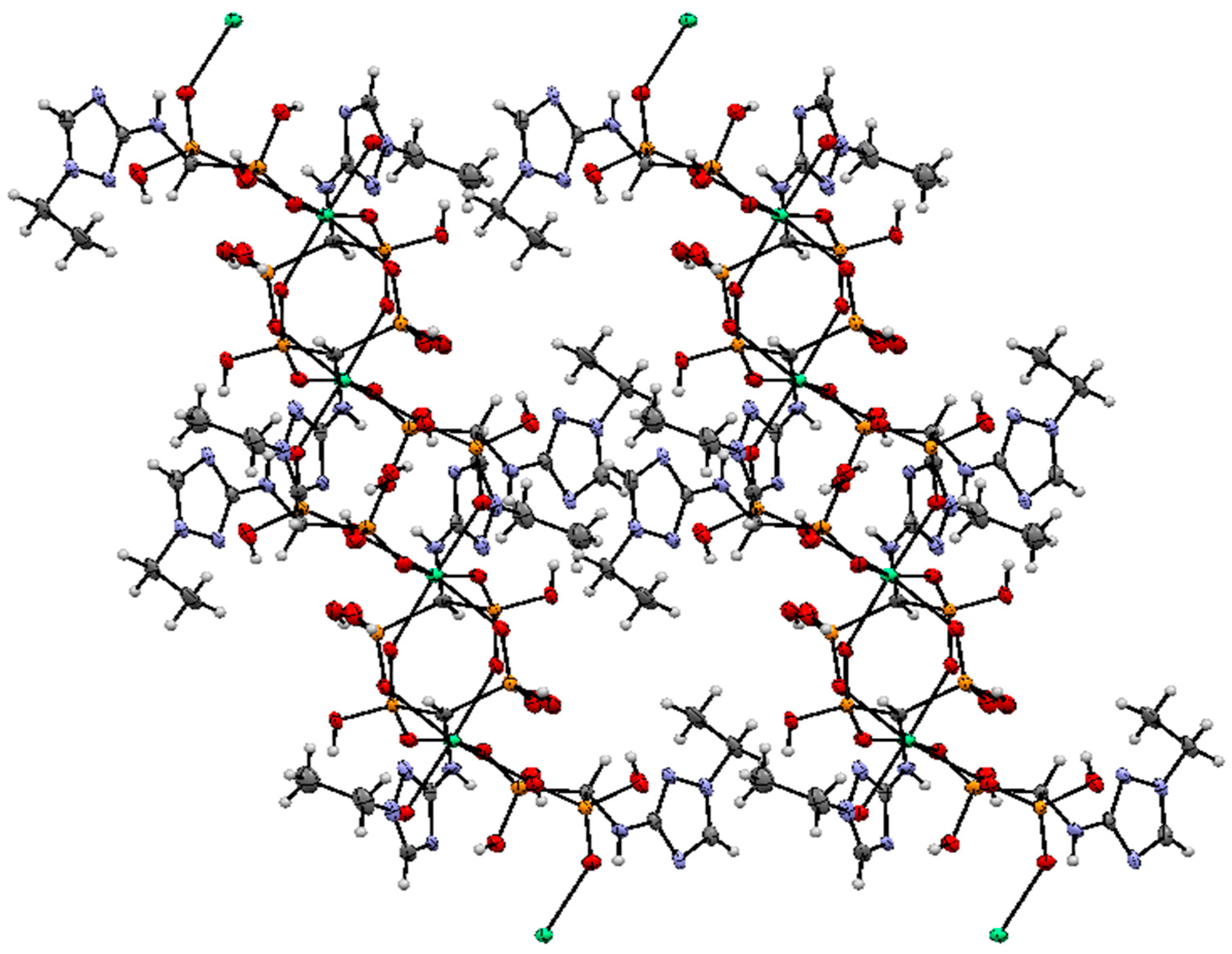
2.2. Crystallography
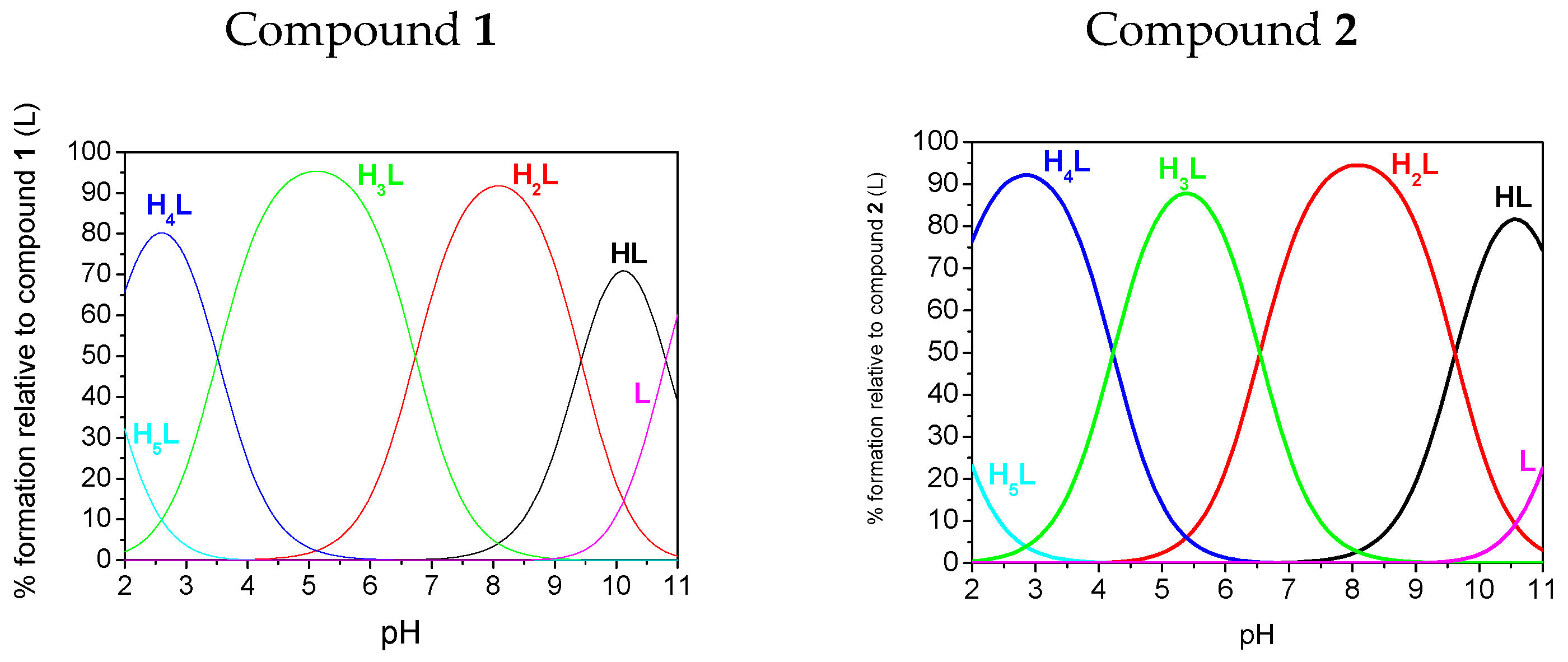
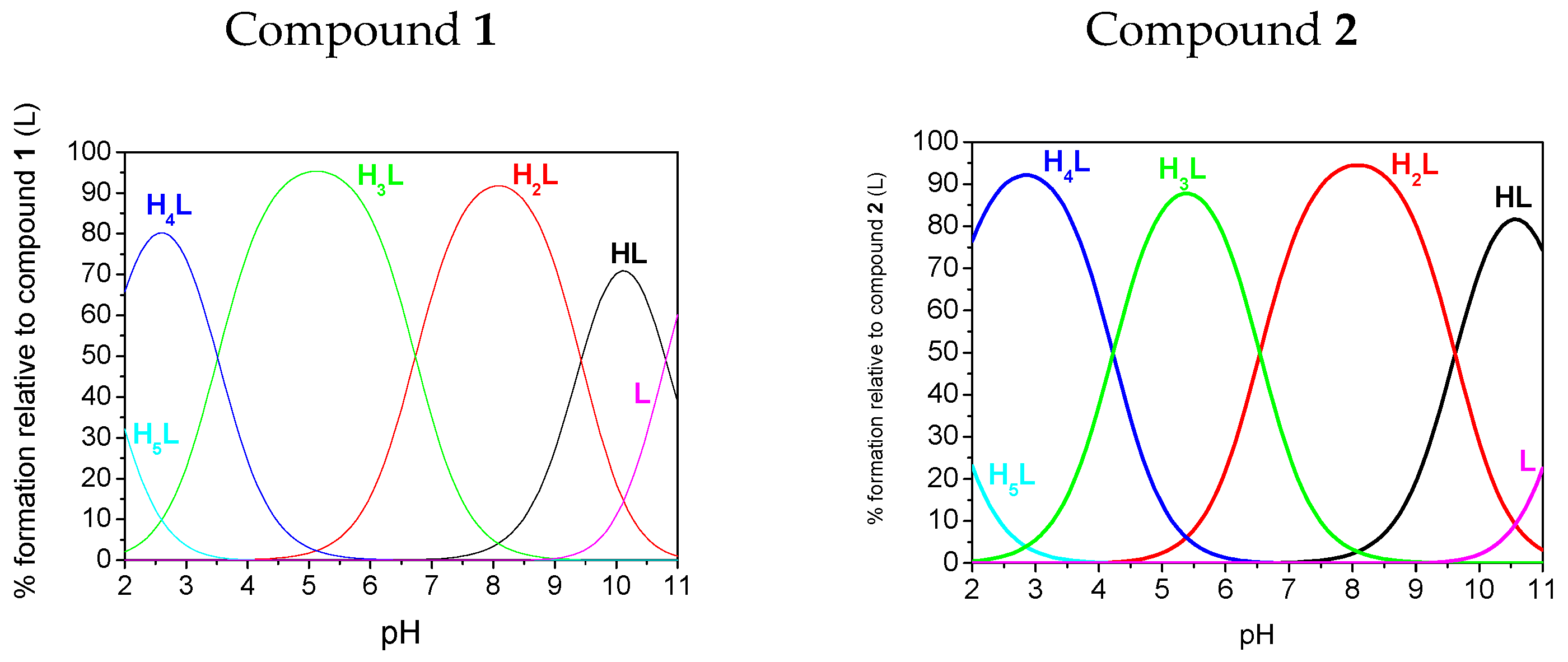
2.3. Potentiometry
2.4. In Vitro Evaluation
3. Experimental Section
3.1. General Information
3.2. Syntheses
3.2.1. Synthesis of 1,2,4-Triazolyl-3-yl-Aminomethylenebisphosphonic Acid (Compound 1)
3.2.2. Synthesis of 1,2,4-Triazolyl-3-yl-(N-Ethyl)aminomethylenebisphosphonic Acid (2)
3.3. Antiproliferative Activity
3.4. NMR Measurements
3.5. UV–Vis Measurements
3.6. Potentiometric Studies
3.7. Crystallography
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chmielewska, E.; Kafarski, P. Physiologic activity of bisphosphonates—Recent advances. Open J. Pharm. Sci. J. 2016, 3, 56–78. [Google Scholar] [CrossRef]
- Ebetino, F.H.; Hogan, A.M.; Sun, S.; Tsoumpra, M.K.; Duan, X.; Triffitt, J.T.; Kwaasi, A.A.; Dunfort, J.E.; Barnett, B.L.; Oppermann, U.; et al. The relationship between the chemistry and biological activity of the bisphosphonates. Bone 2011, 49, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Demkowicz, S.; Rachoń, J.; Dasko, M.; Kozak, W. Selected organophosphorus compounds with biological activity. Applications in medicine. RSC Adv. 2016, 6, 7101–7112. [Google Scholar] [CrossRef]
- Maier, L. Organishe phosphorverbindungen 75: Herstellung und Eigenschaften von Aminomethylendiphosphinaten und -diphosphonaten, RR1NCH[P(O)R2(OR3)]2 und Derivaten. Phosphorus Sulfur Silicon Relat. Elem. 1981, 11, 311–332. [Google Scholar] [CrossRef]
- Kantoci, D.; Denike, J.K.; Wechter, W.J. Synthesis of Aminobisphosphonate. Synth. Commun. 1996, 26, 2037–2043. [Google Scholar] [CrossRef]
- Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. Insight into the mechanism of three component condensation leading to aminomethylenebisphosphonates. J. Organomet. Chem. 2009, 694, 3806–3813. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Dzielak, A.; Hägele, G.; Keglevich, G. Microwave-assisted synthesis of (aminomethylene)bisphosphine oxides and (aminomethylene)bisphosphonates by a three-component condensation. Beilstein J. Org. Chem. 2016, 12, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Gross, H.; Costisella, B. Synthesis of carbocylic acids via PO-activated olefination of tetraethyl dimethyl—Aminomethylenediphosphonate. Angew. Chem. Int. Ed. Engl. 1968, 7, 391–392. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Synthesis of the new types of N-substituted aminomethylenebisorganophosphorus acids and their derivatives. Heteroatom Chem. 2009, 20, 319–324. [Google Scholar] [CrossRef]
- Yokomatsu, T.; Yoshida, Y.; Nakabayashi, N.; Shibuya, S. Simple and efficient method for preparation of conformationally constrained aminomethylene gem-diphosphonate ferivatives via Beckmann rearrangement. J. Org. Chem. 1994, 59, 7562–7564. [Google Scholar] [CrossRef]
- Olive, G.; Le Moigne, F.; Mercier, A.; Rockenbauer, A.; Tordo, P. Synthesis of Tetraalkyl (Pyrrolidine-2,2-diyl)bisphosphonates and 2,2-Bis(diethoxyphosphoryl)-3,4-dihydro-2H-pyrrole 1-Oxide; ESR Study of Derived Nitroxides. J. Org. Chem. 1998, 63, 9095–9099. [Google Scholar] [CrossRef]
- Olive, G.; Le Moigne, F.; Mercier, A.; Tordo, P. One-Step Gem-Diphosphorylation of Amides and Lactams. Synth. Commun. 2000, 30, 619–627. [Google Scholar] [CrossRef]
- Lecouvey, M.; Leroux, I. Synthesis of 1-hydroxy-1,1-bisphosphonates. Heteroatom Chem. 2000, 11, 556–561. [Google Scholar] [CrossRef]
- Abdou, W.M.; Shaddy, A.A. The development of bisphosphonates for therapeutic uses, and bisphosphonate structure-activity consideration. ARKIVOC 2009, IV, 143–182. [Google Scholar]
- Chambers, T.J. Diphosphonates inhibit bone resorption by macrophages in vitro. J. Pathol. 1980, 132, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Balabin, R.M. Tautomeric equilibrium and hydrogen shifts in tetrazole and triazoles: Focal-point analysis and ab initio limit. J. Chem. Phys. 2009, 131, 154307. [Google Scholar] [CrossRef] [PubMed]
- Adrien, A.; Taylor, P.J. The Tautomerism of I,2,3-Triazole in Aqueous Solution. J. Chem. Soc. Perkin Trans. 1989, 11, 1903–1905. [Google Scholar]
- Dolzhenko, A.V.; Chia, H.-S.; Chui, W.-K. Synthesis of 5-amino-3-(het)aryl-1H-1,2,4-triazoles via cyclization of (het)aroylaminoguanidines in queous medium. In Proceedings of the 9th International Electronic Conference on Synthetic Organic Chemistry (ECSOC-9), 1–30 November 2005; MDPI: Basel, Switzerland, 2005. [Google Scholar]
- Shneine, J.K.; Alaraji, Y.H. Chemistry of 1,2,4-triazole: A review article. Int. J. Sci. Res. 2016, 5, 1411–1423. [Google Scholar]
- Mroczek, T.; Plech, T.; Wujec, M. Novel concept of discrimination of 1,2,4-triazole-3-thione and 3-thiol tautomers. J. Chromatogr. Sci. 2016, 55, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Boduszek, B.; Dyba, M.; Jeżowska-Bojczuk, M.; Kiss, T.; Kozłowski, H. Biologically active pyridine mono- and bis-phosphonates: Efficient ligands for co-ordination of Cu2+ ions. J. Chem. Soc. Dalton Trans. 1997, 6, 973–976. [Google Scholar] [CrossRef]
- Kowalik-Jankowska, T.; Pietruszka, M.; Jezierska, J.; Matczak-Jon, E.; Kafarski, P. Copper(II) complexation by (pyridinyl)aminomethane-1,1-diphosphonic acid derivatives; spectroscopic and potentiometric studies. Polyhedron 2011, 30, 1274–1280. [Google Scholar] [CrossRef]
- Hansen, L.D.; West, B.D.; Baca, E.J.; Blank, C.L. Thermodynamics of proton ionization from some substituted 1,2,3-triazoles in dilute aqueous solution. J. Am. Chem. Soc. 1968, 90, 6588–6592. [Google Scholar] [CrossRef]
- Boraei, A.A.A.; Mohamed, N.F.A. Equilibrium studies of ternary systems involving divalent transition metal ions, aliphatic acids, and triazoles. J. Chem. Eng. Data 2002, 47, 987–991. [Google Scholar] [CrossRef]
- Jones, M.R.; Service, E.L.; Thompson, J.R.; Wang, M.C.P.; Kimsey, I.J.; DeToma, A.S.; Ramamoorthy, A.; Lim, M.H.; Storr, T. Dual-function triazole–pyridine derivatives as inhibitors of metal-induced amyloid-β aggregation. Metallomics 2012, 4, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Tiwari, M.; Singh, S.K.; Gangwar, L. Quantum chemical and energy descriptors based qsar studies of triazines inhibiting dihydrofolate reductase. Int. J. Sci. Res. 2013, 2, 1411–1423. [Google Scholar]
- Rogers, T.L.; Holen, I. Tumour macrophages as potential targets of bisphosphonates. J. Transl. Med. 2011, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Luckman, S.P.; Coxon, F.P.; Ebetino, F.H.; Russell, R.G.G.; Rogers, M.J. Heterocycle-containing bisphosphonates cause apoptosis and inhibit bone resorption by preventing protein prenylation: Evidence from structure–activity relationships in J774 macrophages. J. Bone Miner. Res. 1998, 13, 1668–1678. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.J.; Crockett, J.C.; Coxon, F.P.; Mönkkönen, J. Biochemical and molecular mechanisms of action of bisphosphonates. Bone 2011, 49, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [PubMed]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A study of some problems in determining the stoicheiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Gran, G. Determination of the Equivalent Point in Potentiometric Titrations. Acta Chem. Scand. 1950, 4, 559–577. [Google Scholar] [CrossRef]
- Gran, G. Determination of the equivalence point in potentiometric titrations. Part II. Analyst 1952, 77, 661–671. [Google Scholar] [CrossRef]
- Gans, P. Data Fitting in the Chemical Sciences; John Wiley & Sons: Chichester, UK, 1992. [Google Scholar]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad simulation and speciation (HySS): A utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- CrysAlis CCD, Version 1.171.33.57, Oxford Diffraction Ltd.: Abingdon, Oxfordshire, UK, 2005.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Sample Availability: Samples of compounds 1 and 2 are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ratio of Substrates Amine/triethyl orthoformate/diethyl phosphate | 1:1:2 | 1:2:4 | 1:4:6 |
| Temperature of Reaction (°C) | 130 | 130 | 130 |
| Time of Reaction (h) | 15 | 15 | 15 |
| Ratio of Products 1: (2 + 3 + 4 + 5) Determined from 1H-NMR Spectra by Integration of Triazole Protons | 1:0.7 | 1:0.3 | 1:0.1 |
| Yield of Isolated Compounds (%) | 27% (compound 1) | 21% (compound 2) | 37% (compound 1) |
| Assignment |  |  | ||
|---|---|---|---|---|
| 1 | 2 | |||
| Potentiometry | UV | Potentiometry | UV | |
| pK1(HL)amine | 11.52(3) | 11.68(2) | 10.81(1) | nd |
| pK2(H2L)phosphonate | 9.62(3) | 9.32(5) | 9.43(1) | 9.56(2) |
| pK3(H3L)phosphonate | 6.54(4) | 6.15(6) | 6.73(1) | 5.90(2) |
| pK4(H4L)triazole | 4.22(4) | 4.16(6) | 3.51(1) | 3.32(2) |
| pK5(H5L)phosphonate | 1.48(4) | 0.67(6) | 1.69(1) | nd |
| Crystal Data | |
|---|---|
| Chemical formula | C10H30CaN8O16P4 |
| Mr | 682.38 |
| Crystal system, space group | Triclinic, P-1 |
| Temperature (K) | 100.0(1) |
| a, b, c (Å) | 10.6815 (4), 12.5015 (6), 12.6155 (5) |
| α, β, γ (°) | 60.518 (5), 66.986 (4), 69.072 (4) |
| V (Å3) | 1320.90 (12) |
| Z | 2 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.57 |
| Crystal size (mm) | 0.02 × 0.01 × 0.01 |
| Data collection | |
| Diffractometer | Oxford Xcalibur |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 8995, 5080, 2769 |
| Rint | 0.079 |
| (sin θ/λ)max (Å−1) | 0.617 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.0790, 0.1307, 1.031 |
| No. of reflections | 5080 |
| No. of parameters | 420 |
| Δρmax, Δρmin (e Å−3) | 0.66, −0.77 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miszczyk, P.; Wieczorek, D.; Gałęzowska, J.; Dziuk, B.; Wietrzyk, J.; Chmielewska, E. Reaction of 3-Amino-1,2,4-Triazole with Diethyl Phosphite and Triethyl Orthoformate: Acid-Base Properties and Antiosteoporotic Activities of the Products. Molecules 2017, 22, 254. https://doi.org/10.3390/molecules22020254
Miszczyk P, Wieczorek D, Gałęzowska J, Dziuk B, Wietrzyk J, Chmielewska E. Reaction of 3-Amino-1,2,4-Triazole with Diethyl Phosphite and Triethyl Orthoformate: Acid-Base Properties and Antiosteoporotic Activities of the Products. Molecules. 2017; 22(2):254. https://doi.org/10.3390/molecules22020254
Chicago/Turabian StyleMiszczyk, Patrycja, Dorota Wieczorek, Joanna Gałęzowska, Błażej Dziuk, Joanna Wietrzyk, and Ewa Chmielewska. 2017. "Reaction of 3-Amino-1,2,4-Triazole with Diethyl Phosphite and Triethyl Orthoformate: Acid-Base Properties and Antiosteoporotic Activities of the Products" Molecules 22, no. 2: 254. https://doi.org/10.3390/molecules22020254
APA StyleMiszczyk, P., Wieczorek, D., Gałęzowska, J., Dziuk, B., Wietrzyk, J., & Chmielewska, E. (2017). Reaction of 3-Amino-1,2,4-Triazole with Diethyl Phosphite and Triethyl Orthoformate: Acid-Base Properties and Antiosteoporotic Activities of the Products. Molecules, 22(2), 254. https://doi.org/10.3390/molecules22020254