Facile Chemical Access to Biologically Active Norcantharidin Derivatives from Biomass


 ,
, 
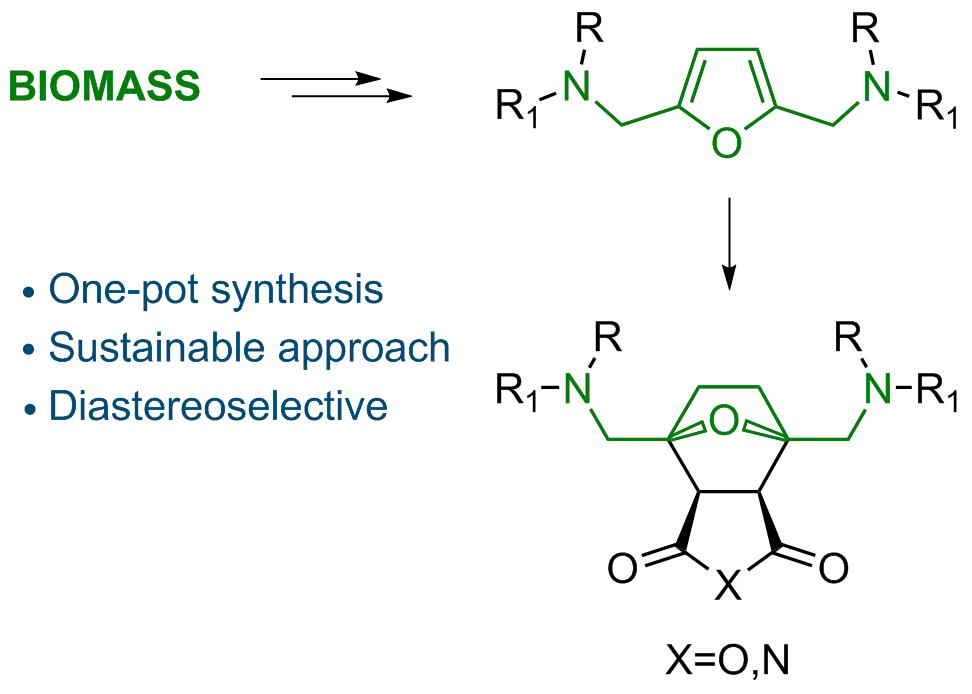
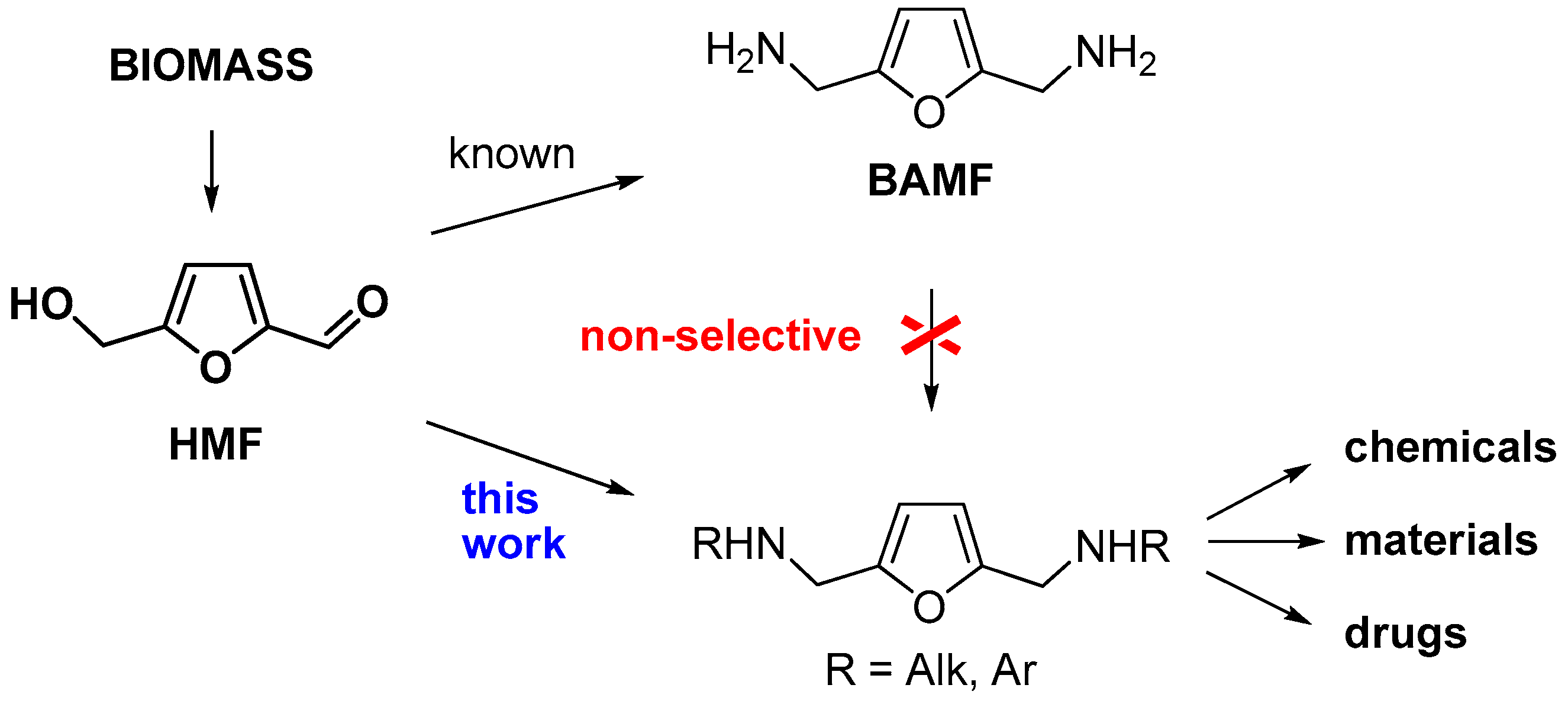
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
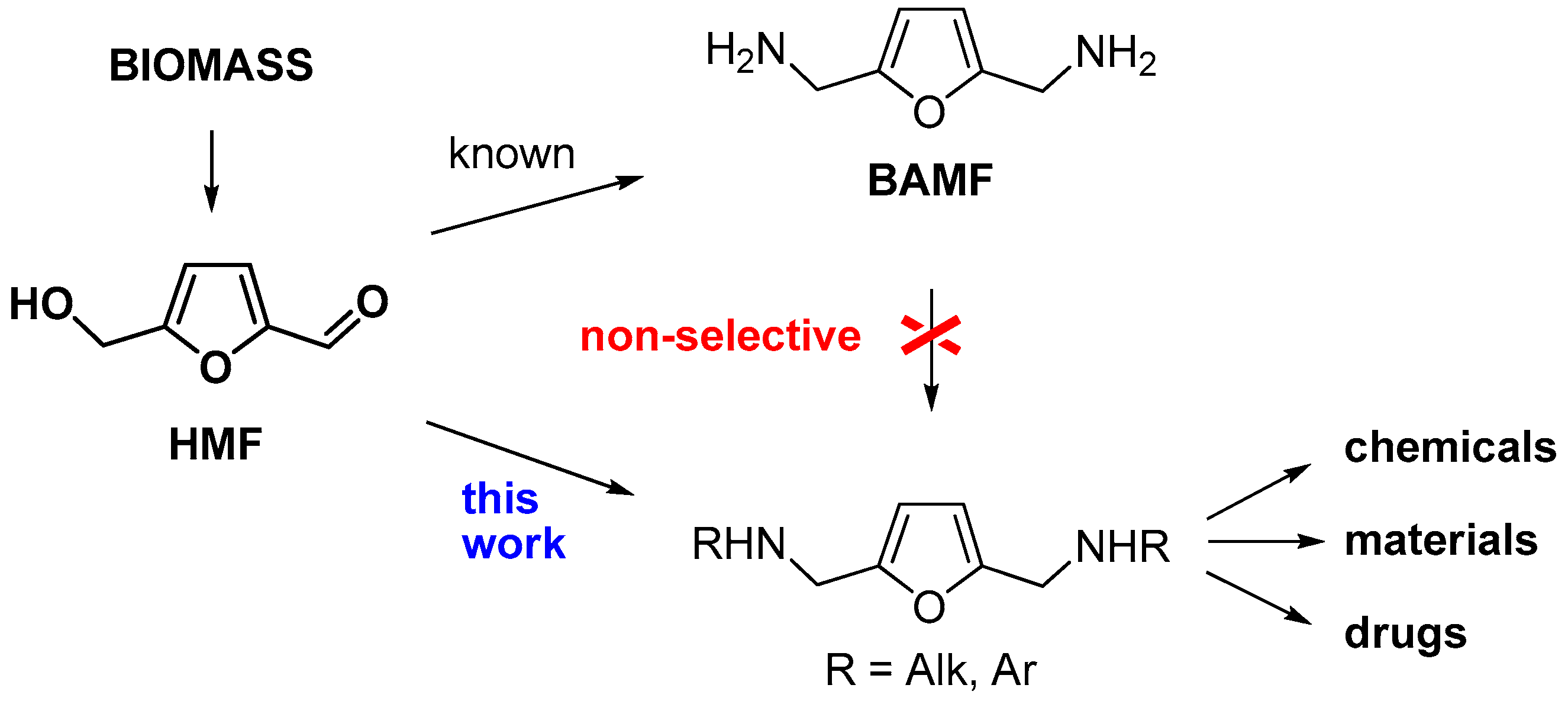
2. Results and Discussion
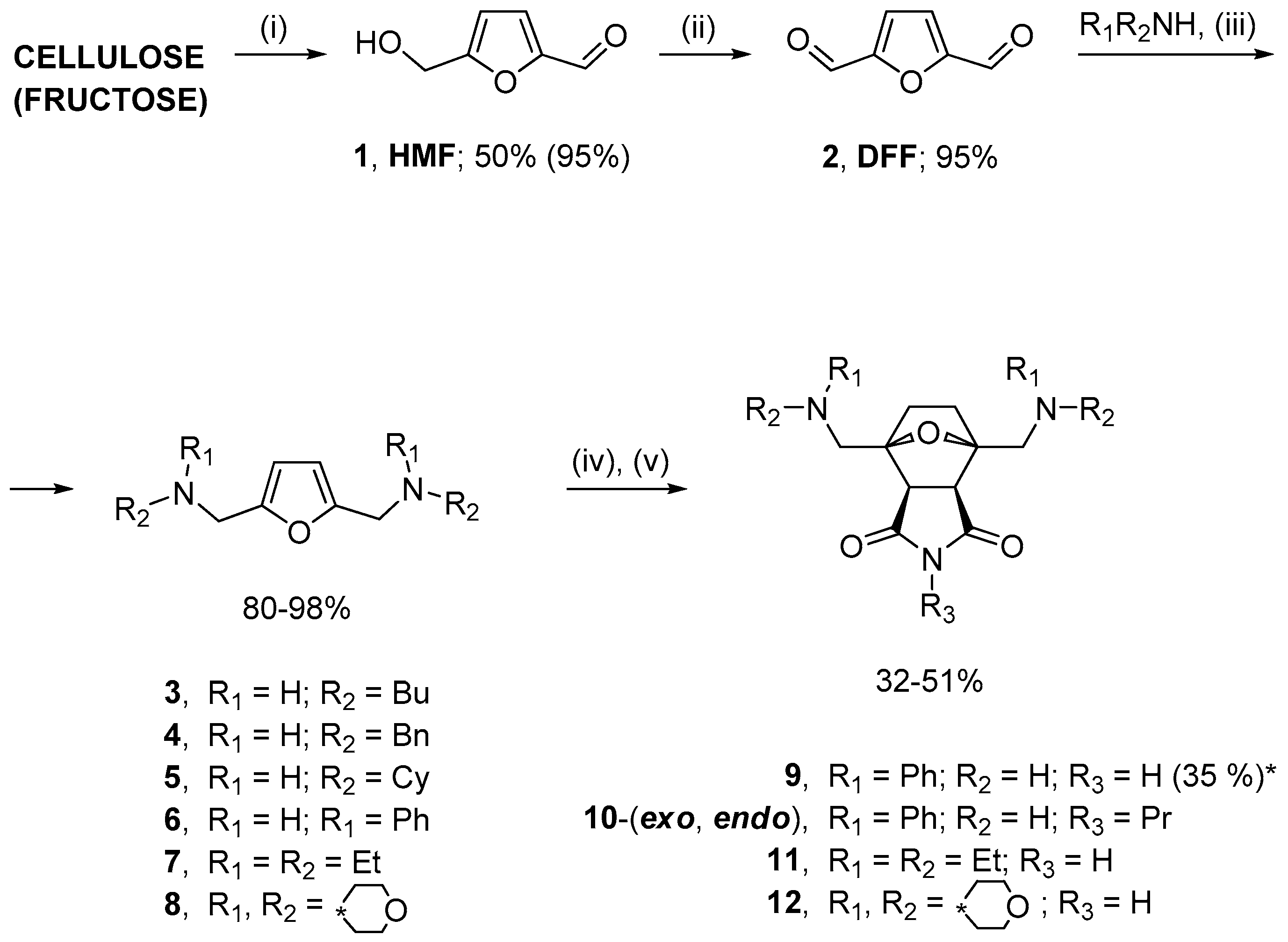
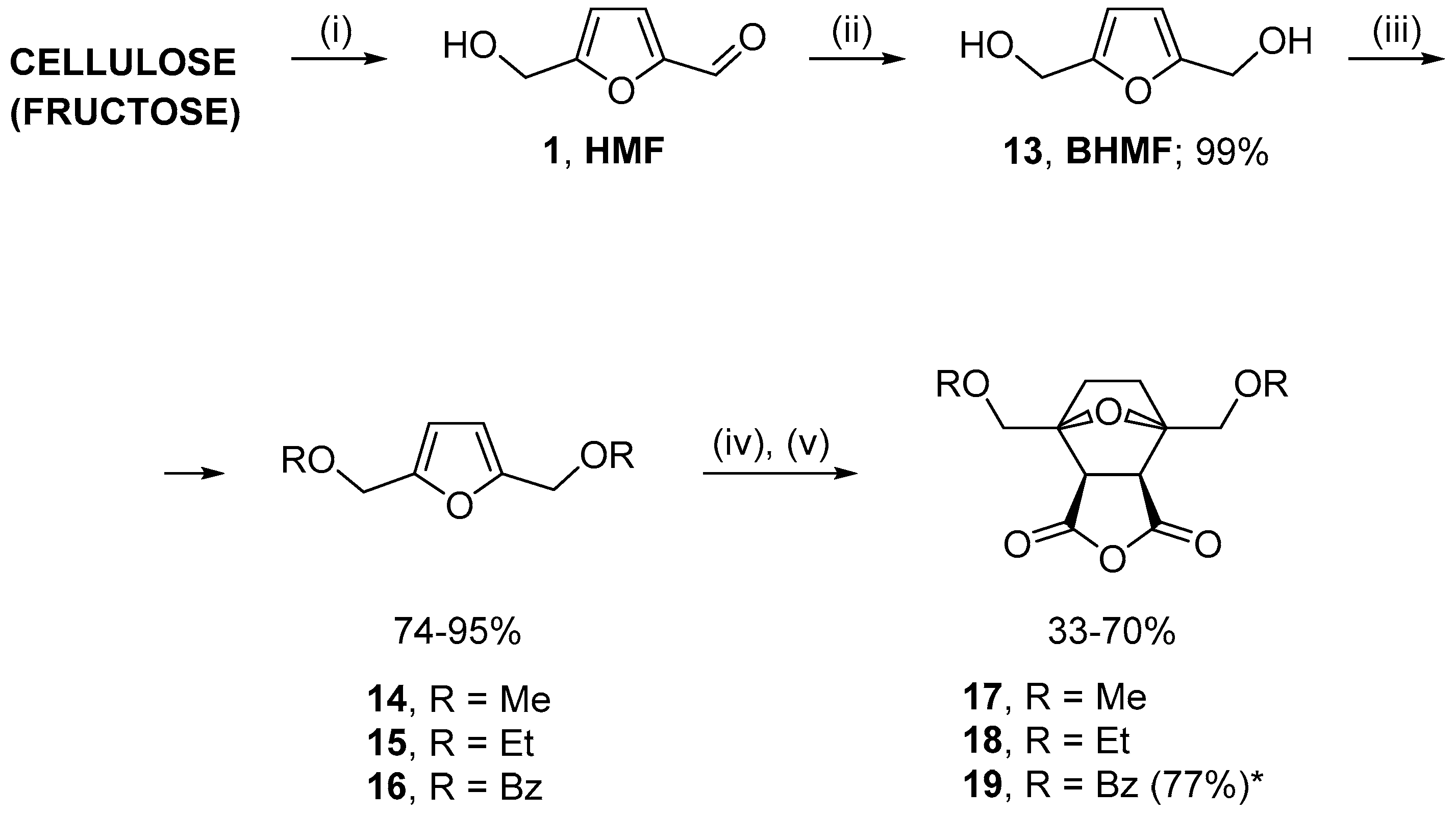
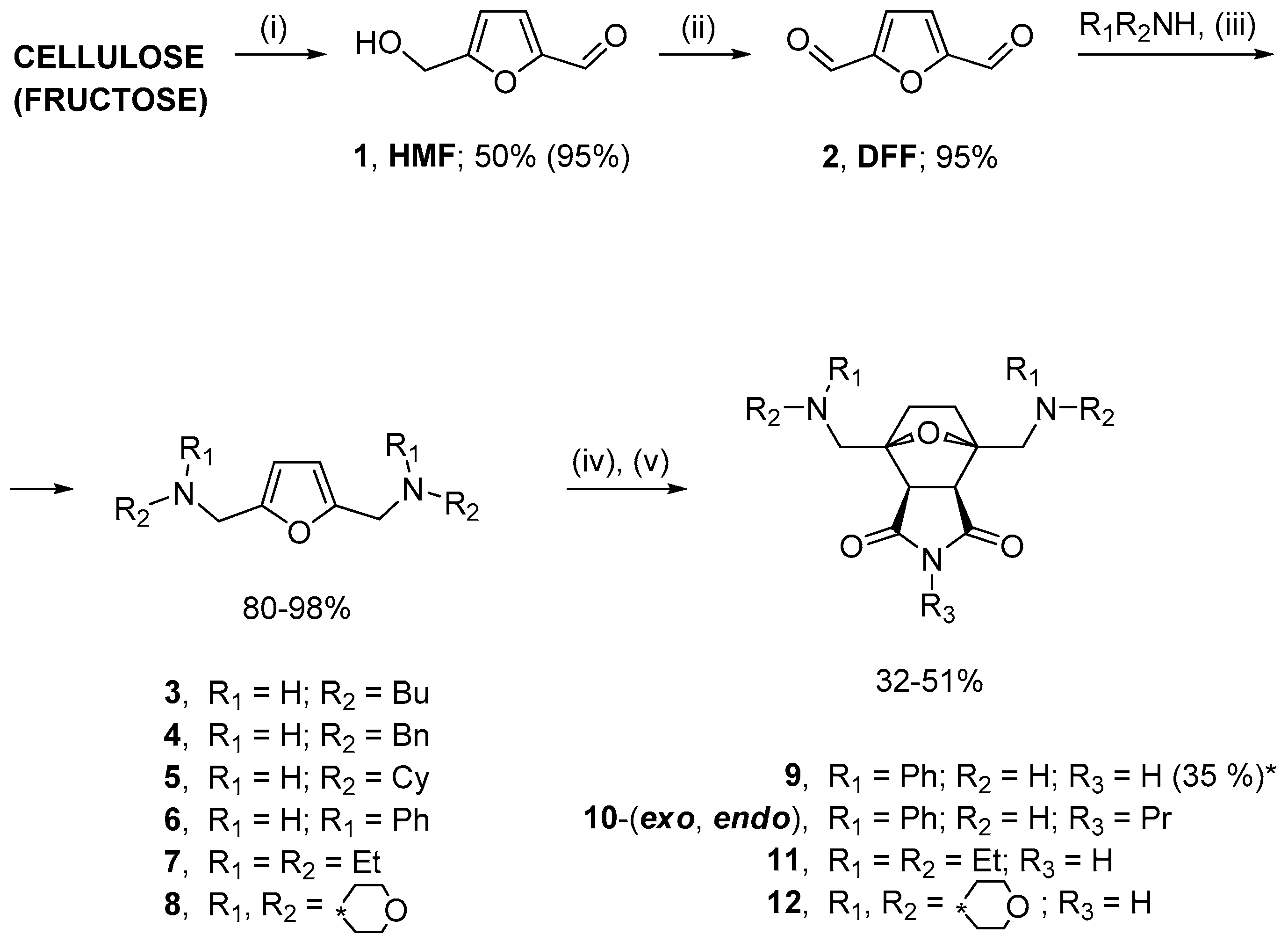
2.1. Synthesis of Norcantharidin Derivatives from HMF
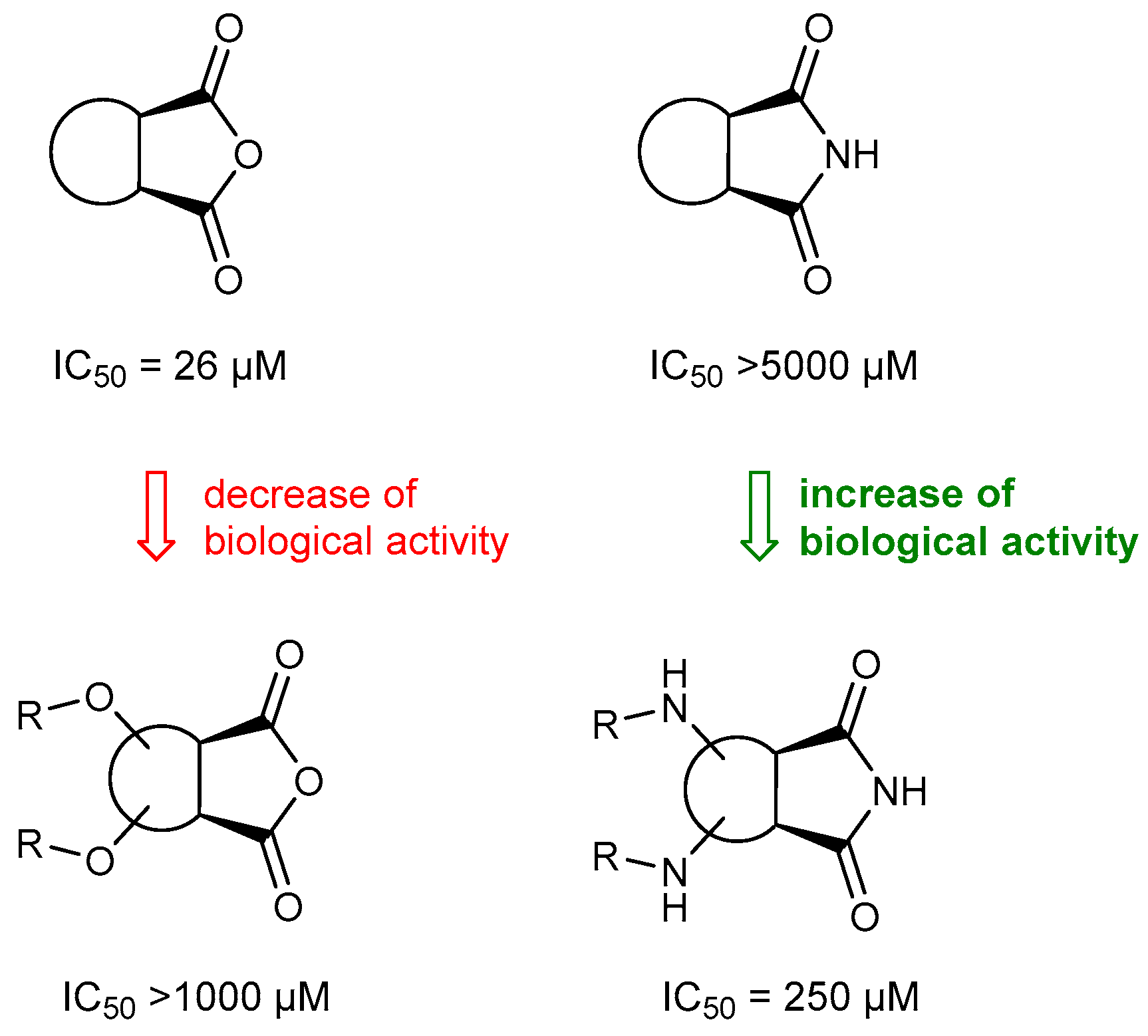
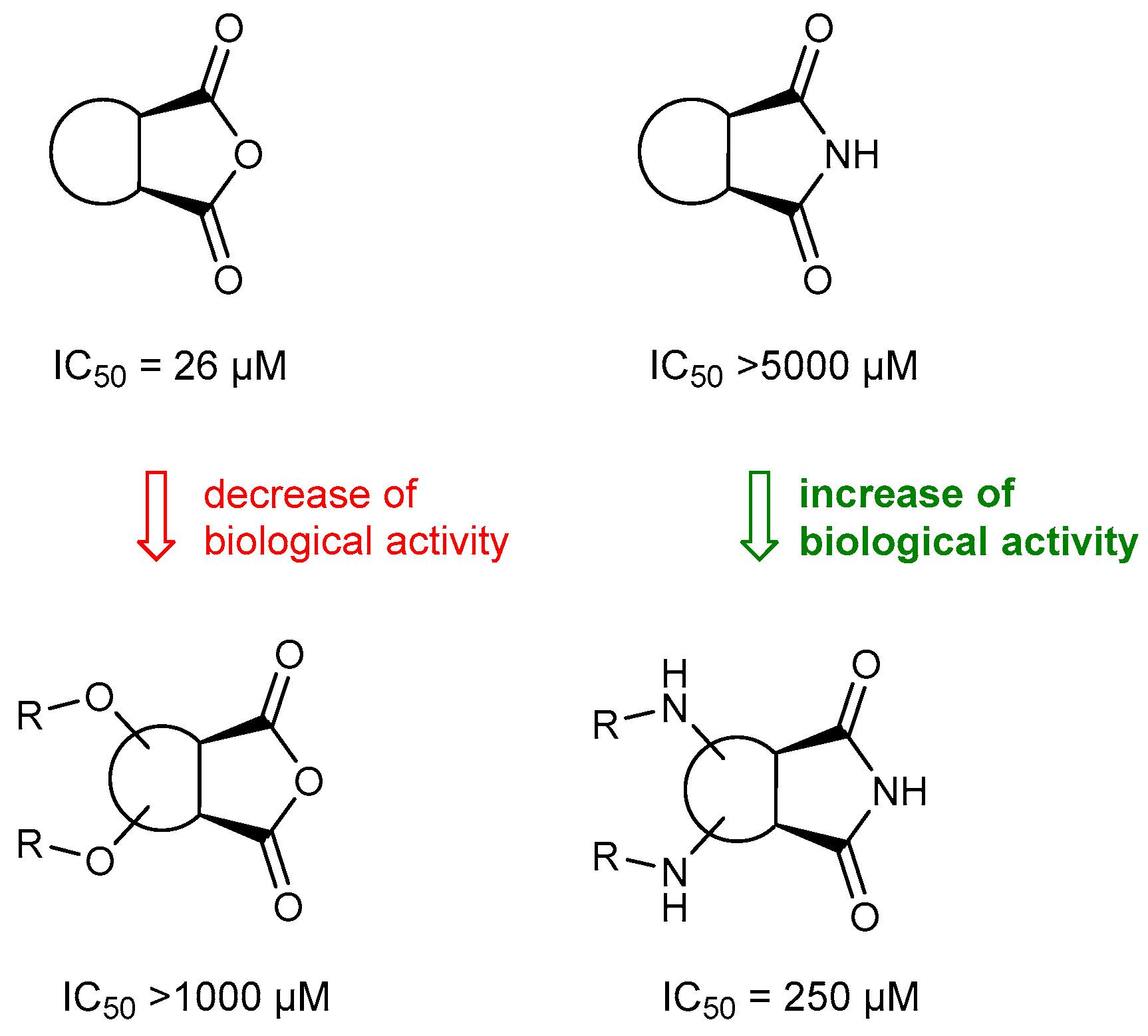
2.2. Biological Activity of Synthesized Compounds
3. Materials and Methods
3.1. Synthesis of Substrates
3.2. General Method for Synthesis of 2,5-Bis(aminomethyl)furans
3.3. General Method for One-Pot Synthesis of Norcantharimide Derivatives
3.4. General Method for One-Pot Synthesis of Norcantharidin Derivatives
3.5. Synthesis of Compounds 9 and 19 Using Step-by-Step Procedure
3.5.1. Preparation of Compound 9
3.5.2. Preparation of Compound 19
3.6. Preliminary Studies of Cytotoxicity of Selected Substances
3.6.1. Cell Culture
3.6.2. MTS Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Field, C.B. Primary Production of the Biosphere: Integrating Terrestrial and Oceanic Components. Science 1998, 281, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Werpy, T.; Petersen, G.; Aden, A.; Bozell, J.; Holladay, J.; White, J.; Manheim, A.; Eliot, D.; Lasure, L.; Jones, S. Top Value Added Chemicals from Biomass. Volume 1—Results of Screening for Potential Candidates from Sugars and Synthesis Gas; Department of Energy: Washington, DC, USA, 2004.
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Román-Leshkov, Y.; Barrett, C.J.; Liu, Z.Y.; Dumesic, J.A. Production of dimethylfuran for liquid fuels from biomass-derived carbohydrates. Nature 2007, 447, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Sutton, A.D.; Waldie, F.D.; Wu, R.; Schlaf, M.; ‘Pete’ Silks, L.A., III; Gordon, J.C. The hydrodeoxygenation of bioderived furans into alkanes. Nat. Chem. 2013, 5, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lin, L.; Wu, Z.; Zhou, S.; Liu, S. Recent advances in catalytic transformation of biomass-derived 5-hydroxymethylfurfural into the innovative fuels and chemicals. Renew. Sustain. Energy Rev. 2017, 74, 230–257. [Google Scholar] [CrossRef]
- Wu, K.; Wu, Y.; Chen, Y.; Chen, H.; Wang, J.; Yang, M. Heterogeneous Catalytic Conversion of Biobased Chemicals into Liquid Fuels in the Aqueous Phase. ChemSusChem 2016, 9, 1355–1385. [Google Scholar] [CrossRef] [PubMed]
- Thananatthanachon, T.; Rauchfuss, T.B. Efficient Production of the Liquid Fuel 2,5-Dimethylfuran from Fructose Using Formic Acid as a Reagent. Angew. Chem. Int. Ed. 2010, 49, 6616–6618. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, A.S. Renewable Polymers: Synthesis, Processing, and Technology; Mittal, V., Ed.; John Wiley & Sons, Scrivener Pub.: Hoboken, NJ, USA, 2012; pp. 398–428. [Google Scholar]
- Liu, W.-J.; Jiang, H.; Yu, H.-Q. Development of Biochar-Based Functional Materials: Toward a Sustainable Platform Carbon Material. Chem. Rev. 2015, 115, 12251–12285. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; De, S.; Saha, B. A Brief Summary of the Synthesis of Polyester Building-Block Chemicals and Biofuels from 5-Hydroxymethylfurfural. ChemPlusChem 2012, 77, 259–272. [Google Scholar] [CrossRef]
- Zhang, X.; Fevre, M.; Jones, G.O.; Waymouth, R.M. Catalysis as an Enabling Science for Sustainable Polymers. Chem. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Van Putten, R.-J.; van der Waal, J.C.; de Jong, E.; Rasrendra, C.B.; Heeres, H.J.; de Vries, J.G. Hydroxymethylfurfural, A Versatile Platform Chemical Made from Renewable Resources. Chem. Rev. 2013, 113, 1499–1597. [Google Scholar] [CrossRef] [PubMed]
- Rosatella, A.A.; Simeonov, S.P.; Frade, R.F.M.; Afonso, C.A.M. 5-Hydroxymethylfurfural (HMF) as a building block platform: Biological properties, synthesis and synthetic applications. Green Chem. 2011, 13, 754. [Google Scholar] [CrossRef]
- Lewkowski, J. Synthesis, chemistry and applications of 5-hydroxymethyl-furfural and its derivatives. ARKIVOC 2005, 2001, 17–54. [Google Scholar] [CrossRef]
- Delidovich, I.; Hausoul, P.J.C.; Deng, L.; Pfützenreuter, R.; Rose, M.; Palkovits, R. Alternative Monomers Based on Lignocellulose and Their Use for Polymer Production. Chem. Rev. 2016, 116, 1540–1599. [Google Scholar] [CrossRef] [PubMed]
- Galbis, J.A.; García-Martín, M.d.G.; de Paz, M.V.; Galbis, E. Synthetic Polymers from Sugar-Based Monomers. Chem. Rev. 2016, 116, 1600–1636. [Google Scholar] [CrossRef] [PubMed]
- Gandini, A.; Lacerda, T.M.; Carvalho, A.J.F.; Trovatti, E. Progress of Polymers from Renewable Resources: Furans, Vegetable Oils, and Polysaccharides. Chem. Rev. 2016, 116, 1637–1669. [Google Scholar] [CrossRef] [PubMed]
- Froidevaux, V.; Negrell, C.; Caillol, S.; Pascault, J.-P.; Boutevin, B. Biobased Amines: From Synthesis to Polymers; Present and Future. Chem. Rev. 2016, 116, 14181–14224. [Google Scholar] [CrossRef] [PubMed]
- Dunbabin, A.; Subrizi, F.; Ward, J.M.; Sheppard, T.D.; Hailes, H.C. Furfurylamines from biomass: transaminase catalysed upgrading of furfurals. Green Chem. 2017, 19, 397–404. [Google Scholar] [CrossRef]
- Komanoya, T.; Kinemura, T.; Kita, Y.; Kamata, K.; Hara, M. Electronic Effect of Ruthenium Nanoparticles on Efficient Reductive Amination of Carbonyl Compounds. J. Am. Chem. Soc. 2017, 139, 11493–11499. [Google Scholar] [CrossRef] [PubMed]
- Roylance, J.J.; Choi, K.-S. Electrochemical reductive amination of furfural-based biomass intermediates. Green Chem. 2016, 18, 5412–5417. [Google Scholar] [CrossRef]
- Sánchez, M.I.; Vázquez, O.; Martínez-Costas, J.; Vázquez, M.E.; Mascareñas, J.L. Straightforward access to bisbenzamidine DNA binders and their use as versatile adaptors for DNA-promoted processes. Chem. Sci. 2012, 3, 2383. [Google Scholar] [CrossRef]
- Galkin, K.I.; Krivodaeva, E.A.; Romashov, L.V.; Zalesskiy, S.S.; Kachala, V.V.; Burykina, J.V.; Ananikov, V.P. Critical Influence of 5-Hydroxymethylfurfural Aging and Decomposition on the Utility of Biomass Conversion in Organic Synthesis. Angew. Chem. Int. Ed. 2016, 55, 8338–8342. [Google Scholar] [CrossRef] [PubMed]
- Kashin, A.S.; Galkin, K.I.; Khokhlova, E.A.; Ananikov, V.P. Direct Observation of Self-Organized Water-Containing Structures in the Liquid Phase and Their Influence on 5-(Hydroxymethyl)furfural Formation in Ionic Liquids. Angew. Chem. Int. Ed. 2016, 55, 2161–2166. [Google Scholar] [CrossRef] [PubMed]
- Kashparova, V.P.; Khokhlova, E.A.; Galkin, K.I.; Chernyshev, V.M.; Ananikov, V.P. The “one-pot” synthesis of 2,5-diformylfuran, a promising synthon for organic materials in the conversion of biomass. Russ. Chem. Bull. 2015, 64, 1069–1073. [Google Scholar] [CrossRef]
- Kucherov, F.A.; Galkin, K.I.; Gordeev, E.G.; Ananikov, V.P. Efficient route for the construction of polycyclic systems from bioderived HMF. Green Chem. 2017, 19, 4858–4864. [Google Scholar] [CrossRef]
- De Graaff, C.; Bensch, L.; van Lint, M.J.; Ruijter, E.; Orru, R.V.A. IBX-mediated oxidation of unactivated cyclic amines: application in highly diastereoselective oxidative Ugi-type and aza-Friedel–Crafts reactions. Org. Biomol. Chem. 2015, 13, 10108–10112. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Hirukawa, N.; Tanohira, N.; Sodeoka, M. Structure-Based Design of a Highly Selective Catalytic Site-Directed Inhibitor of Ser/Thr Protein Phosphatase 2B (Calcineurin). J. Am. Chem. Soc. 2003, 125, 9740–9749. [Google Scholar] [CrossRef] [PubMed]
- Hirai, G.; Sodeoka, M. Focused Library with a Core Structure Extracted from Natural Products and Modified: Application to Phosphatase Inhibitors and Several Biochemical Findings. Acc. Chem. Res. 2015, 48, 1464–1473. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Wu, M.-Y.; Chen, L.-P.; Zhi, Q.; Gong, F.-R.; Chen, K.; Li, D.-M.; Wu, Y.; Tao, M.; Li, W. Cantharidin represses invasion of pancreatic cancer cells through accelerated degradation of MMP2 mRNA. Sci. Rep. 2015, 5, 11836–11848. [Google Scholar] [CrossRef] [PubMed]
- Puerto Galvis, C.E.; Vargas Méndez, L.Y.; Kouznetsov, V.V. Cantharidin-Based Small Molecules as Potential Therapeutic Agents. Chem. Biol. Drug Des. 2013, 82, 477–499. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.A.; Stewart, S.G.; Gordon, C.P.; Ackland, S.P.; Gilbert, J.; Sauer, B.; Sakoff, J.A.; McCluskey, A. Norcantharidin Analogues: Synthesis, Anticancer Activity and Protein Phosphatase 1 and 2A Inhibition. ChemMedChem 2008, 3, 1878–1892. [Google Scholar] [CrossRef] [PubMed]
- Tarleton, M.; Gilbert, J.; Sakoff, J.A.; McCluskey, A. Synthesis and anticancer activity of a series of norcantharidin analogues. Eur. J. Med. Chem. 2012, 54, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Gordon, C.P.; Gilbert, J.; McCluskey, A.; Sakoff, J.A. Norcantharimide analogues possessing terminal phosphate esters and their anti-cancer activity. Bioorg. Med. Chem. 2011, 19, 5734–5741. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.; Walkom, C.; Bowyer, M.C.; Ackland, S.P.; Gardiner, E.; Sakoff, J.A. Cantharimides: A new class of modified cantharidin analogues inhibiting protein phosphatases 1 and 2A. Bioorg. Med. Chem. Lett. 2001, 11, 2941–2946. [Google Scholar] [CrossRef]
- De, S.; Kumar, T.; Bohre, A.; Singh, L.R.; Saha, B. Furan-based acetylating agent for the chemical modification of proteins. Bioorg. Med. Chem. 2015, 23, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Liang, W.; Guan, J.; Wang, L.; Qu, Q.; Zhang, X.; Wang, X.; Mu, X. Catalytic synthesis of 2,5-bis-methoxymethylfuran: A promising cetane number improver for diesel. Appl. Catal. A 2014, 481, 49–53. [Google Scholar] [CrossRef]
- Kuo, Y.-H.; Shieh, C.-J. Sensitized Photooxidation of 2,5-Furanyldimethyl Dibenzoate. Heterocycles 1986, 24, 1271–1274. [Google Scholar] [CrossRef]
- Egorova, K.S.; Seitkalieva, M.M.; Posvyatenko, A.V.; Ananikov, V.P. An unexpected increase of toxicity of amino acid-containing ionic liquids. Toxicol. Res. 2015, 4, 152–159. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galkin, K.I.; Kucherov, F.A.; Markov, O.N.; Egorova, K.S.; Posvyatenko, A.V.; Ananikov, V.P. Facile Chemical Access to Biologically Active Norcantharidin Derivatives from Biomass. Molecules 2017, 22, 2210. https://doi.org/10.3390/molecules22122210
Galkin KI, Kucherov FA, Markov ON, Egorova KS, Posvyatenko AV, Ananikov VP. Facile Chemical Access to Biologically Active Norcantharidin Derivatives from Biomass. Molecules. 2017; 22(12):2210. https://doi.org/10.3390/molecules22122210
Chicago/Turabian StyleGalkin, Konstantin I., Fedor A. Kucherov, Oleg N. Markov, Ksenia S. Egorova, Alexandra V. Posvyatenko, and Valentine P. Ananikov. 2017. "Facile Chemical Access to Biologically Active Norcantharidin Derivatives from Biomass" Molecules 22, no. 12: 2210. https://doi.org/10.3390/molecules22122210