Benzoic Acid Derivatives with Trypanocidal Activity: Enzymatic Analysis and Molecular Docking Studies toward Trans-Sialidase

 ,
,  ,
,  ,
,
Abstract
:
1. Introduction
2. Results and Discussion
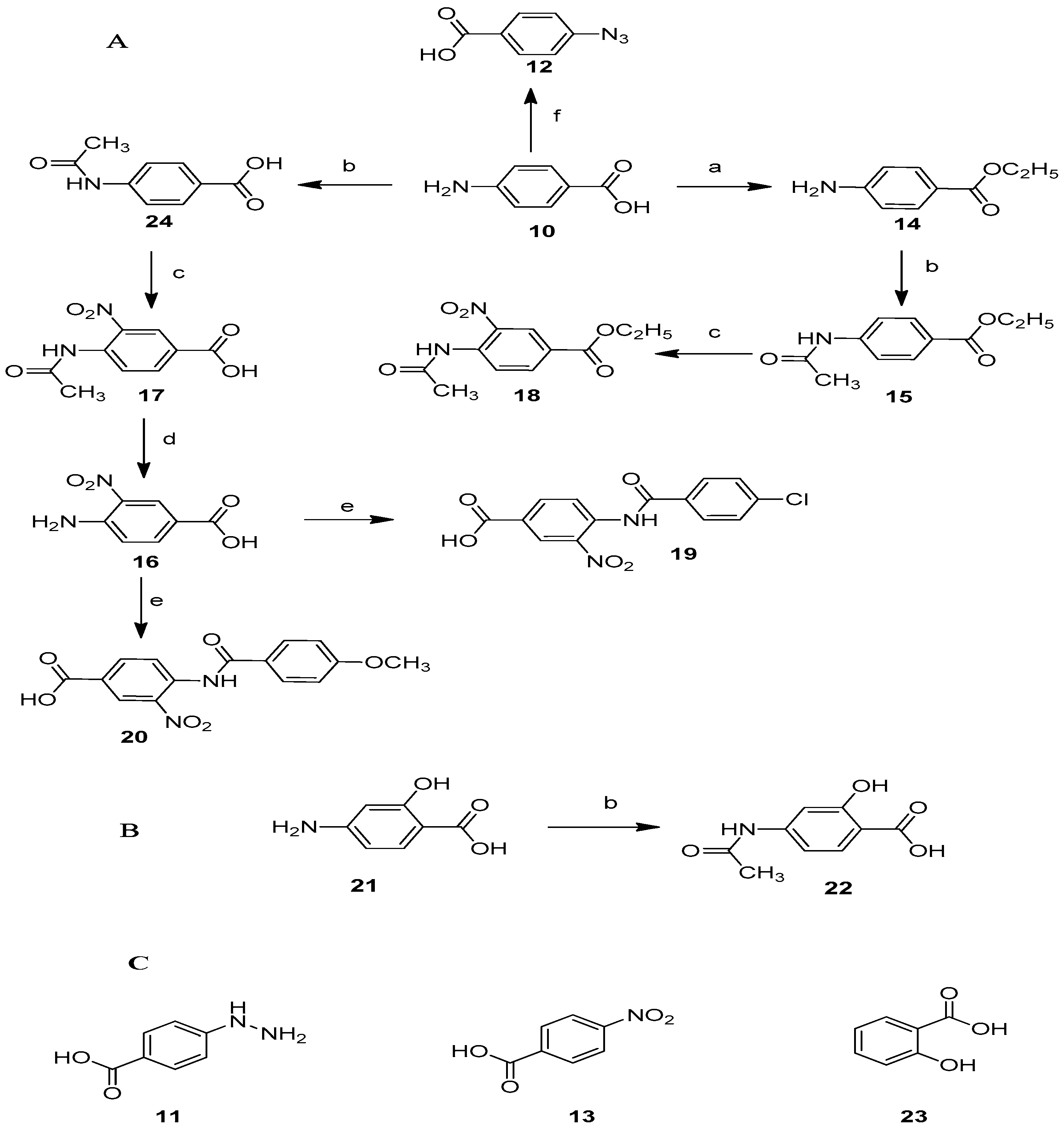
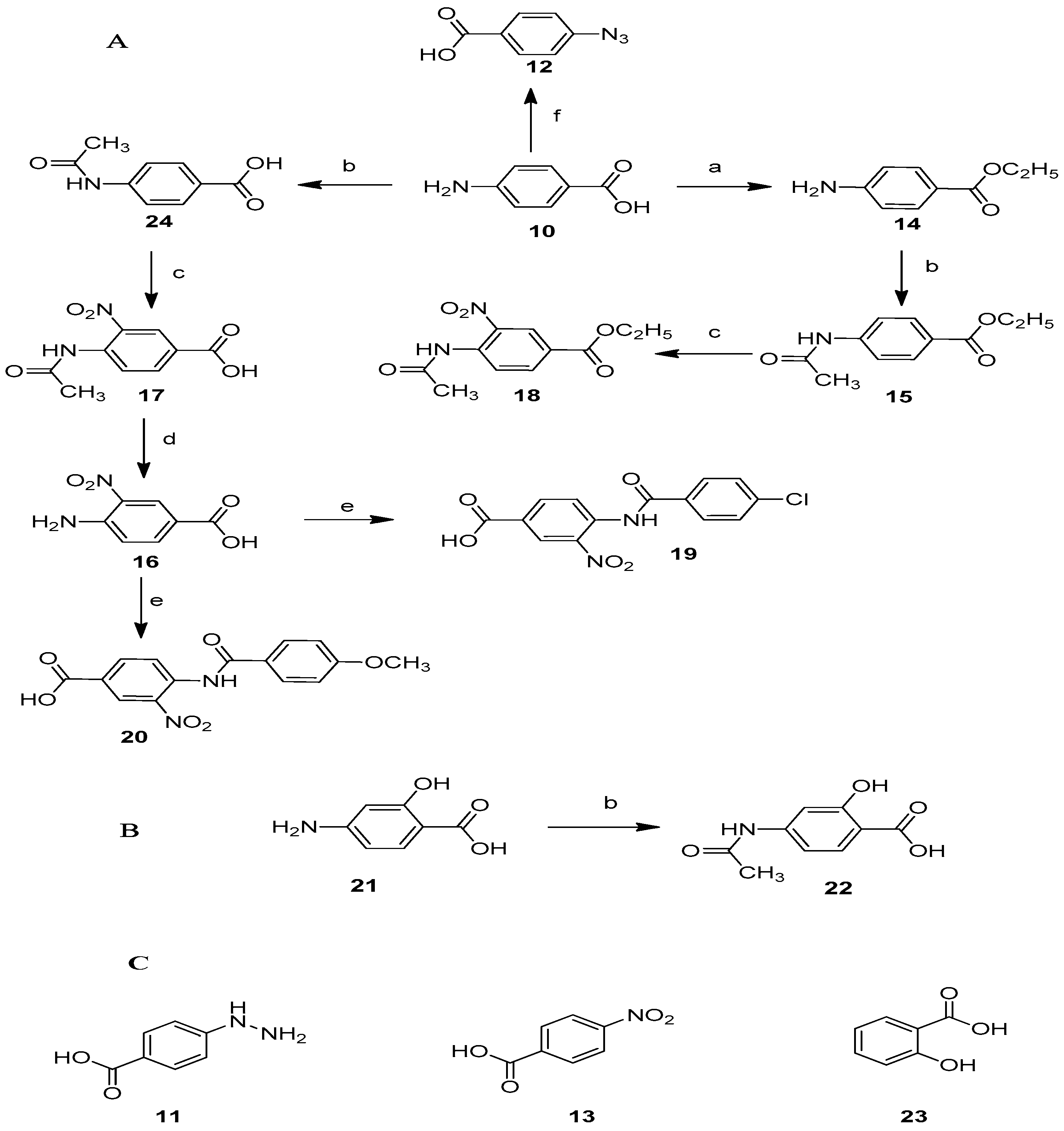
2.1. Synthesis
2.2. Trypanocidal Activity
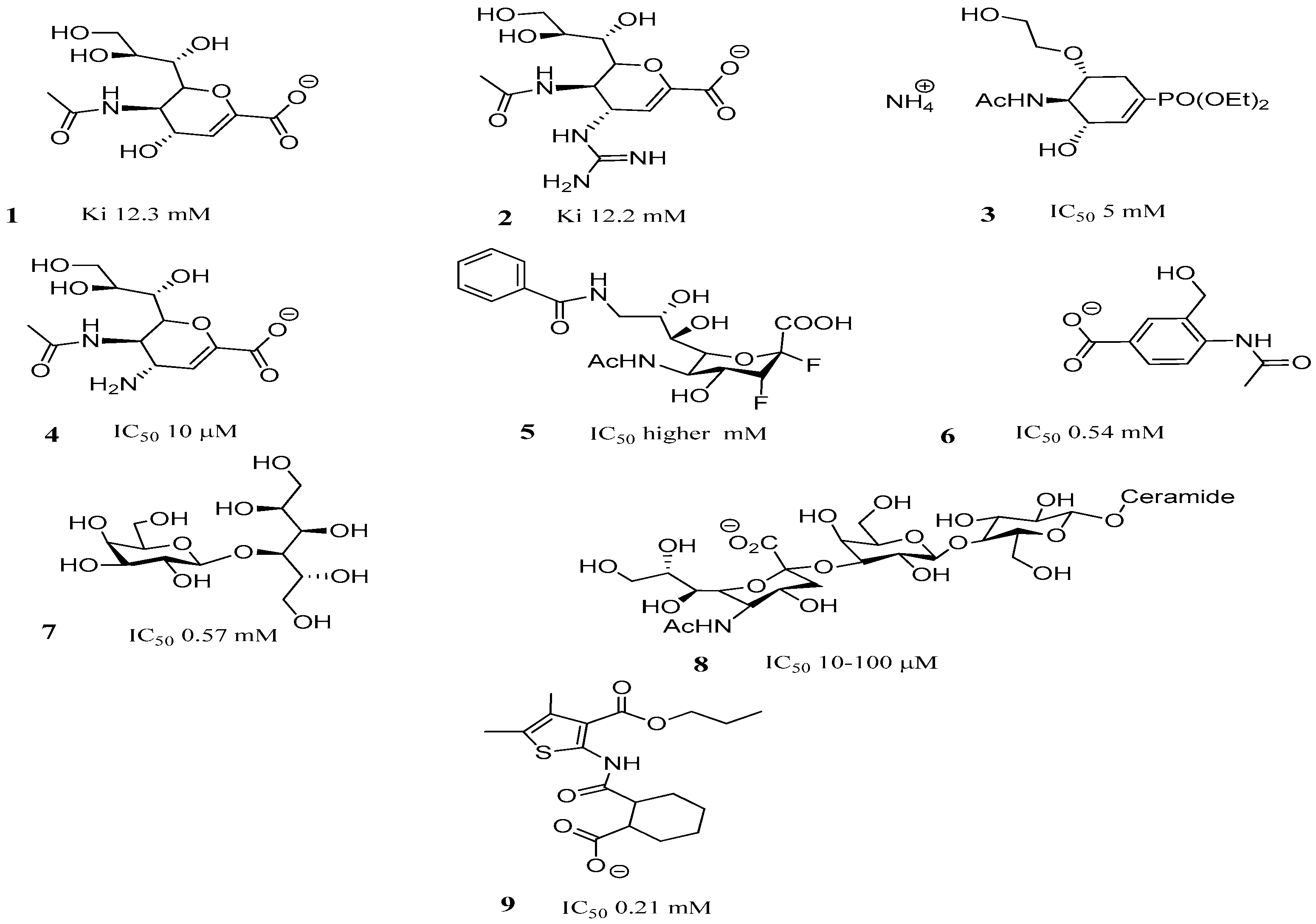
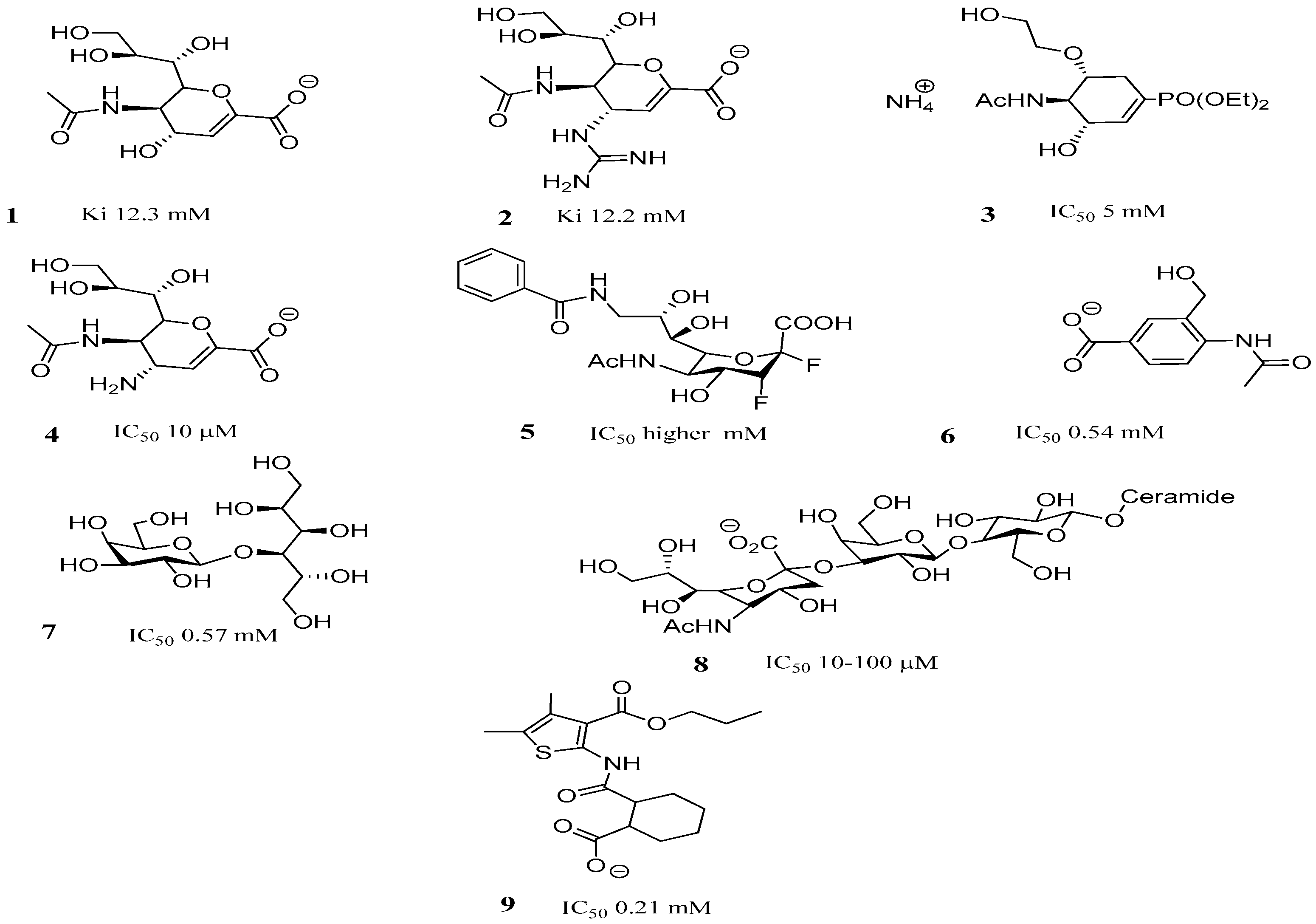
2.3. TcTS Inhibition
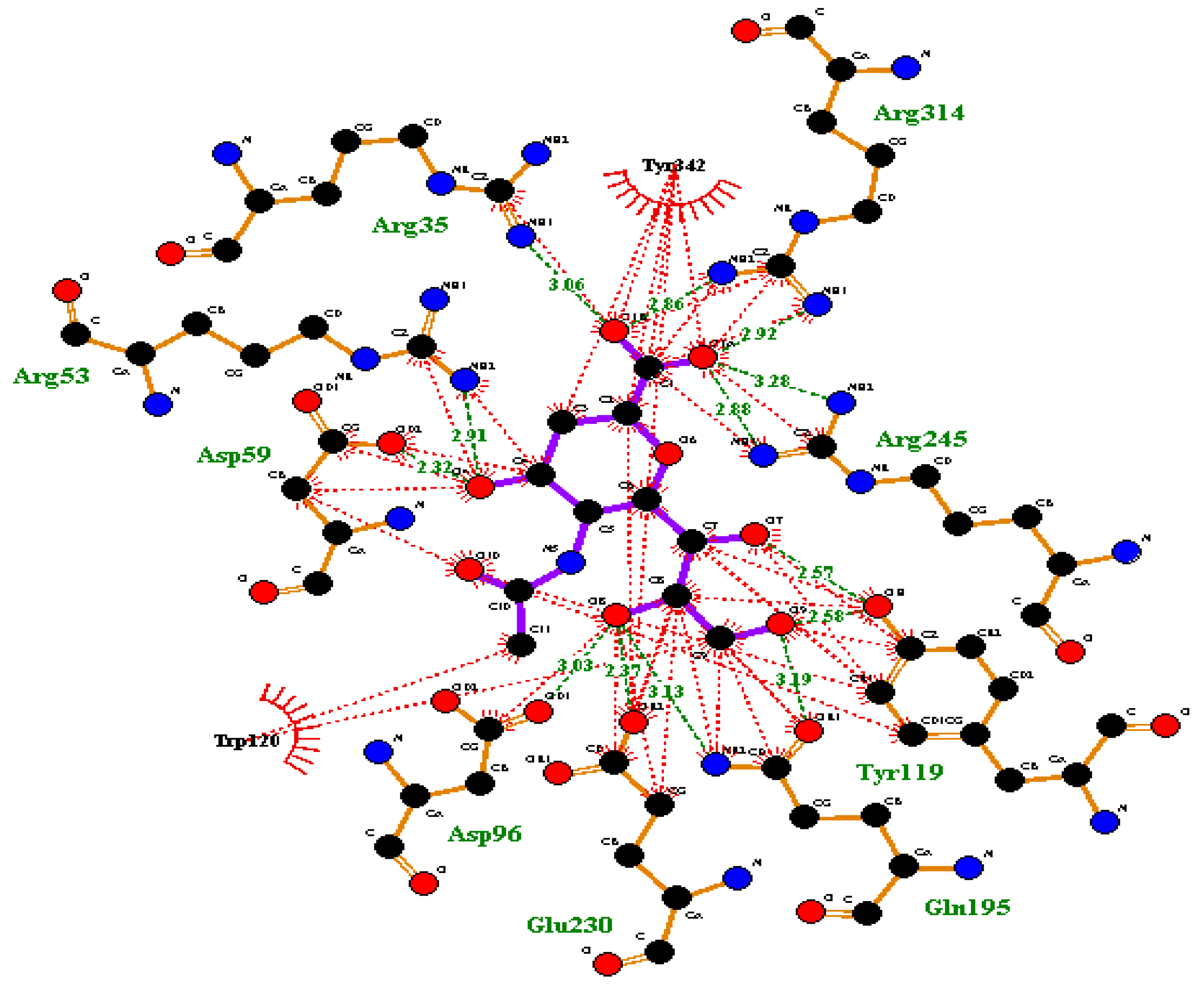
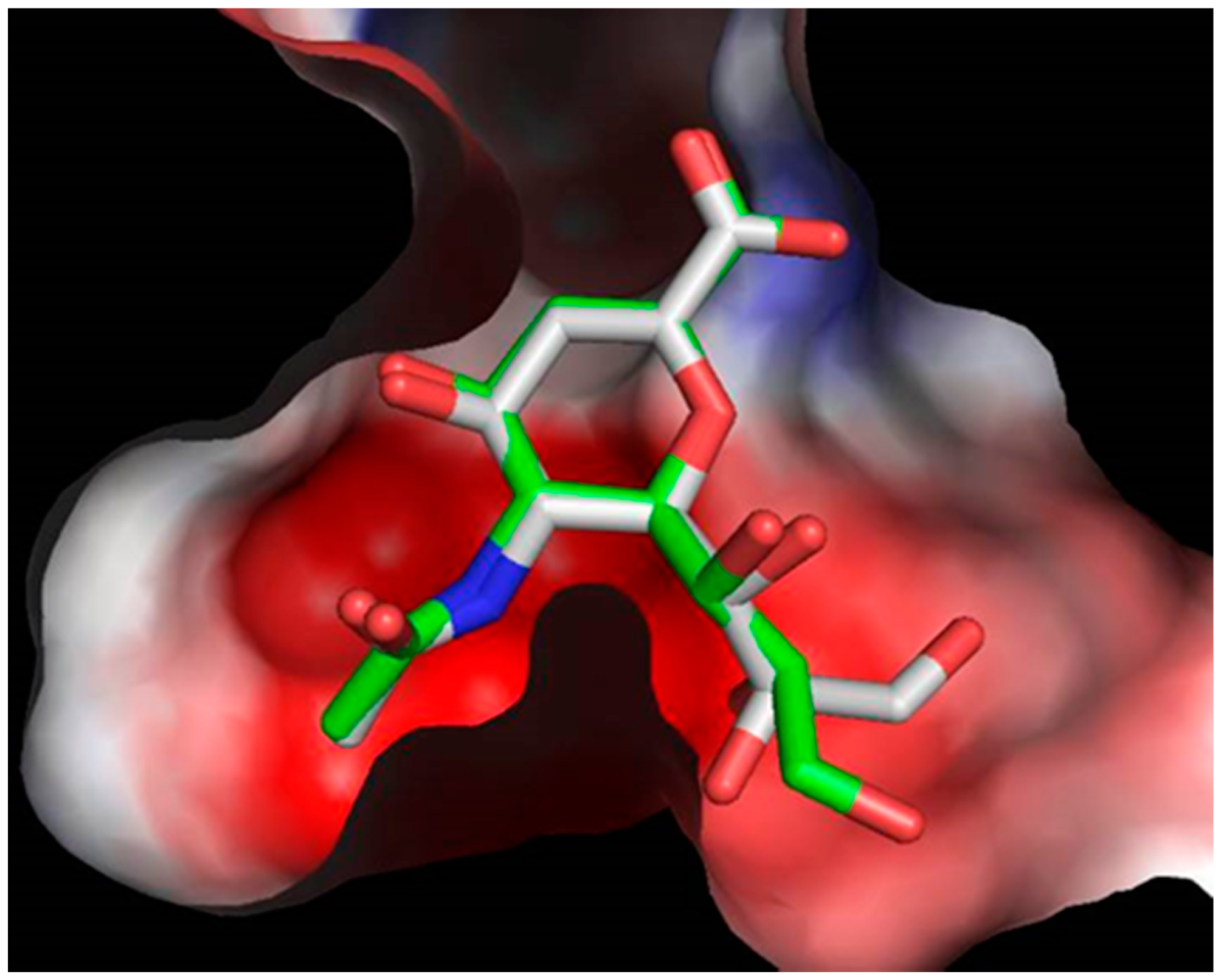
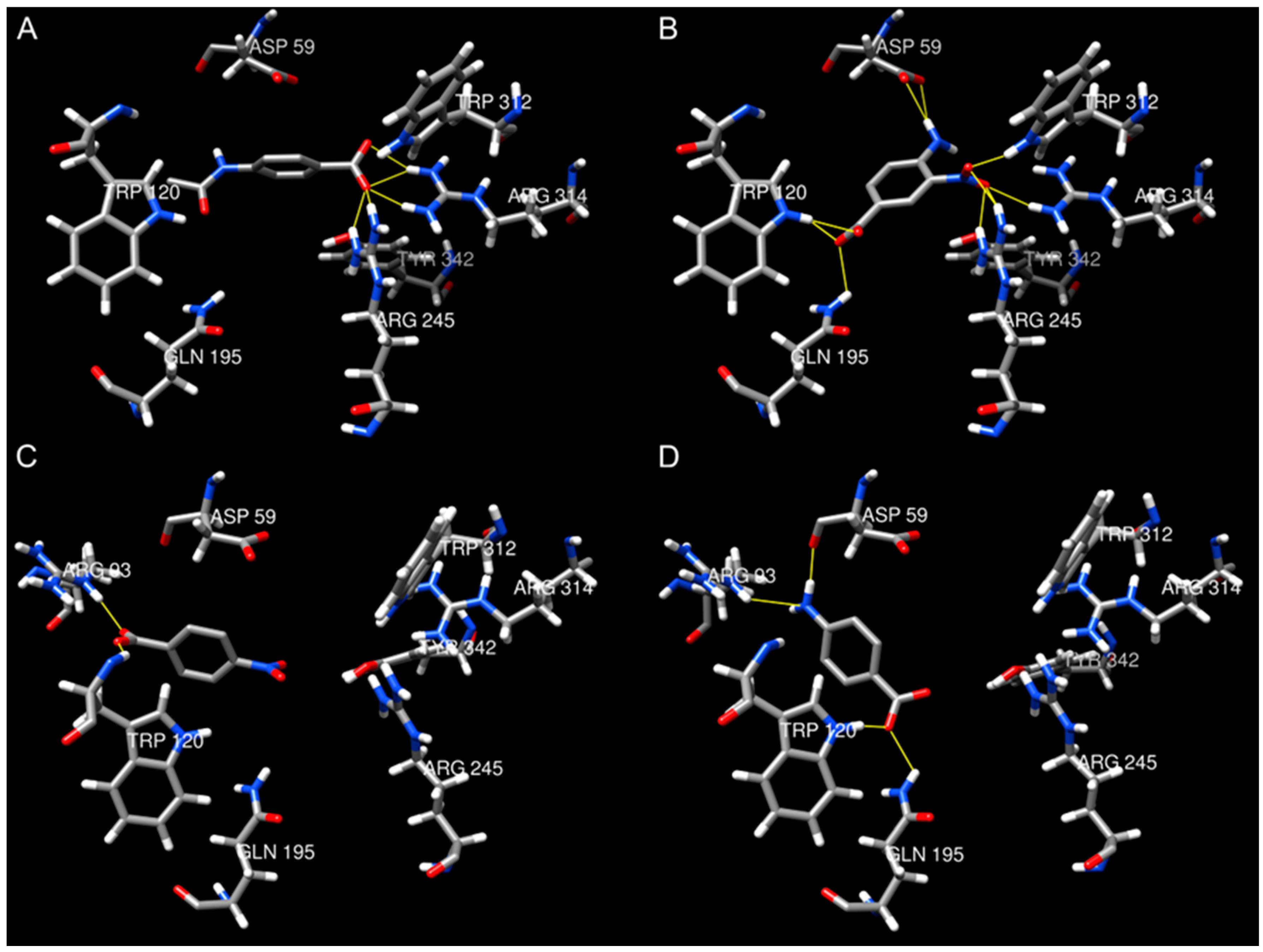
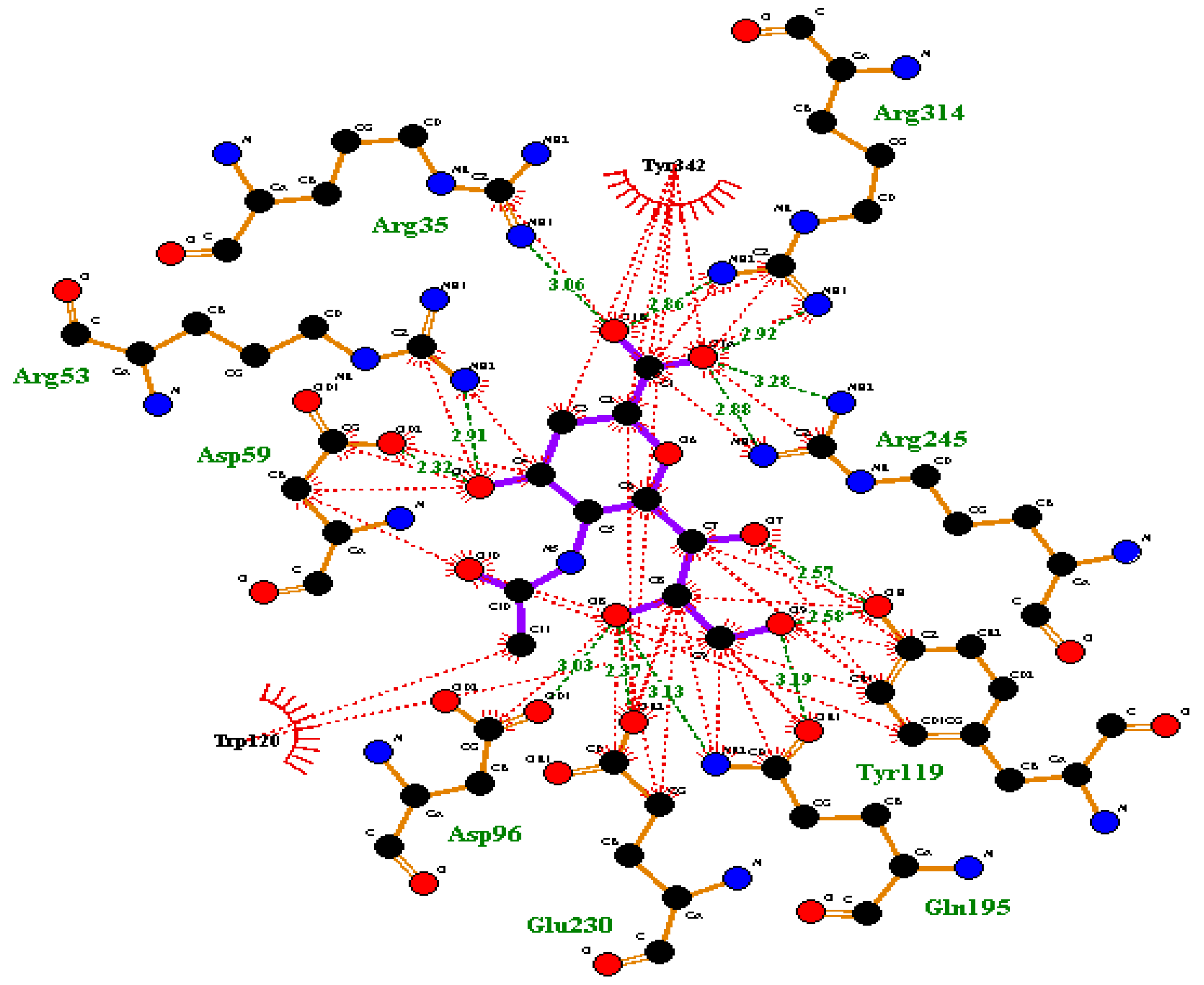
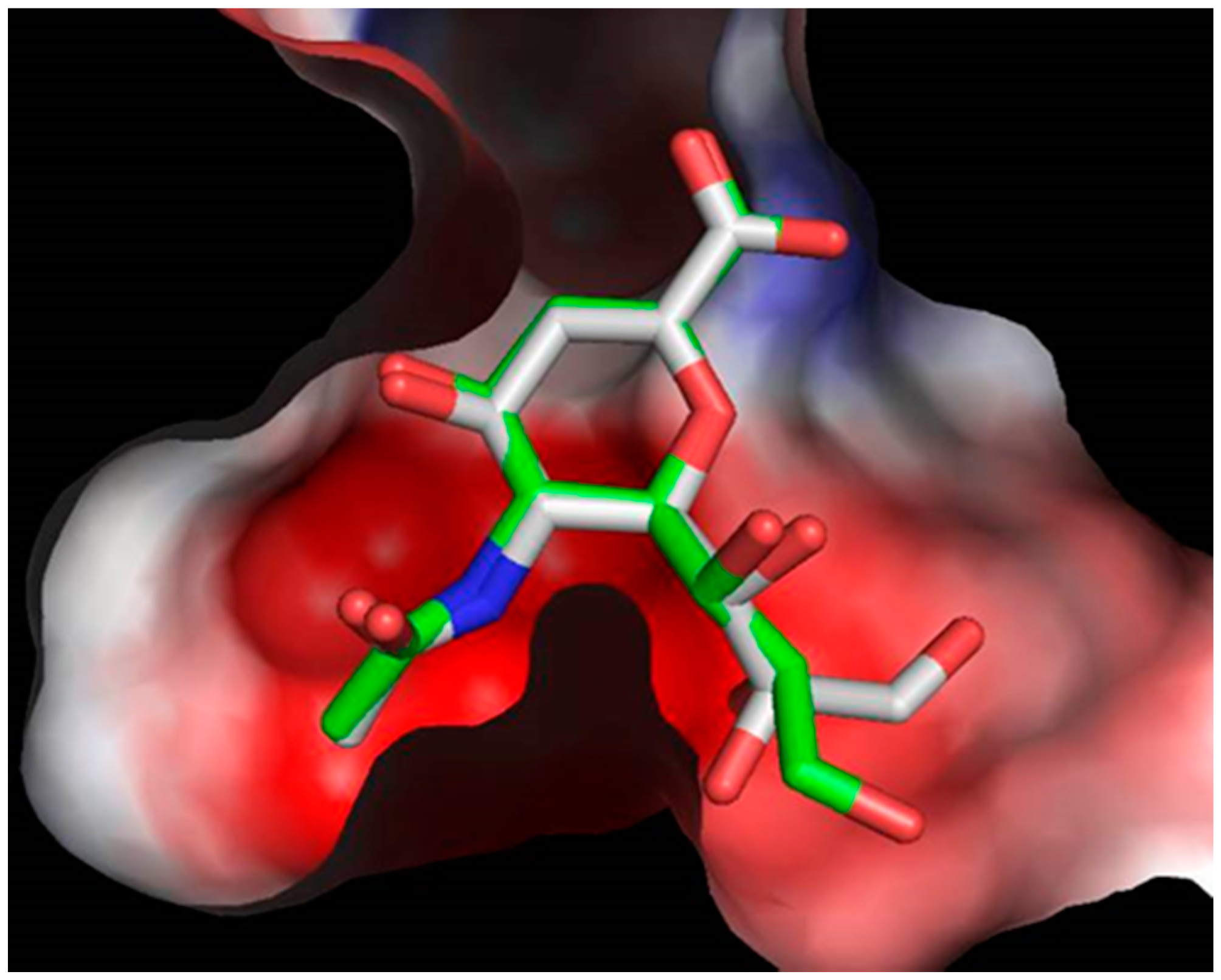
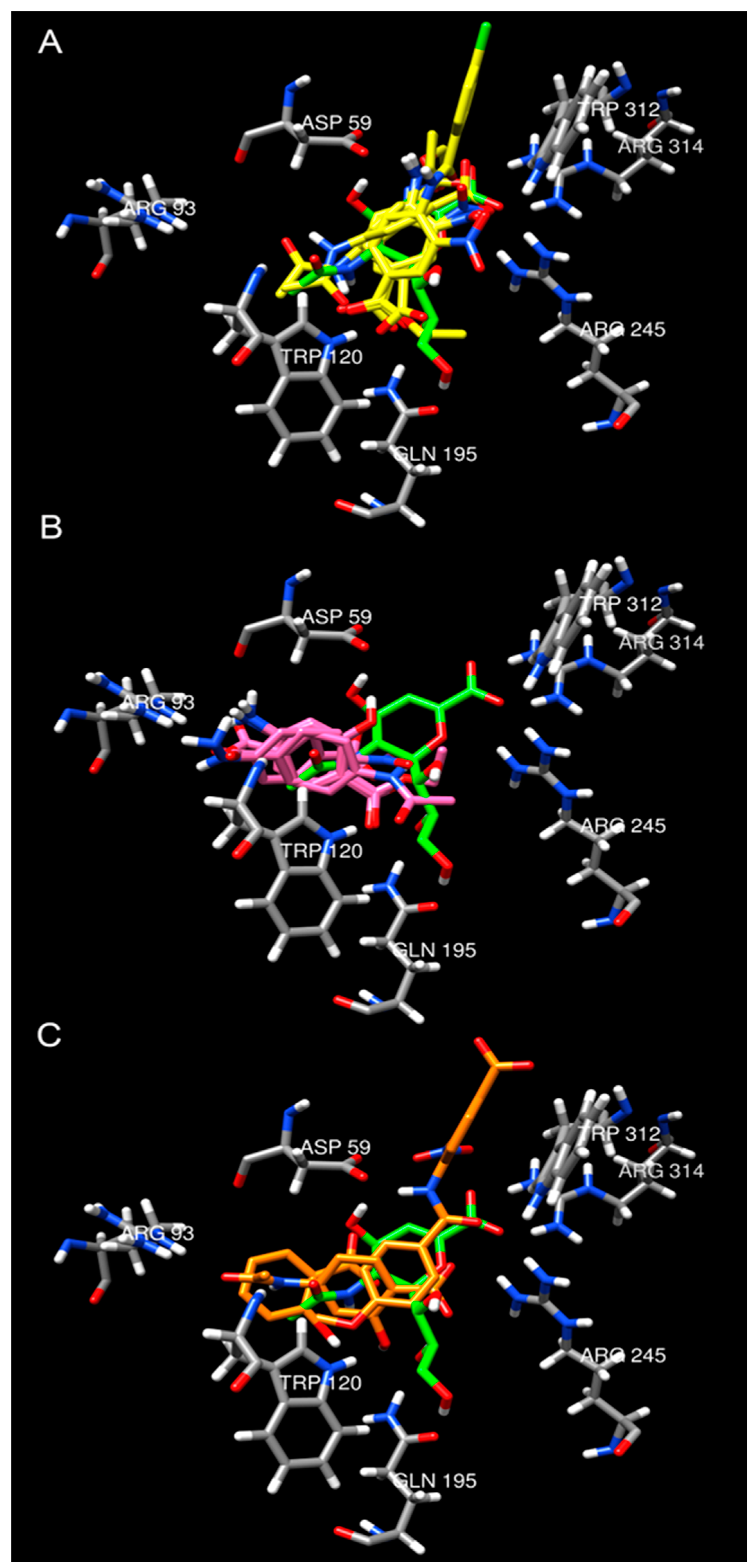
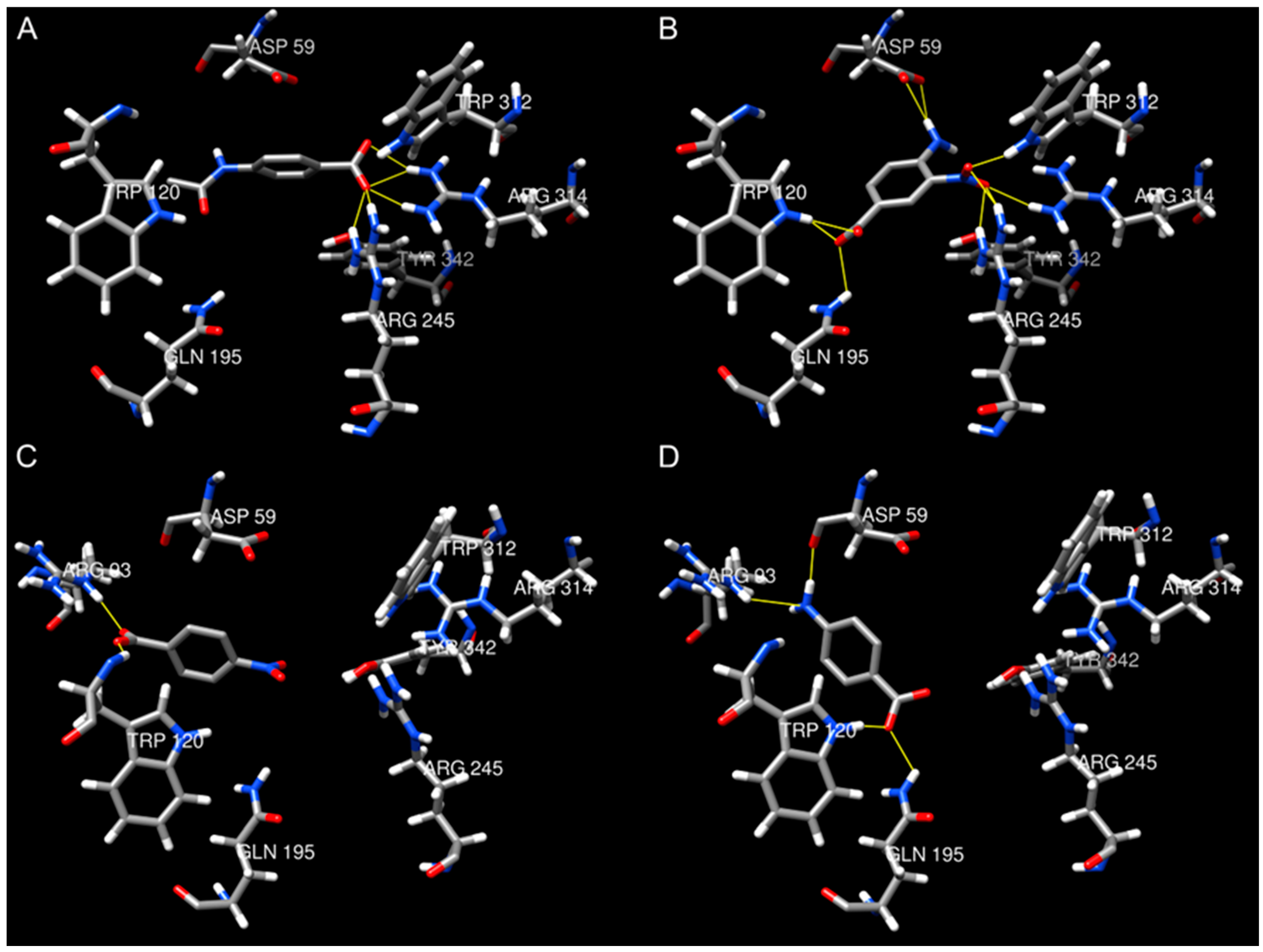
2.4. Molecular Docking
3. Materials and Methods
3.1. Chemistry: General Procedure
3.2. Biological Assays
3.2.1. Trypanocidal Activity
3.2.2. Enzymatic Inhibition Assays
3.2.3. Molecular Docking
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lorca, M.; Soto, F.; Soto, P.; Padilla, G.N.; Rojas, E.J.; Bustamante, M.; Atencio, J.; Raychaudhuri, S. Chagas disease in the rural area of Metropolitan Region (Santiago) and V Region (Aconcagua), Chile. Rev. Med. Chile 2008, 136, 945–946. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Chagas disease after organ transplantation—United States, 2001. Morb. Mortal. Wkly. Rep. 2002, 51, 210–212. [Google Scholar]
- Tarleton, R.L.; Reithinger, R.; Urbina, J.A.; Kitron, U.; Gürtler, R.E. The challenges of Chagas disease—Grim outlook or glimmer of hope. PLoS Med. 2007, 4, e332. [Google Scholar] [CrossRef] [PubMed]
- Toso, M.A.; Vial, U.F.; Galant, N. Oral transmission of Chagas’ disease. Rev. Med. Chile 2011, 139, 258–266. [Google Scholar]
- Gascon, J.; Bern, C.; Pinazo, M.J. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 2010, 115, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Piron, M.; Vergés, M.; Muñoz, J.; Casamitjana, N.; Sanz, S.; Maymó, R.M.; Hernández, J.M.; Puig, L.; Portús, M.; Gascón, J.; et al. Seroprevalence of Trypanosoma cruzi infection in at-risk blood donors in Catalonia (Spain). Transfusion 2008, 48, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Schmunis, G.A. Epidemiology of Chagas disease in non-endemic countries: The role of international migration. Mem. Inst. Oswaldo Cruz 2007, 102, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Girolamo, D.C.; Bodine, C.; Marta, B.L.; Cinnamon, C.; Cacciatore, F. Chagas disease at the crossroad of international migration and public health policies: Why a national screening might not be enough. Euro Surveill. 2011, 16, 57. [Google Scholar]
- Camandaroba, E.L.; Pinheiro, L.C.M.; Andrade, S.G. Oral transmission of Chagas disease: Importance of Trypanosoma cruzi biodeme in the intragastric experimental infection. Rev. Inst. Med. Trop. São Paulo 2002, 44, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Coura, J.R.; Junqueira, A.C.; Fernandes, O.; Valente, S.A.; Miles, M.A. Emerging Chagas disease in Amazonian Brazil. Trends Parasitol. 2002, 18, 171–176. [Google Scholar] [CrossRef]
- Xavier, S.S.C.; Vaz, V.C.; Andrea, P.S.; Herrera, L.; Emperaire, L.; Alves, J.R.; Fernandes, O.; Ferreira, L.F.; Jansen, A.M. Mapping of the distribution of Trypanosoma cruzi infection among small wild mammals in a conservation unit and its surroundings (Northeast-Brazil). Parasitol. Int. 2007, 56, 119–128. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Chagas disease (American trypanosomiasis) fact sheet (revised in June 2010). Wkly. Epidemiol. Rec. 2010, 85, 334–336. [Google Scholar]
- Word Health Organization (WHO). Chagas Disease: Special Programme for Research and Training in Tropical Disease; TDR: Geneva, Switzerland, 2008. [Google Scholar]
- Kashif, M.; Herrera, A.M.; Lara-Ramirez, E.E.; Ramírez-Moreno, E.; García, V.B.; Ashfaq, M.; Rivera, G. Recent developments in trans-sialidase inhibitors of Trypanosoma cruzi. J. Drug Target. 2017, 25, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Yoshioka, K. Review: Surveillance of Chagas. Adv. Parasitol. 2012, 79, 375–428. [Google Scholar] [PubMed]
- Coura, J.R.; Dias, J.C. Epidemiology, control and surveillance of Chagas disease: 100 years after its discovery. Mem. Inst. Oswaldo Cruz 2009, 104, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Salomon, C.J. First century of Chagas’ disease: An overview on novel approaches to nifurtimox and benzonidazole delivery systems. J. Pharm. Sci. 2012, 101, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.A.; de Mecca, M.M.; Bartel, L.C. Toxic side effects of drugs used to treat Chagas’ disease (American trypanosomiasis). Hum. Exp. Toxicol. 2006, 25, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Viotti, R.; Vigliano, C.; Lococo, B.; Alvarez, M.G.; Petti, M.; Bertocchi, G.; Armenti, A. Side effects of benznidazole as treatment in chronic Chagas disease: Fears and realities. Expert Rev. Anti Infect. Ther. 2009, 7, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Altcheh, J.; Moscatelli, G.; Moroni, S.; Bournissen, G.F.; Freilij, H. Adverse events after the use of benznidazole in infants and children with Chagas disease. Pediatrics 2011, 127, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Santos, V.P.; Barrias, E.S.; Santos, J.F.; Barros Moreira, T.L.; De Carvalho, T.M.; Urbina, J.A.; De Souza, W. Effects of amiodarone and posaconazole on the growth and ultrastructure of Trypanosoma cruzi. Int. J. Antimicrob. Agent 2012, 40, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Chamond, N.; Coatnoan, N.; Minoprio, P. Immunotherapy of Trypanosoma cruzi infections. Curr. Drug Targets Immune Endocr. Metab. Disord. 2002, 2, 247–254. [Google Scholar]
- Rivera, G.; Bocanegra-García, V.; Ordaz-Pichardo, C.; Nogueda-Torres, B.; Monge, A. New therapeutic targets for drug design against Trypanosoma cruzi, advances and perspectives. Curr. Med. Chem. 2009, 16, 3286–3293. [Google Scholar] [CrossRef] [PubMed]
- Schenkman, S. The biological role of Trypanosoma cruzi trans-sialidase. Biochem. Soc. Trans. 1999, 27, A86. [Google Scholar] [CrossRef]
- Tomlinson, S.; Raper, J. Natural human immunity to trypanosomes. Parasitol. Today 1998, 14, 354–359. [Google Scholar] [CrossRef]
- Schenkman, S.; Jiang, M.S.; Hart, G.W.; Nussenzweig, V. A novel cell surface trans-sialidase of Trypanosoma cruzi generates a stage-specific epitope required for invasion of mammalian cells. Cell 1991, 65, 1117–1125. [Google Scholar] [CrossRef]
- Todeschini, A.R.; Girard, M.F.; Wieruszeski, J.M.; Nunes, M.P.; DosReis, G.A.; Mendonça-Previato, L.; Previato, J.O. trans-Sialidase from Trypanosoma cruzi binds host T-lymphocytes in a lectin manner. J. Biol. Chem. 2002, 277, 45962–45968. [Google Scholar] [CrossRef] [PubMed]
- Cremona, M.L.; Campetella, O.; Sánchez, D.O.; Frasch, A.C. Enzymically inactive members of the trans-sialidase family from Trypanosoma cruzi display β-galactose binding activity. Glycobiology 1999, 9, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, A.R.; Dias, W.B.; Girard, M.F.; Wieruszeski, J.M.; Mendonça-Previato, L.; Previato, J.O. Enzymatically inactive trans-sialidase from Trypanosoma cruzi binds sialyl and β-galactopyranosyl residues in a sequential ordered mechanism. J. Biol. Chem. 2004, 279, 5323–5328. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Chuenkova, M.; Ortega-Barria, E.; Pereira, M.E. Mediation of Trypanosoma cruzi invasion by sialic acid on the host cell and trans-sialidase on the trypanosome. Mol. Biochem. Parasitol. 1993, 59, 243–252. [Google Scholar] [CrossRef]
- Villalta, F.; Smith, C.M.; Ruiz-Ruano, A.; Lima, M.F. A ligand that Trypanosoma cruzi uses to bind to mammalian cells to initiate infection. FEBS Lett. 2001, 505, 383–388. [Google Scholar] [CrossRef]
- Magdesian, M.H.; Giordano, R.; Ulrich, H.; Juliano, M.A.; Juliano, L.; Schumacher, R.I.; Colli, W.; Alves, M.J. Infection by Trypanosoma cruzi identification of a parasite ligand and its host cell receptor. J. Biol. Chem. 2001, 276, 19382–19389. [Google Scholar] [CrossRef] [PubMed]
- Buschiazzo, A.; Amaya, M.F.; Cremona, M.L.; Frasch, A.C.; Alzari, P.M. The crystal structure and mode of action of trans-sialidase, a key enzyme in Trypanosoma cruzi pathogenesis. Mol. Cell 2002, 10, 757–768. [Google Scholar] [CrossRef]
- Paris, G.; Ratier, L.; Amaya, M.F.; Nguyen, T.; Alzari, P.M.; Frasch, A.C. A sialidase mutant displaying trans-sialidase activity. J. Mol. Biol. 2005, 345, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Neres, J.; Bonnet, P.; Edwards, P.N.; Kotian, P.L.; Buschiazzo, A.; Alzari, P.M.; Bryce, R.A.; Douglas, K.T. Benzoic acid and pyridine derivatives as inhibitors of Trypanosoma cruzi trans-sialidase. Bioorg. Med. Chem. 2007, 15, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Streicher, H.; Busse, H. Building a successful structural motif into sialylmimetics-cyclo-hexenephosphonate monoesters as pseudo-sialosides with promising inhibitory properties. Bioorg. Med. Chem. 2006, 14, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Buchini, S.; Buschiazzo, A.; Withers, S.G. A new generation of specific Trypanosoma cruzi trans-sialidase inhibitors. Angew. Chem. 2008, 47, 2700–2703. [Google Scholar] [CrossRef] [PubMed]
- Agustí, R.; París, G.; Ratier, L.; Frasch, AC.; de Lederkremer, R.M. Lactose derivatives are inhibitors of Trypanosoma cruzi trans-sialidase activity toward conventional substrates in vitro and in vivo. Glycobiology 2004, 14, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Agustí, R.; Giorgi, M.E.; de Lederkremer, R.M. The trans-sialidase from Trypanosoma cruzi efficiently transfers α-(2→3)-linked N-glycolylneuraminic acid to terminal β-galactosyl units. Carbohydr. Res. 2007, 342, 2465–2649. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, F.; Schenkman, S.; de Carvalho, L.P.; Tomlinson, S.; Kiso, M.Y.M.; Hasegawa, A.; Nussenzweig, V. Substrate specificity of the Trypanosoma cruzi trans-sialidase. Glycobiology 1992, 2, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Neres, J.; Brewer, M.L.; Ratier, L.; Botti, H.; Buschiazzo, A.; Edwards, P.N.; Mortenson, P.N.; Charlton, M.H.; Alzari, P.M.; Frasch, A.C.; et al. Discovery of novel inhibitors of Trypanosoma cruzi trans-sialidase from in silico screening. Bioorg. Med. Chem. Lett. 2009, 19, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.D.; Ryu, Y.; Kim, S.C. Facile One-Pot Synthesis of PABA from MFB. Appl. Chem. Eng. 2014, 25, 337–339. [Google Scholar] [CrossRef]
- Jing, L.J.L.; Cheng, Z.L.; Xing, M.Z. Synthesis of 4-hydrazinobenzoic acid. Huaxue Shijie 2012, 53, 681–704. [Google Scholar]
- Doub, L.; Schaefer, J.J.; Bambas, L.L.; Walker, C.T. Some derivatives of 4-amino-2-hydroxybenzoic acid (p-aminosalicylic acid). J. Am. Chem. Soc. 1951, 73, 903–906. [Google Scholar] [CrossRef]
- Siddiki, A.A.; Takale, B.S.; Telvekar, V.N. One pot synthesis of aromatic azide using sodium nitrite and hydrazine hydrate. Tetrahedron Lett. 2013, 54, 1294–1297. [Google Scholar] [CrossRef]
- Mallonee, J.E. Nitration of 4-Acetamidobenzoic Acid. U.S. Patent 3,428,673 A, 18 February 1969. [Google Scholar]
- Toledo, M.J.; Bahia, M.T.; Carneiro, C.M.; Martins-Filho, O.A.; Tibayrenc, M.; Barnabé, C.; Tafuri, W.L.; de Lana, M. Chemotherapy with benznidazole and itraconazole for mice infected with different Trypanosoma cruzi clonal genotypes. Antimicrob. Agents Chemother. 2003, 47, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, G.G.; Carvalho, M.R.; Andrade, P.D.; Lopes, C.D.; Carneiro, Z.A.; Sesti-Costa, R.; Silva, J.S.; Carvalho, I. Synthesis and in vitro Evaluation of Novel Galactosyl-triazolo-benzene sulfonamides Against Trypanosoma cruzi. J. Braz. Chem. Soc. 2014, 25, 1872–1884. [Google Scholar]
- Ferrero García, M.A.; Sánchez, D.O.; Frasch, A.C.; Parodi, A.J. The effect of pyridoxal 5 phosphate and related compounds on Trypanosoma cruzi trans-sialidase. An. Asoc. Quim. Argent. 1993, 8, 127–132. [Google Scholar]
- AutoDock Tools (Version 1.5.6 rc2), Stefano Forte. Molecular Graphics Laboratory, Department of Molecular Biology, The Scripps Research Institute, 1999–2010. Available online: http://mgltools.scripps.edu (accessed on August 2016).
- Brener, Z. Biology of Trypanosoma cruzi. Annu. Rev. Microbiol. 1973, 27, 347–382. [Google Scholar] [CrossRef] [PubMed]
- Neres, J.; Buschiazzo, A.; Alzari, P.M.; Walsh, L.; Douglas, K.T. Continuous fluorimetric assay for high-throughput screening of inhibitors of trans-sialidase from Trypanosoma cruzi. Anal. Biochem. 2006, 357, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Maestro, version 9.1; Schrödinger, LLC: New York, NY, USA, 2010. [Google Scholar]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 30, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 10–24 are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Code | R1 | R2 | R3 | R4 | NINOA LC50 (µM) | INC-5 LC50 (µM) |
|---|---|---|---|---|---|---|
| 10 | NH2 | H | H | OH | 0.52 ± 0.19 | 1.24 ± 1.0 |
| 11 | NHNH2 | H | H | OH | 0.66 ± 0.39 | 0.58 ± 0.4 |
| 12 | N3 | H | H | OH | 0.60 ± 0.46 | 0.47 ± 0.35 |
| 13 | NO2 | H | H | OH | 0.47 ± 0.16 | 0.46 ± 0.38 |
| 14 | NH2 | H | H | OCH2CH3 | 0.10 ± 0.041 | 0.10 ± 0.047 |
| 15 | NHCOCH3 | H | H | OCH2CH3 | 0.34 ± 0.18 | 0.21 ± 0.1 |
| 16 | NH2 | NO2 | H | OH | 1.37 ± 0.56 | 0.63 ± 0.3 |
| 17 | NHCOCH3 | NO2 | H | OH | 1.10 ± 0.58 | 0.21 ± 0.1 |
| 18 | NHCOCH3 | NO2 | H | OCH2CH3 | 0.02 ± 0.012 | 0.22 ± 0.09 |
| 19 | NHCOC6H4-p-Cl | NO2 | H | OH | 0.14 ± 0.08 | 0.0008 ± 0.0001 |
| 20 | NHCOC6H4-OCH3 | NO2 | H | OH | 0.61 ± 0.3 | 0.43 ± 0.28 |
| 21 | NH2 | H | OH | OH | 0.27 ± 0.10 | 0.26 ± 0.09 |
| 22 | NHCOCH3 | H | OH | OH | 1.28 ± 026 | 1.28 ± 0.31 |
| 23 | H | H | OH | OH | 0.576 ± 0.32 | 0.721 ± 0.42 |
| 24 | NHCOCH3 | H | H | OH | 1.39 ± 0.75 | 0.878 ± 0.55 |
| Nfx | 0.213 ± 0.08 | 0.68 ± 0.17 | ||||
| Bzn | 0.292 ± 0.12 | 0.62 ± 0.28 |

| Code | R1 | R2 | R3 | R4 | % Inhib. at 1 mM |
|---|---|---|---|---|---|
| 10 | NH2 | H | H | OH | 30 |
| 11 | NHNH2 | H | H | OH | 61 |
| 12 | N3 | NO2 | H | OH | 40 |
| 13 | NO2 | H | H | OH | 43 |
| 14 | NH2 | H | H | OCH2CH3 | 1 |
| 15 | NHCOCH3 | H | H | OCH2CH3 | 7 |
| 16 | NH2 | NO2 | H | OH | 77 |
| 17 | NHCOCH3 | NO2 | H | OH | 66 |
| 18 | NHCOCH3 | NO2 | H | OCH2CH3 | 47 |
| 19 | NHCOC6H4-p-Cl | NO2 | H | OH | Not tested * |
| 20 | NHCOC6H4-p-OCH3 | NO2 | H | OH | Not tested * |
| 21 | NH2 | H | OH | OH | 32 |
| 22 | NHCOCH3 | H | OH | OH | 34 |
| 23 | H | H | OH | OH | 17 |
| 24 | NHCOCH3 | H | H | OH | 30 |
| Pyr | 64 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kashif, M.; Moreno-Herrera, A.; Villalobos-Rocha, J.C.; Nogueda-Torres, B.; Pérez-Villanueva, J.; Rodríguez-Villar, K.; Medina-Franco, J.L.; De Andrade, P.; Carvalho, I.; Rivera, G. Benzoic Acid Derivatives with Trypanocidal Activity: Enzymatic Analysis and Molecular Docking Studies toward Trans-Sialidase. Molecules 2017, 22, 1863. https://doi.org/10.3390/molecules22111863
Kashif M, Moreno-Herrera A, Villalobos-Rocha JC, Nogueda-Torres B, Pérez-Villanueva J, Rodríguez-Villar K, Medina-Franco JL, De Andrade P, Carvalho I, Rivera G. Benzoic Acid Derivatives with Trypanocidal Activity: Enzymatic Analysis and Molecular Docking Studies toward Trans-Sialidase. Molecules. 2017; 22(11):1863. https://doi.org/10.3390/molecules22111863
Chicago/Turabian StyleKashif, Muhammad, Antonio Moreno-Herrera, Juan Carlos Villalobos-Rocha, Benjamín Nogueda-Torres, Jaime Pérez-Villanueva, Karen Rodríguez-Villar, José Lius Medina-Franco, Peterson De Andrade, Ivone Carvalho, and Gildardo Rivera. 2017. "Benzoic Acid Derivatives with Trypanocidal Activity: Enzymatic Analysis and Molecular Docking Studies toward Trans-Sialidase" Molecules 22, no. 11: 1863. https://doi.org/10.3390/molecules22111863