Emodin Inhibition of Influenza A Virus Replication and Influenza Viral Pneumonia via the Nrf2, TLR4, p38/JNK and NF-kappaB Pathways

 ,
,
Abstract
:1. Introduction
2. Results
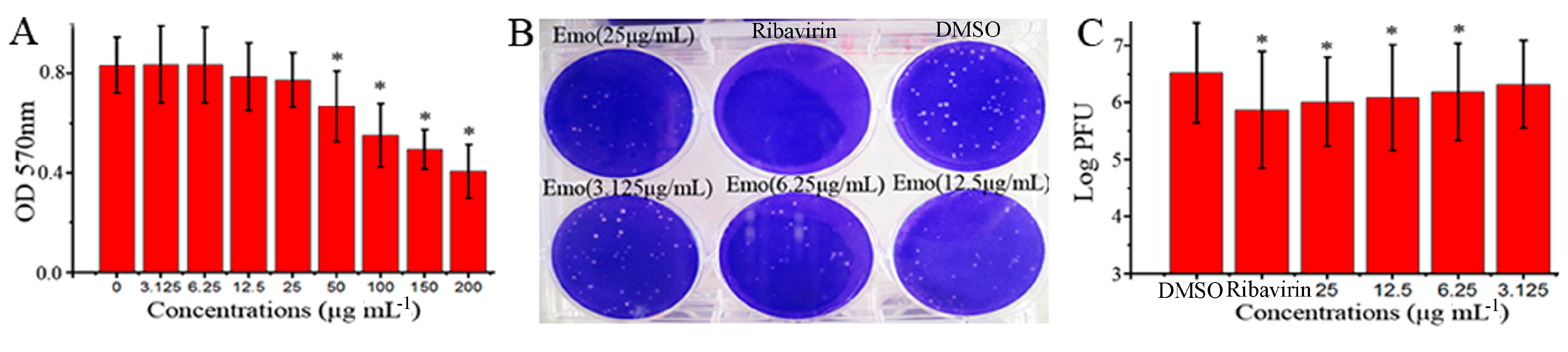
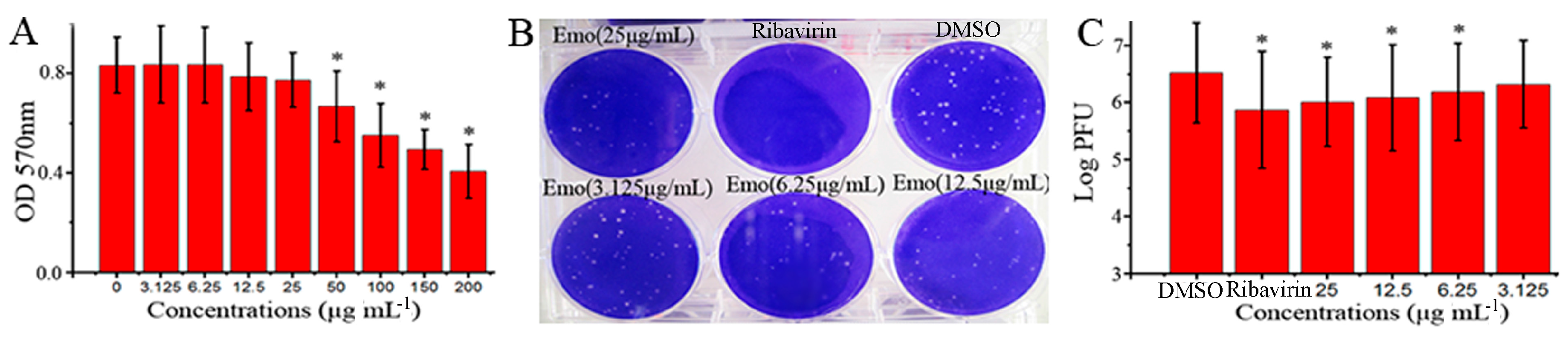
2.1. Emodin Could Inhibit the Replication of IAV In Vitro
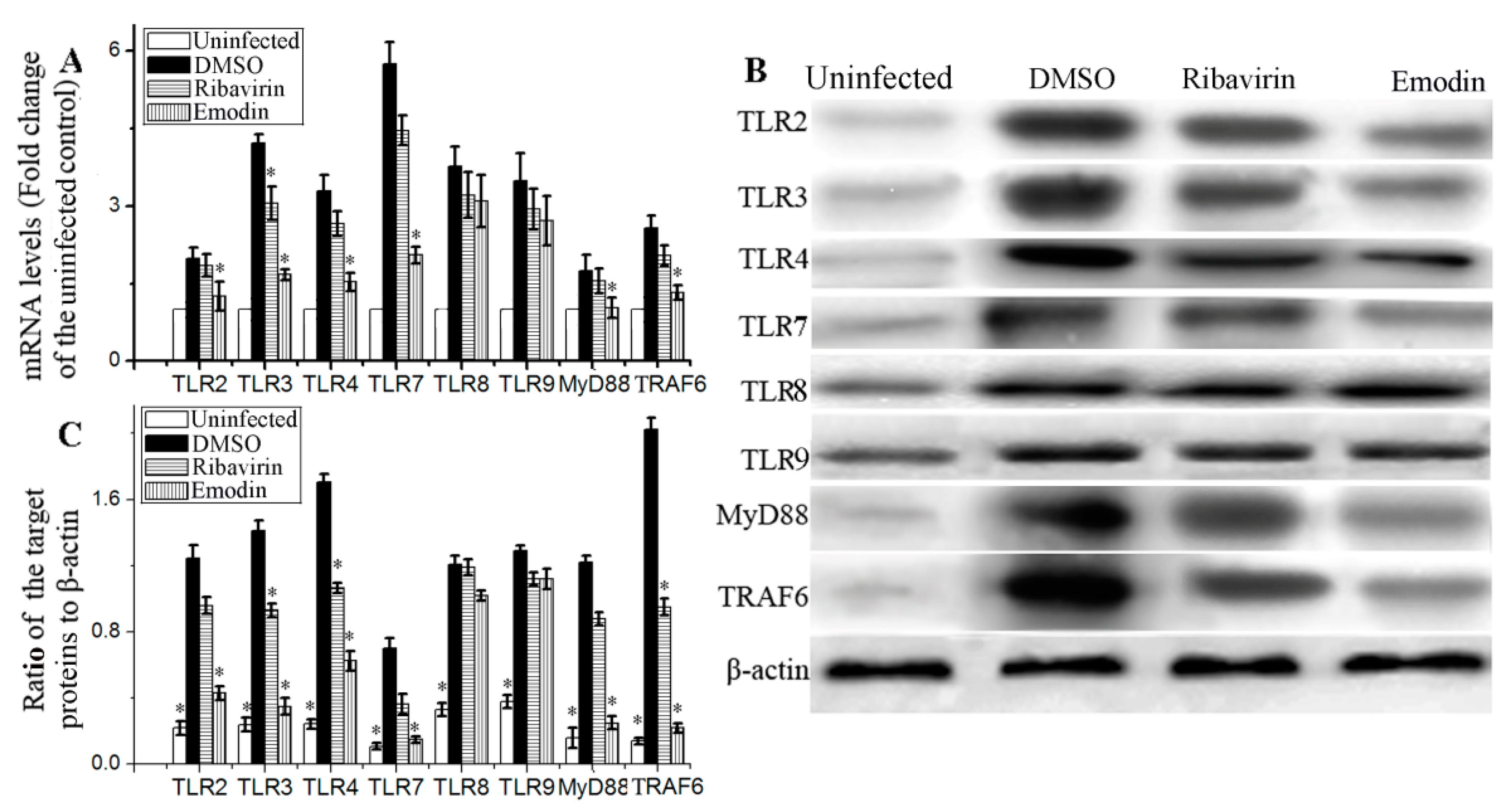
2.2. Emodin Inhibited IAV-Induced Expression of TLR2/3/4/7, MyD88 and TRAF6 In Vitro
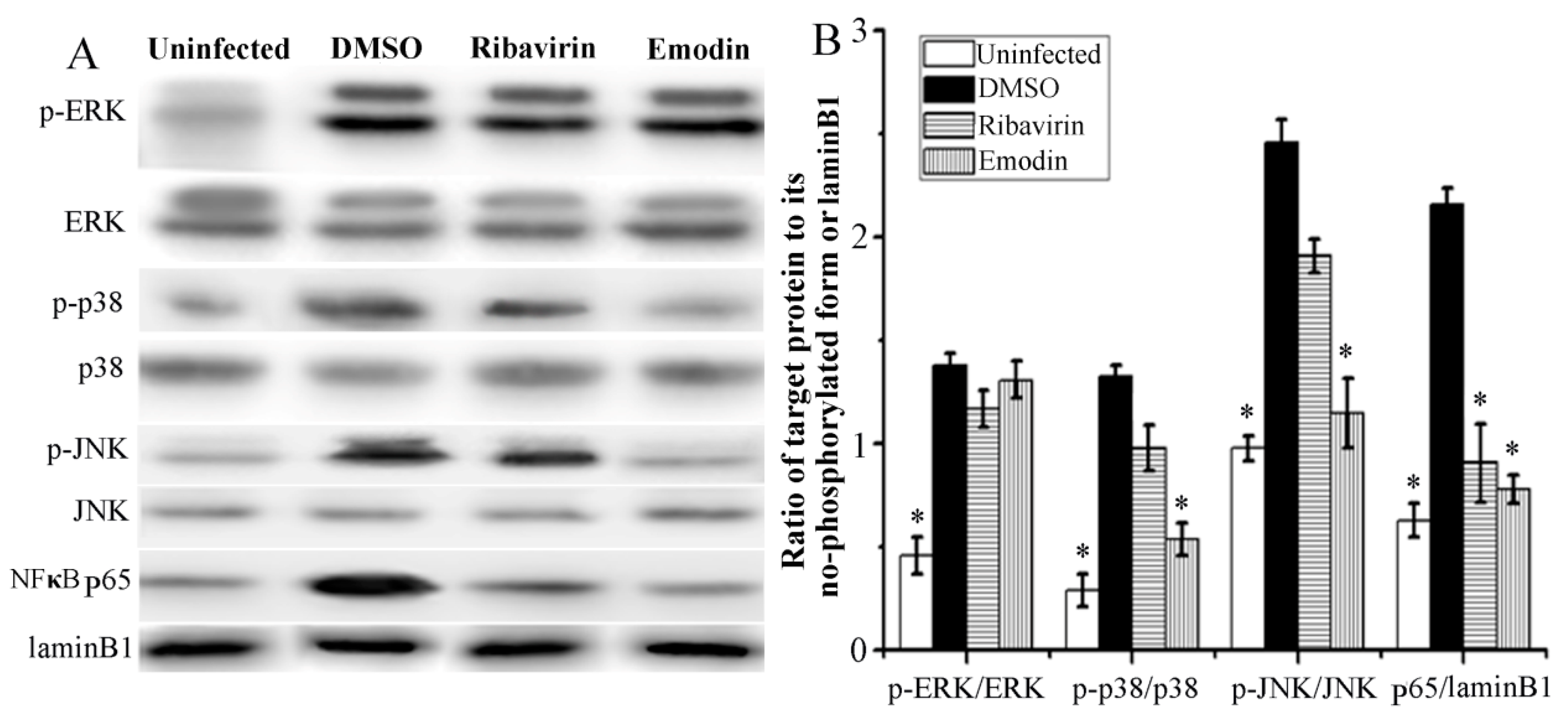
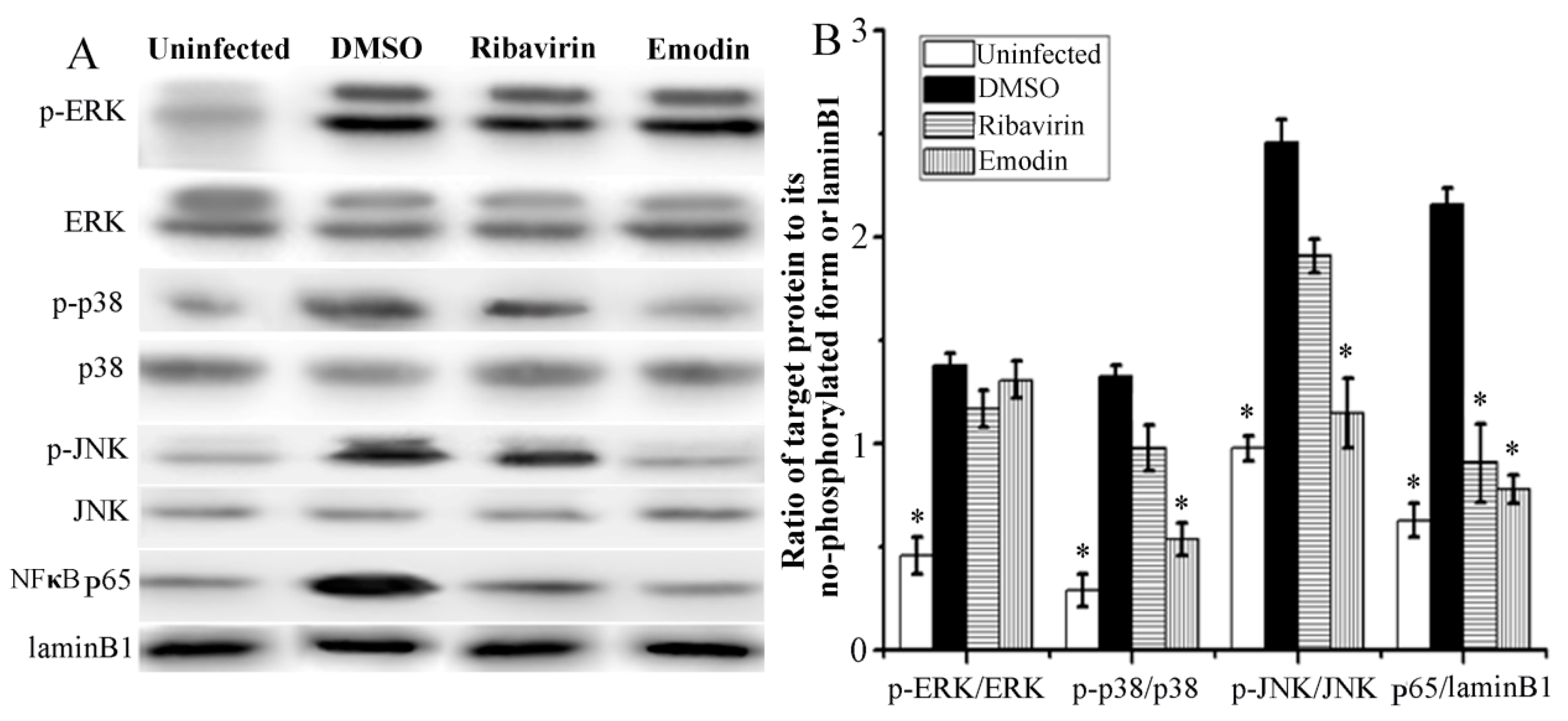
2.3. Emodin Inhibited IAV-Induced Activations of p38/JNK MAPKs and NF-κB Pathways In Vitro
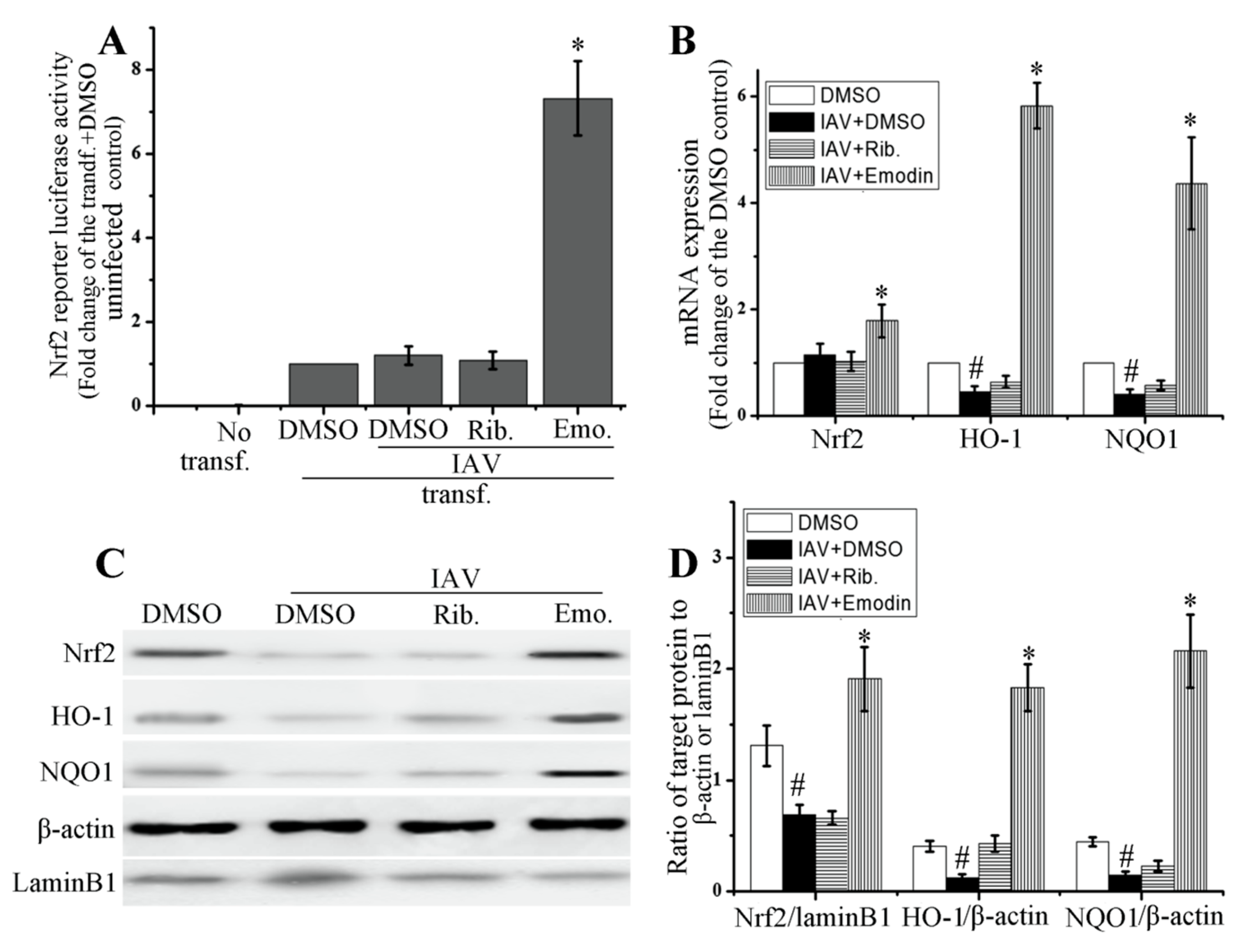
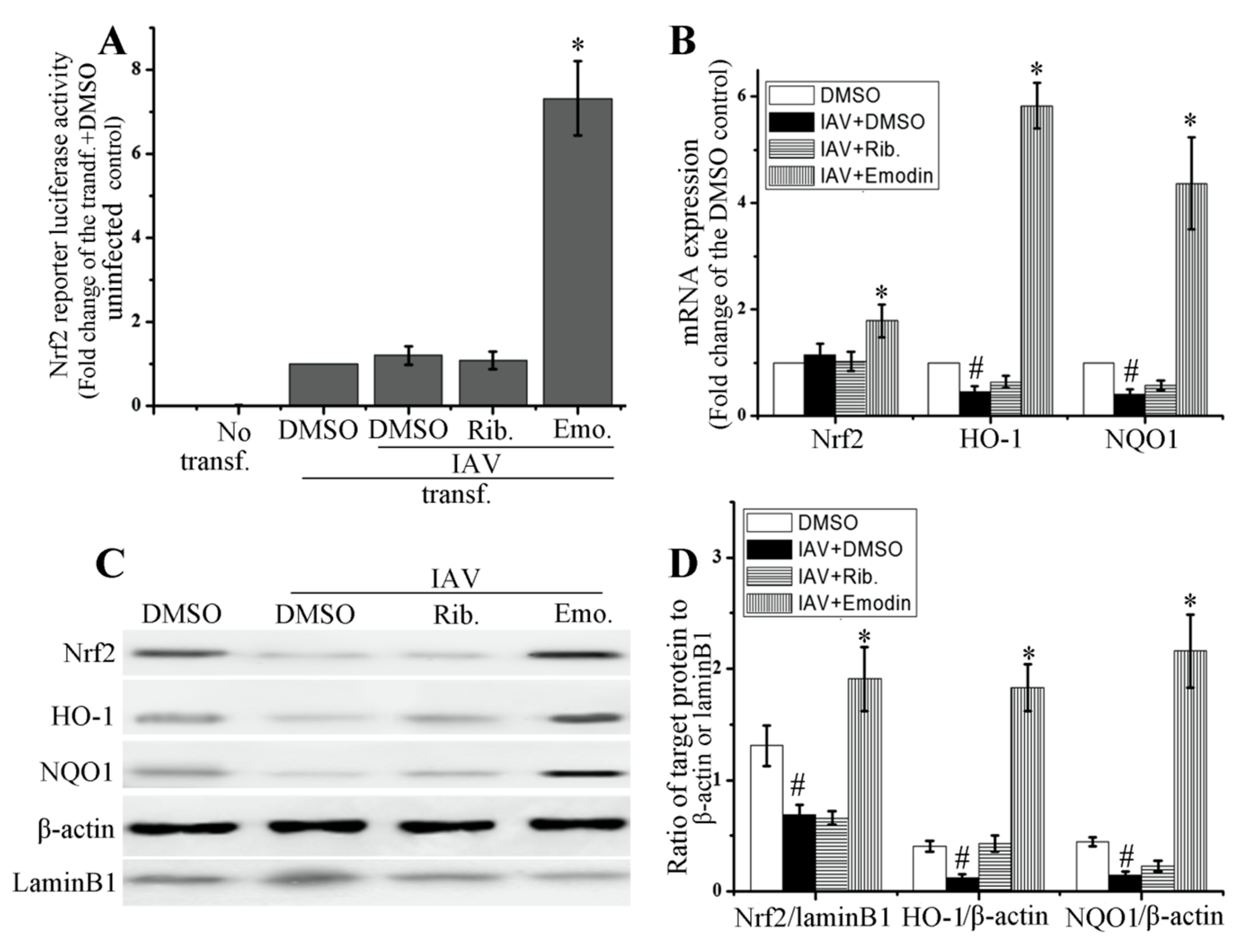
2.4. Emodin Enhanced Nrf2 Signal and Inhibited IAV-Induced Oxidative Stress
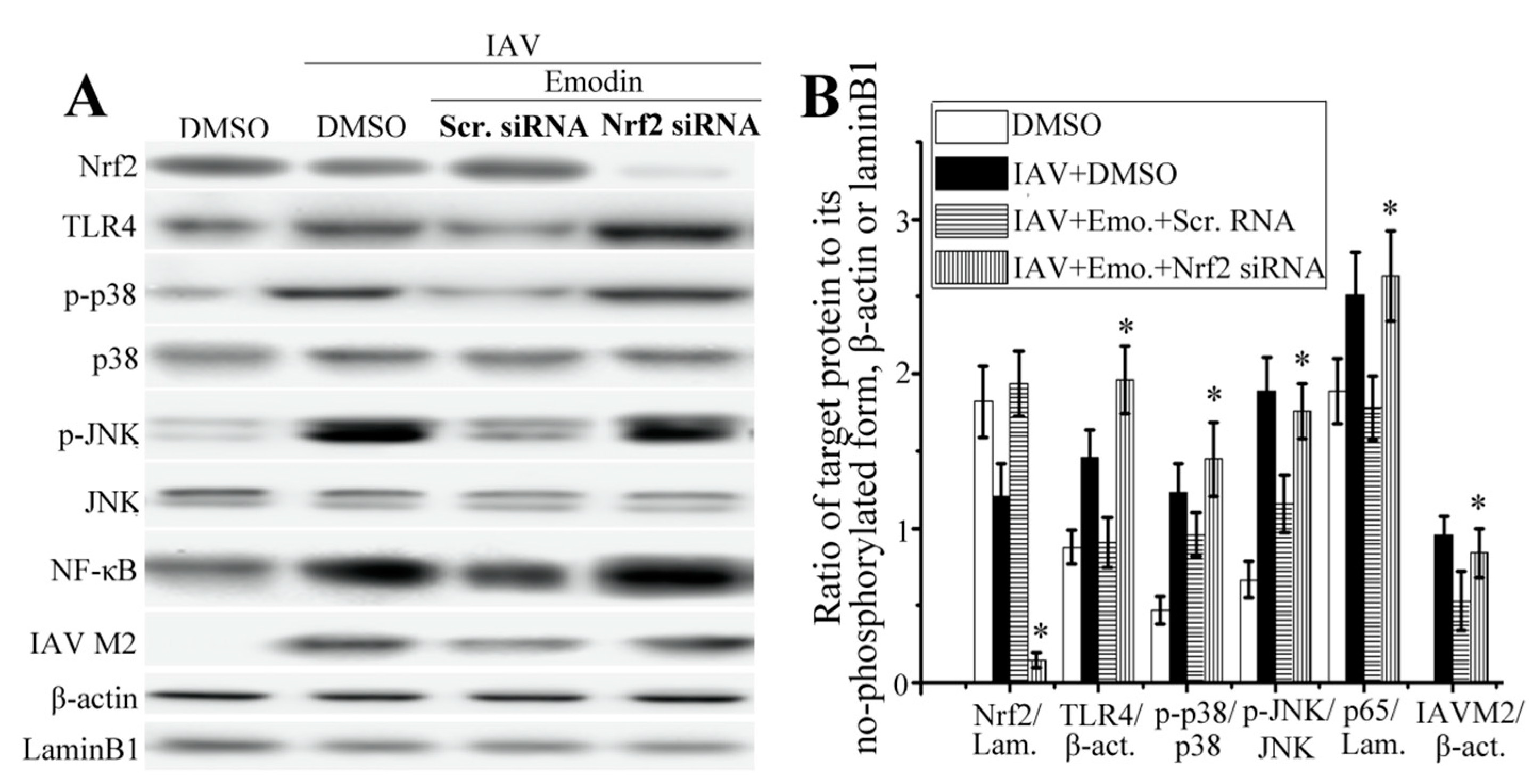
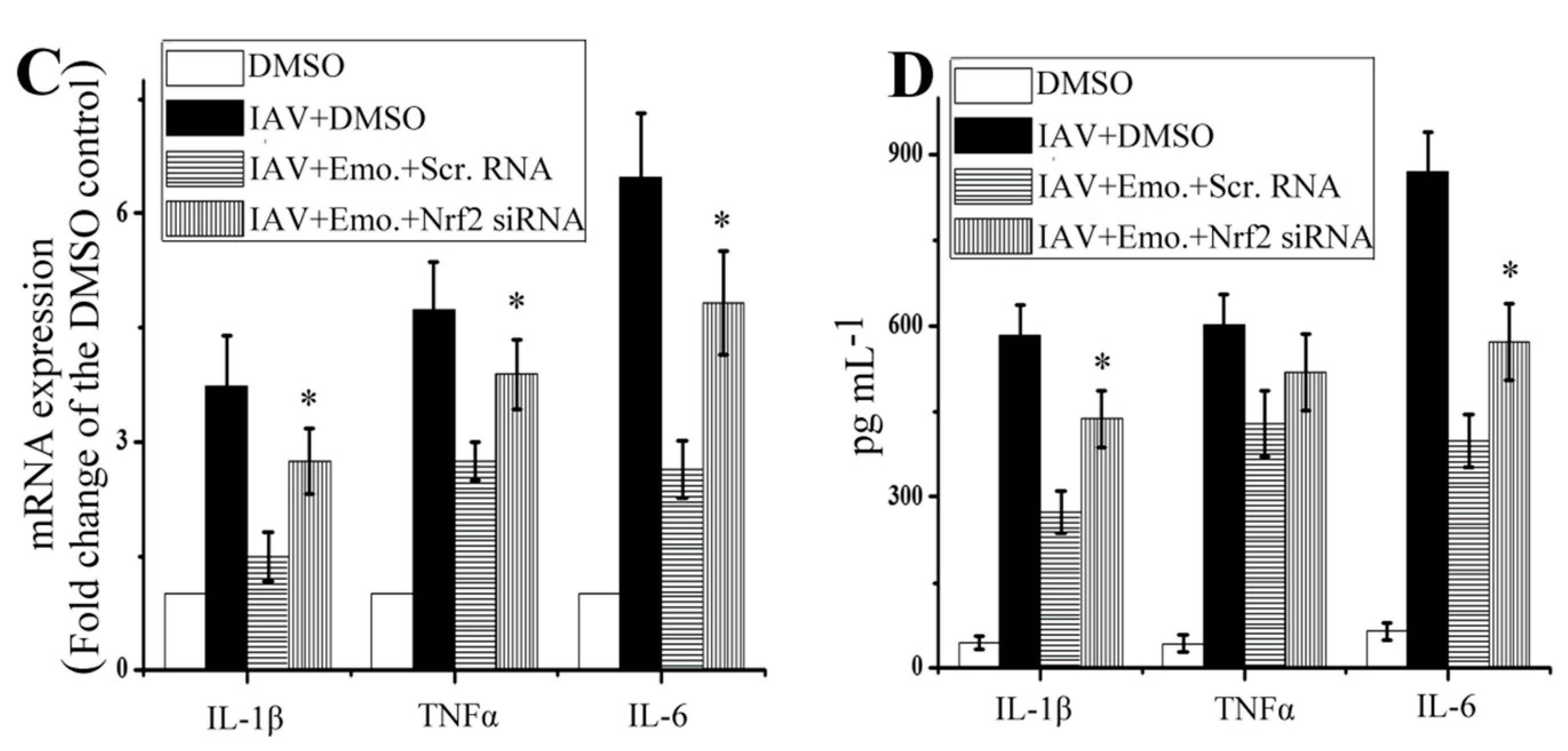
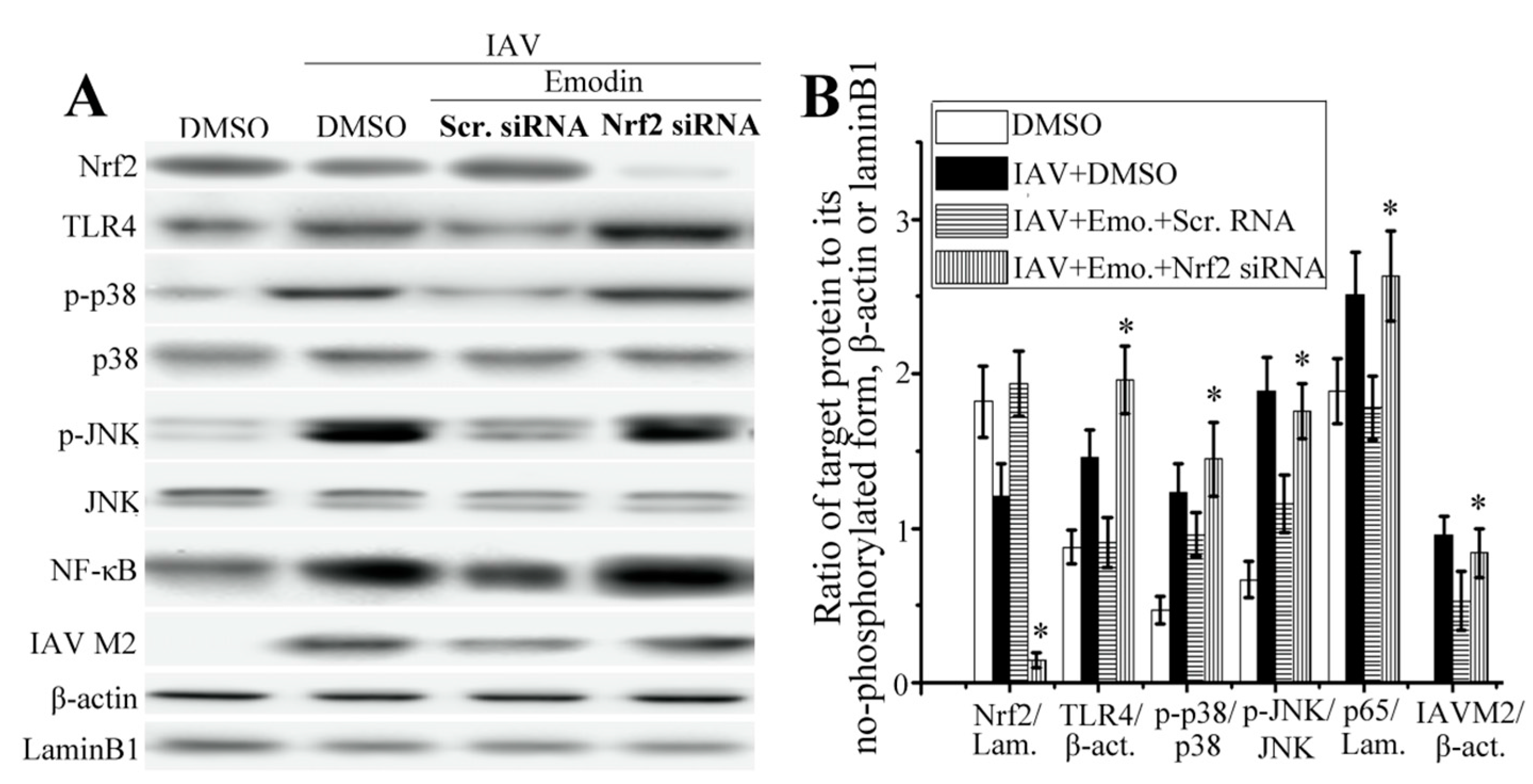
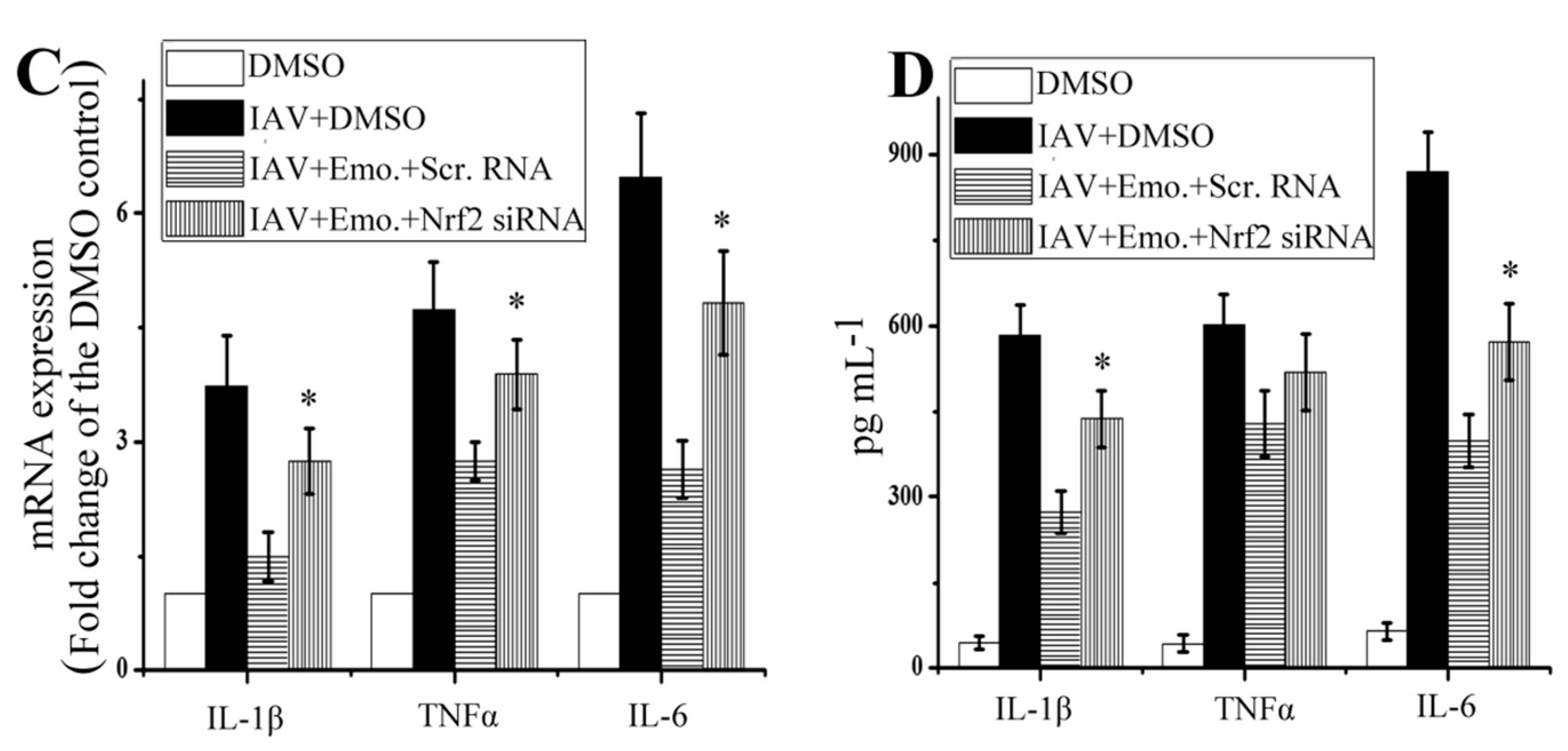
2.5. Nrf2 Signaling Played an Important Role in the Inhibition of Emodin on IAV-Induced Inflammation and IAV Replication
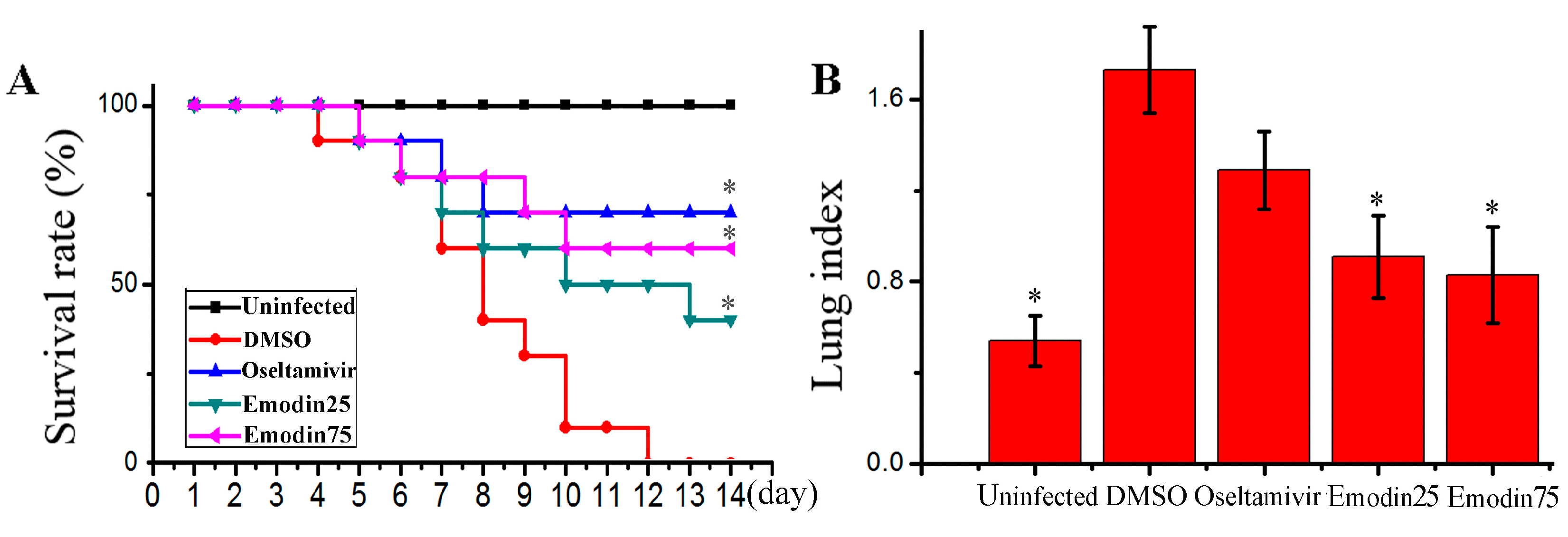
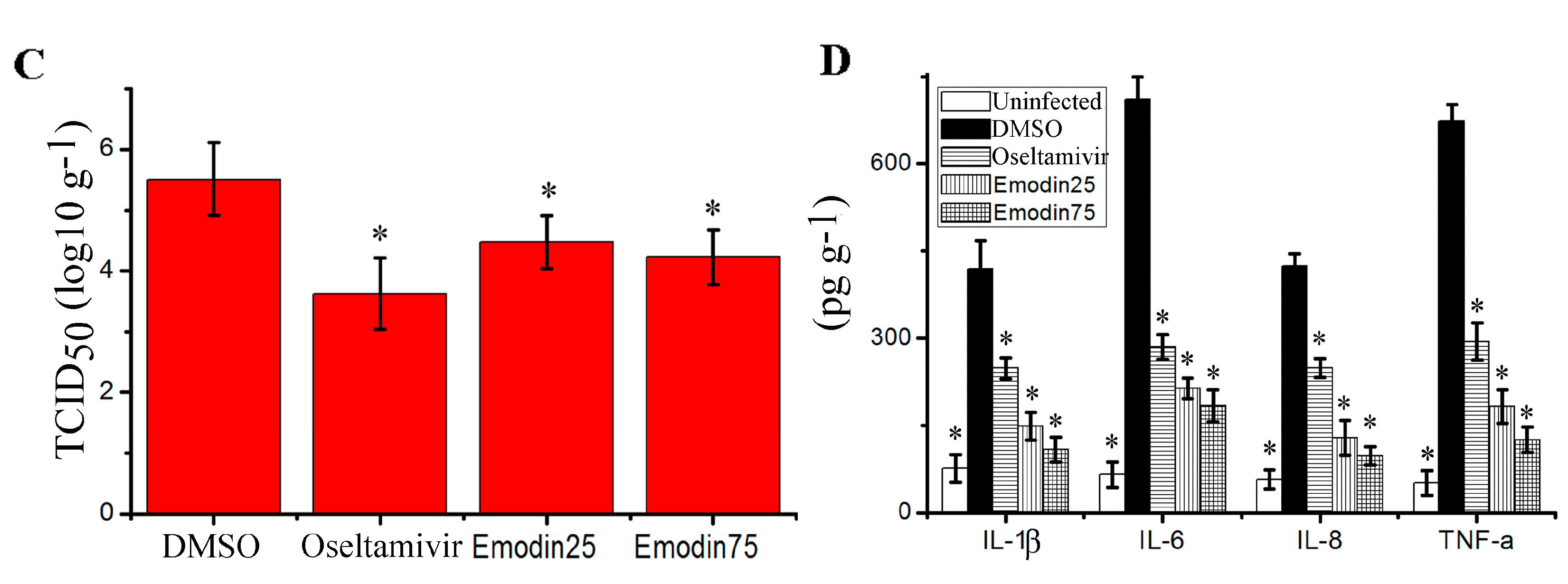
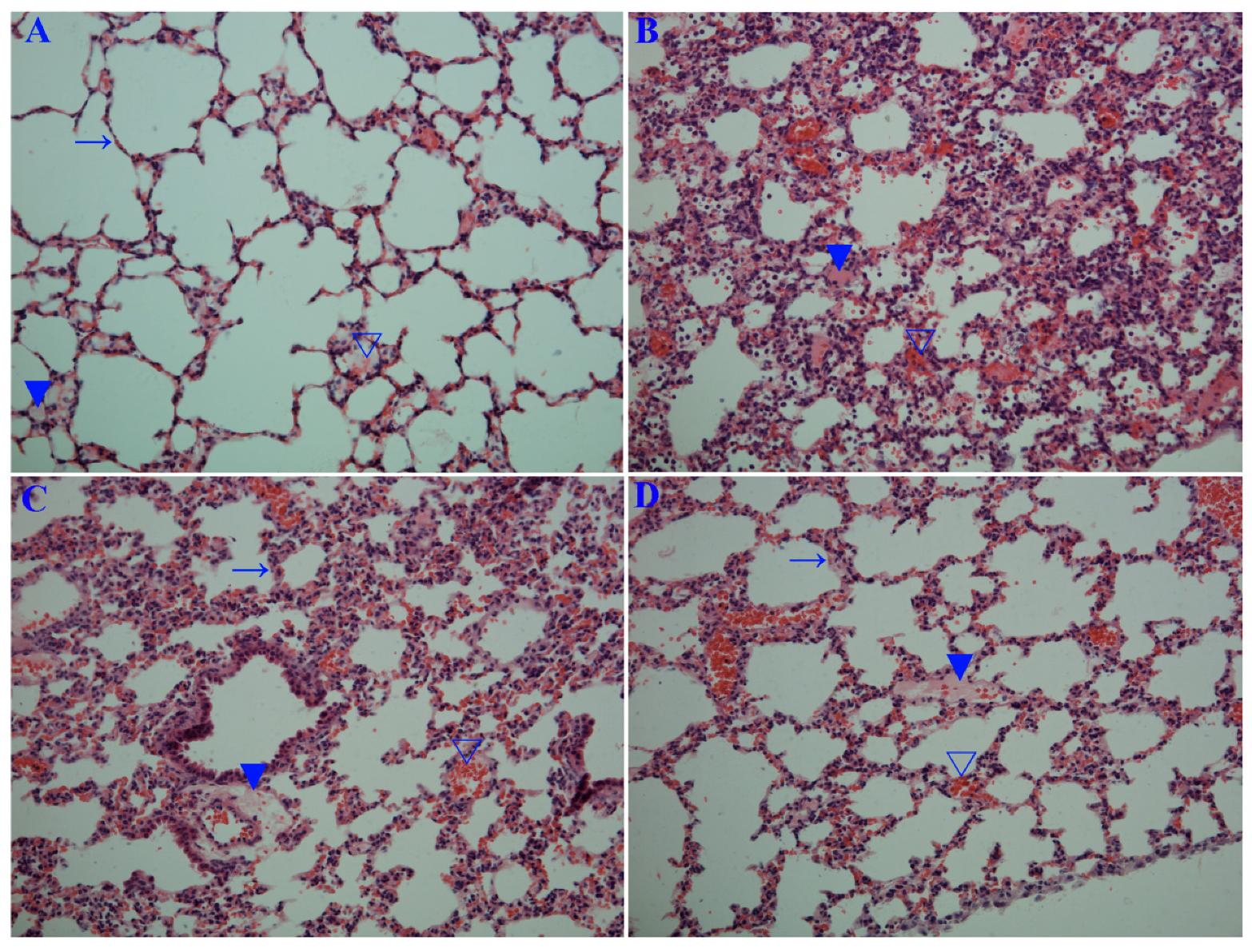
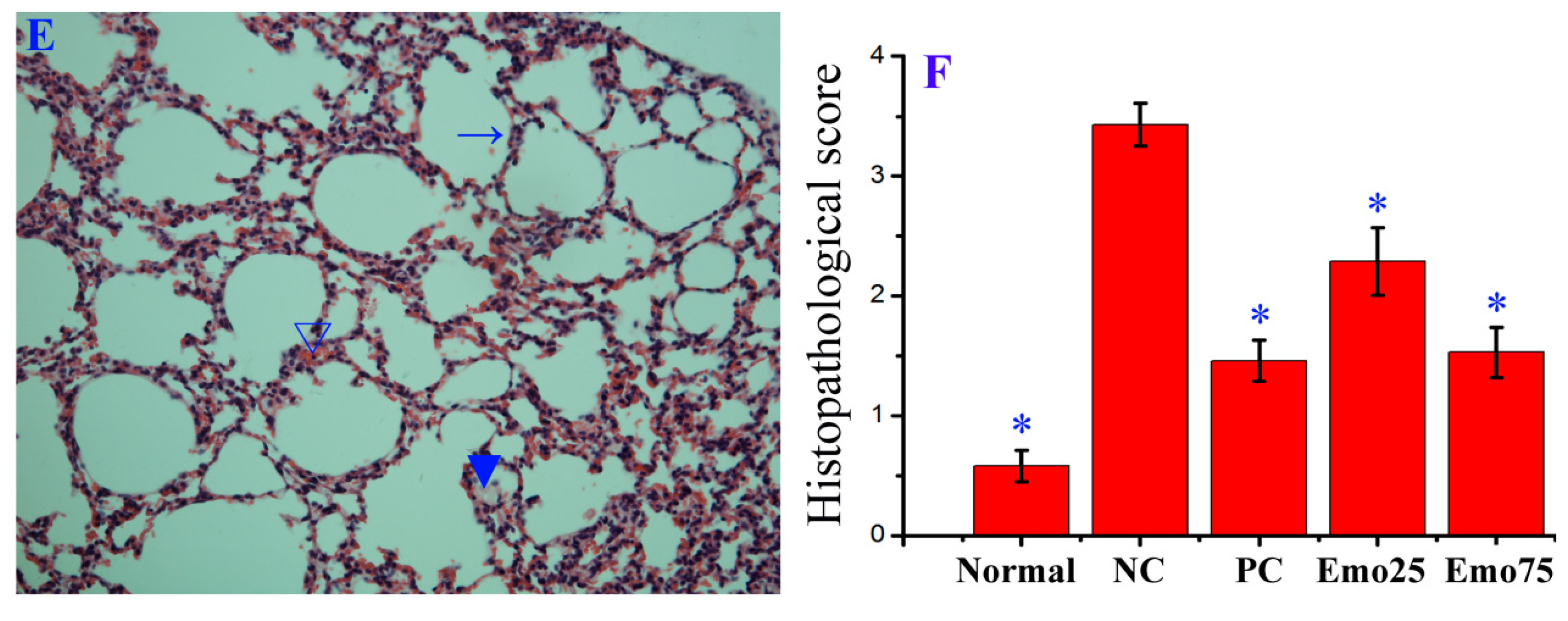
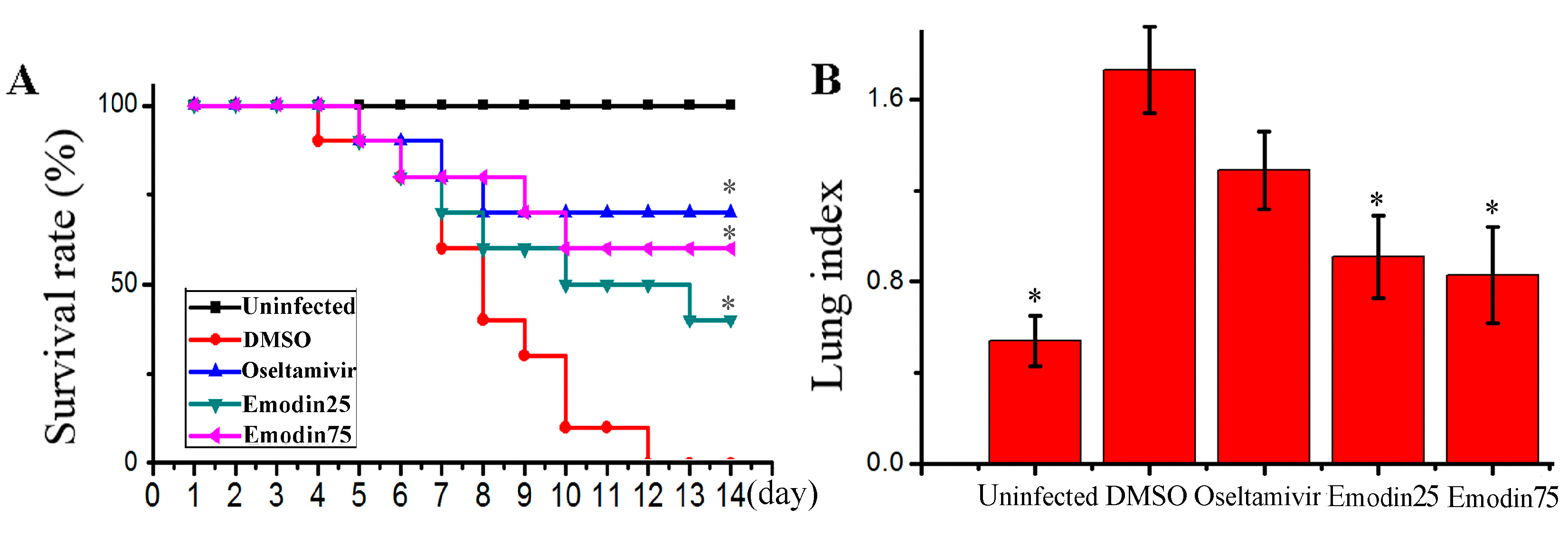
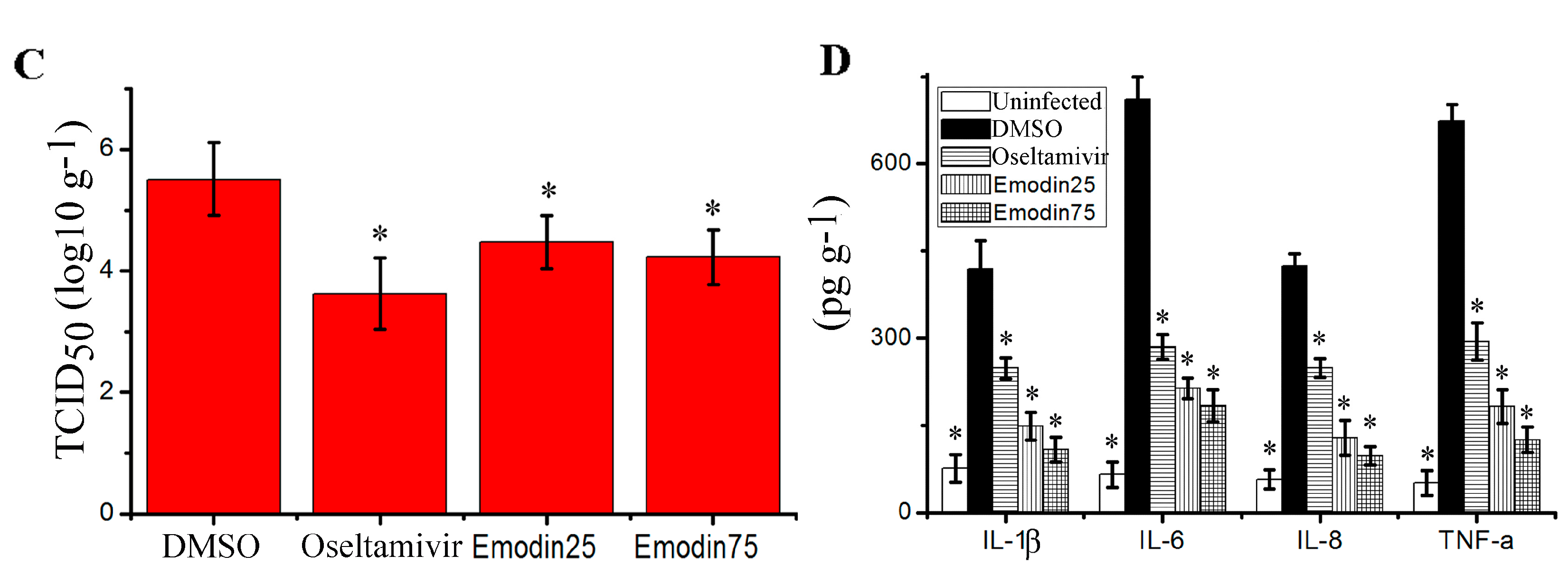
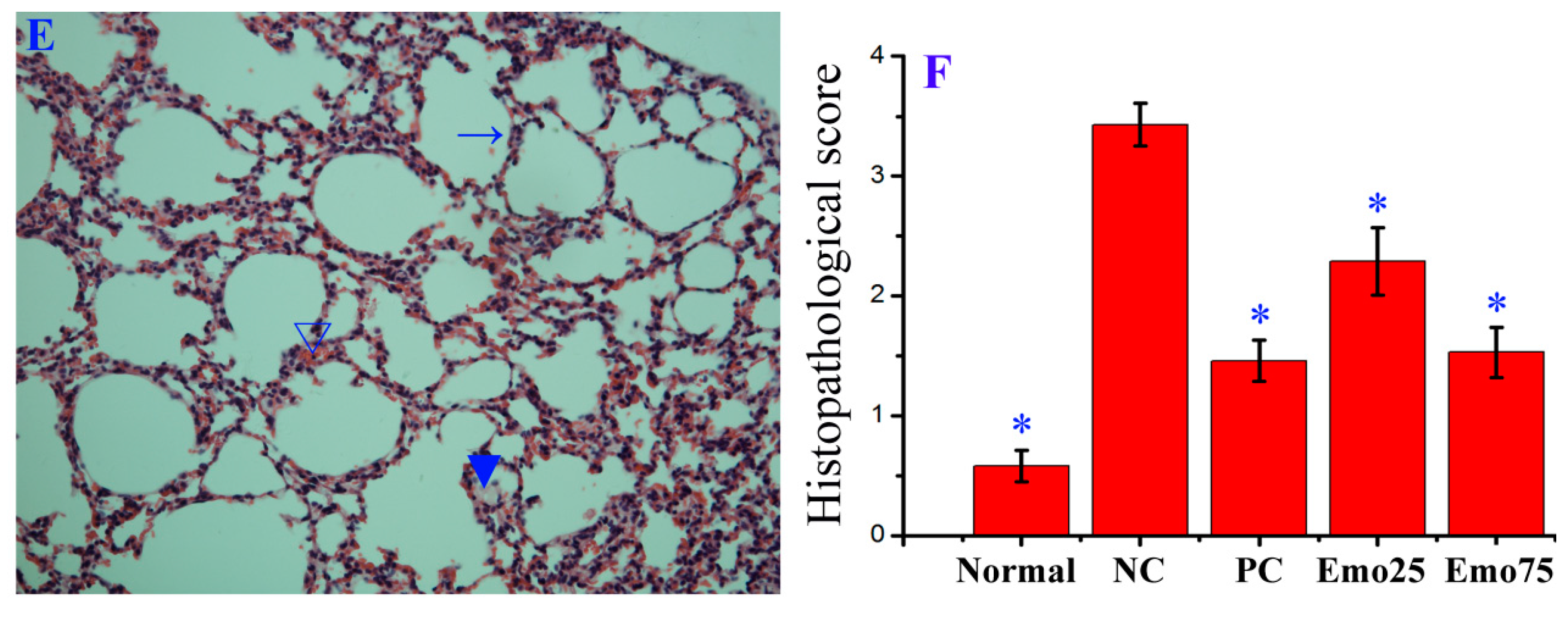
2.6. Emodin Inhibited IAV Replication, Lung Edema and Inflammatory Response In Vivo
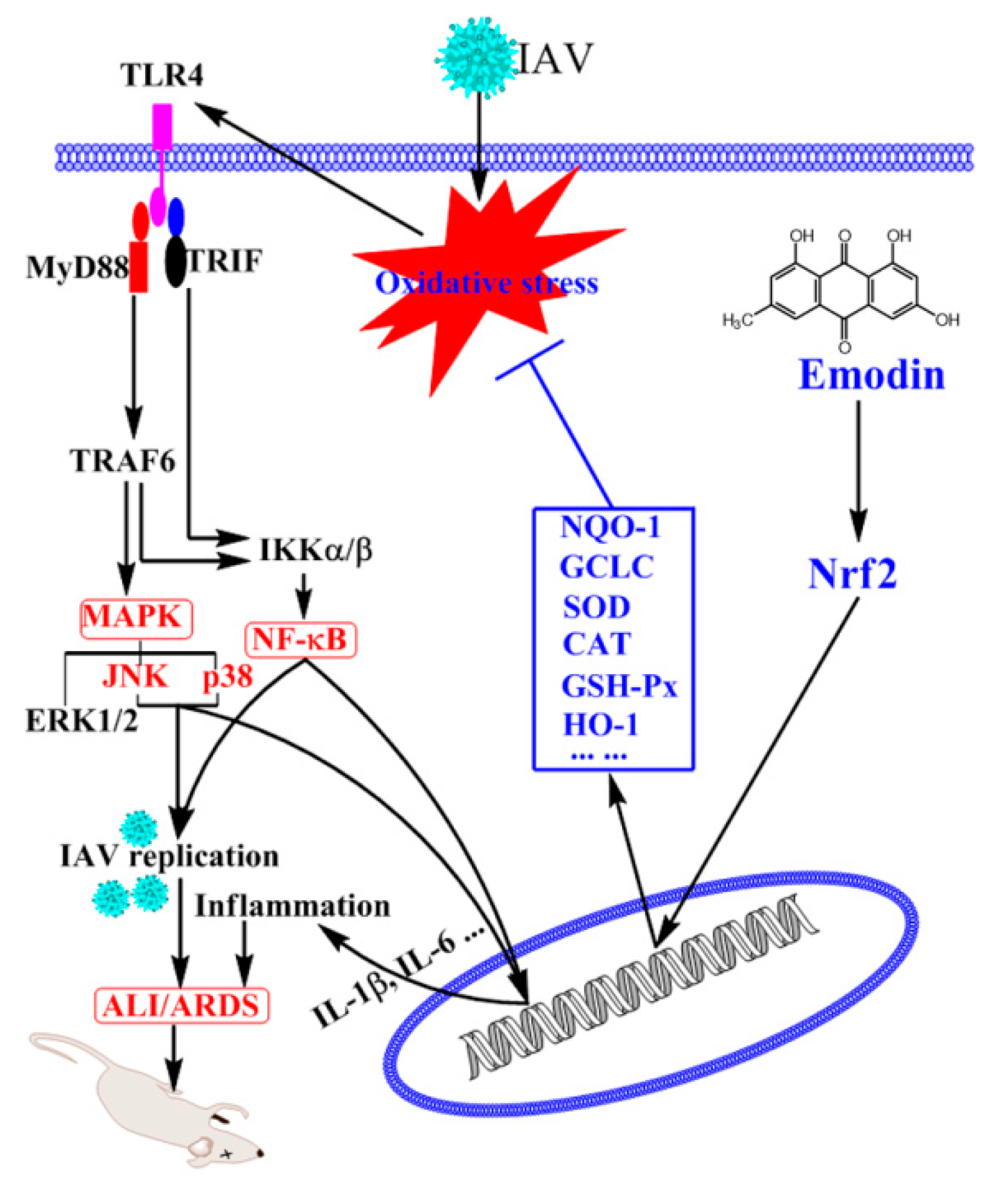
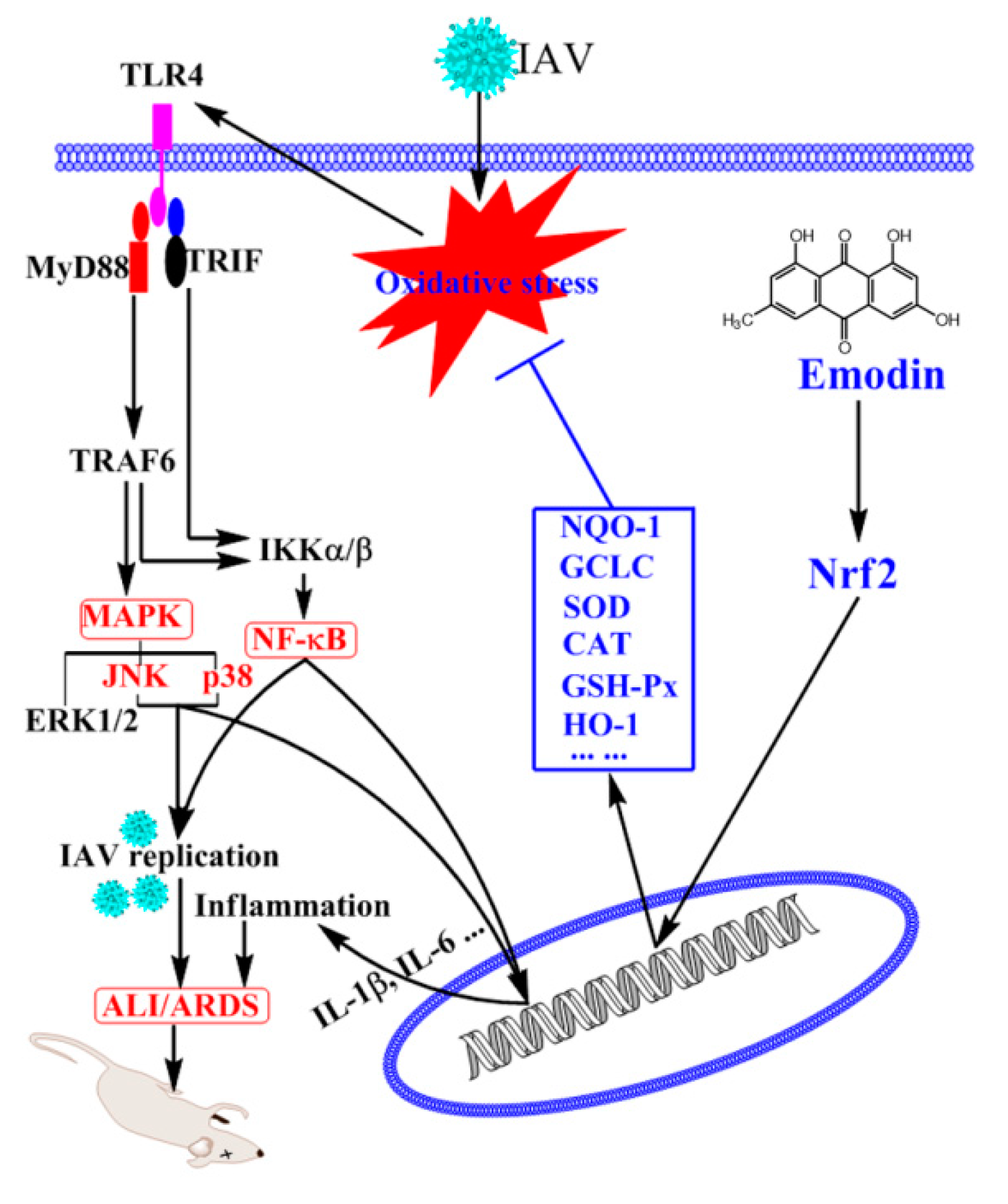
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cells, Viruses and Cytotoxicity Assay
4.3. Plaque Formation and Plaque Inhibition Assays
4.4. TCID50 and Antiviral Assay by the Sulforhodamine B (SRB) Method Using CPE Reduction
4.5. Transfection and Luciferase Assay
4.6. Quantitative Real Time RT-PCR (qRT-PCR)
4.7. Western Blotting
4.8. ELISA Assay
4.9. Antioxidant Assay
4.10. siRNA Assay
4.11. In Vivo Study
- In the uninfected control (Uninfected, n = 16), mice were not infected with IAV (PR8) virus but shamed with VGM medium in a 50 μl volumes intranasally, and treated with DMSO (0.5%) by oral gavage.
- In negative control (DMSO, n = 16), mice were intranasally infected with 10× MLD50 of IAV (PR8) viruses in a 50 μL volumes, and treated with DMSO (0.5%) by oral gavage.
- In positive drug control (Oseltamivir, n = 16), mice were intranasally infected with 10× MLD50 of IAV (PR8) viruses in a 50 μL volumes, and treated with oseltamivir (10 mg kg−1 day−1) by oral gavage.
- In emodin-treated groups (Emodin25 and Emodin75, n = 16 in each group), mice were intranasally infected with 10× MLD50 of IAV (PR8) viruses in a 50 μL volumes, and treated with emodin (25 mg kg−1 day−1 and 75 mg kg−1 day−1, respectively) by oral gavage.
4.12. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sharma, S.; Parida, M.; Shukla, J.; Rao, P.V. Molecular epidemiology of novel swine origin influenza virus (s-oiv) from Gwalior, India, 2009. Virol. J. 2011, 8, 280. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Watanabe, S.; Maher, E.A.; Neumann, G.; Kawaoka, Y. Pandemic potential of avian influenza a (h7n9) viruses. Trends Microbiol. 2014, 22, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Van der Vries, E.; Schutten, M.; Fraaij, P.; Boucher, C.; Osterhaus, A. Influenza virus resistance to antiviral therapy. Adv. Pharmacol. 2013, 67, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Guillot, L.; Le Goffic, R.; Bloch, S.; Escriou, N.; Akira, S.; Chignard, M.; Si-Tahar, M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza a virus. J. Boil. Chem. 2005, 280, 5571–5580. [Google Scholar] [CrossRef] [PubMed]
- Le Goffic, R.; Balloy, V.; Lagranderie, M.; Alexopoulou, L.; Escriou, N.; Flavell, R.; Chignard, M.; Si-Tahar, M. Detrimental contribution of the toll-like receptor (tlr)3 to influenza a virus-induced acute pneumonia. PLoS Pathog. 2006, 2, e53. [Google Scholar] [CrossRef] [PubMed]
- Marchant, D.; Singhera, G.K.; Utokaparch, S.; Hackett, T.L.; Boyd, J.H.; Luo, Z.; Si, X.; Dorscheid, D.R.; McManus, B.M.; Hegele, R.G. Toll like receptor 4 mediated p38 mitogen activated protein kinase activation is a determinant of respiratory virus entry and tropism. J. Virol. 2010, 84, 11359–11373. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Xin, Z.T.; Liang, Y.; Ly, H.; Liang, Y. Nf-kappab signaling differentially regulates influenza virus RNA synthesis. J. Virol. 2008, 82, 9880–9889. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Dudziak, D.; Dirmeier, U.; Hobom, G.; Riedel, A.; Schlee, M.; Staudt, L.M.; Rosenwald, A.; Behrends, U.; Bornkamm, G.W.; et al. Active nf-kappab signalling is a prerequisite for influenza virus infection. J. Gen. Virol. 2004, 85, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Marjuki, H.; Yen, H.L.; Franks, J.; Webster, R.G.; Pleschka, S.; Hoffmann, E. Higher polymerase activity of a human influenza virus enhances activation of the hemagglutinin-induced raf/mek/erk signal cascade. Virol. J. 2007, 4, 134. [Google Scholar] [CrossRef] [PubMed]
- Nacken, W.; Ehrhardt, C.; Ludwig, S. Small molecule inhibitors of the c-jun n-terminal kinase (jnk) possess antiviral activity against highly pathogenic avian and human pandemic influenza a viruses. Biol. Chem. 2012, 393, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Geiler, J.; Naczk, P.; Sithisarn, P.; Leutz, A.; Doerr, H.W.; Cinatl, J., Jr. Glycyrrhizin exerts antioxidative effects in h5n1 influenza a virus-infected cells and inhibits virus replication and pro-inflammatory gene expression. PLoS ONE 2011, 6, e19705. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Kashiwakura, I. Involvement of reactive oxygen species in ionizing radiation-induced upregulation of cell surface toll-like receptor 2 and 4 expression in human monocytic cells. J. Radiat. Res. 2017, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Mendoza, C.; Layunta, E.; Alcalde, A.I.; Mesonero, J.E. Tlr2, tlr3, and tlr4 activation specifically alters the oxidative status of intestinal epithelial cells. Cell Stress Chaperones 2014, 19, 289–293. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Dong, C.; Luan, Z.; McAllan, B.M.; Xu, T.; Zhao, L.; Qiao, J. Oxygen free radical involvement in acute lung injury induced by h5n1 virus in mice. Influenza Other Respir. Viruses 2013, 7, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Deramaudt, T.B.; Dill, C.; Bonay, M. Regulation of oxidative stress by nrf2 in the pathophysiology of infectious diseases. Med. Mal. Infect. 2013, 43, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.F.; Zeng, Z.; Wang, K.H.; Zhang, H.Y.; Wang, S.; Zhou, W.X.; Wang, Z.B.; Xu, W.G.; Duan, J. Heme oxygenase-1 protects rat liver against warm ischemia/reperfusion injury via tlr2/tlr4-triggered signaling pathways. World J. Gastroenterol. 2015, 21, 2937–2948. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Thimmulappa, R.; Craciun, F.; Harvey, C.; Singh, A.; Kombairaju, P.; Reddy, S.P.; Remick, D.; Biswal, S. Enhancing nrf2 pathway by disruption of keap1 in myeloid leukocytes protects against sepsis. Am. J. Respir. Crit. Care Med. 2011, 184, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.M.; Kim, Y.R.; Kim, H.N.; Shin, H.K.; Choi, B.T. Beneficial effects of polygonum multiflorum on hippocampal neuronal cells and mouse focal cerebral ischemia. Am. J. Chin. Med. 2015, 43, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Chen, F.; Wang, J.; Wu, S.; Zheng, M.; Zhu, H.; Liu, Y.; He, J.; Chen, Z. Emodin protects against concanavalin a-induced hepatitis in mice through inhibiting activation of the p38 mapk-nf-kappab signaling pathway. Cell. Physiol. Biochem. 2015, 35, 1557–1570. [Google Scholar] [CrossRef] [PubMed]
- Yiu, C.Y.; Chen, S.Y.; Yang, T.H.; Chang, C.J.; Yeh, D.B.; Chen, Y.J.; Lin, T.P. Inhibition of epstein-barr virus lytic cycle by an ethyl acetate subfraction separated from polygonum cuspidatum root and its major component, emodin. Molecules 2014, 19, 1258–1272. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, N.; Zhong, Y.; Yang, Z.Q. Antiviral effect of emodin from rheum palmatum against coxsakievirus b5 and human respiratory syncytial virus in vitro. J. Huazhong Univ. Sci. Technol. Med. Sci. 2015, 35, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.R.; Luo, J.; Hou, W.; Xiao, H.; Yang, Z.Q. The effect of emodin, an anthraquinone derivative extracted from the roots of rheum tanguticum, against herpes simplex virus in vitro and in vivo. J. Ethnopharmacol. 2011, 133, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Dang, S.S.; Jia, X.L.; Song, P.; Cheng, Y.A.; Zhang, X.; Sun, M.Z.; Liu, E.Q. Inhibitory effect of emodin and astragalus polysaccharide on the replication of hbv. World J. Gastroenterol. 2009, 15, 5669–5673. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Liao, W.; Kuang, X. Effects of emodin on il-23/il-17 inflammatory axis, th17 cells and viral replication in mice with viral myocarditis. Nan Fang Yi Ke Da Xue Xue Bao 2014, 34, 373–378. [Google Scholar] [PubMed]
- Lin, T.J.; Lin, C.F.; Chiu, C.H.; Lee, M.C.; Horng, J.T. Inhibition of endosomal fusion activity of influenza virus by rheum tanguticum (da-huang). Sci. Rep. 2016, 6, 27768. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Lin, H.J.; Chen, T.H.; Hsu, Y.A.; Liu, C.S.; Hwang, G.Y.; Wan, L. Polygonum cuspidatum and its active components inhibit replication of the influenza virus through toll-like receptor 9-induced interferon beta expression. PLoS ONE 2015, 10, e0117602. [Google Scholar] [CrossRef]
- Li, S.W.; Yang, T.C.; Lai, C.C.; Huang, S.H.; Liao, J.M.; Wan, L.; Lin, Y.J.; Lin, C.W. Antiviral activity of aloe-emodin against influenza a virus via galectin-3 up-regulation. Eur. J. Pharmacol. 2014, 738, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Heo, J.; Yi, C.M.; Ban, J.; Lee, N.J.; Lee, N.R.; Kim, S.W.; Kim, N.J.; Inn, K.S. A novel p38 mitogen activated protein kinase (mapk) specific inhibitor suppresses respiratory syncytial virus and influenza a virus replication by inhibiting virus-induced p38 mapk activation. Biochem. Biophys. Res. Commun. 2016, 477, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Dong, L.; Duan, M.L.; Sun, K.; Liu, Y.Y.; Wang, M.X.; Deng, J.N.; Fan, J.Y.; Wang, B.E.; Han, J.Y. Emodin improves lipopolysaccharide-induced microcirculatory disturbance in rat mesentery. Microcirculation 2013, 20, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jeong, Y.T.; Li, X.; Kim, M.J.; Park, P.H.; Hwang, S.L.; Son, J.K.; Chang, H.W. Emodin isolated from polygoni cuspidati radix inhibits tnf-alpha and il-6 release by blockading nf-kappab and map kinase pathways in mast cells stimulated with pma plus a23187. Biomol. Ther. 2013, 21, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, N.; Cao, Y.; Zhang, W.; Su, G.; Sun, Y.; Liu, Z.; Li, F.; Liang, D.; Liu, B.; et al. Emodin ameliorates lipopolysaccharide-induced mastitis in mice by inhibiting activation of nf-kappab and mapks signal pathways. Eur. J. Pharmacol. 2013, 705, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.S.; Tsai, H.D.; Cheung, W.M.; Hsu, C.Y.; Lin, T.N. Ppar-gamma ameliorates neuronal apoptosis and ischemic brain injury via suppressing nf-kappab-driven p22phox transcription. Molecul. Neurobiol. 2016, 53, 3626–3645. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yue, S.; Ke, B.; Zhu, J.; Shen, X.D.; Zhai, Y.; Yamamoto, M.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Nuclear factor erythroid 2-related factor 2 regulates toll-like receptor 4 innate responses in mouse liver ischemia-reperfusion injury through akt-forkhead box protein o1 signaling network. Transplantation 2014, 98, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Kim, H.P.; Geng, X.H.; Nakao, A.; Wang, X.; Murase, N.; Drain, P.F.; Wang, X.; Sasidhar, M.; Nabel, E.G.; et al. Carbon monoxide differentially inhibits tlr signaling pathways by regulating ros-induced trafficking of tlrs to lipid rafts. J. Exp. Med. 2006, 203, 2377–2389. [Google Scholar] [CrossRef] [PubMed]
- Kesic, M.J.; Simmons, S.O.; Bauer, R.; Jaspers, I. Nrf2 expression modifies influenza a entry and replication in nasal epithelial cells. Free Radic. Biol. Med. 2011, 51, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Kosmider, B.; Messier, E.M.; Janssen, W.J.; Nahreini, P.; Wang, J.; Hartshorn, K.L.; Mason, R.J. Nrf2 protects human alveolar epithelial cells against injury induced by influenza a virus. Respir. Res. 2012, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.H.; Shi, X.Q.; Liang, S.H.; Zhou, L.; Liu, K.F.; Zhao, J. Emodin attenuates cigarette smoke induced lung injury in a mouse model via suppression of reactive oxygen species production. J. Biochem. Mol. Toxicol. 2015, 29, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Jin, M.L.; Ko, M.J.; Park, G.; Choi, Y.W. Anti-neuroinflammatory effect of emodin in lps-stimulated microglia: Involvement of ampk/nrf2 activation. Neurochem. Res. 2016, 41, 2981–2992. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.T.; Wan, B.; Liu, D.D.; Wan, S.X.; Fu, H.Y.; Wan, Y.; Zhang, H.; Chen, Y. Emodin alleviates lung injury in rats with sepsis. J. Surg. Res. 2016, 202, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.P.; Zhao, X.F.; Zeng, J.; Wan, Q.Y.; Yang, J.C.; Li, W.Z.; Chen, X.X.; Wang, G.F.; Li, K.S. Drug screening for autophagy inhibitors based on the dissociation of beclin1-bcl2 complex using bifc technique and mechanism of eugenol on anti-influenza a virus activity. PLoS ONE 2013, 8, e61026. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.P.; Chen, J.; Bei, Y.F.; Han, B.X.; Wang, S. Influence of borneol on primary mice oral fibroblasts: A penetration enhancer may be used in oral submucous fibrosis. J. Oral Pathol. Med. 2009, 38, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.P.; Wu, L.Q.; Li, R.; Zhao, X.F.; Wan, Q.Y.; Chen, X.X.; Li, W.Z.; Wang, G.F.; Li, K.S. Identification of 23-(s)-2-amino-3-phenylpropanoyl-silybin as an antiviral agent for influenza a virus infection in vitro and in vivo. Antimicrob. Agents Chemother. 2013, 57, 4433–4443. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, L.; Zhou, H.; Zeng, L.; Chen, T.; Chen, Q.; Zhou, B.; Wang, Y.; Chen, Q.; Hu, P.; et al. Radix isatidis polysaccharides inhibit influenza a virus and influenza a virus-induced inflammation via suppression of host tlr3 signaling in vitro. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Wang, G.; Li, W.; Zhang, L.; Yang, J.; Zhao, X.; Chen, X.; Xu, Y.; Li, K. High-throughput screening for antiinfluenza a virus drugs and study of the mechanism of procyanidin on influenza a virusinduced autophagy. J. Biomol. Screen. 2011, 17, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Lim, C.H.; Song, J.H.; Baek, S.H.; Kwon, D.H. Antiviral activity of raoulic acid from raoulia australis against picornaviruses. Phytomedicine 2009, 16, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.P.; Zhu, D.X.; Sheng, J.T.; Chen, X.X.; Li, W.Z.; Wang, G.F.; Li, K.S.; Su, Y. Inhibition of tanshinone iia, salvianolic acid a and salvianolic acid b on areca nut extract-induced oral submucous fibrosis in vitro. Molecules 2015, 20, 6794–6807. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.C.; Lilley, E. Implementing guidelines on reporting research using animals (arrive etc.): New requirements for publication in bjp. Br. J. Pharmacol. 2015, 172, 3189–3193. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.T.; He, W.Y.; Ma, L.L.; Wang, H.Q.; Wang, B.; Zheng, G.H.; Ji, X.Y.; Zhang, T.; Li, Y.H.; Jiang, J.D.; et al. Synthesis and anti-influenza virus activities of a novel class of gastrodin derivatives. Molecules 2013, 18, 3789–3805. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Zhao, X.; Wang, X.; Song, N.; Guo, Y.; Yan, X.; Jiang, L.; Cheng, W.; Shen, L. Emodin alleviates bleomycin-induced pulmonary fibrosis in rats. Toxicol. Lett. 2016, 262, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, T.F.; Lu, H.J.; Liou, S.S.; Chang, C.J.; Liu, I.M. Emodin, a naturally occurring anthraquinone derivative, ameliorates dyslipidemia by activating amp-activated protein kinase in high-fat-diet-fed rats. Evid. Based Complement. Altern. Med. 2012, 2012, 781812. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Winn, R.K.; Jonas, M.; Chi, E.Y.; Martin, T.R.; Liles, W.C. Fas (cd95) induces alveolar epithelial cell apoptosis in vivo: Implications for acute pulmonary inflammation. Am. J. Pathol. 2001, 158, 153–161. [Google Scholar] [CrossRef]
- Curtis, M.J.; Bond, R.A.; Spina, D.; Ahluwalia, A.; Alexander, S.P.; Giembycz, M.A.; Gilchrist, A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; et al. Experimental design and analysis and their reporting: New guidance for publication in bjp. Br. J. Pharmacol. 2015, 172, 3461–3471. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Sample of the compound emodin is available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | GSH (nmol mg Protein−1) | GSSG (nmol mg Protein−1) | GSH/GSSG (Ratio) | ROS (Fold Change of Uninfected Group) |
| Uninfected | 45.42 ± 5.33 * | 3.07 ± 0.93 * | 14.79 ± 0.93 * | 1.00 ± 0.00 |
| DMSO | 20.48 ± 4.63 | 4.22 ± 0.81 | 4.85 ± 0.51 | 1.92 ± 0.17 |
| Ribavirin | 25.78 ± 5.46 | 3.78 ± 0.72 | 6.82 ± 0.62 * | 1.81 ± 0.11 |
| Emodin | 33.32 ± 4.59 * | 3.87 ± 0.83 | 8.61 ± 0.74 * | 1.46 ± 0.15 * |
| Groups | T-SOD (U mg Protein−1) | GR (U mg Protein−1) | CAT (U mg Protein−1) | GSH-Px (mU mg Protein−1) |
| Uninfected | 11.65 ± 2.63 * | 11.41 ± 1.24 * | 41.38 ± 5.52 * | 5.21 ± 0.84 * |
| DMSO | 4.93 ± 0.82 | 2.03 ± 0.52 | 19.43± 2.63 | 1.93 ± 0.27 |
| Ribavirin | 4.91 ± 0.73 | 2.47 ± 0.59 | 27.92 ± 3.16 * | 2.28 ± 0.42 |
| Emodin | 7.82 ± 0.92 * | 5.27 ± 0.63 * | 33.64 ± 3.27 * | 3.94 ± 0.49 * |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, J.-P.; Wang, Q.-W.; Su, Y.; Gu, L.-M.; Zhao, Y.; Chen, X.-X.; Chen, C.; Li, W.-Z.; Wang, G.-F.; Li, K.-S. Emodin Inhibition of Influenza A Virus Replication and Influenza Viral Pneumonia via the Nrf2, TLR4, p38/JNK and NF-kappaB Pathways. Molecules 2017, 22, 1754. https://doi.org/10.3390/molecules22101754
Dai J-P, Wang Q-W, Su Y, Gu L-M, Zhao Y, Chen X-X, Chen C, Li W-Z, Wang G-F, Li K-S. Emodin Inhibition of Influenza A Virus Replication and Influenza Viral Pneumonia via the Nrf2, TLR4, p38/JNK and NF-kappaB Pathways. Molecules. 2017; 22(10):1754. https://doi.org/10.3390/molecules22101754
Chicago/Turabian StyleDai, Jian-Ping, Qian-Wen Wang, Yun Su, Li-Ming Gu, Ying Zhao, Xiao-Xua Chen, Cheng Chen, Wei-Zhong Li, Ge-Fei Wang, and Kang-Sheng Li. 2017. "Emodin Inhibition of Influenza A Virus Replication and Influenza Viral Pneumonia via the Nrf2, TLR4, p38/JNK and NF-kappaB Pathways" Molecules 22, no. 10: 1754. https://doi.org/10.3390/molecules22101754
APA StyleDai, J.-P., Wang, Q.-W., Su, Y., Gu, L.-M., Zhao, Y., Chen, X.-X., Chen, C., Li, W.-Z., Wang, G.-F., & Li, K.-S. (2017). Emodin Inhibition of Influenza A Virus Replication and Influenza Viral Pneumonia via the Nrf2, TLR4, p38/JNK and NF-kappaB Pathways. Molecules, 22(10), 1754. https://doi.org/10.3390/molecules22101754