BACE1 Function and Inhibition: Implications of Intervention in the Amyloid Pathway of Alzheimer’s Disease Pathology

Abstract
:1. Introduction
2. BACE1 Characteristics
2.1. Structural Attributes of BACE1
2.2. BACE1 Activity and Native Substrates
2.3. BACE1 Substrate Specificity: Implications for A673T
3. The Prospect of Therapeutic Inhibition of BACE1 Activity to Block Aβ Production
3.1. Aβ
3.2. Stragetic Approaches to BACE1 Inhibition
3.3. Impact of BACE1 Inhibition on Other Biologic Functions
3.4. Magnitude of Aβ Reduction to Prevent the Onset of AD
4. Discussion
Acknowledgments
Conflicts of Interest
References
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 6, a006262. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar]
- Weiner, M.W.; Veitch, D.P.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Cedarbaum, J.; Donohue, M.C.; Green, R.C.; Harvey, D.; Jack, C.R.; et al. Impact of the Alzheimer’s Disease Neuroimaging Initiative, 2004 to 2014. Alzheimers Dement. 2015, 11, 865–884. [Google Scholar]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar]
- Reiman, E.M.; Quiroz, Y.T.; Fleisher, A.S.; Chen, K.; Velez-Pardo, C.; Jimenez-Del-Rio, M.; Fagan, A.M.; Shah, A.R.; Alvarez, S.; Arbelaez, A.; et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: A case-control study. Lancet Neurol. 2012, 11, 1048–1056. [Google Scholar]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid beta-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 7, a006338. [Google Scholar]
- Ono, K. Alzheimer’s disease as oligomeropathy. Neurochem. Int. 2017. [Google Scholar] [CrossRef]
- Parihar, M.S.; Brewer, G.J. Amyloid-beta as a modulator of synaptic plasticity. J. Alzheimers Dis. 2010, 22, 741–763. [Google Scholar]
- Pearson, H.A.; Peers, C. Physiological roles for amyloid beta peptides. J. Physiol. 2006, 575, 5–10. [Google Scholar]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar]
- Cheng, X.; Wu, J.; Geng, M.; Xiong, J. Role of synaptic activity in the regulation of amyloid beta levels in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1217–1232. [Google Scholar]
- Tomita, T. Aberrant proteolytic processing and therapeutic strategies in Alzheimer disease. Adv. Biol. Regul. 2017, 64, 33–38. [Google Scholar]
- Vassar, R.; Kuhn, P.H.; Haass, C.; Kennedy, M.E.; Rajendran, L.; Wong, P.C.; Lichtenthaler, S.F. Function, therapeutic potential and cell biology of BACE proteases: Current status and future prospects. J. Neurochem. 2014, 130, 4–28. [Google Scholar]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar]
- Ohno, M. Alzheimer’s therapy targeting the beta-secretase enzyme BACE1: Benefits and potential limitations from the perspective of animal model studies. Brain Res. Bull. 2016, 126, 183–198. [Google Scholar]
- Dunn, B.M. Introduction to the aspartic proteinase family. In Aspartic Acid Proteases as Therapeutic Targets; Ghosh, A.K., Ed.; WILEY-VCH Verlag GmbH & Co.: Weinheim, Germany, 2010; pp. 3–21. [Google Scholar]
- Hartsuck, J.A.; Tang, J. The carboxylate ion in the active center of pepsin. J. Biol. Chem. 1972, 247, 2575–2580. [Google Scholar]
- Ghosh, A.K.; Brindisi, M.; Yen, Y.C.; Cardenas, E.L.; Ella-Menye, J.R.; Kumaragurubaran, N.; Huang, X.; Tang, J.; Mesecar, A.D. Design, synthesis, and X-ray structural studies of BACE-1 inhibitors containing substituted 2-oxopiperazines as P1’–P2’ ligands. Bioorg. Med. Chem. Lett. 2017, 27, 2432–2438. [Google Scholar]
- Hamada, Y.; Kiso, Y. New directions for protease inhibitors directed drug discovery. Biopolymers 2016, 106, 563–579. [Google Scholar]
- Ghosh, A.K.; Osswald, H.L. BACE1 (beta-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6765–6813. [Google Scholar]
- Yan, R.; Vassar, R. Targeting the beta secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar]
- Yuan, J.; Venkatraman, S.; Zheng, Y.; McKeever, B.M.; Dillard, L.W.; Singh, S.B. Structure-based design of beta-site APP cleaving enzyme 1 (BACE1) inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2013, 56, 4156–4180. [Google Scholar]
- Stamford, A.; Strickland, C. Inhibitors of BACE for treating Alzheimer’s disease: A fragment-based drug discovery story. Curr. Opin. Chem. Biol. 2013, 17, 320–328. [Google Scholar]
- Butini, S.; Gabellieri, E.; Brindisi, M.; Casagni, A.; Guarino, E.; Huleatt, P.B.; Relitti, N.; La Pietra, V.; Marinelli, L.; Giustiniano, M.; et al. Novel peptidomimetics as BACE-1 inhibitors: Synthesis, molecular modeling, and biological studies. Bioorg. Med. Chem. Lett. 2013, 23, 85–89. [Google Scholar]
- Kacker, P.; Bottegoni, G.; Cavalli, A. Computational methods in the discovery and design of BACE-1 inhibitors. Curr. Med. Chem. 2012, 19, 6095–6111. [Google Scholar]
- Ghosh, A.K.; Brindisi, M.; Tang, J. Developing beta-secretase inhibitors for treatment of Alzheimer’s disease. J. Neurochem. 2012, 120, 71–83. [Google Scholar]
- Ghosh, A.K.; Gemma, S.; Tang, J. Beta-Secretase as a therapeutic target for Alzheimer’s disease. Neurotherapeutics 2008, 5, 399–408. [Google Scholar]
- Kennedy, M.E.; Stamford, A.W.; Chen, X.; Cox, K.; Cumming, J.N.; Dockendorf, M.F.; Egan, M.; Ereshefsky, L.; Hodgson, R.A.; Hyde, L.A.; et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS beta-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016, 8, 363ra150. [Google Scholar] [CrossRef]
- Hong, L.; Koelsch, G.; Lin, X.; Wu, S.; Terzyan, S.; Ghosh, A.K.; Zhang, X.C.; Tang, J. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 2000, 290, 150–153. [Google Scholar]
- Li, X.; Bo, H.; Zhang, X.C.; Hartsuck, J.A.; Tang, J. Predicting memapsin 2 (beta-secretase) hydrolytic activity. Protein Sci. 2010, 19, 2175–2185. [Google Scholar]
- Turner, R.T.; Koelsch, G.; Hong, L.; Castanheira, P.; Ermolieff, J.; Ghosh, A.K.; Tang, J. Subsite specificity of memapsin 2 (beta-secretase): Implications for inhibitor design. Biochemistry 2001, 40, 10001–10006. [Google Scholar]
- Wang, W.; Liu, Y.; Lazarus, R.A. Allosteric inhibition of BACE1 by an exosite-binding antibody. Curr. Opin. Struct. Biol. 2013, 23, 797–805. [Google Scholar]
- Benjannet, S.; Elagoz, A.; Wickham, L.; Mamarbachi, M.; Munzer, J.S.; Basak, A.; Lazure, C.; Cromlish, J.A.; Sisodia, S.; Checler, F.; et al. Post-translational processing of beta-secretase (beta-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-beta production. J. Biol. Chem. 2001, 276, 10879–10887. [Google Scholar]
- Bennett, B.D.; Denis, P.; Haniu, M.; Teplow, D.B.; Kahn, S.; Louis, J.C.; Citron, M.; Vassar, R. A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer’s beta-secretase. J. Biol. Chem. 2000, 275, 37712–37717. [Google Scholar]
- Ermolieff, J.; Loy, J.A.; Koelsch, G.; Tang, J. Proteolytic activation of recombinant pro-memapsin 2 (Pro-beta-secretase) studied with new fluorogenic substrates. Biochemistry 2000, 39, 16263. [Google Scholar]
- Costantini, C.; Ko, M.H.; Jonas, M.C.; Puglielli, L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem. J. 2007, 407, 383–395. [Google Scholar]
- Hussain, I.; Powell, D.; Howlett, D.R.; Tew, D.G.; Meek, T.D.; Chapman, C.; Gloger, I.S.; Murphy, K.E.; Southan, C.D.; Ryan, D.M.; et al. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell. Neurosci. 1999, 14, 419–427. [Google Scholar]
- Kizuka, Y.; Kitazume, S.; Fujinawa, R.; Saito, T.; Iwata, N.; Saido, T.C.; Nakano, M.; Yamaguchi, Y.; Hashimoto, Y.; Staufenbiel, M.; et al. An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer’s disease. EMBO Mol. Med. 2015, 7, 175–189. [Google Scholar]
- Pastorino, L.; Ikin, A.F.; Nairn, A.C.; Pursnani, A.; Buxbaum, J.D. The carboxyl-terminus of BACE contains a sorting signal that regulates BACE trafficking but not the formation of total A(beta). Mol. Cell. Neurosci. 2002, 19, 175–185. [Google Scholar]
- Kang, E.L.; Biscaro, B.; Piazza, F.; Tesco, G. BACE1 protein endocytosis and trafficking are differentially regulated by ubiquitination at lysine 501 and the Di-leucine motif in the carboxyl terminus. J. Biol. Chem. 2012, 287, 42867–42880. [Google Scholar]
- Kang, E.L.; Cameron, A.N.; Piazza, F.; Walker, K.R.; Tesco, G. Ubiquitin regulates GGA3-mediated degradation of BACE1. J. Biol. Chem. 2010, 285, 24108–24119. [Google Scholar]
- Vetrivel, K.S.; Meckler, X.; Chen, Y.; Nguyen, P.D.; Seidah, N.G.; Vassar, R.; Wong, P.C.; Fukata, M.; Kounnas, M.Z.; Thinakaran, G. Alzheimer disease Abeta production in the absence of S-palmitoylation-dependent targeting of BACE1 to lipid rafts. J. Biol. Chem. 2009, 284, 3793–3803. [Google Scholar]
- Ko, M.H.; Puglielli, L. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J. Biol. Chem. 2009, 284, 2482–2492. [Google Scholar]
- Araki, W. Post-translational regulation of the beta-secretase BACE1. Brain Res. Bull. 2016, 126, 170–177. [Google Scholar]
- Fujinaga, M.; Chernaia, M.M.; Tarasova, N.I.; Mosimann, S.C.; James, M.N. Crystal structure of human pepsin and its complex with pepstatin. Protein Sci. 1995, 4, 960–972. [Google Scholar]
- Moreland, J.L.; Gramada, A.; Buzko, O.V.; Zhang, Q.; Bourne, P.E. The Molecular Biology Toolkit (MBT): A modular platform for developing molecular visualization applications. BMC Bioinform. 2005, 6, 21. [Google Scholar]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar]
- Zhang, X.; Zhou, K.; Wang, R.; Cui, J.; Lipton, S.A.; Liao, F.F.; Xu, H.; Zhang, Y.W. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 2007, 282, 10873–10880. [Google Scholar]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G.; et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1alpha. J. Neurochem. 2009, 108, 1045–1056. [Google Scholar]
- Koritzinsky, M.; Levitin, F.; van den Beucken, T.; Rumantir, R.A.; Harding, N.J.; Chu, K.C.; Boutros, P.C.; Braakman, I.; Wouters, B.G. Two phases of disulfide bond formation have differing requirements for oxygen. J. Cell Biol. 2013, 203, 615–627. [Google Scholar]
- He, X.; Zhu, G.; Koelsch, G.; Rodgers, K.K.; Zhang, X.C.; Tang, J. Biochemical and structural characterization of the interaction of memapsin 2 (beta-secretase) cytosolic domain with the VHS domain of GGA proteins. Biochemistry 2003, 42, 12174–12180. [Google Scholar]
- He, X.; Chang, W.P.; Koelsch, G.; Tang, J. Memapsin 2 (beta-secretase) cytosolic domain binds to the VHS domains of GGA1 and GGA2: Implications on the endocytosis mechanism of memapsin 2. FEBS Lett. 2002, 524, 183–187. [Google Scholar]
- He, X.; Li, F.; Chang, W.P.; Tang, J. GGA proteins mediate the recycling pathway of memapsin 2 (BACE). J. Biol. Chem. 2005, 280, 11696–11703. [Google Scholar]
- Shiba, T.; Kametaka, S.; Kawasaki, M.; Shibata, M.; Waguri, S.; Uchiyama, Y.; Wakatsuki, S. Insights into the phosphoregulation of beta-secretase sorting signal by the VHS domain of GGA1. Traffic 2004, 5, 437–448. [Google Scholar]
- Von Arnim, C.A.; Tangredi, M.M.; Peltan, I.D.; Lee, B.M.; Irizarry, M.C.; Kinoshita, A.; Hyman, B.T. Demonstration of BACE (beta-secretase) phosphorylation and its interaction with GGA1 in cells by fluorescence-lifetime imaging microscopy. J. Cell Sci. 2004, 117, 5437–5445. [Google Scholar]
- Capell, A.; Steiner, H.; Willem, M.; Kaiser, H.; Meyer, C.; Walter, J.; Lammich, S.; Multhaup, G.; Haass, C. Maturation and pro-peptide cleavage of beta-secretase. J. Biol. Chem. 2000, 275, 30849–30854. [Google Scholar]
- Huse, J.T.; Pijak, D.S.; Leslie, G.J.; Lee, V.M.; Doms, R.W. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease beta-secretase. J. Biol. Chem. 2000, 275, 33729–33737. [Google Scholar]
- Koelsch, G.; Mares, M.; Metcalf, P.; Fusek, M. Multiple functions of pro-parts of aspartic proteinase zymogens. FEBS Lett. 1994, 343, 6–10. [Google Scholar]
- Horimoto, Y.; Dee, D.R.; Yada, R.Y. Multifunctional aspartic peptidase prosegments. New Biotechnol. 2009, 25, 318–324. [Google Scholar]
- Shimizu, H.; Tosaki, A.; Kaneko, K.; Hisano, T.; Sakurai, T.; Nukina, N. Crystal structure of an active form of BACE1, an enzyme responsible for amyloid beta protein production. Mol. Cell. Biol. 2008, 28, 3663–3671. [Google Scholar]
- Arbor, S.C.; LaFontaine, M.; Cumbay, M. Amyloid-beta Alzheimer targets—Protein processing, lipid rafts, and amyloid-beta pores. Yale J. Biol. Med. 2016, 89, 5–21. [Google Scholar]
- Kinoshita, A.; Fukumoto, H.; Shah, T.; Whelan, C.M.; Irizarry, M.C.; Hyman, B.T. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J. Cell Sci. 2003, 116, 3339–3346. [Google Scholar]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar]
- Schneider, A.; Rajendran, L.; Honsho, M.; Gralle, M.; Donnert, G.; Wouters, F.; Hell, S.W.; Simons, M. Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J. Neurosci. 2008, 28, 2874–2882. [Google Scholar]
- Kalvodova, L.; Kahya, N.; Schwille, P.; Ehehalt, R.; Verkade, P.; Drechsel, D.; Simons, K. Lipids as modulators of proteolytic activity of BACE: Involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 2005, 280, 36815–36823. [Google Scholar]
- Udayar, V.; Buggia-Prevot, V.; Guerreiro, R.L.; Siegel, G.; Rambabu, N.; Soohoo, A.L.; Ponnusamy, M.; Siegenthaler, B.; Bali, J.; Simons, M.; et al. A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of beta-amyloid production. Cell Rep. 2013, 5, 1536–1551. [Google Scholar]
- Buggia-Prevot, V.; Thinakaran, G. Sorting the role of SORLA in Alzheimer’s disease. Sci. Transl. Med. 2014, 6, 223fs8. [Google Scholar] [CrossRef]
- Koh, Y.H.; von Arnim, C.A.; Hyman, B.T.; Tanzi, R.E.; Tesco, G. BACE is degraded via the lysosomal pathway. J. Biol. Chem. 2005, 280, 32499–32504. [Google Scholar]
- Toh, W.H.; Gleeson, P.A. Dysregulation of intracellular trafficking and endosomal sorting in Alzheimer’s disease: Controversies and unanswered questions. Biochem. J. 2016, 473, 1977–1993. [Google Scholar]
- Ye, X.; Feng, T.; Tammineni, P.; Chang, Q.; Jeong, Y.Y.; Margolis, D.J.; Cai, H.; Kusnecov, A.; Cai, Q. Regulation of Synaptic Amyloid-beta Generation through BACE1 Retrograde Transport in a Mouse Model of Alzheimer’s Disease. Neuroscience 2017, 37, 2639–2655. [Google Scholar]
- Lee, E.B.; Zhang, B.; Liu, K.; Greenbaum, E.A.; Doms, R.W.; Trojanowski, J.Q.; Lee, V.M. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J. Cell Biol. 2005, 168, 291–302. [Google Scholar]
- Yan, R. Physiological Functions of the beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 and 2. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef]
- Kitazume, S.; Tachida, Y.; Oka, R.; Shirotani, K.; Saido, T.C.; Hashimoto, Y. Alzheimer’s beta-secretase, beta-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc. Natl. Acad. Sci. USA 2001, 98, 13554–13559. [Google Scholar]
- Lichtenthaler, S.F.; Dominguez, D.I.; Westmeyer, G.G.; Reiss, K.; Haass, C.; Saftig, P.; De Strooper, B.; Seed, B. The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J. Biol. Chem. 2003, 278, 48713–48719. [Google Scholar]
- Von Arnim, C.A.; Kinoshita, A.; Peltan, I.D.; Tangredi, M.M.; Herl, L.; Lee, B.M.; Spoelgen, R.; Hshieh, T.T.; Ranganathan, S.; Battey, F.D.; et al. The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J. Biol. Chem. 2005, 280, 17777–17785. [Google Scholar]
- Willem, M.; Garratt, A.N.; Novak, B.; Citron, M.; Kaufmann, S.; Rittger, A.; DeStrooper, B.; Saftig, P.; Birchmeier, C.; Haass, C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science 2006, 314, 664–666. [Google Scholar]
- Hu, X.; Hicks, C.W.; He, W.; Wong, P.; Macklin, W.B.; Trapp, B.D.; Yan, R. Bace1 modulates myelination in the central and peripheral nervous system. Nat. Neurosci. 2006, 9, 1520–1525. [Google Scholar]
- Kim, D.Y.; Carey, B.W.; Wang, H.; Ingano, L.A.; Binshtok, A.M.; Wertz, M.H.; Pettingell, W.H.; He, P.; Lee, V.M.; Woolf, C.J.; et al. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat. Cell Biol. 2007, 9, 755–764. [Google Scholar]
- Hemming, M.L.; Elias, J.E.; Gygi, S.P.; Selkoe, D.J. Identification of beta-secretase (BACE1) substrates using quantitative proteomics. PLoS ONE 2009, 4, e8477. [Google Scholar]
- Kuhn, P.H.; Koroniak, K.; Hogl, S.; Colombo, A.; Zeitschel, U.; Willem, M.; Volbracht, C.; Schepers, U.; Imhof, A.; Hoffmeister, A.; et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012, 31, 3157–3168. [Google Scholar]
- Dislich, B.; Lichtenthaler, S.F. The Membrane-Bound Aspartyl Protease BACE1: Molecular and Functional Properties in Alzheimer’s Disease and Beyond. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef]
- Muller, S.A.; Scilabra, S.D.; Lichtenthaler, S.F. Proteomic Substrate Identification for Membrane Proteases in the Brain. Front. Mol. Neurosci. 2016, 9. [Google Scholar] [CrossRef]
- Pigoni, M.; Wanngren, J.; Kuhn, P.H.; Munro, K.M.; Gunnersen, J.M.; Takeshima, H.; Feederle, R.; Voytyuk, I.; De Strooper, B.; Levasseur, M.D.; et al. Seizure protein 6 and its homolog seizure 6-like protein are physiological substrates of BACE1 in neurons. Mol. Neurodegener. 2016, 11. [Google Scholar] [CrossRef]
- Munro, K.M.; Nash, A.; Pigoni, M.; Lichtenthaler, S.F.; Gunnersen, J.M. Functions of the Alzheimer’s Disease Protease BACE1 at the Synapse in the Central Nervous System. J. Mol. Neurosci. 2016, 60, 305–315. [Google Scholar]
- Michailov, G.V.; Sereda, M.W.; Brinkmann, B.G.; Fischer, T.M.; Haug, B.; Birchmeier, C.; Role, L.; Lai, C.; Schwab, M.H.; Nave, K.A. Axonal neuregulin-1 regulates myelin sheath thickness. Science 2004, 304, 700–703. [Google Scholar]
- Taveggia, C.; Zanazzi, G.; Petrylak, A.; Yano, H.; Rosenbluth, J.; Einheber, S.; Xu, X.; Esper, R.M.; Loeb, J.A.; Shrager, P.; et al. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron 2005, 47, 681–694. [Google Scholar]
- Hu, X.; He, W.; Diaconu, C.; Tang, X.; Kidd, G.J.; Macklin, W.B.; Trapp, B.D.; Yan, R. Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J. 2008, 22, 2970–2980. [Google Scholar]
- Van Bebber, F.; Hruscha, A.; Willem, M.; Schmid, B.; Haass, C. Loss of Bace2 in zebrafish affects melanocyte migration and is distinct from Bace1 knock out phenotypes. J. Neurochem. 2013, 127, 471–481. [Google Scholar]
- Luo, X.; Prior, M.; He, W.; Hu, X.; Tang, X.; Shen, W.; Yadav, S.; Kiryu-Seo, S.; Miller, R.; Trapp, B.D.; et al. Cleavage of neuregulin-1 by BACE1 or ADAM10 protein produces differential effects on myelination. J. Biol. Chem. 2011, 286, 23967–23974. [Google Scholar]
- Hu, X.; Hou, H.; Bastian, C.; He, W.; Qiu, S.; Ge, Y.; Yin, X.; Kidd, G.J.; Brunet, S.; Trapp, B.D.; et al. BACE1 regulates the proliferation and cellular functions of Schwann cells. Glia 2017, 65, 712–726. [Google Scholar]
- Wong, H.K.; Sakurai, T.; Oyama, F.; Kaneko, K.; Wada, K.; Miyazaki, H.; Kurosawa, M.; De Strooper, B.; Saftig, P.; Nukina, N. beta Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J. Biol. Chem. 2005, 280, 23009–23017. [Google Scholar]
- Lehnert, S.; Hartmann, S.; Hessler, S.; Adelsberger, H.; Huth, T.; Alzheimer, C. Ion channel regulation by beta-secretase BACE1-enzymatic and non-enzymatic effects beyond Alzheimer’s disease. Channels 2016, 10, 365–378. [Google Scholar]
- Huth, T.; Schmidt-Neuenfeldt, K.; Rittger, A.; Saftig, P.; Reiss, K.; Alzheimer, C. Non-proteolytic effect of beta-site APP-cleaving enzyme 1 (BACE1) on sodium channel function. Neurobiol. Dis. 2009, 33, 282–289. [Google Scholar]
- Hessler, S.; Zheng, F.; Hartmann, S.; Rittger, A.; Lehnert, S.; Volkel, M.; Nissen, M.; Edelmann, E.; Saftig, P.; Schwake, M.; et al. Beta-Secretase BACE1 regulates hippocampal and reconstituted M-currents in a beta-subunit-like fashion. J. Neurosci. 2015, 35, 3298–3311. [Google Scholar]
- Mulley, J.C.; Iona, X.; Hodgson, B.; Heron, S.E.; Berkovic, S.F.; Scheffer, I.E.; Dibbens, L.M. The Role of Seizure-Related SEZ6 as a Susceptibility Gene in Febrile Seizures. Neurol. Res. Int. 2011, 2011, 917565. [Google Scholar] [CrossRef]
- Hitt, B.D.; Jaramillo, T.C.; Chetkovich, D.M.; Vassar, R. BACE1-mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Mol. Neurodegener. 2010, 5. [Google Scholar] [CrossRef]
- Hu, X.; Zhou, X.; He, W.; Yang, J.; Xiong, W.; Wong, P.; Wilson, C.G.; Yan, R. BACE1 deficiency causes altered neuronal activity and neurodegeneration. J. Neurosci. 2010, 30, 8819–8829. [Google Scholar]
- Shaftel, S.S.; Griffin, W.S.; O’Banion, M.K. The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J. Neuroinflamm. 2008, 5. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Marjaux, E.; Imhof, A.; De Strooper, B.; Haass, C.; Lichtenthaler, S.F. Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J. Biol. Chem. 2007, 282, 11982–11995. [Google Scholar]
- Lindberg, C.; Chromek, M.; Ahrengart, L.; Brauner, A.; Schultzberg, M.; Garlind, A. Soluble interleukin-1 receptor type II, IL-18 and caspase-1 in mild cognitive impairment and severe Alzheimer’s disease. Neurochem. Int. 2005, 46, 551–557. [Google Scholar]
- Garlind, A.; Brauner, A.; Hojeberg, B.; Basun, H.; Schultzberg, M. Soluble interleukin-1 receptor type II levels are elevated in cerebrospinal fluid in Alzheimer’s disease patients. Brain Res. 1999, 826, 112–116. [Google Scholar]
- Peters, V.A.; Joesting, J.J.; Freund, G.G. IL-1 receptor 2 (IL-1R2) and its role in immune regulation. Brain Behav. Immun. 2013, 32, 1–8. [Google Scholar]
- Maloney, J.A.; Bainbridge, T.; Gustafson, A.; Zhang, S.; Kyauk, R.; Steiner, P.; van der Brug, M.; Liu, Y.; Ernst, J.A.; Watts, R.J.; et al. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J. Biol. Chem. 2014, 289, 30990–31000. [Google Scholar]
- Vassar, R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6. [Google Scholar] [CrossRef]
- Liu, X.; Wong, H.; Scearce-Levie, K.; Watts, R.J.; Coraggio, M.; Shin, Y.G.; Peng, K.; Wildsmith, K.R.; Atwal, J.K.; Mango, J.; et al. Mechanistic pharmacokinetic-pharmacodynamic modeling of BACE1 inhibition in monkeys: Development of a predictive model for amyloid precursor protein processing. Drug Metab. Dispos. 2013, 41, 1319–1328. [Google Scholar]
- Martiskainen, H.; Herukka, S.K.; Stancakova, A.; Paananen, J.; Soininen, H.; Kuusisto, J.; Laakso, M.; Hiltunen, M. Decreased plasma beta-amyloid in the Alzheimer’s disease APP A673T variant carriers. Ann. Neurol. 2017, 82, 128–132. [Google Scholar]
- Kokawa, A.; Ishihara, S.; Fujiwara, H.; Nobuhara, M.; Iwata, M.; Ihara, Y.; Funamoto, S. The A673T mutation in the amyloid precursor protein reduces the production of beta-amyloid protein from its beta-carboxyl terminal fragment in cells. Acta Neuropathol. Commun. 2015, 3. [Google Scholar] [CrossRef]
- Saido, T.C. Alzheimer’s disease as proteolytic disorders: Anabolism and catabolism of beta-amyloid. Neurobiol. Aging 1998, 19, S69–75. [Google Scholar]
- Schilling, S.; Zeitschel, U.; Hoffmann, T.; Heiser, U.; Francke, M.; Kehlen, A.; Holzer, M.; Hutter-Paier, B.; Prokesch, M.; Windisch, M.; et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Aβ and Alzheimer’s disease-like pathology. Nat. Med. 2008, 14, 1106–1111. [Google Scholar]
- Morawski, M.; Schilling, S.; Kreuzberger, M.; Waniek, A.; Jager, C.; Koch, B.; Cynis, H.; Kehlen, A.; Arendt, T.; Hartlage-Rubsamen, M.; et al. Glutaminyl cyclase in human cortex: Correlation with (pGlu)-amyloid-beta load and cognitive decline in Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 385–400. [Google Scholar]
- Saido, T.C.; Iwatsubo, T.; Mann, D.M.; Shimada, H.; Ihara, Y.; Kawashima, S. Dominant and differential deposition of distinct beta-amyloid peptide species, Aβ N3(pE), in senile plaques. Neuron 1995, 14, 457–466. [Google Scholar]
- Wirths, O.; Erck, C.; Martens, H.; Harmeier, A.; Geumann, C.; Jawhar, S.; Kumar, S.; Multhaup, G.; Walter, J.; Ingelsson, M.; et al. Identification of low molecular weight pyroglutamate Aβ oligomers in Alzheimer disease: A novel tool for therapy and diagnosis. J. Biol. Chem. 2010, 285, 41517–41524. [Google Scholar]
- Walsh, D.M.; Selkoe, D.J. Aβ oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar]
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra372. [Google Scholar]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE 2010, 5, e9505. [Google Scholar]
- Welling, M.M.; Nabuurs, R.J.; van der Weerd, L. Potential role of antimicrobial peptides in the early onset of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2015, 11, 51–57. [Google Scholar]
- Izzo, N.J.; Xu, J.; Zeng, C.; Kirk, M.J.; Mozzoni, K.; Silky, C.; Rehak, C.; Yurko, R.; Look, G.; Rishton, G.; et al. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity. PLoS ONE 2014, 9, e111899. [Google Scholar]
- Wang, H.Y.; Stucky, A.; Liu, J.; Shen, C.; Trocme-Thibierge, C.; Morain, P. Dissociating beta-amyloid from alpha 7 nicotinic acetylcholine receptor by a novel therapeutic agent, S 24795, normalizes alpha 7 nicotinic acetylcholine and NMDA receptor function in Alzheimer’s disease brain. J. Neurosci. 2009, 29, 10961–10973. [Google Scholar]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar]
- Wu, J.; Kuo, Y.P.; George, A.A.; Xu, L.; Hu, J.; Lukas, R.J. Beta-Amyloid directly inhibits human alpha4beta2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J. Biol. Chem. 2004, 279, 37842–37851. [Google Scholar]
- Li, X.D.; Buccafusco, J.J. Effect of beta-amyloid peptide 1-42 on the cytoprotective action mediated by alpha7 nicotinic acetylcholine receptors in growth factor-deprived differentiated PC-12 cells. J. Pharmacol. Exp. Ther. 2003, 307, 670–675. [Google Scholar]
- Wang, H.Y.; Lee, D.H.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. Beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar]
- Oz, M.; Lorke, D.E.; Yang, K.H.; Petroianu, G. On the interaction of beta-amyloid peptides and alpha7-nicotinic acetylcholine receptors in Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 618–630. [Google Scholar]
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar]
- Umezawa, H.; Aoyagi, T.; Morishima, H.; Matsuzaki, M.; Hamada, M. Pepstatin, a new pepsin inhibitor produced by Actinomycetes. J. Antibiot. 1970, 23, 259–262. [Google Scholar]
- Marciniszyn, J., Jr.; Hartsuck, J.A.; Tang, J. Mode of inhibition of acid proteases by pepstatin. J. Biol. Chem. 1976, 251, 7088–7094. [Google Scholar]
- Nguyen, J.T.; Hamada, Y.; Kimura, T.; Kiso, Y. Design of potent aspartic protease inhibitors to treat various diseases. Arch. Pharm. (Weinh.) 2008, 341, 523–535. [Google Scholar]
- Ghosh, A.K.; Bilcer, G.; Harwood, C.; Kawahama, R.; Shin, D.; Hussain, K.A.; Hong, L.; Loy, J.A.; Nguyen, C.; Koelsch, G.; et al. Structure-based design: Potent inhibitors of human brain memapsin 2 (beta-secretase). J. Med. Chem. 2001, 44, 2865–2868. [Google Scholar]
- Chang, W.P.; Koelsch, G.; Wong, S.; Downs, D.; Da, H.; Weerasena, V.; Gordon, B.; Devasamudram, T.; Bilcer, G.; Ghosh, A.K.; et al. In vivo inhibition of Aβ production by memapsin 2 (beta-secretase) inhibitors. J. Neurochem. 2004, 89, 1409–1416. [Google Scholar]
- Ghosh, A.K.; Kumaragurubaran, N.; Tang, J. Recent developments of structure based beta-secretase inhibitors for Alzheimer’s disease. Curr. Top. Med. Chem. 2005, 5, 1609–1622. [Google Scholar]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar]
- Yan, R.; Bienkowski, M.J.; Shuck, M.E.; Miao, H.; Tory, M.C.; Pauley, A.M.; Brashier, J.R.; Stratman, N.C.; Mathews, W.R.; Buhl, A.E.; et al. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 1999, 402, 533–537. [Google Scholar]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar]
- Solans, A.; Estivill, X.; de La Luna, S. A new aspartyl protease on 21q22.3, BACE2, is highly similar to Alzheimer’s amyloid precursor protein beta-secretase. Cytogenet. Cell Genet. 2000, 89, 177–184. [Google Scholar]
- Szecsi, P.B. The aspartic proteases. Scand. J. Clin. Lab. Investig. Suppl. 1992, 210, 5–22. [Google Scholar]
- Chang, W.P.; Downs, D.; Huang, X.P.; Da, H.; Fung, K.M.; Tang, J. Amyloid-beta reduction by memapsin 2 (beta-secretase) immunization. FASEB J. 2007, 21, 3184–3196. [Google Scholar]
- Gadkar, K.; Yadav, D.B.; Zuchero, J.Y.; Couch, J.A.; Kanodia, J.; Kenrick, M.K.; Atwal, J.K.; Dennis, M.S.; Prabhu, S.; Watts, R.J.; et al. Mathematical PKPD and safety model of bispecific TfR/BACE1 antibodies for the optimization of antibody uptake in brain. Eur. J. Pharm. Biopharm. 2016, 101, 53–61. [Google Scholar]
- Atwal, J.K.; Chen, Y.; Chiu, C.; Mortensen, D.L.; Meilandt, W.J.; Liu, Y.; Heise, C.E.; Hoyte, K.; Luk, W.; Lu, Y.; et al. A therapeutic antibody targeting BACE1 inhibits amyloid-beta production in vivo. Sci. Transl. Med. 2011, 3, 84ra43. [Google Scholar]
- Zhou, L.; Chavez-Gutierrez, L.; Bockstael, K.; Sannerud, R.; Annaert, W.; May, P.C.; Karran, E.; De Strooper, B. Inhibition of beta-secretase in vivo via antibody binding to unique loops (D and F) of BACE1. J. Biol. Chem. 2011, 286, 8677–8687. [Google Scholar]
- Rabinovich-Nikitin, I.; Solomon, B. Inhibition of amyloid precursor protein processing leads to downregulation of apoptotic genes in Alzheimer’s disease animal models. Neurodegener. Dis. 2014, 13, 107–109. [Google Scholar]
- Arbel, M.; Yacoby, I.; Solomon, B. Inhibition of amyloid precursor protein processing by beta-secretase through site-directed antibodies. Proc. Natl. Acad. Sci. USA 2005, 102, 7718–7723. [Google Scholar]
- Dulin, F.; Leveille, F.; Ortega, J.B.; Mornon, J.P.; Buisson, A.; Callebaut, I.; Colloc’h, N. P3 peptide, a truncated form of Aβ devoid of synaptotoxic effect, does not assemble into soluble oligomers. FEBS Lett. 2008, 582, 1865–1870. [Google Scholar]
- Gowing, E.; Roher, A.E.; Woods, A.S.; Cotter, R.J.; Chaney, M.; Little, S.P.; Ball, M.J. Chemical characterization of Aβ 17-42 peptide, a component of diffuse amyloid deposits of Alzheimer disease. J. Biol. Chem. 1994, 269, 10987–10990. [Google Scholar]
- Patton, R.L.; Kalback, W.M.; Esh, C.L.; Kokjohn, T.A.; Van Vickle, G.D.; Luehrs, D.C.; Kuo, Y.M.; Lopez, J.; Brune, D.; Ferrer, I.; et al. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: A biochemical analysis. Am. J. Pathol. 2006, 169, 1048–1063. [Google Scholar]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Azimova, R.; Kagan, B.L.; Nussinov, R.; Lal, R. Truncated beta-amyloid peptide channels provide an alternative mechanism for Alzheimer’s Disease and Down syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 6538–6543. [Google Scholar]
- Marchesi, V.T. An alternative interpretation of the amyloid Aβ hypothesis with regard to the pathogenesis of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 9093–9098. [Google Scholar]
- Wei, W.; Norton, D.D.; Wang, X.; Kusiak, J.W. Aβ 17-42 in Alzheimer’s disease activates JNK and caspase-8 leading to neuronal apoptosis. Brain 2002, 125, 2036–2043. [Google Scholar]
- Hoe, H.S.; Lee, H.K.; Pak, D.T. The upside of APP at synapses. CNS Neurosci. Ther. 2012, 18, 47–56. [Google Scholar]
- Sosa, L.J.; Caceres, A.; Dupraz, S.; Oksdath, M.; Quiroga, S.; Lorenzo, A. The physiological role of the amyloid precursor protein as an adhesion molecule in the developing nervous system. J. Neurochem. 2017, 143, 11–29. [Google Scholar]
- Breen, K.C.; Bruce, M.; Anderton, B.H. Beta amyloid precursor protein mediates neuronal cell-cell and cell-surface adhesion. J. Neurosci. Res. 1991, 28, 90–100. [Google Scholar]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8. [Google Scholar] [CrossRef]
- Bates, K.A.; Verdile, G.; Li, Q.X.; Ames, D.; Hudson, P.; Masters, C.L.; Martins, R.N. Clearance mechanisms of Alzheimer’s amyloid-beta peptide: Implications for therapeutic design and diagnostic tests. Mol. Psychiatry 2009, 14, 469–486. [Google Scholar]
- Baranello, R.J.; Bharani, K.L.; Padmaraju, V.; Chopra, N.; Lahiri, D.K.; Greig, N.H.; Pappolla, M.A.; Sambamurti, K. Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 32–46. [Google Scholar]
- Holsinger, R.M.; McLean, C.A.; Beyreuther, K.; Masters, C.L.; Evin, G. Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann. Neurol. 2002, 51, 783–786. [Google Scholar]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar]
- Johnston, J.A.; Liu, W.W.; Todd, S.A.; Coulson, D.T.; Murphy, S.; Irvine, G.B.; Passmore, A.P. Expression and activity of beta-site amyloid precursor protein cleaving enzyme in Alzheimer’s disease. Biochem. Soc. Trans. 2005, 33, 1096–1100. [Google Scholar]
- Karran, E.; Hardy, J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann. Neurol. 2014, 76, 185–205. [Google Scholar]
- Yan, R. Stepping closer to treating Alzheimer’s disease patients with BACE1 inhibitor drugs. Transl. Neurodegener. 2016, 5. [Google Scholar] [CrossRef]
- Golde, T.E.; Schneider, L.S.; Koo, E.H. Anti-abeta therapeutics in Alzheimer’s disease: The need for a paradigm shift. Neuron 2011, 69, 203–213. [Google Scholar]
- McDade, E.; Bateman, R.J. Stop Alzheimer’s before it starts. Nature 2017, 547, 153–155. [Google Scholar]
- Barao, S.; Moechars, D.; Lichtenthaler, S.F.; De Strooper, B. BACE1 Physiological Functions May Limit Its Use as Therapeutic Target for Alzheimer’s Disease. Trends Neurosci. 2016, 39, 158–169. [Google Scholar]
- Zuhl, A.M.; Nolan, C.E.; Brodney, M.A.; Niessen, S.; Atchison, K.; Houle, C.; Karanian, D.A.; Ambroise, C.; Brulet, J.W.; Beck, E.M.; et al. Chemoproteomic profiling reveals that cathepsin D off-target activity drives ocular toxicity of beta-secretase inhibitors. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Hefti, F.F. Requirements for a lead compound to become a clinical candidate. BMC Neurosci. 2008, 9. [Google Scholar] [CrossRef]
- Steinmetz, K.L.; Spack, E.G. The basics of preclinical drug development for neurodegenerative disease indications. BMC Neurol. 2009, 9. [Google Scholar] [CrossRef]
- Hu, X.; He, W.; Luo, X.; Tsubota, K.E.; Yan, R. BACE1 regulates hippocampal astrogenesis via the Jagged1-Notch pathway. Cell Rep. 2013, 4, 40–49. [Google Scholar]
- Cheret, C.; Willem, M.; Fricker, F.R.; Wende, H.; Wulf-Goldenberg, A.; Tahirovic, S.; Nave, K.A.; Saftig, P.; Haass, C.; Garratt, A.N.; et al. Bace1 and Neuregulin-1 cooperate to control formation and maintenance of muscle spindles. EMBO J. 2013, 32, 2015–2028. [Google Scholar]
- Rajapaksha, T.W.; Eimer, W.A.; Bozza, T.C.; Vassar, R. The Alzheimer’s beta-secretase enzyme BACE1 is required for accurate axon guidance of olfactory sensory neurons and normal glomerulus formation in the olfactory bulb. Mol. Neurodegener. 2011, 6. [Google Scholar] [CrossRef]
- Cao, L.; Rickenbacher, G.T.; Rodriguez, S.; Moulia, T.W.; Albers, M.W. The precision of axon targeting of mouse olfactory sensory neurons requires the BACE1 protease. Sci. Rep. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Hitt, B.; Riordan, S.M.; Kukreja, L.; Eimer, W.A.; Rajapaksha, T.W.; Vassar, R. Beta-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J. Biol. Chem. 2012, 287, 38408–38425. [Google Scholar]
- Savonenko, A.V.; Melnikova, T.; Laird, F.M.; Stewart, K.A.; Price, D.L.; Wong, P.C. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc. Natl. Acad. Sci. USA 2008, 105, 5585–5590. [Google Scholar]
- Filser, S.; Ovsepian, S.V.; Masana, M.; Blazquez-Llorca, L.; Brandt Elvang, A.; Volbracht, C.; Muller, M.B.; Jung, C.K.; Herms, J. Pharmacological inhibition of BACE1 impairs synaptic plasticity and cognitive functions. Biol. Psychiatry 2015, 77, 729–739. [Google Scholar]
- Laird, F.M.; Cai, H.; Savonenko, A.V.; Farah, M.H.; He, K.; Melnikova, T.; Wen, H.; Chiang, H.C.; Xu, G.; Koliatsos, V.E.; et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 2005, 25, 11693–11709. [Google Scholar]
- Kobayashi, D.; Zeller, M.; Cole, T.; Buttini, M.; McConlogue, L.; Sinha, S.; Freedman, S.; Morris, R.G.; Chen, K.S. BACE1 gene deletion: impact on behavioral function in a model of Alzheimer’s disease. Neurobiol. Aging 2008, 29, 861–873. [Google Scholar]
- Wang, H.; Song, L.; Laird, F.; Wong, P.C.; Lee, H.K. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J. Neurosci. 2008, 28, 8677–8681. [Google Scholar]
- Cai, J.; Qi, X.; Kociok, N.; Skosyrski, S.; Emilio, A.; Ruan, Q.; Han, S.; Liu, L.; Chen, Z.; Bowes Rickman, C.; et al. Beta-Secretase (BACE1) inhibition causes retinal pathology by vascular dysregulation and accumulation of age pigment. EMBO Mol. Med. 2012, 4, 980–991. [Google Scholar]
- Weber, M.; Wu, T.; Meilandt, W.J.; Dominguez, S.L.; Solanoy, H.O.; Maloney, J.A.; Ngu, H.; Baca, M.; Kung, C.; Lima, L.; et al. BACE1 across species: A comparison of the in vivo consequences of BACE1 deletion in mice and rats. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Dominguez, D.; Tournoy, J.; Hartmann, D.; Huth, T.; Cryns, K.; Deforce, S.; Serneels, L.; Camacho, I.E.; Marjaux, E.; Craessaerts, K.; et al. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol. Chem. 2005, 280, 307906. [Google Scholar]
- Harrison, S.M.; Harper, A.J.; Hawkins, J.; Duddy, G.; Grau, E.; Pugh, P.L.; Winter, P.H.; Shilliam, C.S.; Hughes, Z.A.; Dawson, L.A.; et al. BACE1 (beta-secretase) transgenic and knockout mice: Identification of neurochemical deficits and behavioral changes. Mol. Cell. Neurosci. 2003, 24, 646–655. [Google Scholar]
- Ghosh, A.K.; Tang, J. Prospects of beta-Secretase Inhibitors for the Treatment of Alzheimer’s Disease. ChemMedChem 2015, 10, 1463–1466. [Google Scholar]
- Yan, R.; Fan, Q.; Zhou, J.; Vassar, R. Inhibiting BACE1 to reverse synaptic dysfunctions in Alzheimer’s disease. Neurosci. Biobehav. Rev. 2016, 65, 326–340. [Google Scholar]
- Cebers, G.; Alexander, R.C.; Haeberlein, S.B.; Han, D.; Goldwater, R.; Ereshefsky, L.; Olsson, T.; Ye, N.; Rosen, L.; Russell, M.; et al. AZD3293: Pharmacokinetic and Pharmacodynamic Effects in Healthy Subjects and Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 55, 1039–1053. [Google Scholar]
- Sakamoto, K.; Matsuki, S.; Matsuguma, K.; Yoshihara, T.; Uchida, N.; Azuma, F.; Russell, M.; Hughes, G.; Haeberlein, S.B.; Alexander, R.C.; et al. BACE1 Inhibitor Lanabecestat (AZD3293) in a Phase 1 Study of Healthy Japanese Subjects: Pharmacokinetics and Effects on Plasma and Cerebrospinal Fluid Aβ Peptides. J. Clin. Pharmacol. 2017. [Google Scholar] [CrossRef]
- Albala, B.; Kaplow, J.M.; Lai, R.; Matijevic, M.; Aluri, J.; Satlin, A. CSF amyloid lowering in human volunteers after 14 days’ oral administration of the novel BACE1 inhibitor E2609. Alzheimer’s Dement. 2012, 8, S743. [Google Scholar]
- Timmers, M.; Van Broeck, B.; Ramael, S.; Slemmon, J.; De Waepenaert, K.; Russu, A.; Bogert, J.; Stieltjes, H.; Shaw, L.M.; Engelborghs, S.; et al. Profiling the dynamics of CSF and plasma Aβ reduction after treatment with JNJ-54861911, a potent oral BACE inhibitor. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2016, 2, 202–212. [Google Scholar]
- Ufer, M.; Rouzade-Dominguez, M.-L.; Huledal, G.; Pezous, N.; Avrameas, A.; David, O.; Kretz, S.; Kucher, K.; Neumann, U.; Cha, J.-H.; et al. Results from a First-in-Human Study with the Bace Inhibitor Cnp520. Alzheimer’s Dement. 2016, 12, P200. [Google Scholar]
- Rozga, M.; Bittner, T.; Hoglund, K.; Blennow, K. Accuracy of cerebrospinal fluid Abeta1-42 measurements: Evaluation of pre-analytical factors using a novel Elecsys immunosassay. Clin. Chem. Lab. Med. 2017, 55, 1545–1554. [Google Scholar]
- Cicognola, C.; Chiasserini, D.; Eusebi, P.; Andreasson, U.; Vanderstichele, H.; Zetterberg, H.; Parnetti, L.; Blennow, K. No diurnal variation of classical and candidate biomarkers of Alzheimer’s disease in CSF. Mol. Neurodegener. 2016, 11. [Google Scholar] [CrossRef]
- Kumar, D.K.; Eimer, W.A.; Tanzi, R.E.; Moir, R.D. Alzheimer’s disease: The potential therapeutic role of the natural antibiotic amyloid-beta peptide. Neurodegener. Dis. Manag. 2016, 6, 345–348. [Google Scholar]
- Cummings, J. Lessons learned from Alzheimer disease: Clinical trials with negative outcomes. Clin. Transl. Sci. 2017. [Google Scholar] [CrossRef]
- Bamne, M.N.; Demirci, F.Y.; Berman, S.; Snitz, B.E.; Rosenthal, S.L.; Wang, X.; Lopez, O.L.; Kamboh, M.I. Investigation of an amyloid precursor protein protective mutation (A673T) in a North American case-control sample of late-onset Alzheimer’s disease. Neurobiol. Aging 2014, 35. [Google Scholar] [CrossRef]
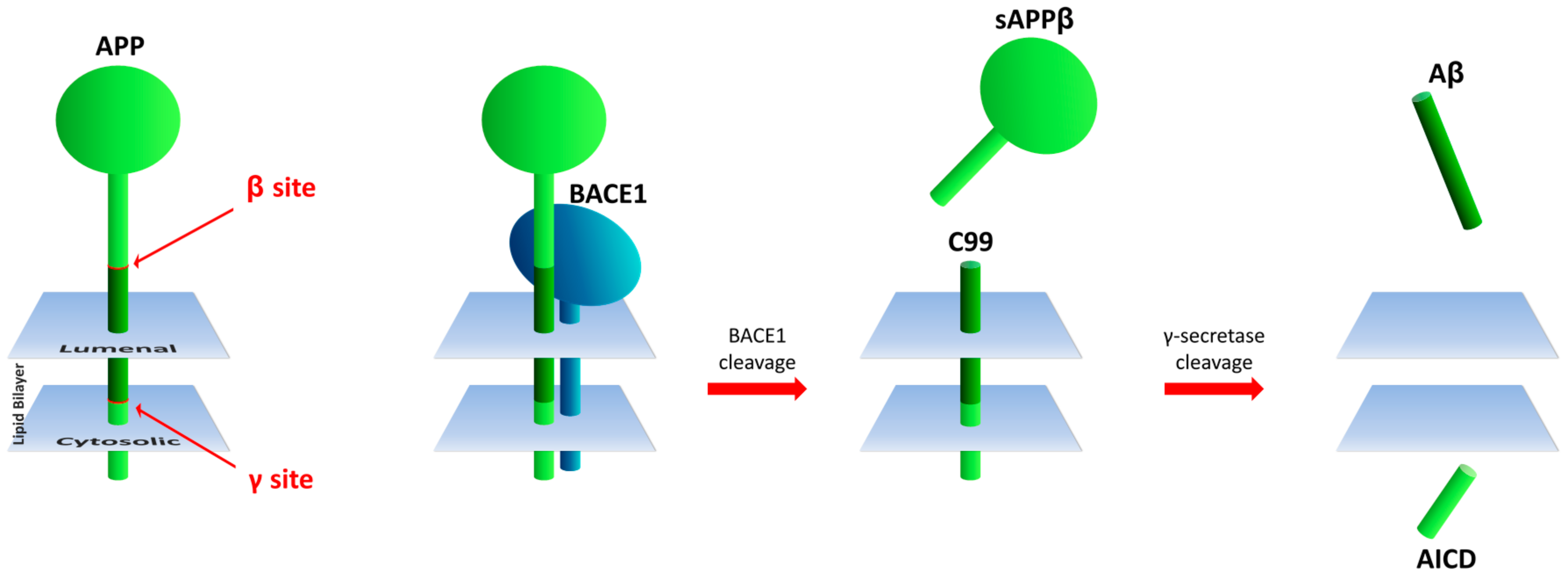


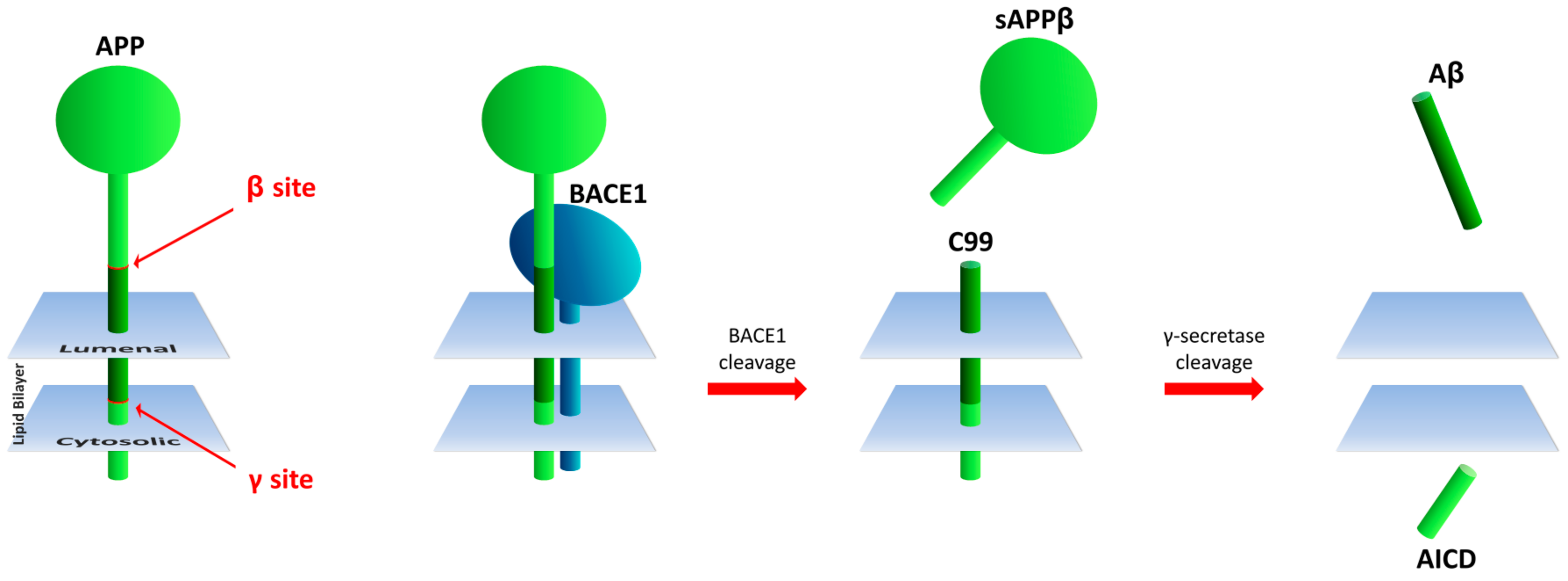{kind=link}
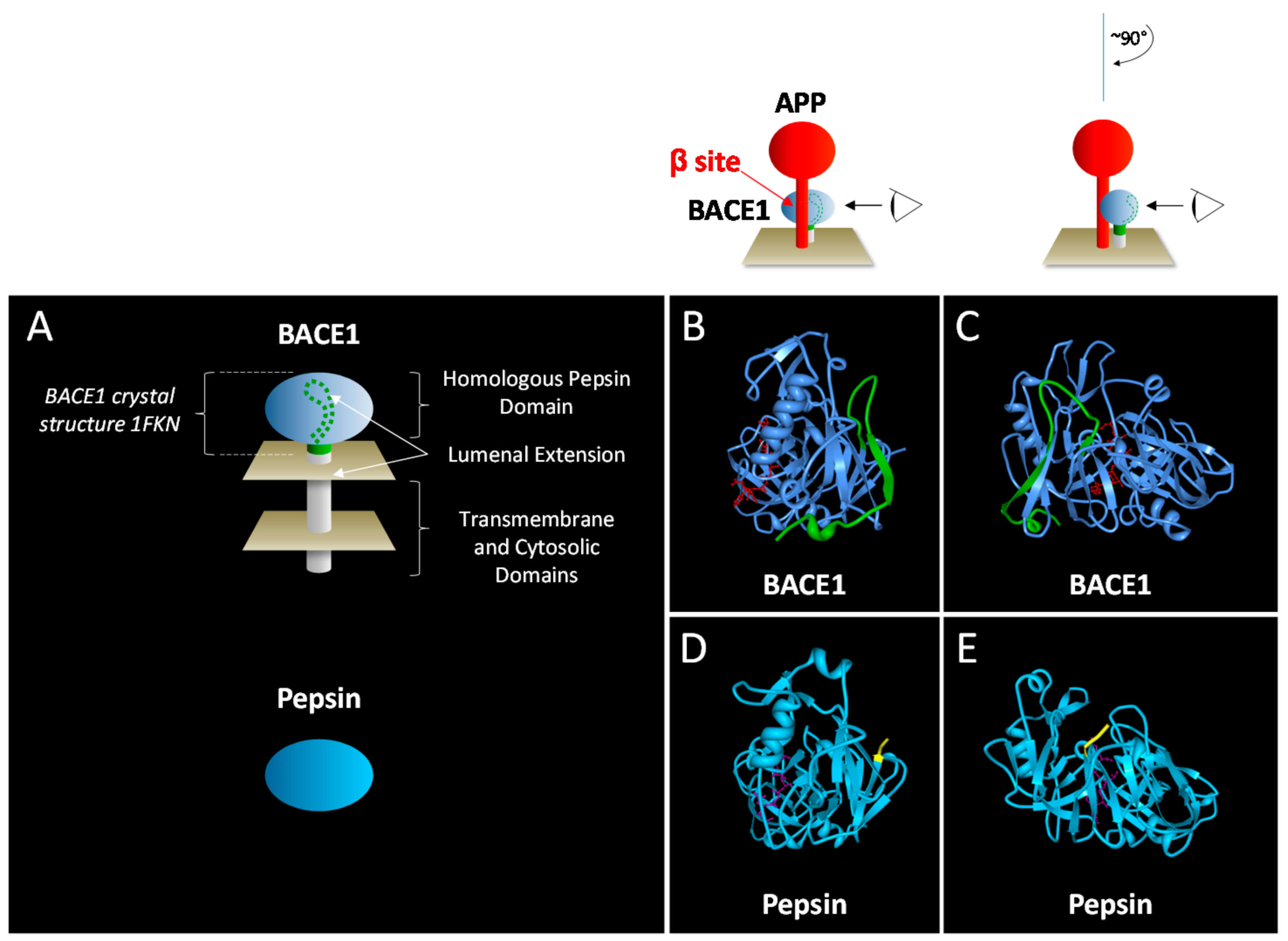{kind=link}
| Feature | Position in Amino Acid Sequence 2 | Function | References |
|---|---|---|---|
| Propeptide | 22–45 | Folding, enhancement of activity | [34,35,36] |
| Disulfide bonds | 216–420, 278–443, 330–380 | Structural stability | [30] |
| Glycosylation | 153, 172, 223, 354 | Lysosomal targeting, degradation | [37,38,39] |
| Phosphorylation | 498 | Endosomal-lysosomal trafficking | [40] |
| Ubuiquitination | 501 | Trafficking, degradation | [41,42] |
| S-Palmitoylation | 474, 478, 483, 485 | Lipid raft, amyloidogenesis | [43] |
| Lysine acetylation | 126, 275, 279, 285, 299, 300, 307 | Stability; transit from ER; enhancement of intracellular activity | [37,44] |
| ↓ | ||||||||
|---|---|---|---|---|---|---|---|---|
| Substrate 2 | P4 | P3 | P2 | P1 | P1’ | P2’ | P3’ | P4’ |
| APP, β site | E | V | K | M | D | A | E | F |
| APP A673T, β site | E | V | K | M | D | E | F | |
| APP, β’ site | D | S | G | Y | E | V | H | H |
| Nrg1 type I & III-β1α | G | I | E | F | M | E | A | E |
| Nrg3 | G | I | E | F | M | E | S | E |
| IL-1R2 | T | L | S | F | Q | L | R | |
| Navβ2, major site | L | Q | V | L | M | E | E | P |
| Navβ2, minor site | K | I | H | L | Q | V | L | M |
| Nrg1 type III-β1α | E | T | N | L | Q | A | P | |
| Sez6 | G | R | S | L | D | V | A | K |
| Delta-1 | V | V | D | L | T | E | K | L |
| PSGL-1 | A | S | N | L | S | V | N | Y |
| Jag2 | S | L | L | L | A | V | T | E |
| Sez6L | A | L | E | A | E | A | A | A |
| Jag1 | S | L | I | A | A | V | A | E |
| ST6Gal | E | K | A | Q | L | L | A | |
| CHL-l | S | I | F | Q | D | V | I | E |
| Function/Dysfunction | Degree of BACE1 Inhibition 2 | Reference | |
|---|---|---|---|
| Pharmacologic, Subchronic | Gene Deletion | ||
| Neurogenesis, Astrogenesis | 100% | [170] | |
| Growth cone collapse | 100% 3 | [171] | |
| Axonal growth | 100% | 50–100% 4 | [172,173,174] |
| Spine density | 60% 5 | 100% | [175,176] |
| Muscle Spindle | 68% 6 | 100% | [97] |
| Myelination | 100% 7 | [77,78,88] | |
| Synaptic dysfunction | 50% 5 | 100% 7 | [176,177,178,179] |
| Retinopathy | 100% | [180] | |
| Muscle coordination | 100% | [171,178,181] | |
| Memory impairment | 60% 5 | 100% 7 | [176,177,178] |
| Lethality, growth impairment | 50–100% | [98,181,182] | |
| Seizures | 100% | [97,98,178] | |
| Social/emotional | 100% 6 | [177,182,183] | |
| Psychosis | 100% | [175,178] | |
| Compound | Sponsor(s) | Patient Population and Identifier 1 | Dose, Target Aβ Reduction 2 | Reference |
|---|---|---|---|---|
| Verubecestat (MK-8931) | Merck | Prodromal AD (NCT01953601) | 12 mg: 50% 40 mg: 75% | [29] |
| Lanabecestat (LY3314814, AZD3293) | AstraZeneca, Eli Lilly | Early AD (NCT02245737) | 20 mg: 60% 3 50 mg: 75% 3 | [186,187] |
| Elenbecestat (E2609) | Biogen Idec, Eisai | Early AD (NCT03036280, NCT02956486) | 50 mg: 60% 4 | [188] |
| JNJ-54861911 | Shionogi, Janssen | Asymptomatic, at risk for AD (NCT02569398) | 5 mg: 50% 25 mg: 75–85% | [189] |
| CNP520 | Novartis, Amgen | At risk for AD (NCT03131453) | 15 mg: N.D. 5 50 mg: N.D. 5 | [190] |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koelsch, G. BACE1 Function and Inhibition: Implications of Intervention in the Amyloid Pathway of Alzheimer’s Disease Pathology. Molecules 2017, 22, 1723. https://doi.org/10.3390/molecules22101723
Koelsch G. BACE1 Function and Inhibition: Implications of Intervention in the Amyloid Pathway of Alzheimer’s Disease Pathology. Molecules. 2017; 22(10):1723. https://doi.org/10.3390/molecules22101723
Chicago/Turabian StyleKoelsch, Gerald. 2017. "BACE1 Function and Inhibition: Implications of Intervention in the Amyloid Pathway of Alzheimer’s Disease Pathology" Molecules 22, no. 10: 1723. https://doi.org/10.3390/molecules22101723