A Theoretical Study of the N to O Linkage Photoisomerization Efficiency in a Series of Ruthenium Mononitrosyl Complexes

 ,
,
Abstract
:1. Introduction
2. Results
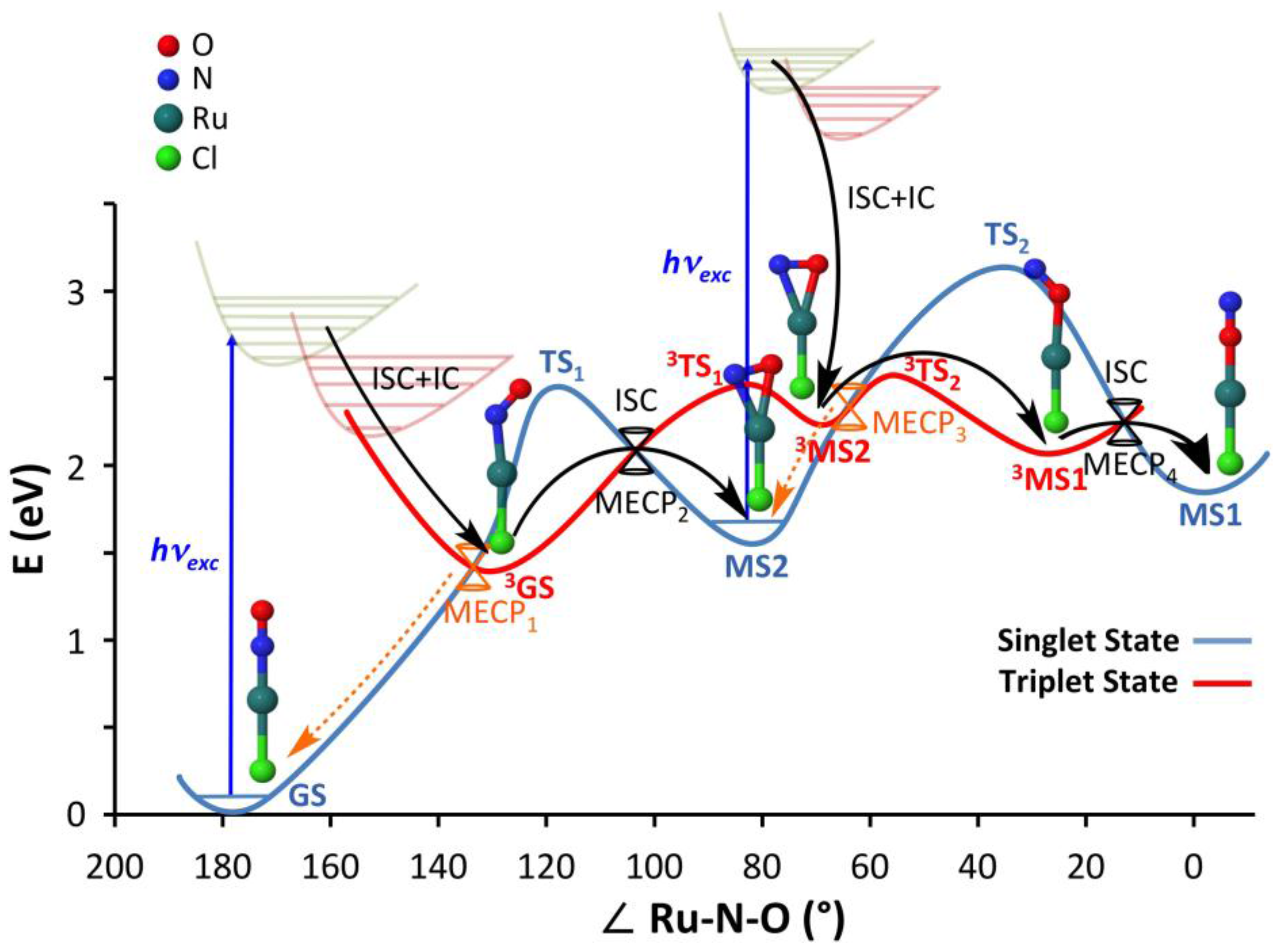
2.1. Overview on the Mechanistic Picture for the trans-[RuCl(NO)(py)4]2+ Complex
2.2. Comparison with the trans-[RuBr(NO)(py)4]2+ and trans-(Cl,Cl)[RuCl2(NO)(tpy)]+ Complexes
2.2.1. Thermal Isomerization Pathway
2.2.2. Adiabatic Photoisomerization Pathway
2.2.3. Nonadiabatic Photoisomerization Pathway
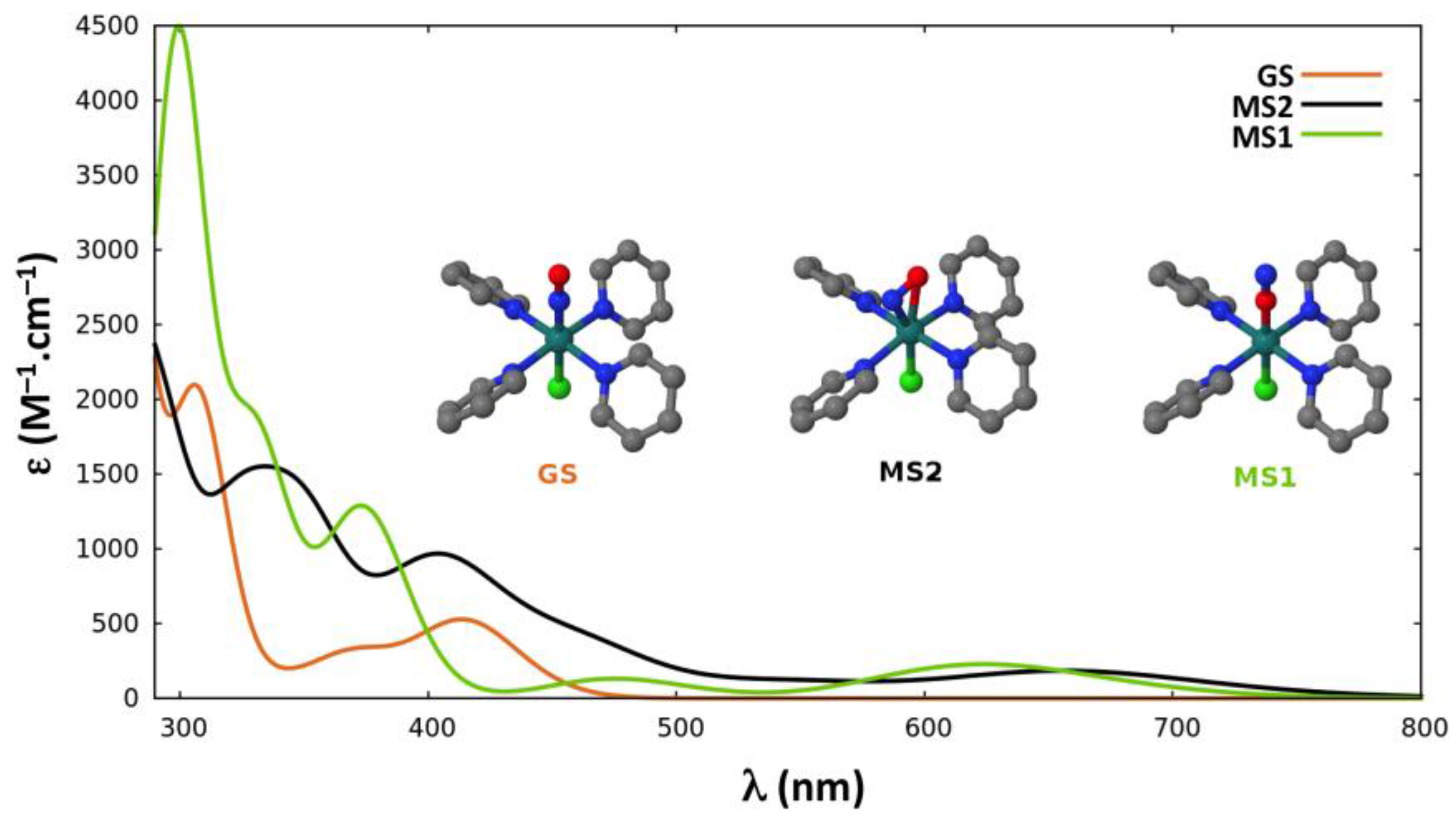
2.2.4. Absorption Properties
3. Discussion
4. Computational Details
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Photochromism, Molecules and Systems; Dürr, H.; Bouas-Laurent, H. (Eds.) Elsevier: Amsterdam, The Netherlands, 1990. [Google Scholar]
- Bouas-Laurent, H.; Dürr, H. Organic photochromism. Pure Appl. Chem. 2001, 73, 639–665. [Google Scholar] [CrossRef]
- Crano, J.C.; Guglielmetti, R.J. Organic Photochromic and Thermochromic Compound; Plenum Press: New York, NY, USA; London, UK, 1998. [Google Scholar]
- Irie, M. Diarylethenes for memories and switches. Chem. Rev. 2000, 100, 1685–1716. [Google Scholar] [CrossRef] [PubMed]
- Berkovic, G.; Krongauz, V.; Weiss, V. Spiropyrans and spirooxazines for memories and switches. Chem. Rev. 2000, 100, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y. Fulgides for memories and switches. Chem. Rev. 2000, 100, 1717–1739. [Google Scholar] [CrossRef] [PubMed]
- Görner, H.; Fischer, C.; Gierisch, S.; Daub, J. Dihydroazulene/vinylheptafulvene photochromism: Effects of substituents, solvent, and temperature in the photorearrangement of dihydroazulenes to vinylheptafulvenes. J. Phys. Chem. 1993, 97, 4110–4117. [Google Scholar] [CrossRef]
- Jockusch, S.; Turro, N.J.; Blackburn, F.R. Photochromism of 2H-naphto[1,2-b]pyrans: A spectroscopic investigation. J. Phys. Chem. A 2002, 106, 9236–9241. [Google Scholar] [CrossRef]
- Mitchell, R.H. The metacyclophanediene-dihydropyrene photochromic π switch. Eur. J. Org. Chem. 1999, 2695–2703. [Google Scholar] [CrossRef]
- Coppens, P.; Novozhilova, I.; Kovalevsky, A. Photoinduced linkage isomers of transition-metal nitrosyl compounds and related complexes. Chem. Rev. 2002, 102, 861–883. [Google Scholar] [CrossRef] [PubMed]
- Bitterwolf, T.E. Photochemical nitrosyl linkage isomerism/metastable states. Coord. Chem. Rev. 2006, 250, 1196–1207. [Google Scholar] [CrossRef]
- Rack, J.J. Electron transfer triggered sulfoxide isomerization in ruthenium and osmium complexes. Coord. Chem. Rev. 2009, 253, 78–85. [Google Scholar] [CrossRef]
- McClure, B.A.; Rack, J.J. Isomerization in photochromic ruthenium sulfoxide complexes. Eur. J. Inorg. Chem. 2010, 3895–3904. [Google Scholar] [CrossRef]
- Sylvester, S.O.; Cole, J.M.; Waddell, P.G. Photoconversion bonding mechanism in ruthenium sulfur dioxide linkage photoisomers revealed by in situ diffraction. J. Am. Chem. Soc. 2012, 134, 11860–11863. [Google Scholar] [CrossRef] [PubMed]
- Feringa, B.L. The art of building small: From molecular switches to molecular motors. J. Org. Chem. 2007, 72, 6635–6652. [Google Scholar] [CrossRef] [PubMed]
- Szaciłowski, K. Digital Information Processing in Molecular Systems. Chem. Rev. 2008, 108, 3481–3548. [Google Scholar] [CrossRef] [PubMed]
- Andréasson, J.; Pischel, U. Smart Molecules at Work-Mimicking Advanced Logic Operations. Chem. Soc. Rev. 2010, 39, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, W.; Beierle, J.M.; Kistemaker, H.A.V.; Velema, W.A.; Feringa, B.L. Reversible photocontrol of biological systems by the incorporation of molecular photoswitches. Chem. Rev. 2013, 113, 6114–6178. [Google Scholar] [CrossRef] [PubMed]
- Pischel, U.; Andréasson, J.; Gust, D.; Pais, V. Information processing with molecules—Quo vadis? ChemPhysChem 2013, 14, 28–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Zhu, L.; Al-Kaysi, R.O.; Bardeen, C. Organic photomechanical materials. ChemPhysChem 2014, 15, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Asadirad, A.M.; Boutault, S.; Erno, Z.; Branda, N.R. Controlling a polymer adhesive using light and a molecular switch. J. Am. Chem. Soc. 2014, 136, 3024–3027. [Google Scholar] [CrossRef] [PubMed]
- Boggio-Pasqua, M.; Bearpark, M.J.; Hunt, P.A.; Robb, M.A. Dihydroazulene/vinylheptafulvene photochromism: A model for one-way photochemistry via a conical intersection. J. Am. Chem. Soc. 2002, 124, 1456–1470. [Google Scholar] [CrossRef] [PubMed]
- Boggio-Pasqua, M.; Ravaglia, M.; Bearpark, M.J.; Garavelli, M.; Robb, M.A. Can diarylethene photochromism be explained by a reaction path alone? A CASSCF study with model MMVB dynamics. J. Phys. Chem. A 2003, 107, 11139–11152. [Google Scholar] [CrossRef]
- Migani, A.; Gentili, P.L.; Negri, F.; Olivucci, M.; Romani, A.; Favaro, G.; Becker, R.S. The ring-opening reaction of chromenes: A photochemical mode-dependent transformation. J. Phys. Chem. A 2005, 109, 8684–8692. [Google Scholar] [CrossRef] [PubMed]
- Maurel, F.; Aubard, J.; Millie, P.; Dognon, J.P.; Rajzmann, M.; Guglielmetti, R.; Samat, A. Quantum chemical study of the photocoloration reaction in the napthoxazine series. J. Phys. Chem. A 2006, 110, 4759–4771. [Google Scholar] [CrossRef] [PubMed]
- Boggio-Pasqua, M.; Bearpark, M.J.; Ogliaro, F.; Robb, M.A. Photochemical reactivity of 2-vinylbiphenyl and 2-vinyl-1,3-terphenyl: The balance between nonadiabatic and adiabatic photocyclization. J. Am. Chem. Soc. 2006, 128, 10533–10540. [Google Scholar] [CrossRef] [PubMed]
- Boggio-Pasqua, M.; Bearpark, M.J.; Robb, M.A. Towards a mechanistic understanding of the photochromism of dimethyldihydropyrenes. J. Org. Chem. 2007, 72, 4497–4503. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, G.; Ogliaro, F.; Bearpark, M.J.; Robb, M.A.; Garavelli, M. Modeling the photophysics and photochromic potential of 1,2-dihydronaphthalene (DHN): A combined CASPT2//CASSCF-topological and MMVB-dynamical investigation. J. Phys. Chem. A 2008, 112, 10096–10107. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, G.; Bearpark, M.J.; Robb, M.A.; Orlandi, G.; Garavelli, M. Significance of a zwitterionic state for fulgide photochromism: Implications for the design of mimics. Angew. Chem. Int. Ed. 2010, 49, 2913–2916. [Google Scholar] [CrossRef] [PubMed]
- Nenov, A.; Schreier, W.J.; Koller, F.O.; Braun, M.; de Vivie-Riedle, R.; Zinth, W.; Pugliesi, I. Molecular model of the ring-opening and ring-closure reaction of a fluorinated indolylfulgide. J. Phys. Chem. A 2012, 116, 10518–10528. [Google Scholar] [CrossRef] [PubMed]
- Raymo, F.M. Computational insights on the isomerization of photochromic oxazines. J. Phys. Chem. A 2012, 116, 11888–11895. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Morokuma, K. Multiple pathways for the primary step of the spiropyran photochromic reaction: A CASPT2//CASSCF study. J. Am. Chem. Soc. 2013, 135, 10693–10702. [Google Scholar] [CrossRef] [PubMed]
- Boggio-Pasqua, M.; Garavelli, M. Rationalization and design of enhanced photoinduced cycloreversion in photochromic dimethyldihydropyrenes by theoretical calculations. J. Phys. Chem. A 2015, 119, 6024–6032. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Gourlaouen, C. Chemical Bonding Alteration Upon Electronic Excitation in Transition Metal Complexes. Cood. Chem. Rev. 2017, 344, 131–149. [Google Scholar] [CrossRef]
- Ciofini, I.; Daul, C.A.; Adamo, C. Phototriggered Linkage Isomerization in Ruthenium-Dimethylsulfoxyde Complexes: Insights from Theory. J. Phys. Chem. A 2003, 107, 11182–11190. [Google Scholar] [CrossRef]
- Göttle, A.J.; Dixon, I.M.; Alary, F.; Heully, J.-L.; Boggio-Pasqua, M. Adiabatic versus nonadiabatic photoisomerization in photochromic ruthenium sulfoxide complexes: a mechanistic picture from density functional theory calculations. J. Am. Chem. Soc. 2011, 133, 9172–9174. [Google Scholar] [CrossRef] [PubMed]
- Vieuxmaire, O.P.J.; Piau, R.E.; Alary, F.; Heully, J.-L.; Sutra, P.; Igau, A.; Boggio-Pasqua, M. Theoretical investigation of phosphinidene oxide polypyridine ruthenium(II) complexes: Toward the design of a new class of photochromic compounds. J. Phys. Chem. A 2013, 117, 12821–12830. [Google Scholar] [CrossRef] [PubMed]
- Göttle, A.J.; Alary, F.; Dixon, I.M.; Heully, J.-L.; Boggio-Pasqua, M. Unravelling the S→O linkage photoisomerization mechanisms in cis- and trans-[Ru(bpy)2(DMSO)2]2+ using density functional theory. Inorg. Chem. 2014, 53, 6752–6760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, L.; Wang, Y.; Fan, X. Theoretical Studies on the Photoisomerization Mechanism of Osmium(II) Sulfoxide Complexes. RSC Adv. 2015, 5, 58580–58586. [Google Scholar] [CrossRef]
- Li, H.; Zhang, L.; Zheng, I.; Li, X.; Fan, X.; Zhao, Y. Photoisomerization Mechanism of Ruthenium Sulfoxide Complexes: Role of the Metal-Centered Excited State in the Bond Rupture and Bond Construction Processes. Chem. Eur. J. 2016, 22, 14285–14292. [Google Scholar] [CrossRef] [PubMed]
- Sanz García, J.; Alary, F.; Boggio-Pasqua, M.; Dixon, I.M.; Malfant, I.; Heully, J.-L. Establishing the Two-Photon Linkage Isomerization Mechanism in the Nitrosyl Complex trans-[RuCl(NO)(py)4]2+ by DFT and TDDFT. Inorg. Chem. 2015, 54, 8310–8318. [Google Scholar] [CrossRef] [PubMed]
- Sanz García, J.; Alary, F.; Boggio-Pasqua, M.; Dixon, I.M.; Heully, J.-L. Is photoisomerization required for NO photorelease in ruthenium nitrosyl complexes? J. Mol. Model. 2016, 22, 284. [Google Scholar] [CrossRef] [PubMed]
- Woike, Th.; Haussühl, S. Infrared-Spectroscopic and Differential Scanning Calorimetric Studies of the Two Light-Induced Metastable States in K2[Ru(NO2)4(OH)(NO)]. Solid State Commun. 1993, 86, 333–337. [Google Scholar] [CrossRef]
- Fomitchev, D.V.; Coppens, P. X-ray Diffraction Analysis of Geometry Changes upon Excitation: The Ground-State and Metastable-State Structures of K2[Ru(NO2)4(OH)(NO)]. Inorg. Chem. 1996, 35, 7021–7026. [Google Scholar] [CrossRef] [PubMed]
- Fomitchev, D.V.; Coppens, P.; Li, T.; Bagley, K.A.; Chen, L.; Richter-Addo, G.B. Photo-induced metastable linkage isomers of ruthenium nitrosyl porphyrins. Chem. Commun. 1999, 2013–2014. [Google Scholar] [CrossRef]
- Da Silva, S.C.; Franco, D.W. Metastable Excited State and Electronic Structure of [Ru(NH3)5NO]3+ and [Ru(NH3)4(OH)NO]2+. Spectrochim. Acta A 1999, 55, 1515–1525. [Google Scholar] [CrossRef]
- Gorelsky, S.I.; Lever, A.B.P. Metastable States of Ruthenium (II) Nitrosyl Complexes and Comparison with [Fe(CN)5NO]2−. Int. J. Quantum Chem. 2000, 80, 636–645. [Google Scholar] [CrossRef]
- Ferlay, S.; Schmalle, H.W.; Francese, G.; Stoeckli-Evans, H.; Imlau, M.; Schaniel, D.; Woike, T. Light-Induced Metastable States in Oxalatenitrosylruthenium(II) and Terpyridinenitrosylruthenium(II) Complexes. Inorg. Chem. 2004, 43, 3500–3506. [Google Scholar] [CrossRef] [PubMed]
- Schaniel, D.; Woike, T.; Boskovic, C.; Güdel, H.-U. Evidence for two light-induced metastable states in Cl3[Ru(NH3)5NO]H2O. Chem. Phys. Lett. 2004, 390, 347–351. [Google Scholar] [CrossRef]
- Zangl, A.; Klüfers, P.; Schaniel, D.; Woike, T. Photoinduced Linkage Isomerism of {RuNO}6 Complexes with Bioligands and Related Chelators. Dalton Trans. 2009, 6, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Schaniel, D.; Cormary, B.; Malfant, I.; Valade, L.; Woike, T.; Delley, B.; Krämer, K.W.; Güdel, H.-U. Photogeneration of Two Metastable NO Linkage Isomers with High Populations of up to 76% in trans-[RuCl(py)4(NO)][PF6]2·1/2H2O. Phys. Chem. Chem. Phys. 2007, 9, 3717–3724. [Google Scholar] [CrossRef] [PubMed]
- Cormary, B.; Malfant, I.; Buron-Le Cointe, M.; Toupet, L.; Delley, B.; Schaniel, D.; Mockus, N.; Woike, T.; Fejfarová, K.; Petříček, V.; Dušek, M. [Ru(py)4Cl(NO)](PF6)2·0.5H2O: A Model System for Structural Determination and Ab Initio Calculations of Photo-Induced Linkage NO Isomers. Acta Cryst. B 2009, 65, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Cormary, B.; Ladeira, S.; Jacob, K.; Lacroix, P.G.; Woike, T.; Schaniel, D.; Malfant, I. Structural Influence on the Photochromic Response of a Series of Ruthenium Mononitrosyl Complexes. Inorg. Chem. 2012, 51, 7492–7501. [Google Scholar] [CrossRef] [PubMed]
- Khadeeva, L.; Kaszub, W.; Lorenc, M.; Malfant, I.; Buron-Le Cointe, M. Two-Step Photon Absorption Driving the Chemical Reaction in the Model Ruthenium Nitrosyl System [Ru(py)4Cl(NO)](PF6)2·1/2H2O. Inorg. Chem. 2016, 55, 4117–4123. [Google Scholar] [CrossRef] [PubMed]
- Tassé, M.; Mohammed, H.S.; Sabourdy, C.; Mallet-Ladeira, S.; Lacroix, P.G.; Malfant, I. Synthesis, Crystal Structure, Spectroscopic, and Photoreactive Properties of a Ruthenium(II)-Mononitrosyl Complex. Polyhedron 2016, 119, 350–358. [Google Scholar] [CrossRef]
- Ford, P.C.; Bourassa, J.; Miranda, K.; Lee, B.; Lorkovic, I.; Boggs, S.; Kudo, S.; Laverman, L. Photochemistry of Metal Nitrosyl Complexes. Delivery of Nitric Oxide to Biological Targets. Coord. Chem. Rev. 1998, 171, 185–202. [Google Scholar] [CrossRef]
- Tfouni, E.; Krieger, M.; McGarvey, B.R.; Franco, D.W. Structure, Chemical and Photochemical Reactivity and Biological Activity of Some Ruthenium Amine Nitrosyl Complexes. Coord. Chem. Rev. 2003, 236, 57–69. [Google Scholar] [CrossRef]
- Szundi, I.; Rose, M.J.; Sen, I.; Eroy-Reveles, A.A.; Mascharak, P.K.; Einarsdóttir, Ó. A New Approach for Studying Fast Biological Reactions Involving Nitric Oxide: Generation of NO Using Photolabile Ruthenium and Manganese NO Donors. Photochem. Photobiol. 2006, 82, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Bitterwolf, T.E. Photolysis of [Ru(bipy)2(NO)Cl](PF6)2 in Frozen Ionic Glass Matrices. Evidence for Nitrosyl Linkage Isomerism and NO-Loss in a Physiologically Relevant Nitric Oxide Source. Inorg. Chem. Commun. 2008, 11, 772–773. [Google Scholar] [CrossRef]
- Rose, M.J.; Mascharak, P.K. Photoactive Ruthenium Nitrosyls: Effects of Light and Potential Application as NO Donors. Coord. Chem. Rev. 2008, 252, 2093–2114. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.J.; Fry, N.L.; Marlow, R.; Hink, L.; Mascharak, P.K. Sensitization of Ruthenium Nitrosyls to Visible Light via Direct Coordination of the Dye Resorufin: Trackable NO Donors for Light-Triggered NO Delivery to Cellular Targets. J. Am. Chem. Soc. 2008, 130, 8834–8846. [Google Scholar] [CrossRef] [PubMed]
- Giglmeier, H.; Kerscher, T.; Klüfers, P.; Schaniel, D.; Woike, T. Nitric-Oxide Photorelease and Photoinduced Linkage Isomerism on Solid [Ru(NO)(terpy)(L)]BPh4 (L = glycolate dianion). Dalton Trans. 2009, 9113–9116. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, A.D.; Ford, P.C. Metal Complexes as Photochemical Nitric Oxide Precursors: Potential Applications in the Treatment of Tumors. Dalton Trans. 2009, 10660–10669. [Google Scholar] [CrossRef] [PubMed]
- Fry, N.L.; Mascharak, P.K. Photoactive Ruthenium Nitrosyls as NO Donors: How to Sensitize Them toward Visible Light. Acc. Chem. Res. 2011, 44, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Akl, J.; Sasaki, I.; Lacroix, P.G.; Malfant, I.; Mallet-Ladeira, S.; Vicendo, P.; Farfán, N.; Santillan, R. Comparative Photo-Release of Nitric Oxide from Isomers of Substituted Terpyridinenitrosyl-Ruthenium(II) Complexes: Experimental and Computational Investigations. Dalton Trans. 2014, 43, 12721–12733. [Google Scholar] [CrossRef] [PubMed]
- deBoer, T.R.; Mascharak, P.K. Recent Progress in Photoinduced NO Delivery With Designed Ruthenium Nitrosyl Complexes. Adv. Inorg. Chem. 2015, 67, 145–170. [Google Scholar]
- Talotta, F.; Heully, J.-L.; Alary, F.; Dixon, I.M.; González, L.; Boggio-Pasqua, M. Linkage Photoisomerization Mechanism in a Photochromic Ruthenium Nitrosyl Complex: New Insights from a MS-CASPT2 Study. J. Chem. Theory Comput. in press.
- Freitag, L.; González, L. Theoretical Spectroscopy and Photodynamics of a Ruthenium Nitrosyl Complex. Inorg. Chem. 2014, 53, 6415–6426. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Weinan, E.; Ren, W.; Vanden-Eijnden, E. Simplified and improved string method for computing the minimum energy paths in barrier-crossing events. J. Chem. Phys. 2007, 126, 164103. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Sundermann, A. Correlation consistent valence basis sets for use with the Stuttgart–Dresden–Bonn relativistic effective core potentials: The atoms Ga–Kr and In–Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-Dependent Density Functional Theory Within the Tamm−Dancoff Approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J. Comput. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A 'chain-of-spheres' algorithm for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
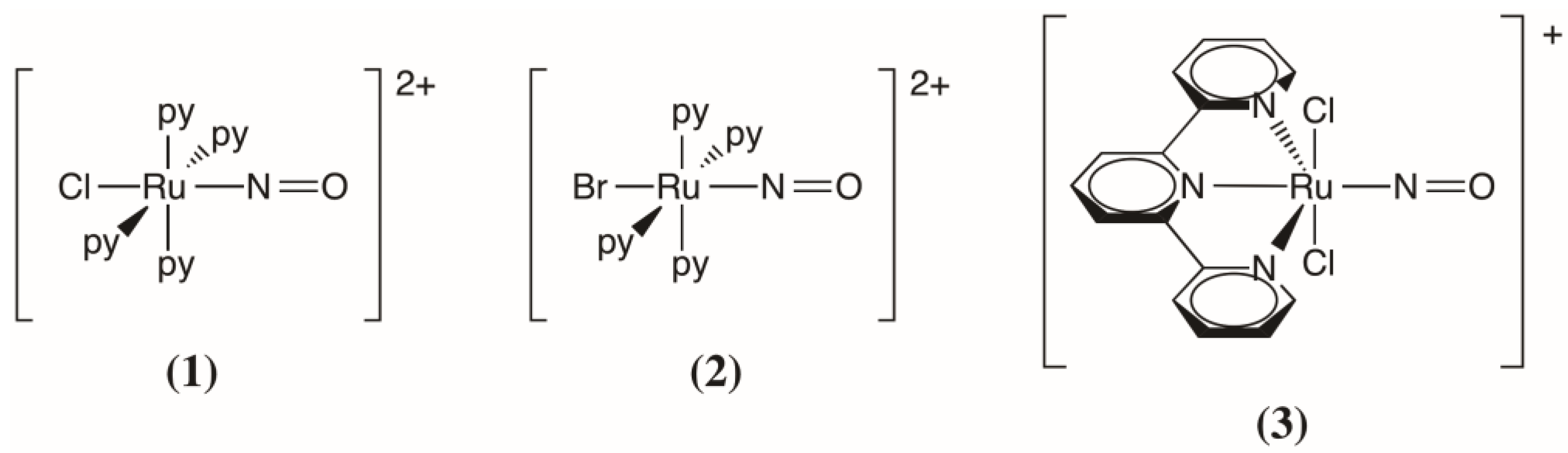




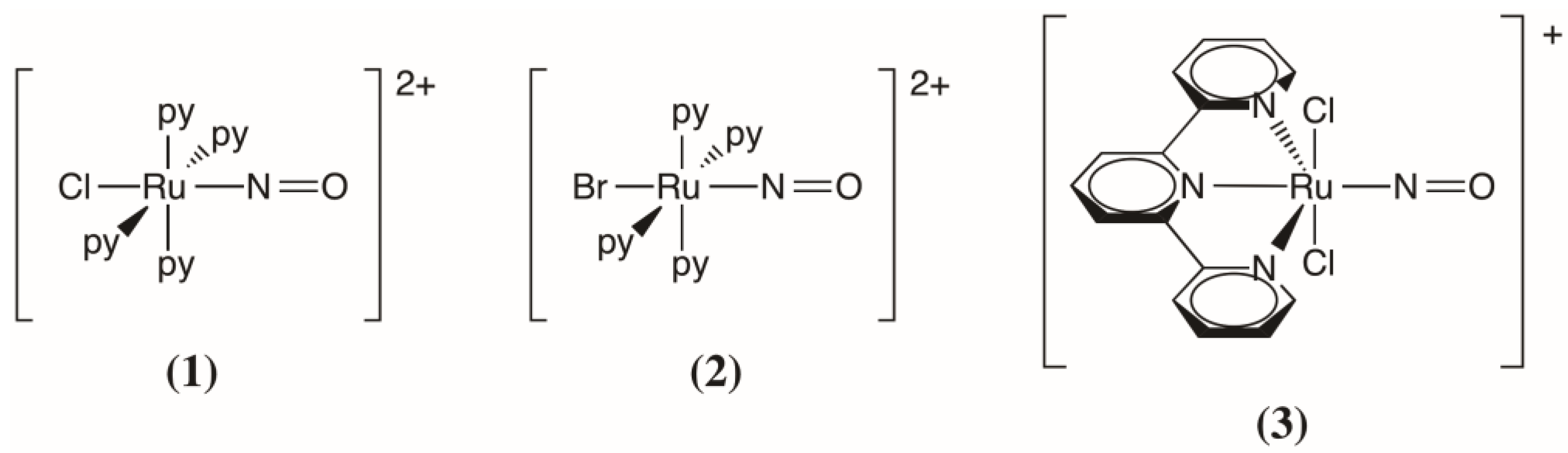{kind=link}
{kind=link}
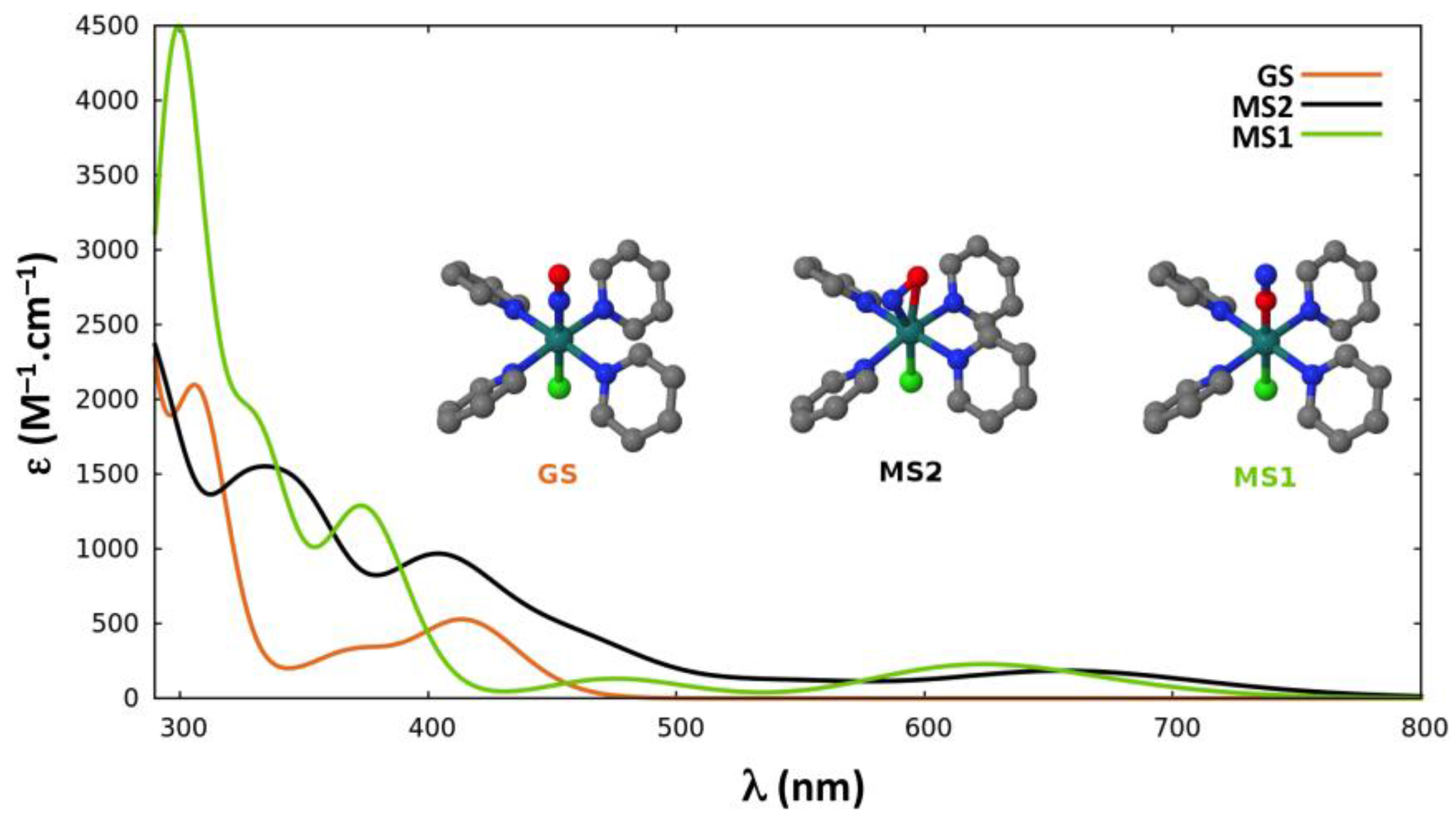{kind=link}
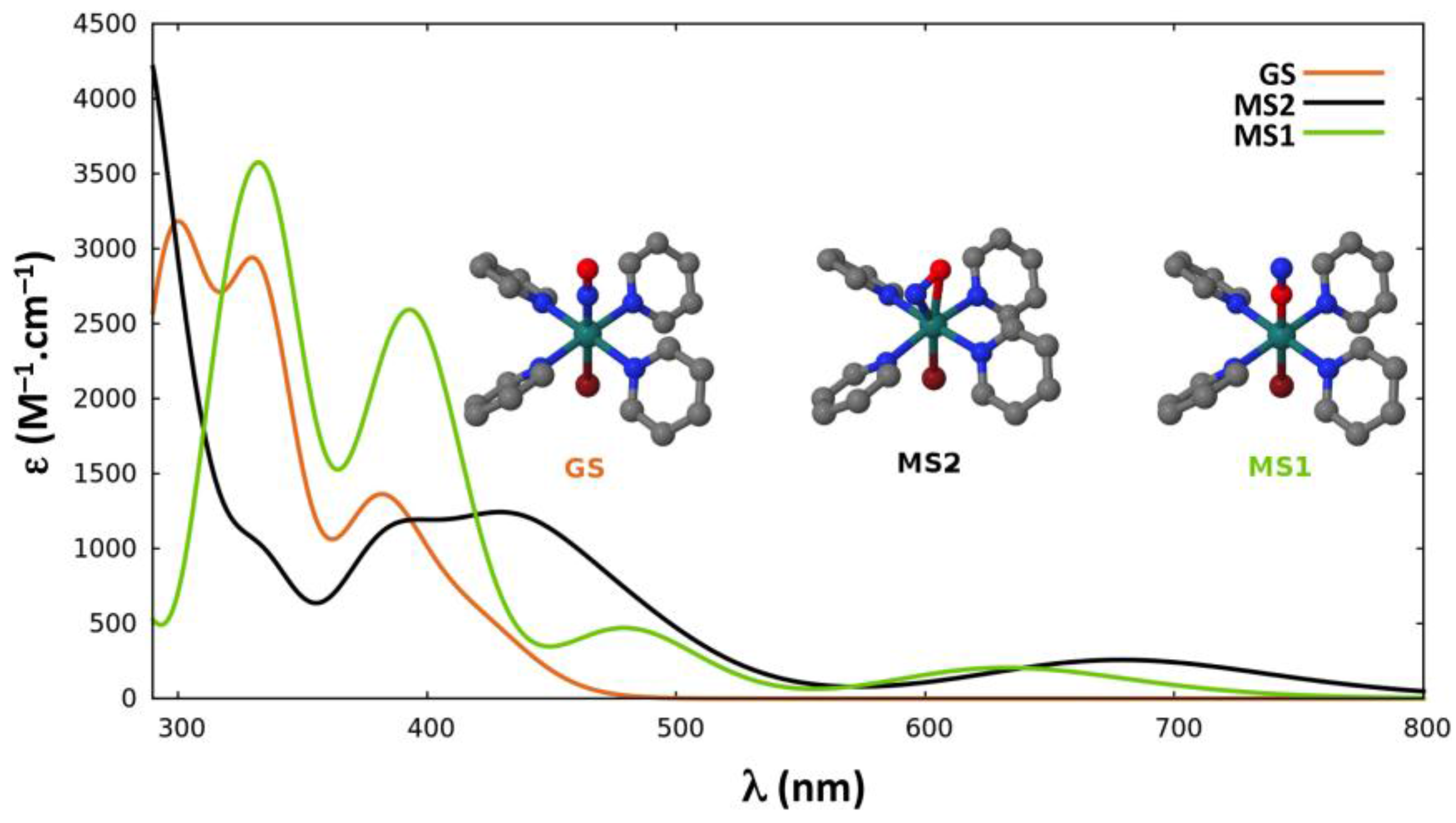{kind=link}
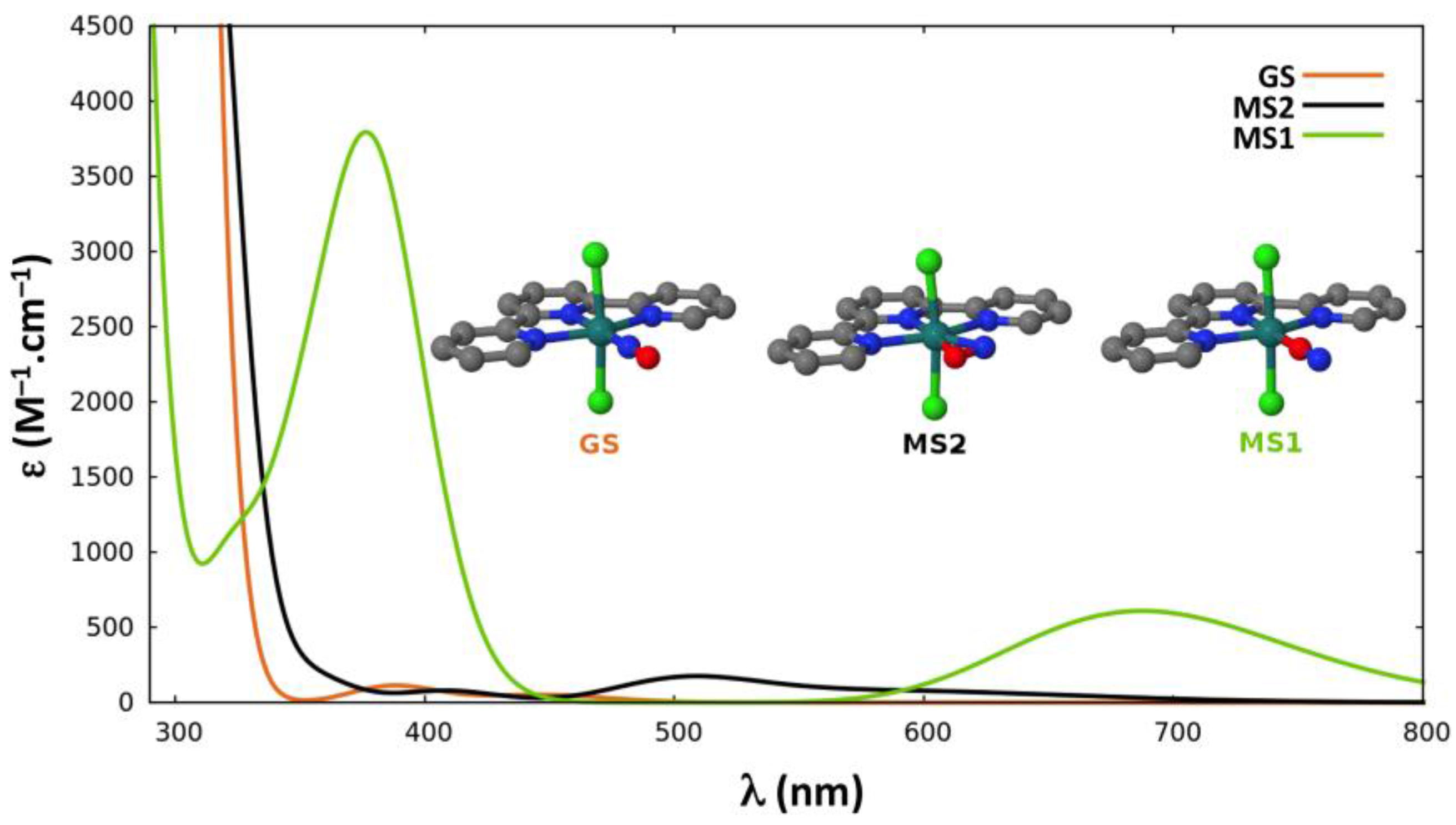{kind=link}
| Relative Energy | trans-[RuCl(NO)(py)4]2+ (1) | trans-[RuBr(NO)(py)4]2+ (2) | trans-(Cl,Cl)[RuCl2(NO)(tpy)]+ (3) |
|---|---|---|---|
| ΔE(MS2–GS) | 1.56 | 1.51 | 1.48 |
| ΔE(MS1–GS) | 1.86 | 1.82 | 1.91 |
| ΔE(TS1–GS) | 2.44 | 2.30 | 2.30 1 |
| ΔE(TS2–MS2) | 1.57 | 1.49 | 1.44 1 |
| Relative Energy | trans-[RuCl(NO)(py)4]2+ (1) | trans-[RuBr(NO)(py)4]2+ (2) | trans-(Cl,Cl)[RuCl2(NO)(tpy)]+ (3) |
|---|---|---|---|
| ΔE(3GS–GS) | 1.41 | 1.32 | 0.98 |
| ΔE(3MS2–GS) | 2.25 | 2.19 | 1.85 |
| ΔE(3MS1–GS) | 2.08 | 1.98 | 1.70 |
| ΔE(3TS1–3GS) | 1.06 | 1.05 | 0.98 2 |
| ΔE(3TS2–3MS2) | 0.25 | 0.20 1 | 0.16 2 |
| Relative Energy | trans-[RuCl(NO)(py)4]2+ (1) | trans-[RuBr(NO)(py)4]2+ (2) | trans-(Cl,Cl)[RuCl2(NO)(tpy)]+ (3) |
|---|---|---|---|
| ΔE(MECP1–3GS) | 0.00 | 0.00 | 0.01 |
| ΔE(MECP2–3GS) | 0.67 | 0.65 | 0.90 |
| ΔE(MECP3–3MS2) | 0.03 | 0.03 | 0.02 |
| ΔE(MECP4–3MS1) | 0.17 | 0.18 | 0.34 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanz García, J.; Talotta, F.; Alary, F.; Dixon, I.M.; Heully, J.-L.; Boggio-Pasqua, M. A Theoretical Study of the N to O Linkage Photoisomerization Efficiency in a Series of Ruthenium Mononitrosyl Complexes. Molecules 2017, 22, 1667. https://doi.org/10.3390/molecules22101667
Sanz García J, Talotta F, Alary F, Dixon IM, Heully J-L, Boggio-Pasqua M. A Theoretical Study of the N to O Linkage Photoisomerization Efficiency in a Series of Ruthenium Mononitrosyl Complexes. Molecules. 2017; 22(10):1667. https://doi.org/10.3390/molecules22101667
Chicago/Turabian StyleSanz García, Juan, Francesco Talotta, Fabienne Alary, Isabelle M. Dixon, Jean-Louis Heully, and Martial Boggio-Pasqua. 2017. "A Theoretical Study of the N to O Linkage Photoisomerization Efficiency in a Series of Ruthenium Mononitrosyl Complexes" Molecules 22, no. 10: 1667. https://doi.org/10.3390/molecules22101667