Selective O-Alkylation of the Crown Conformer of Tetra(4-hydroxyphenyl)calix[4]resorcinarene to the Corresponding Tetraalkyl Ether

Abstract
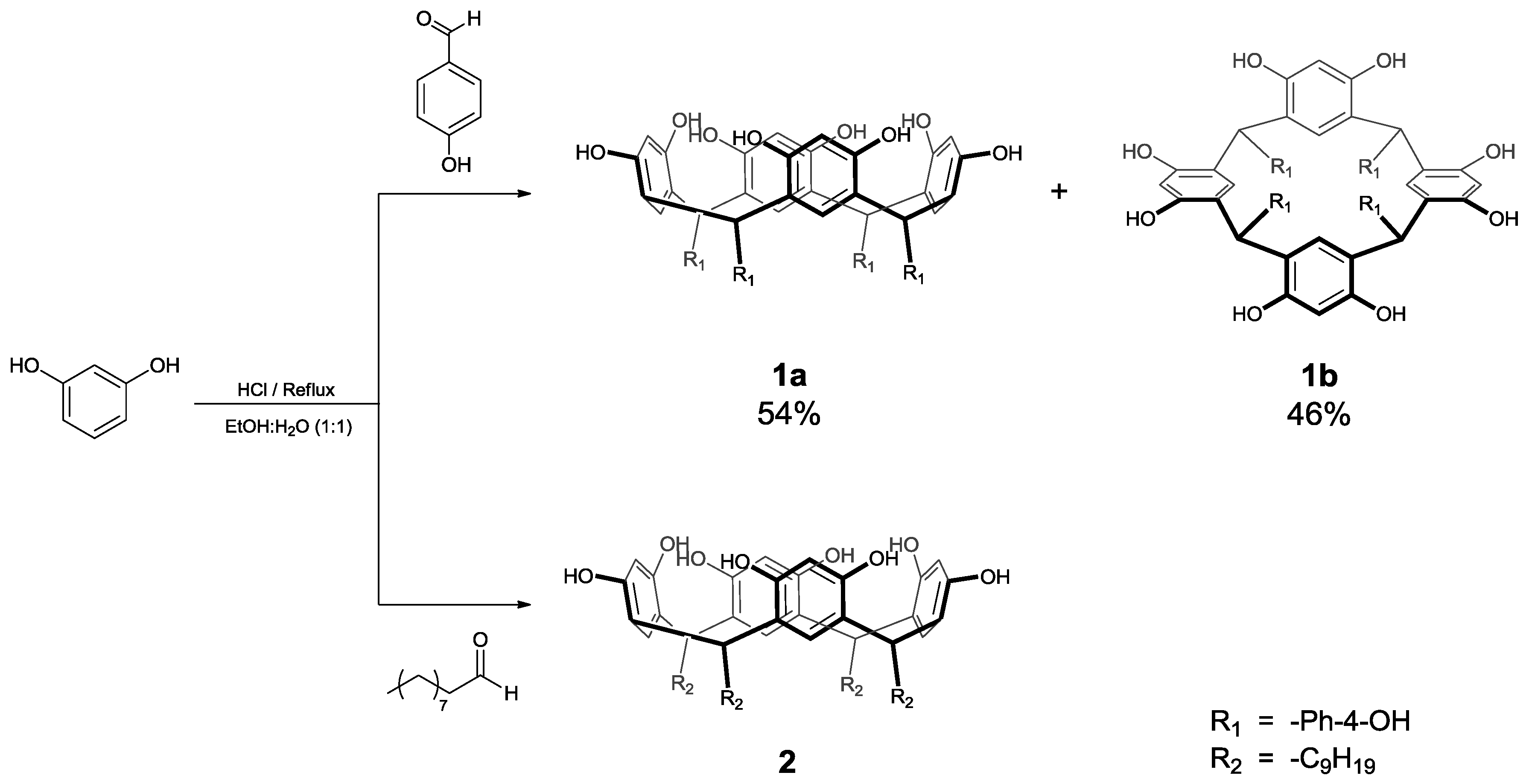
:1. Introduction
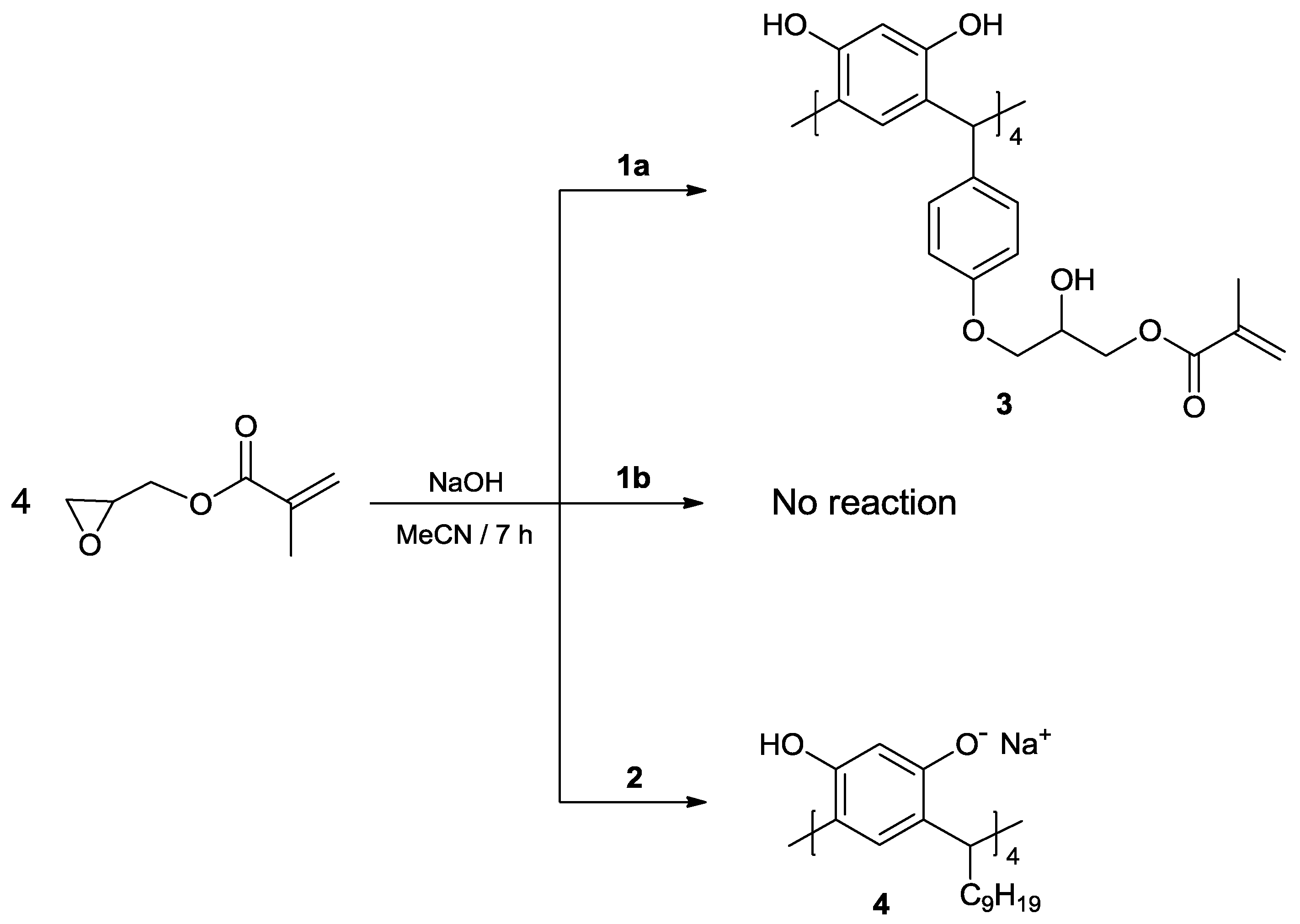
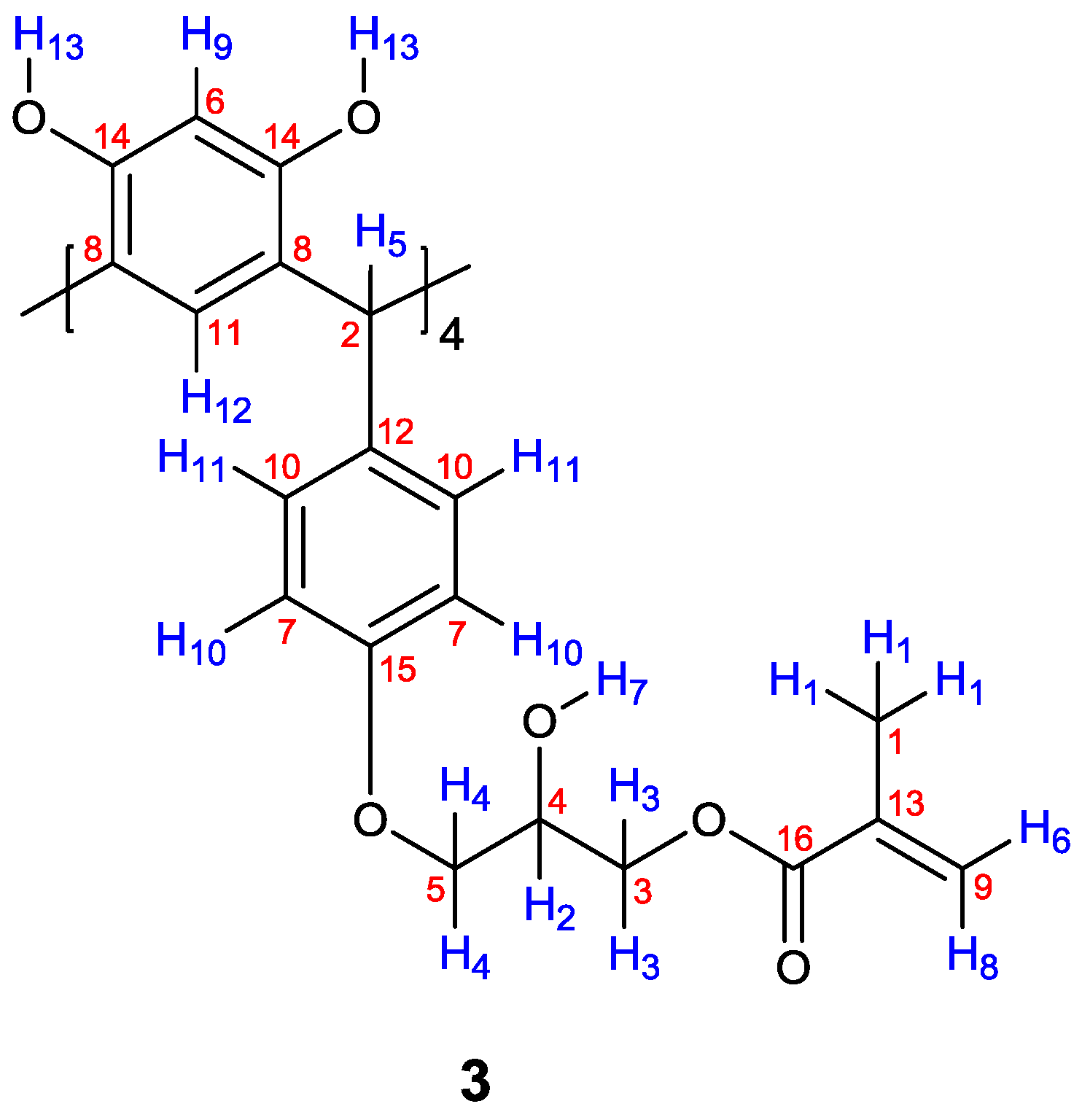
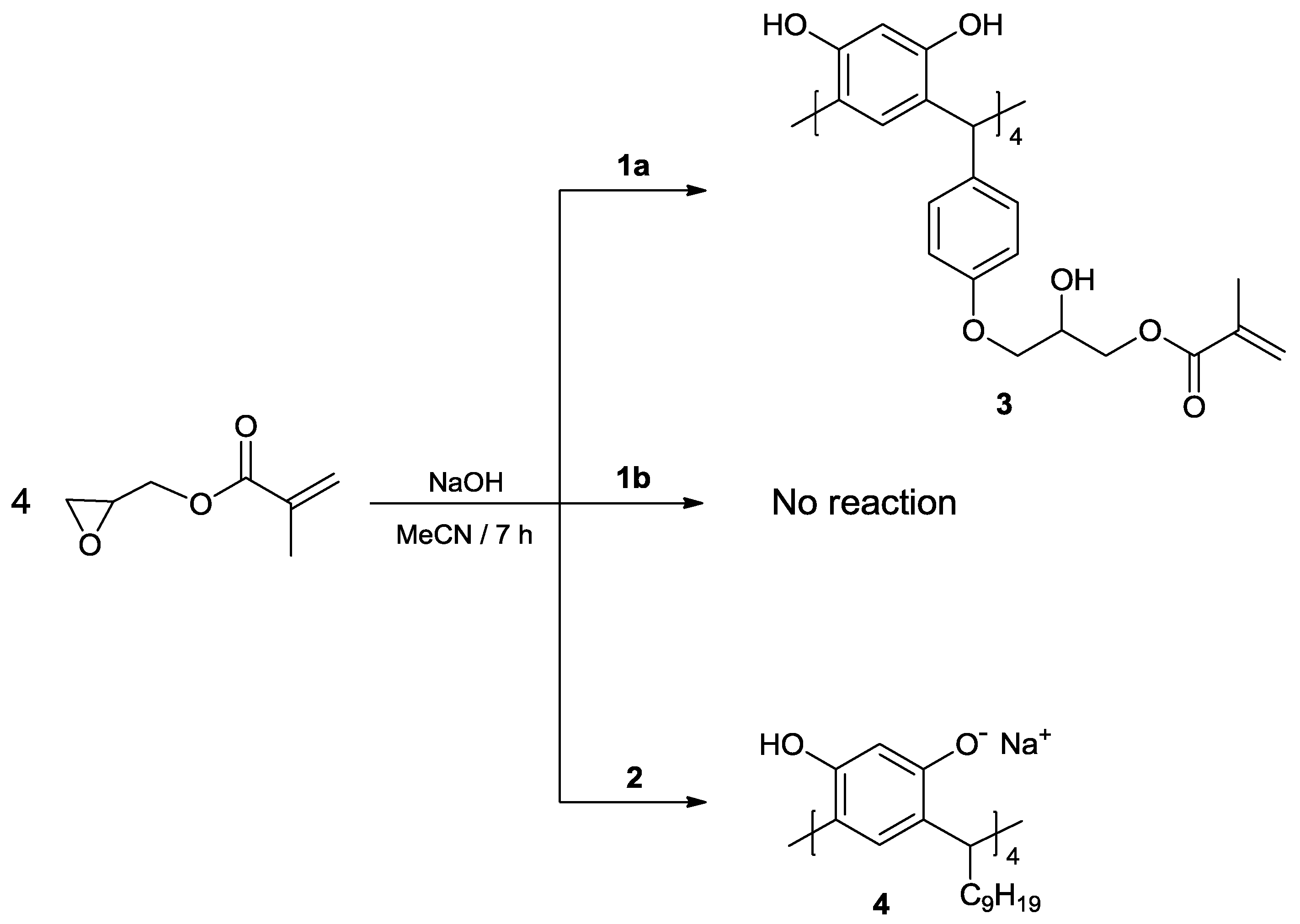
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Information
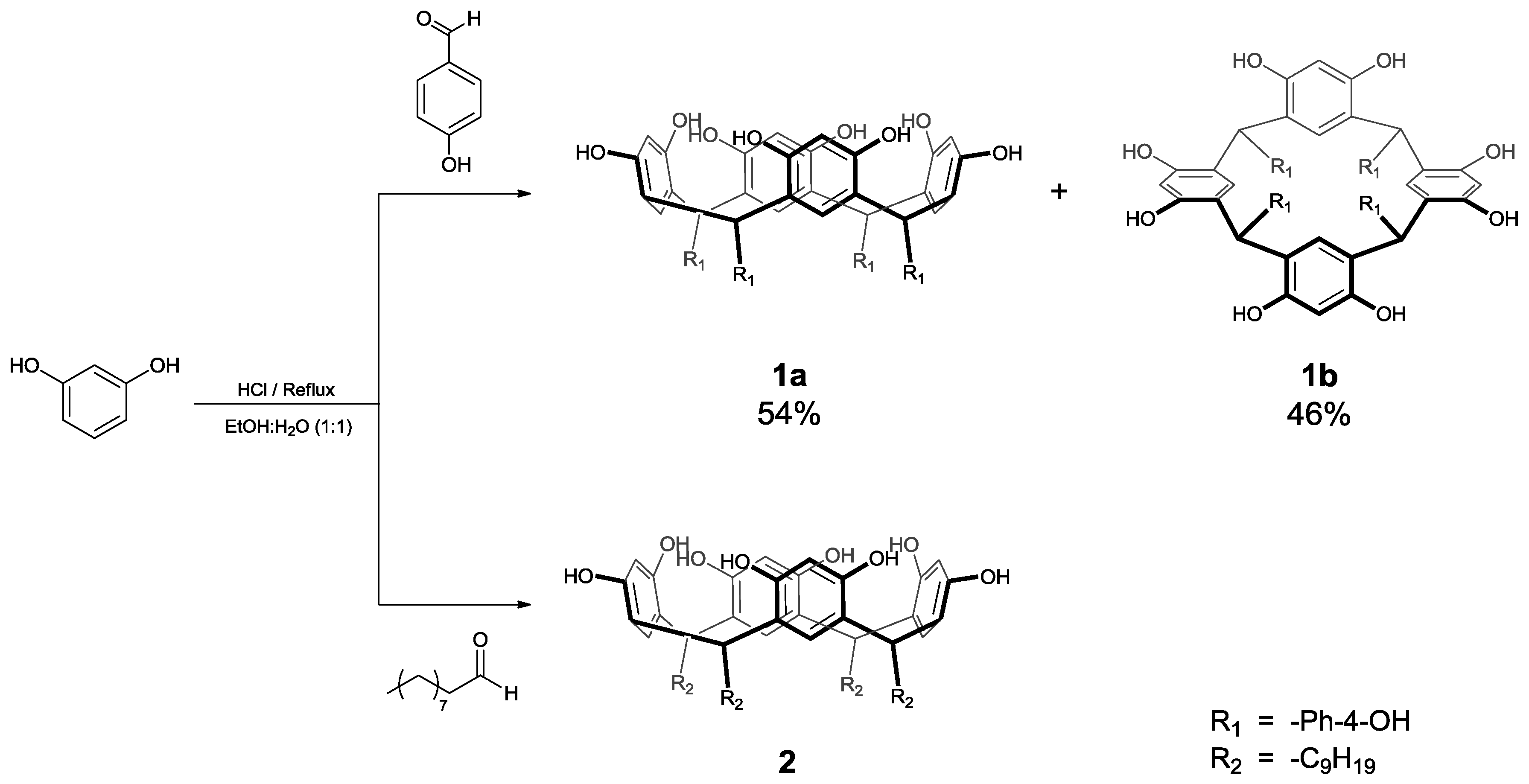
3.2. Synthesis of Calix[4]resorcinarenes
3.3. Reaction of calix[4]resorcinarenes 1a, 1b and 2 with GMA
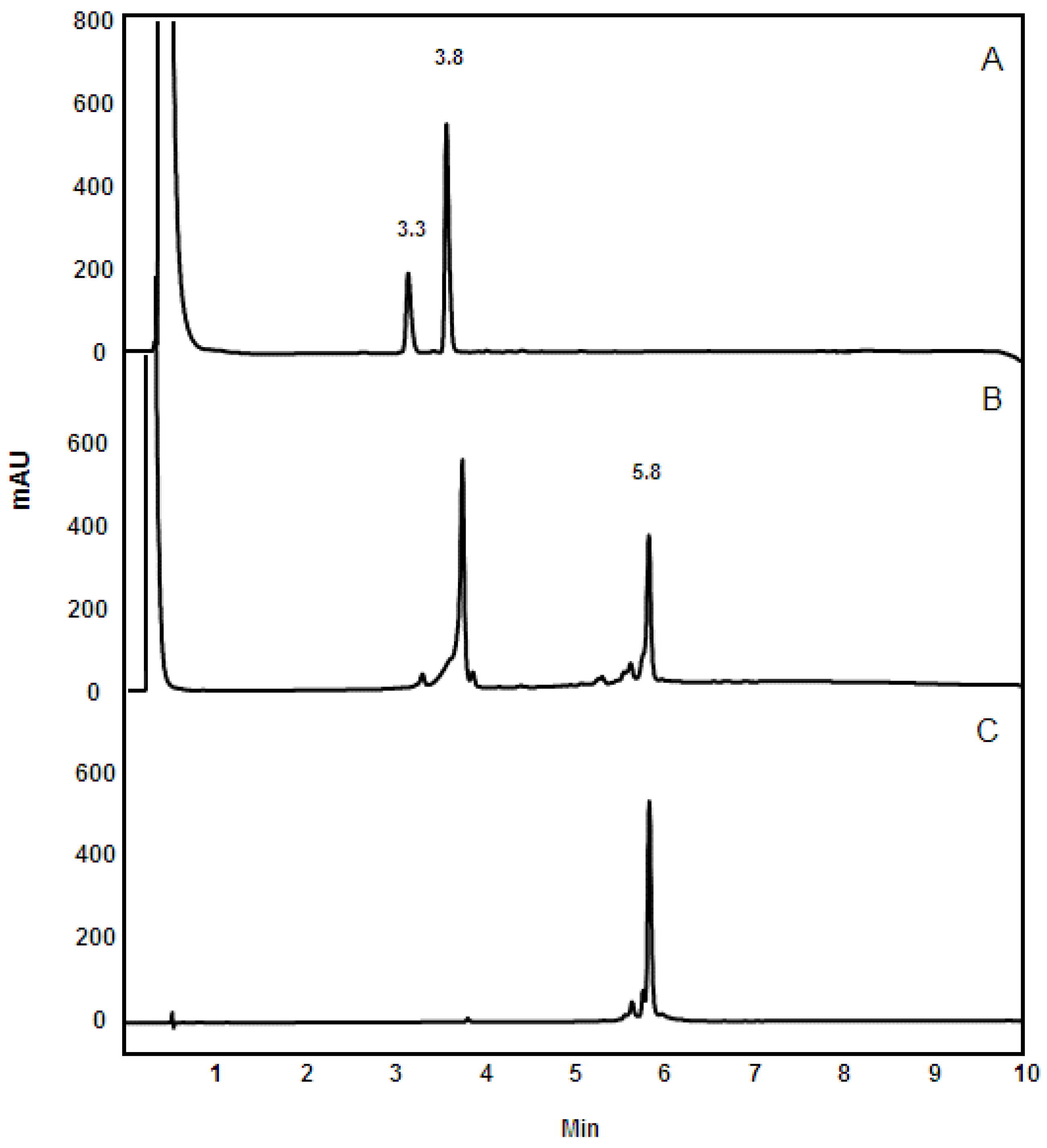
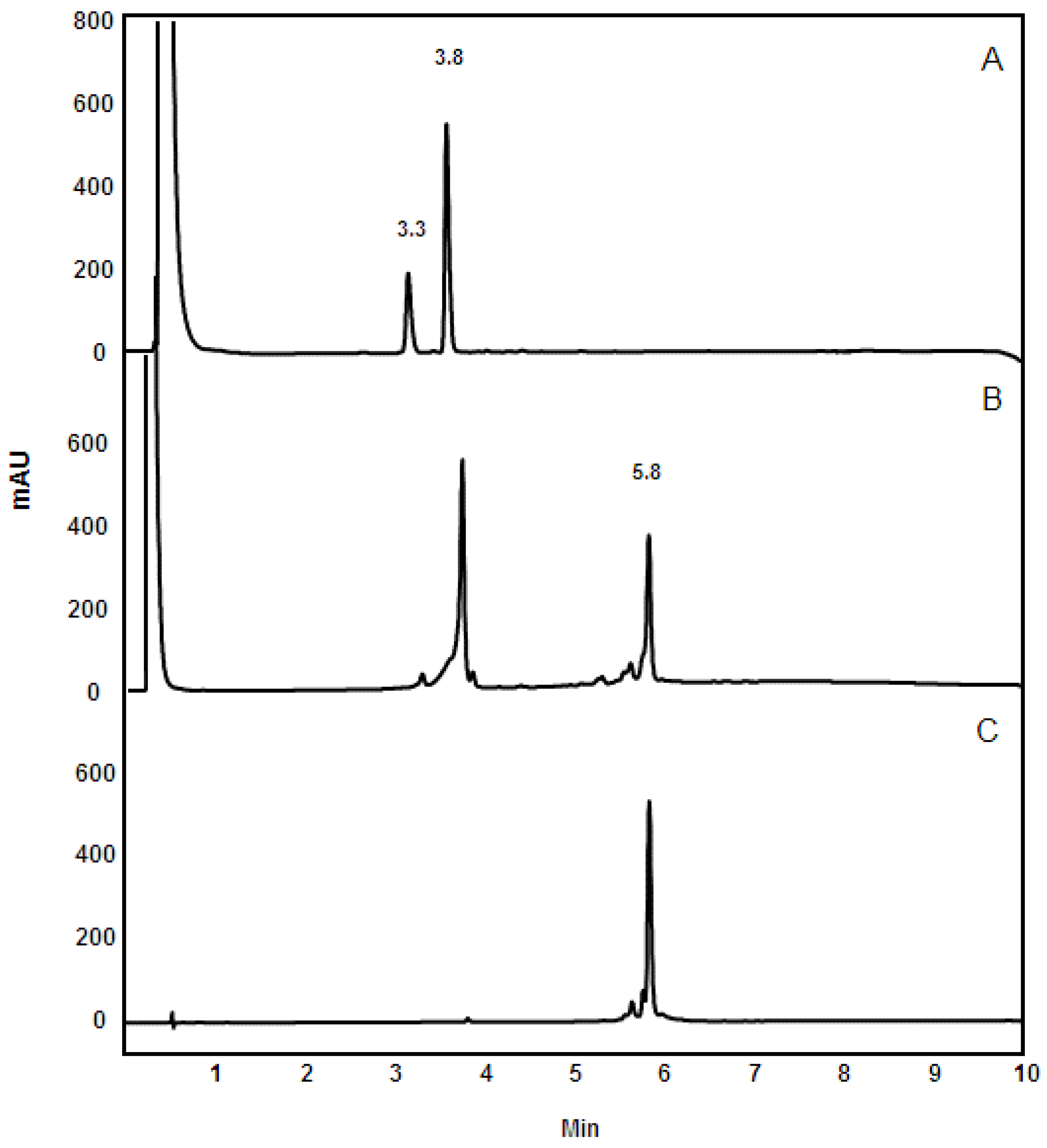
3.4. Reversed-Phase HPLC Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Högberg, A.G. Two stereoisomeric macrocyclic resorcinol-acetaldehyde condensation products. J. Org. Chem. 1980, 45, 4498–4500. [Google Scholar] [CrossRef]
- Tero, T.; Nissinen, M. A perspective to resorcinarene crowns. Tetrahedron 2014, 70, 1111–1123. [Google Scholar] [CrossRef]
- Wang, F.; Wu, Y.; Lu, K.; Ye, B. A simple but highly sensitive and selective calixarene-based voltammetric sensor for serotonin. Electrochim. Acta 2013, 87, 756–762. [Google Scholar] [CrossRef]
- Cortez-Maya, S.; Hernández-Ortega, S.; Ramírez-Apan, T.; Lijanova, I.V.; Martínez-García, M. Synthesis of 5-aryl-1,4-benzodiazepine derivatives attached in resorcinaren-PAMAM dendrimers and their anti-cancer activity. Bioorg. Med. Chem. 2012, 20, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Lijanova, I.V.; Moggio, I.; Arias, E.; Klimova, T.; Martínez-García, M. Resorcinarene-dendrimers with stilbene moieties for optoelectronics. Tetrahedron 2008, 64, 10258–10266. [Google Scholar] [CrossRef]
- Jain, V.K.; Kanaiya, P.H.; Bhojak, N. Synthesis, spectral characterization of azo dyes derived from calix[4]resorcinarene and their application in dyeing of fibers. Fibers Polym. 2008, 9, 720–726. [Google Scholar] [CrossRef]
- Kazakova, E.K.; Morozova, J.E.; Mironova, D.A.; Konovalov, A.I. Sorption of azo dyes from aqueous solutions by tetradodecyloxybenzylcalix[4]resorcinarene derivatives. J. Incl. Phenom. Macrocycl. Chem. 2012, 74, 467–472. [Google Scholar] [CrossRef]
- O’Farrell, C.M.; Chudomel, J.M.; Collins, J.M.; Dignam, C.F.; Wenzel, T.J. Water-soluble calix[4]resorcinarenes with hydroxyproline groups as chiral NMR solvating agents. J. Org. Chem. 2008, 73, 2843–2851. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, T.J. Calixarenes and calix[4]resorcinarenes as chiral NMR solvating agents. J. Incl. Phenom. Macrocycl. Chem. 2014, 78, 1–14. [Google Scholar] [CrossRef]
- Beyeh, N.K.; Weimann, D.P.; Kaufmann, L.; Schalley, C.A.; Rissanen, K. Ion-pair recognition of tetramethylammonium salts by halogenated resorcinarenes. Chem. A Eur. J. 2012, 18, 5552–5557. [Google Scholar] [CrossRef] [PubMed]
- Salorinne, K.; Weimann, D.P.; Schalley, C.A.; Nissinen, M. Resorcinarene podand with amine-functionalized side arms-Synthesis, structure, and binding properties of a neutral anion receptor. Eur. J. Org. Chem. 2009, 2009, 6151–6159. [Google Scholar] [CrossRef]
- Hayashida, O.; Uchiyama, M. Cyclophane-based tetra(resorcinarene) as a host for both histone and hydrophobic molecular guests. Tetrahedron Lett. 2006, 47, 4091–4094. [Google Scholar] [CrossRef]
- Jumina; Sarjono, R.E.; Siswanta, D.; Santosa, S.J.; Ohto, K. Adsorption characteristics of Pb(II) and Cr(III) onto C-Methylcalix[4]resorcinarene. J. Korean Chem. Soc. 2011, 55, 454–462. [Google Scholar]
- Gramage-Doria, R.; Armspach, D.; Matt, D. Metallated cavitands (calixarenes, resorcinarenes, cyclodextrins) with internal coordination sites. Coord. Chem. Rev. 2013, 257, 776–816. [Google Scholar] [CrossRef]
- Ruderisch, A.; Iwanek, W.; Pfeiffer, J.; Fischer, G.; Albert, K.; Schurig, V. Synthesis and characterization of a novel resorcinarene-based stationary phase bearing polar headgroups for use in reversed-phase high-performance liquid chromatography. J. Chromatogr. A 2005, 1095, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Soh, S.; Zhao, J.; Yong, E.; Gong, Y. Preparation and application of methylcalix[4]resorcinarene-bonded silica particles as chiral stationary phase in high-performance liquid chromatography. Chirality 2011, 23, E91–E97. [Google Scholar] [CrossRef] [PubMed]
- Thondorf, I.; Brenn, J.; Böhmer, V. Conformational properties of methylene bridged resorcarenes. Tetrahedron 1998, 54, 12823–12828. [Google Scholar] [CrossRef]
- Morinaga, H.; Tsuneishi, F.; Taniguchi, S.; Kawakami, G. Metal-free synthesis of reactive oligomers by ring-opening oligomerization of glycidyl phenyl ether initiated with tetra-n-butylammonium fluoride in the presence of various protic compounds. Tetrahedron Lett. 2014, 55, 3768–3770. [Google Scholar] [CrossRef]
- Velásquez-Silva, A.; Cortés, B.; Rivera-Monroy, Z.; Pérez-Redondo, A.; Maldonado, M. Crystal structure and dynamic NMR studies of octaacetyl-tetra(propyl)calix[4]resorcinarene. J. Mol. Struct. 2017, 1137, 380–386. [Google Scholar] [CrossRef]
- Hong, M.; Zhang, Y.-M.; Liu, Y. Selective binding affinity between quaternary ammonium cations and water-soluble calix[4]resorcinarene. J. Org. Chem. 2015, 80, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Kashapov, R.; Zakharova, L.; Saifutdinova, M.; Kochergin, Y.; Gavrilova, E.; Sinyashin, O. Construction of a water-soluble form of amino acid C-methylcalix[4]resorcinarene. J. Mol. Liq. 2015, 208, 58–62. [Google Scholar] [CrossRef]
- Safa, K.D.; Nasirtabrizi, M.H. One-pot, novel chemical modification of glycidyl methacrylate copolymers with very bulky organosilicon side chain substituents. Eur. Polym. J. 2005, 41, 2310–2319. [Google Scholar] [CrossRef]
- Lv, Y.; Mei, D.; Pan, X.; Tan, T. Preparation of novel b-cyclodextrin functionalized monolith and its application in chiral separation. J. Chromatogr. B 2010, 878, 2461–2464. [Google Scholar] [CrossRef] [PubMed]
- Ge, F.; Huang, Y.; Luo, Y.; Jiang, L.; Dan, Y. Macromolecular chain structure design, synthesis and analysis of poly(L-lactide) linking ultraviolet absorbing groups. RSC Adv. 2014, 4, 63118–63127. [Google Scholar] [CrossRef]
- Maldonado, M.; Sanabria, E.; Batanero, B.; Esteso, M. Apparent molal volume and viscosity values for a new synthesized diazoted resorcin[4]arene in DMSO at several temperatures. J. Mol. Liq. 2017, 231, 142–148. [Google Scholar] [CrossRef]
- Jain, V.K.; Kanaiya, P.H. Chemistry of calix[4]resorcinarenes. Russ. Chem. Rev. 2011, 80, 75–102. [Google Scholar] [CrossRef]
- Tunstad, L.M.; Tucker, J.A.; Dalcanale, E.; Weiser, J.; Bryant, J.A.; Sherman, J.C.; Helgeson, R.C.; Knobler, C.B.; Cram, D.J. Host-guest complexation. Octol building blocks for cavitands and carcerands. J. Org. Chem. 1989, 54, 1305–1312. [Google Scholar]
- Aoyama, Y.; Tanaka, Y.; Sugahara, S. Molecular Recognition. Molecular recognition of sugars via hydrogen-bonding interaction with a synthetic polyhydroxy macrocycle. J. Am. Chem. Soc. 1989, 111, 5397–5404. [Google Scholar]
- Yamakawa, Y.; Ueda, M.; Nagahata, R.; Takeuchi, K.; Asai, M. Rapid synthesis of dendrimers based on calix[4]resorcinarenes. J. Chem. Soc. Perkin Trans. 1998, 1, 4135–4139. [Google Scholar] [CrossRef]
- Kudo, H.; Mitani, K.; Nishikubo, T.; Mitsuishi, M.; Miyashita, T. The synthesis and photo-induced deprotection reaction of calix[4]resorcinarene derivatives containing t-Butyl ester moieties. Bull. Chem. Soc. Jpn. 2004, 77, 819–826. [Google Scholar] [CrossRef]
- Weinelt, F.; Schneider, H.-J. Mechanisms of macrocycle genesis. The condensation of resorcinol with aldehydes. J. Org. Chem. 1991, 56, 5527–5535. [Google Scholar]
- Patil, R.S.; Zhang, C.; Atwood, J.L. Process development for separation of conformers from derivatives of resorcin[4]arenes and pyrogallol[4]arenes. Chem. A Eur. J. 2016, 22, 15202–15207. [Google Scholar] [CrossRef] [PubMed]
- Darvish, F.; Khazraee, S. Molecular iodine: An efficient and environment-friendly catalyst for the synthesis of calix[4]resorcinarenes. C. R. Chim. 2014, 17, 890–893. [Google Scholar] [CrossRef]
- Vlakh, E.G.; Tennikova, T.B. Preparation of methacrylate monoliths. J. Sep. Sci. 2007, 30, 2801–2813. [Google Scholar] [CrossRef] [PubMed]
- Shivanyuk, A.; Paulus, E.F.; Böhmer, V.; Vogt, W. Quasi-complete solvation of organic molecules in the crystalline state. Angew. Chem. Int. Ed. 1997, 36, 1301–1303. [Google Scholar] [CrossRef]
- Pfeiffer, C.R.; Feaster, K.A.; Dalgarno, S.J.; Atwood, J.L. Syntheses and characterization of aryl-substituted pyrogallol[4]arenes and resorcin[4]arenes. CrystEngComm 2016, 18, 222–229. [Google Scholar] [CrossRef]
- Kundrat, O.; Dvorakova, H.; Böhm, S.; Eigner, V.; Lhotak, P. S-alkylation of thiacalixarenes: How the regio- and stereoselectivities depend on the starting conformation. J. Org. Chem. 2012, 77, 2272–2278. [Google Scholar] [CrossRef] [PubMed]
- Lukášek, J.; Böhm, S.; Dvořáková, H.; Eigner, V.; Lhoták, P. Regioselective halogenation of thiacalix[4]arenes in the cone and 1,3-alternate conformations. Org. Lett. 2014, 16, 5100–5103. [Google Scholar] [CrossRef] [PubMed]
- Slavík, P.; Eigner, V.; Lhoták, P. A general method for obtaining calix[4]arene derivatives in the 1,2-alternate conformation. Tetrahedron 2016, 72, 6348–6355. [Google Scholar] [CrossRef]
- Cicco, L.; Sblendorio, S.; Mansueto, R.; Perna, F.M.; Salomone, A.; Florio, S.; Capriati, V. Water opens the door to organolithiums and Grignard reagents: Exploring and comparing the reactivity of highly polar organometallic compounds in unconventional reaction media towards the synthesis of tetrahydrofurans. Chem. Sci. 2016, 7, 1192–1199. [Google Scholar] [CrossRef]
- Dilauro, G.; Dell’Aera, M.; Vitale, P.; Capriati, V.; Perna, F.M. Unprecedented nucleophilic additions of highly polar organometallic compounds to imines and nitriles using water as a non-innocent reaction medium. Angew. Chem. 2017, 56, 10200–10203. [Google Scholar] [CrossRef] [PubMed]
- Sanabria, E.; Esteso, M.; Pérez-Redondo, A.; Vargas, E.; Maldonado, M. Synthesis and characterization of two sulfonated resorcinarenes: A new example of a linear array of sodium centers and macrocycles. Molecules 2015, 20, 9915–9928. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.; Salamanca, Y.; Maldonado, M.; Vargas, E. Solubility of calix[4]resorcinarene in water from (278.15 to 308.15) K. J. Chem. Eng. Data 2010, 55, 1042–1044. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
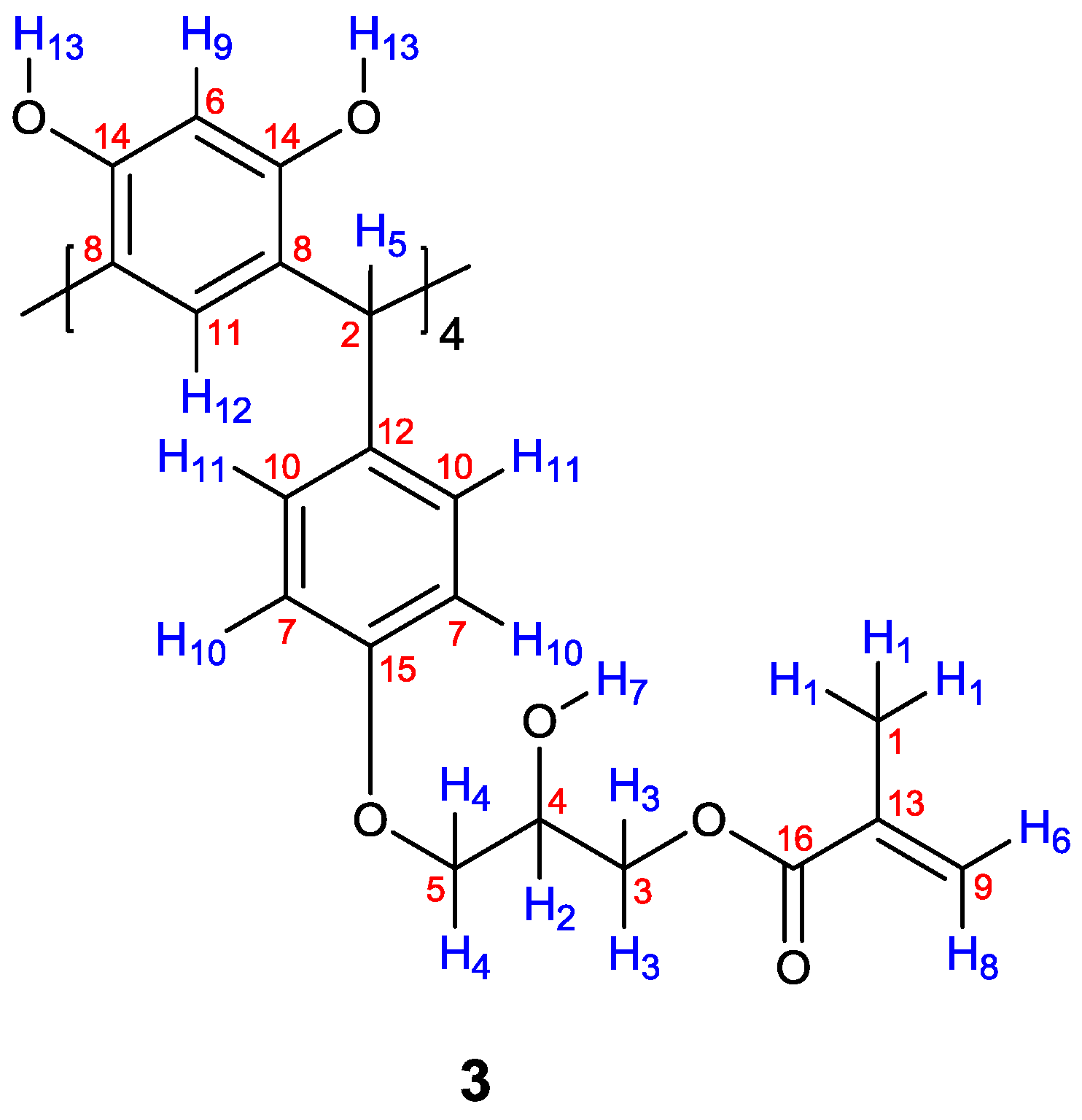
| Proton | δ (ppm) | Correlation |
|---|---|---|
| 1 | 1.91 | 6 (5.69) |
| 1 | 1.91 | 8 (6.11) |
| 2 | 3.88 | 3 (4.09) |
| 2 | 3.88 | 4 (4.23) |
| 6 | 5.69 | 8 (6.11) |
| 10 | 6.31 | 11 (6.42) |
| Carbon | δ (ppm) | Correlation HMQC | Correlation HMBC |
|---|---|---|---|
| 1 | 18.0 | 1 (1.91) | 6 (5.69), 8 (6.11) |
| 2 | 41.2 | 5 (5.42) | 11 (6.42), 12 (6.43) |
| 3 | 66.3 | 3 (4.09) | 2 (3.88) |
| 4 | 68.8 | 2 (3.88) | 4 (4.23) |
| 5 | 71.4 | 4 (4.23) | 3 (4.09) |
| 6 | 101.6 | 9 (6.27) | 13 (8.40) |
| 7 | 113.8 | 10 (6.31) | 11 (6.42) |
| 8 | 120.9 | - | 5 (5.42), 9 (6.27), 13 (8.40) |
| 9 | 125.9 | 6 (5.69), 8 (6.11) | 1 (1.91) |
| 10 | 129.7 | 11 (6.42) | 5 (5.42), 10 (6.31) |
| 11 | 129.8 | 12 (6.43) | 5 (5.42) |
| 12 | 134.4 | - | 5 (5.42), 10 (6.31), 11 (6.42) |
| 13 | 135.8 | - | 1 (1.91) |
| 14 | 152.3 | - | 5 (5.42), 9 (6.27), 13 (8.40) |
| 15 | 154.2 | - | 10 (6.31), 11 (6.42) |
| 16 | 167.4 | - | 1 (1.91), 3 (4.09), 6 (5.69), 8 (6.11) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillo-Aguirre, A.; Rivera-Monroy, Z.; Maldonado, M. Selective O-Alkylation of the Crown Conformer of Tetra(4-hydroxyphenyl)calix[4]resorcinarene to the Corresponding Tetraalkyl Ether. Molecules 2017, 22, 1660. https://doi.org/10.3390/molecules22101660
Castillo-Aguirre A, Rivera-Monroy Z, Maldonado M. Selective O-Alkylation of the Crown Conformer of Tetra(4-hydroxyphenyl)calix[4]resorcinarene to the Corresponding Tetraalkyl Ether. Molecules. 2017; 22(10):1660. https://doi.org/10.3390/molecules22101660
Chicago/Turabian StyleCastillo-Aguirre, Alver, Zuly Rivera-Monroy, and Mauricio Maldonado. 2017. "Selective O-Alkylation of the Crown Conformer of Tetra(4-hydroxyphenyl)calix[4]resorcinarene to the Corresponding Tetraalkyl Ether" Molecules 22, no. 10: 1660. https://doi.org/10.3390/molecules22101660