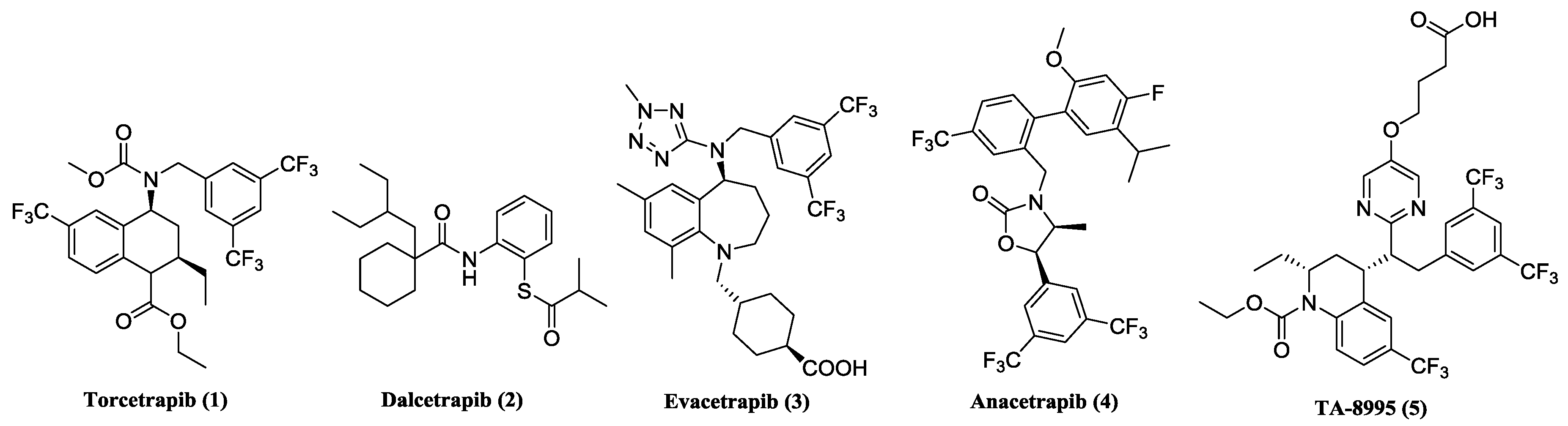
3.1. General Information
All chemicals were obtained from commercial sources and were used without purification unless otherwise specified. Solvents were distilled and dried using standard methods. TLC was performed on silica gel plates with F-254 indicator and visualized by UV-light. The purities of target compounds (all ≥95%). were detected by HPLC, performed on a Waters 1525–2489 system (Waters, Milford, MA, USA). The method conditions were as follows: 100% CH3OH or a mixture of solvents H2O (A) and MeOH (B) (VA:VB = 5:95) as eluent, flow rate at 1.0 mL/min. Peaks were detected at λ = 254 nm. NMR spectra were recorded on 400 MHz and 600 MHz instruments (Bruker, Karlsruhe, Germany) and the chemical shifts were reported in terms of parts per million with TMS as the internal reference. High-resolution accurate mass determinations (HRMS) for all final target compounds were obtained on a Bruker Micromass Time of Flight mass spectrometer equipped with electrospray ionisation (ESI). Column chromatography was performed with silica gel (200–300 mesh) purchased from Qingdao Haiyang Chemical Co. Ltd. (Qingdao, China)
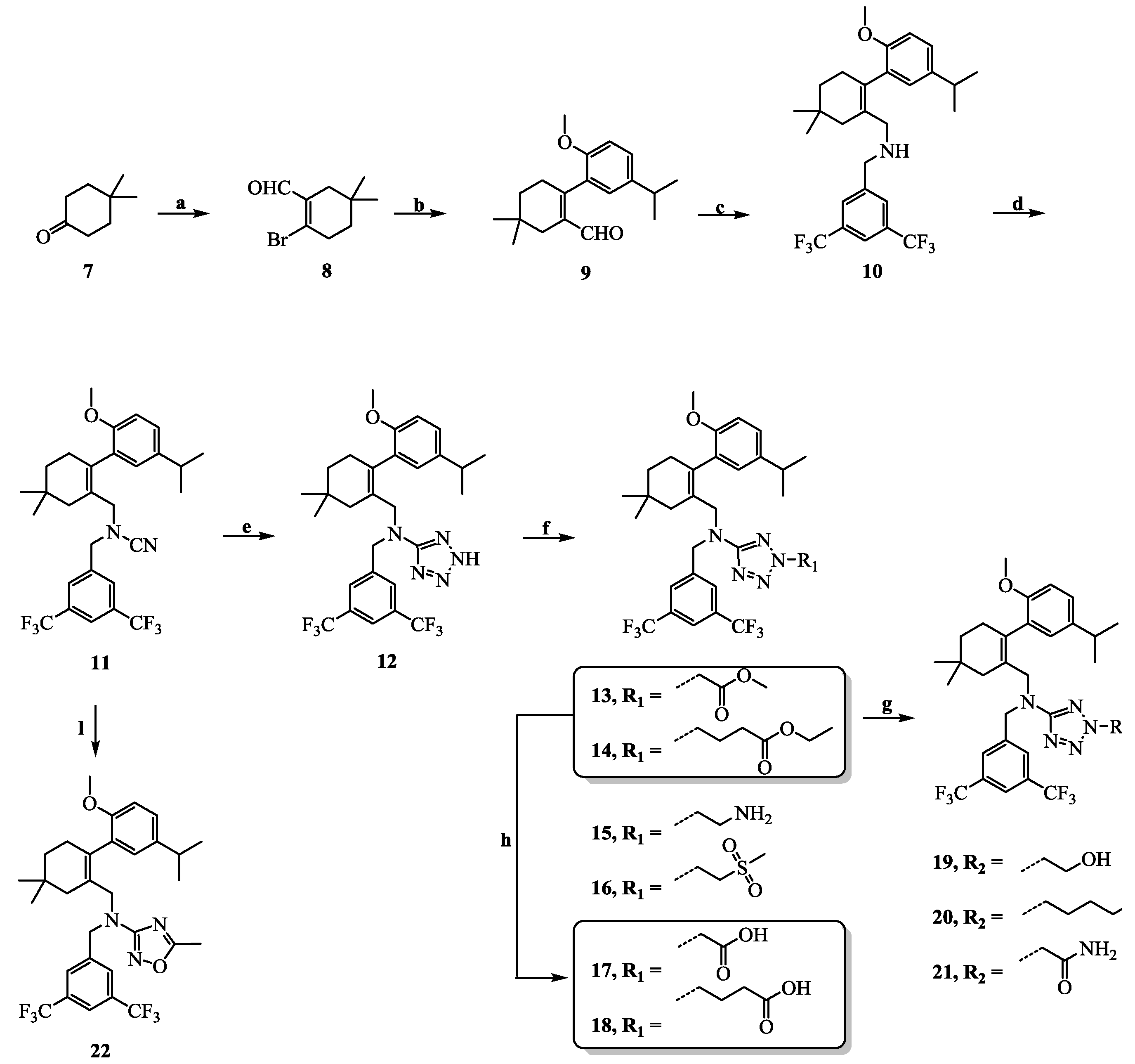
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)cyanamide (11): Intermediate 10 (0.3 g, 0.6 mmol) was dissolved in THF (5 mL) and cyanogen bromide (0.2 g, 1.8 mmol) and N,N-diisopropylethylamine (0.4 mL, 2.4 mmol) were added. The reaction mixture was stirred at room temperature for 5 h and then poured into H2O (10 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 15:1) to give 11 (0.3 g, 92.8%) as a pale yellow oil. 1H-NMR (400 MHz, DMSO-d6) δ 8.06 (s, 1H), 7.90 (s, 2H), 7.05 (dd, J = 8.5, 2.2 Hz, 1H), 6.91–6.77 (m, 2H), 4.29 (s, 2H), 3.64 (s, 3H), 3.49–3.35 (m, 2H), 2.72 (dt, J = 13.7, 6.9 Hz, 1H), 2.40–2.28 (m, 1H), 2.11–1.99 (m, 1H), 1.90 (m, 2H), 1.40 (t, J = 6.4 Hz, 2H), 1.08 (dd, J = 6.9, 1.6 Hz, 6H), 0.98 (d, J = 2.8 Hz, 6H). HPLC: tR = 13.710 min, 99.27%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)-2H-tetrazol-5-amine (12): Intermediate 11 (0.1 g, 0.2 mmol) was dissolved in DMF (5 mL) and ammonium chloride (0.01 g, 0.8 mmol) and sodium azide (0.05 g, 0.2 mmol) were added. After being stirred at 100 °C for 5 h, the reaction mixture was cooled to room temperature, and H2O (20 mL) was added. The mixture was extracted with CH2Cl2 (20 mL × 3) and the combined organic layers were washed with water (20 mL × 3) and brine (20 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 2:1) to give 12 (0.1 g, 96.3%) as a white solid. Mp 150.6–152.6 °C. 1H-NMR (600 MHz, DMSO-d6) δ 14.88 (s, 1H), 7.94 (s, 1H), 7.66 (s, 2H), 6.99 (dd, J = 8.5, 2.2 Hz, 1H), 6.77 (d, J = 8.5 Hz, 1H), 6.71 (d, J = 2.2 Hz, 1H), 4.55 (s, 2H), 3.91 (m, 2H), 3.61 (s, 3H), 2.61 (dt, J = 13.8, 6.9 Hz, 1H), 2.36–2.27 (m, 1H), 2.06–2.03 (m, 1H), 1.77–1.69 (m, 2H), 1.38 (s, 2H), 1.36 (t, J = 6.4 Hz, 2H), 1.00 (d, J = 6.9 Hz, 6H), 0.91 (d, J = 10.3 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 151.95, 142.58, 139.79, 136.20, 132.01(×2), 129.76, 128.36, 127.92(×2), 127.27, 126.44, 123.95, 122.14, 121.79, 111.11(×2), 55.84(×2), 53.23, 39.75, 34.93, 33.16, 30.00, 28.75, 28.44, 27.24, 24.07, 23.99. HRMS calcd for C29H34F6N5O, [M + H]+, 582.2589; found 582.2668. HPLC: tR = 14.640 min, 96.47%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)-2H-tetrazol-5-amine-2-carboxylic acid methyl ester (13): Compound 12 (0.1 g, 0.2 mmol) and triethylamine (0.1 mL, 0.8 mmol) were dissolved in acetonitrile (2 mL) followed by the addition of methyl 2-bromoacetate (0.03 mL, 0.4 mmol). After being stirred at 80 °C for 2 h, the reaction mixture was cooled to room temperature, and H2O (10 mL) was added. The aqueous layer was extracted with EtOAc (5 mL × 3) and the combined organic layers were washed with H2O (5 mL × 3) and brine (5 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 4:1) to give 13 (0.09 g, 68.4%) as a colourless oil. 1H-NMR (400 MHz, CDCl3) δ 7.67 (s, 1H), 7.54 (s, 2H), 7.03 (dd, J = 8.4, 2.3 Hz, 1H), 6.76 (d, J = 2.3 Hz, 1H), 6.72 (d, J = 8.5 Hz, 1H), 5.18 (s, 2H), 4.58–4.38 (m, 2H), 4.19 (d, J = 14.5 Hz, 1H), 4.00 (d, J = 14.4 Hz, 1H), 3.76 (s, 3H), 3.68 (s, 3H), 2.76 (dt, J = 13.8, 6.9 Hz, 1H), 2.54–2.37 (m, 1H), 2.16–2.00 (m, 1H), 1.83 (s, 2H), 1.50–1.33 (m, 2H), 1.15 (d, J = 6.9 Hz, 6H), 0.94 (d, J = 11.9 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 169.64, 165.70, 154.12, 141.11, 140.87, 135.34, 131.28(×2), 130.52, 128.06(×2), 127.76(×2), 127.63, 125.66(×2), 110.64(×2), 55.24, 53.01, 52.91, 51.69, 49.38, 40.57, 35.42, 33.03, 29.10, 28.99, 28.03(×2), 23.99(×2). HRMS calcd for C32H38F6N5O3, [M + H]+, 654.2801; found 654.2877. HPLC: tR = 8.753 min, 95.8%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)-2H-tetrazol-5-amine-2-butyric acid ethyl ester (14): Colourless oil; yield 71.3%; 1H-NMR (400 MHz, CDCl3) δ 7.66 (s, 1H), 7.54 (s, 2H), 7.03 (d, J = 7.6 Hz, 1H), 6.82–6.65 (m, 2H), 4.46 (d, J = 3.5 Hz, 4H), 4.18–4.11(m, 3H), 3.98 (d, J = 14.5 Hz, 1H), 3.67 (s, 3H), 2.76 (s, 1H), 2.46 (d, J = 19.3 Hz, 1H), 2.30 (d, J = 4.5 Hz, 2H), 2.23 (s, 2H), 2.12–2.03 (m, 1H), 1.82 (s, 2H), 1.42 (s, 2H), 1.25–1.24 (m, 3H), 1.14 (s, 6H), 0.94 (d, J = 11.1 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 172.11, 169.37, 154.14, 141.34, 140.85, 135.18, 131.24(×2), 130.57, 128.08, 127.77(×2), 125.63, 124.16, 122.35, 120.60, 110.62(×2), 60.54, 55.22, 51.77, 51.65, 49.36, 40.61, 35.43, 33.03, 30.60, 29.10, 29.00, 28.04, 28.02, 24.12, 23.99, 23.98, 14.04. HRMS calcd for C35H44F6N5O3, [M + H]+, 696.3270; found 656.3361. HPLC: tR = 7.653 min, 96.4%.
2-(2-Aminoethyl)-N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethyl-cyclohex-1-enyl)methyl)-2H-tetrazol-5-amine (15): Compound 12 (0.5 g, 0.9 mmol) and triethylamine (1.8 mL, 13.0 mmol) were dissolved in acetonitrile (10 mL), followed by the addition of tert-butyl 2-bromoethylcarbamate (0.6 mL, 2.6 mmol). After being stirred at 80 °C for 2 h, the reaction mixture was cooled to room temperature, and H2O (10 mL) was added. The aqueous layer was extracted with EtOAc (5 mL × 3) and the combined organic layers were washed with H2O (5 mL × 3) and brine (5 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was immediately dissolved in a trifluoroacetic acid–dichloromethane (1:1) solution (2 mL) and stirred at room temperature overnight. After concentration, the residue was dissolved in EtOAc (5 mL), washed with H2O (5 mL × 3) and brine (5 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 2:1) to give 15 (0.35 g, 62.2%) as a colourless oil. 1H-NMR (400 MHz, DMSO-d6) δ 7.93 (s, 1H), 7.66 (s, 2H), 7.00 (dd, J = 8.4, 2.2 Hz, 1H), 6.79–6.76 (m, 2H), 4.49 (s, 2H), 4.35 (t, J = 6.1 Hz, 2H), 4.02–3.89 (m, 2H), 3.62 (s, 3H), 2.94 (t, J = 6.2 Hz, 2H), 2.67 (dt, J = 13.8, 6.9 Hz, 1H), 2.54–2.37 (m, 1H), 2.16–2.00 (m, 1H), 1.76 (s, 2H), 1.35 (t, J = 6.3 Hz, 2H), 1.04 (d, J = 6.9 Hz, 6H), 0.89 (d, J = 10.2 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 169.41, 154.13, 141.31, 140.86, 135.29, 131.25(×2), 130.55, 128.07, 127.80, 127.70, 125.65, 124.14, 122.34, 120.63, 110.62(×2), 55.81, 55.22, 51.72, 49.39, 40.93, 40.64, 35.42, 33.03, 29.11, 29.01, 28.06, 28.03, 24.00, 23.98. HRMS calcd for C31H39F6N6O, [M + H]+, 625.3011; found 625.3070. HPLC: tR = 18.895 min, 96.6%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)-2-(2-(methylsulfonyl)ethyl)-2H-tetrazol-5-amine (16): 2-(methylsulfonyl)ethanol (0.04 mL, 0.4 mmol) was added to a solution of compound 12 (0.1 g, 0.2 mmol) in THF (2 mL) and cooled to 0 °C, followed by the addition of Ph3P (0.07 g, 0.3 mmol) and DIAD (0.05 mL, 0.3 mmol). After being stirred at room temperature for 4 h, the reaction mixture was poured into H2O (10 mL) and extracted with EtOAc (5 mL × 3), and the combined organic layers were washed with H2O (5 mL × 3) and brine (5 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 1:1) to give 16 (0.09 g, 65.3%) as a colourless oil. 1H-NMR (400 MHz, CDCl3) δ 7.68 (s, 1H), 7.50 (s, 2H), 7.03 (dd, J = 8.4, 2.2 Hz, 1H), 6.76 (d, J = 2.3 Hz, 1H), 6.71 (d, J = 8.5 Hz, 1H), 4.90 (t, J = 6.9 Hz, 2H), 4.48 (s, 2H), 4.17 (d, J = 15.4 Hz, 1H), 3.99 (d, J = 14.4 Hz, 1H), 3.70–3.60 (m, 5H), 2.82–2.71 (m, 4H), 2.46 (d, J = 18.1 Hz, 1H), 2.10 (d, J = 18.4 Hz, 1H), 1.79 (s, 2H), 1.43 (dd, J = 9.7, 6.4 Hz, 2H), 1.15 (d, J = 6.9 Hz, 6H), 0.94 (d, J = 11.3 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 169.58, 154.08,140.96, 135.68, 131.36(×2), 130.40, 128.01, 127.69, 127.33, 125.74(×2), 124.10, 122.29, 120.77, 110.66(×2), 55.25, 52.90, 51.66, 49.20, 46.20, 41.37, 40.66, 35.36, 33.03, 29.10, 29.02, 28.08, 28.03, 24.00, 23.98. HRMS calcd for C32H40F6N5O3S, [M + H]+, 688.2678; found 688.2754. HPLC: tR = 13.667 min, 95.6%.
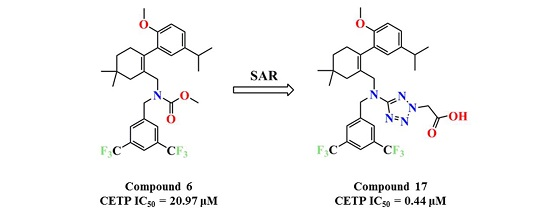
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)-2H-tetrazol-5-amine-2-acetic acid (17): Compound 13 (0.2 g, 0.3 mmol) was dissolved in MeOH (5 mL) and 1 mol/L NaOH (5 mL) was added and the mixture was stirred at room temperature for 2 h. After concentration, the residue was dissolved in H2O (10 mL). Then, 1 mol/L HCl (5 mL) was added to the mixture, the mixture was extracted with EtOAc (10 mL × 3), and the combined organic layers were washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (DCM:MeOH = 5:1) to give 17 (0.17 g, 86.4%) as a colourless oil. 1H-NMR (600 MHz, DMSO-d6) δ 7.92 (s, 1H), 7.65 (s, 2H), 7.00 (dd, J = 8.5, 2.2 Hz, 1H), 6.77 (dd, J = 7.9, 5.4 Hz, 2H), 5.35 (s, 2H), 4.57–4.45 (m, 2H), 3.97 (t, J = 10.4 Hz, 2H), 3.63 (s, 3H), 2.67 (dt, J = 13.7, 6.9 Hz, 1H), 2.33 (d, J = 17.9 Hz, 1H), 2.02 (d, J = 18.0 Hz, 1H), 1.76 (s, 2H), 1.36 (t, J = 6.4 Hz, 2H), 1.04 (d, J = 6.9 Hz, 6H), 0.90 (d, J = 14.3 Hz, 6H). 13C-NMR (150 MHz, DMSO-d6) δ: 169.21, 167.87, 154.37, 142.28, 140.54, 134.58, 130.63(×2), 130.29, 127.81, 127.74, 127.62, 125.85, 124.50, 122.69, 120.98, 111.32(×2), 55.70, 53.85, 51.65, 49.55, 40.57, 35.36, 32.75, 29.24, 29.07, 28.46, 28.09, 24.27, 24.21. HRMS calcd for C31H36F6N5O3, [M + H]+, 640.2644; found 640.2709. HPLC: tR = 19.013 min, 97.3%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)-2H-tetrazol-5-amine-2-butyric acid (18): Colourless oil; yield 81.3%; 1H-NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1H), 7.90 (s, 1H), 7.65 (s, 2H), 6.99 (d, J = 8.5 Hz, 1H), 6.78 (d, J = 8.5 Hz, 2H), 4.50 (s, 2H), 4.44 (t, J = 6.8 Hz, 2H), 4.00–3.90 (m, 2H), 3.62 (s, 3H), 2.66 (dt, J = 13.7, 6.8 Hz, 1H), 2.32 (d, J = 17.7 Hz, 1H), 2.19–2.15(m, 2H), 2.07–1.98 (m, 3H), 1.75 (s, 2H), 1.35 (t, J = 5.9 Hz, 2H), 1.03 (d, J = 6.9 Hz, 6H), 0.88 (d, J = 7.5 Hz, 6H). 13C-NMR (150 MHz, DMSO-d6) δ: 173.76, 169.19, 154.39, 142.35, 140.54, 134.46, 130.61(×2), 127.88, 127.74, 127.72(×2), 125.84, 124.51, 122.70, 120.94, 111.33(×2), 55.69, 51.88, 51.70, 49.66, 40.63, 35.36, 32.76, 30.40, 29.25, 29.05, 28.40, 28.13, 24.44, 24.25, 24.21. HRMS calcd for C33H40F6N5O3, [M + H]+, 668.2957; found 668.3050. HPLC: tR = 18.767 min, 97.1%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)-2H-tetrazol-5-amine-2-ethyl alcohol (19): Compound 13 (0.4 g, 0.6 mmol) was dissolved in THF (5 mL) and NaBH4 (0.05 g, 1.2 mmol) and 2 drops of MeOH were added and the mixture was stirred at 70 °C for 2 h and then cooled to room temperature. After concentration, the residue was dissolved in EtOAc (10 mL), washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 4:1) to give 19 (0.3 g, 79.8%) as a colourless oil. 1H-NMR (600 MHz, DMSO-d6) δ 7.92 (s, 1H), 7.67 (s, 2H), 7.00 (dd, J = 8.5, 2.2 Hz, 1H), 6.78 (d, J = 8.5 Hz, 1H), 6.77 (d, J = 2.3 Hz, 1H), 4.95 (t, J = 5.6 Hz, 1H), 4.49 (s, 2H), 4.42 (t, J = 5.4 Hz, 2H), 3.97 (q, J = 14.4 Hz, 2H), 3.78 (dd, J = 10.8, 5.5 Hz, 2H), 3.62 (s, 3H), 2.67 (dt, J = 13.8, 6.9 Hz, 1H), 2.33 (d, J = 18.0 Hz, 1H), 2.02 (d, J = 18.0 Hz, 1H), 1.76 (s, 2H), 1.35 (t, J = 6.4 Hz, 2H), 1.04 (d, J = 6.9 Hz, 6H), 0.89 (d, J = 16.3 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 169.30, 154.11, 141.18, 140.89, 135.43, 131.32(×2), 130.51, 128.04, 127.72, 127.55, 125.67, 124.13, 122.32, 120.71, 110.63(×2), 60.30, 55.23, 54.95, 51.80, 49.41, 40.68, 35.40, 33.03, 29.12, 29.02, 28.04(×2), 23.99, 23.98. HRMS calcd for C31H38F6N5O2, [M + H]+, 626.2851; found 626.2920. HPLC: tR = 16.776 min, 95.8%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)-2H-tetrazol-5-amine-2-butanol (20): Colourless oil; yield 73.8%; 1H-NMR (400 MHz, DMSO-d6) δ 7.92 (s, 1H), 7.67 (s, 2H), 7.02 (dd, J = 8.5, 2.1 Hz, 1H), 6.83–6.76 (m, 2H), 4.52 (s, 2H), 4.48–4.39 (m, 3H), 4.03–3.92 (m, 2H), 3.64 (s, 3H), 3.37 (d, J = 5.4 Hz, 2H), 2.68 (dt, J = 13.7, 6.8 Hz, 1H), 2.40–2.29 (m, 1H), 2.06–2.00 (m, 1H), 1.89–1.80 (m, 2H), 1.77 (s, 2H), 1.40–1.28 (m, 4H), 1.05 (d, J = 6.9 Hz, 6H), 0.90 (d, J = 7.5 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 169.27, 154.14, 141.37, 140.85, 135.19, 131.22(×2), 130.58, 128.09, 127.77(×2), 125.63, 124.16, 122.36, 120.59, 110.62(×2), 61.78, 55.22, 52.52, 51.64, 49.35, 40.60, 35.43, 33.03, 29.12, 29.10, 29.00, 28.04(×2), 25.53, 23.99, 23.98. HRMS calcd for C33H42F6N5O2, [M + H]+, 654.3164; found 654.3257. HPLC: tR = 15.755 min, 96.4%.
2-(5-((3,5-Bis(trifluoromethyl)benzyl)((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)amino)-2H-tetrazol-2-yl)acetamide (21): Compound 13 (0.2 g, 0.3 mmol) was dissolved in a saturated NH3–EtOH solution (5 mL). After being stirred at room temperature for 12 h, the reaction mixture was concentrated in vacuo. The residue was dissolved in EtOAc (10 mL), washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 4:1) to give 21 (0.12 g, 62.6%) as a colourless oil. 1H-NMR (400 MHz, DMSO-d6) δ 7.91 (s, 1H), 7.70 (s, 1H), 7.66 (s, 2H), 7.42 (s, 1H), 6.99 (dd, J = 8.5, 2.2 Hz, 1H), 6.78–6.75(m, 2H), 5.12 (s, 2H), 4.50 (s, 2H), 3.97 (d, J = 4.6 Hz, 2H), 3.62 (s, 3H), 2.66 (dt, J = 13.7, 6.8 Hz, 1H), 2.33 (d, J = 18.0 Hz, 1H), 2.01 (d, J = 17.7 Hz, 1H), 1.76 (s, 2H), 1.35 (t, J = 6.2 Hz, 2H), 1.04 (d, J = 6.9 Hz, 6H), 0.89 (d, J = 10.9 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 169.77, 154.08, 140.96, 140.88, 131.43(×2), 130.42, 127.99(×2), 127.73, 127.29, 125.77(×2), 124.07, 122.26, 120.84, 110.68(×2), 55.24, 54.83, 51.87, 49.43, 40.79, 35.35, 33.04, 29.13, 29.03, 28.10, 28.00, 24.01, 23.98. HRMS calcd for C31H37F6N6O2, [M + H]+, 639.2804; found 639.2909. HPLC: tR = 12.757 min, 98.2%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)-5-methyl-1,2,4-oxadiazol-3-amine (22): Intermediate 11 (0.3 g, 0.6 mmol) was dissolved in EtOH (5 mL) and triethylamine (0.08 mL, 0.6 mmol) and hydroxylamine hydrochloride (0.04 g, 0.6 mmol) were added. After being stirred at 80 °C for 2 h, the reaction mixture was cooled to room temperature and concentrated in vacuo. Then, the residue was added pyridine (5 mL) and acetic anhydride (0.07 mL, 0.7 mmol). After being stirred at 80 °C for 12 h, the reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was dissolved in EtOAc (10 mL), washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 4:1) to give 22 (0.15 g, 41.9%) as a colourless oil. 1H-NMR (600 MHz, CDCl3) δ 7.66 (s, 1H), 7.50 (s, 2H), 7.00 (dd, J = 8.4, 2.1 Hz, 1H), 6.72 (d, J = 2.0 Hz, 1H), 6.67 (d, J = 8.4 Hz, 1H), 4.43 (q, J = 16.2 Hz, 2H), 4.06 (d, J = 14.5 Hz, 1H), 3.90 (d, J = 14.5 Hz, 1H), 3.65 (s, 3H), 2.73 (dt, J = 13.8, 6.9 Hz, 1H), 2.47–2.43 (m, 4H), 2.08 (d, J = 18.2 Hz, 1H), 1.81 (s, 2H), 1.42 (dq, J = 13.0, 6.5 Hz, 2H), 1.13 (d, J = 6.9 Hz, 6H), 0.95 (d, J = 13.1 Hz, 6H). 13C- NMR (150 MHz, CDCl3) δ: 175.39, 170.36, 154.07, 140.92, 140.85, 135.59, 131.31(×2), 130.37, 128.00, 127.54, 127.35, 125.66, 124.13, 122.32, 120.70, 110.62(×2), 55.22, 51.08, 48.61, 40.40, 35.41, 33.01, 29.04, 29.01, 28.10, 27.97, 23.96(×2), 12.63. HRMS calcd for C31H36F6N3O2, [M + H]+, 596.2633; found 596.2716. HPLC: tR = 9.953 min, 95.8%.
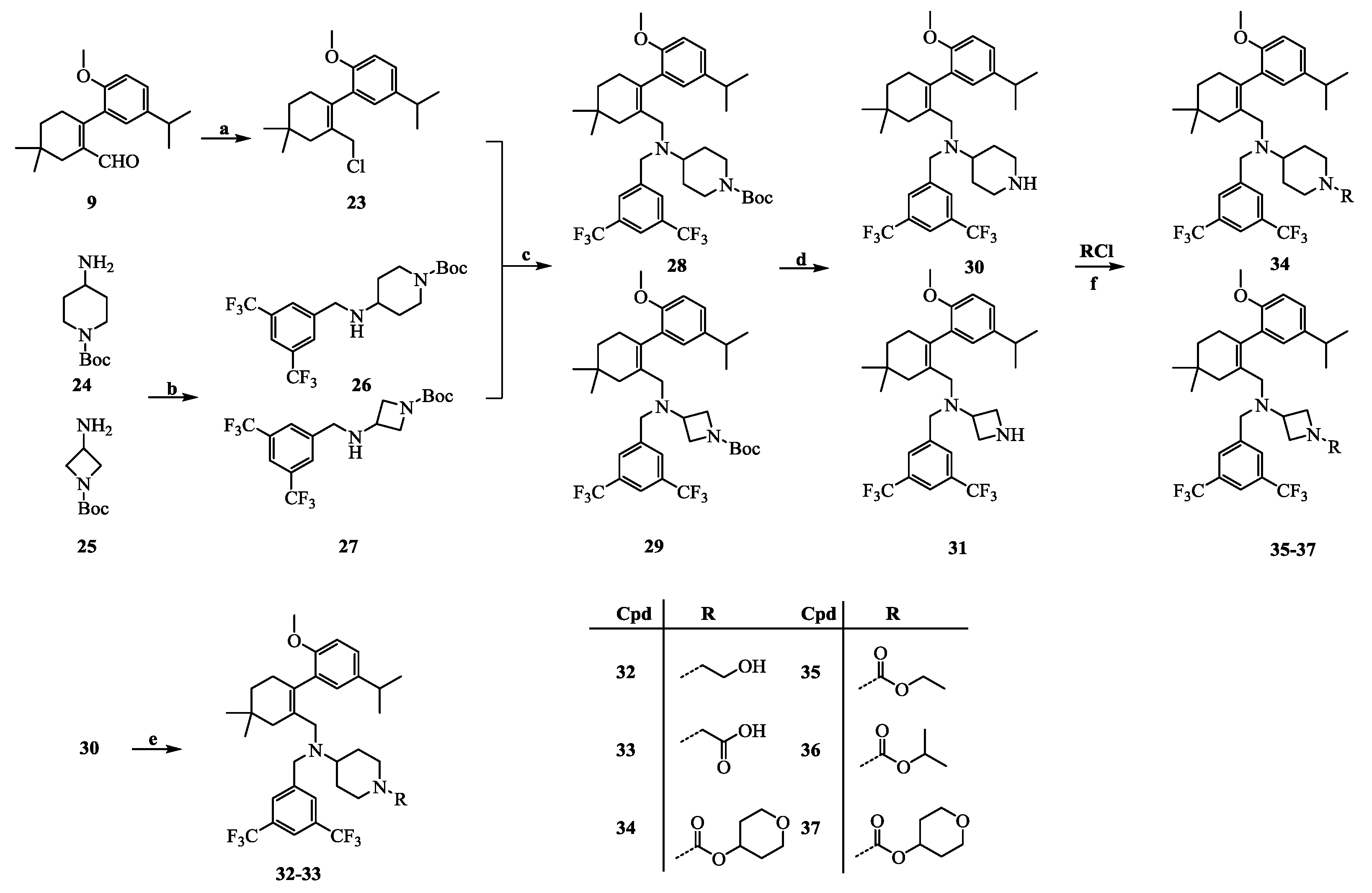
2-(2-(Chloromethyl)-4,4-dimethylcyclohex-1-enyl)-4-isopropyl-1-methoxybenzene (23): Intermediate 9 (114.5 mg, 0.4 mmol) was dissolved in ethanol (5 mL). Sodium borohydride (19.0 mg, 0.5 mmol) was added to the mixture. After being stirred at room temperature for 30 min, the reaction mixture was poured into H2O (20 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. SOCl2 (0.1 mL, 1.4 mmol) was added to a solution of the above residue in DMF (2 mL) cooled to 0 °C. After being stirred at room temperature for 1 h, the reaction mixture was poured into H2O (10 mL) and was extracted with EtOAc (5 mL × 3). The combined organic layers were washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 10:1) to give 23 (92.5 mg, 75.3% in two steps), which was used immediately for the next step because of its instability. HPLC: tR = 7.706 min, 96.8%.
tert-Butyl 4-(3,5-bis(trifluoromethyl)benzylamino)piperidine-1-carboxylate (26): tert-butyl 4-amino-piperidine-1-carboxylate (1.0 g, 5.0 mmol) and 3,5-bis(trifluoromethyl) benzaldehyde (1.0 g, 4.1 mmol) were dissolved in MeOH (15 mL). After stirring for 3 h at room temperature, NaBH4 (0.3 g, 7.0 mmol) was added to the mixture. The reaction mixture was stirred at room temperature for 30 min and then poured into a saturated sodium bicarbonate solution (20 mL). The mixture was extracted with CH2Cl2 (20 mL × 3) and the combined organic layers were washed with water (20 mL × 3) and brine (20 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 4:1) to give 26 (1.4 g, 80.0%) as a pale yellow oil. 1H-NMR (600 MHz, CDCl3) δ 7.83 (s, 2H), 7.76 (s, 1H), 4.04 (s, 2H), 3.96 (s, 2H), 2.82 (s, 2H), 2.71–2.63 (m, 1H), 1.88 (d, J = 11.2 Hz, 2H), 1.46 (s, 9H), 1.35–1.26 (m, 2H). HPLC: tR = 13.657 min, 95.7%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)- methyl)piperidine-4-amine-1-carboxylic acid tert-butyl ester (28): NaH (30.0 mg, 0.7 mmol, 60% in oil) was added to a solution of intermediate 26 (213.1 mg, 0.5 mmol) in DMF (5 mL) cooled to 0 °C. After stirring at 0 °C for 30 min, a solution of intermediate 23 (153.4 mg, 0.5 mmol) in DMF (5 mL) was added. The reaction mixture was allowed to warm to room temperature and stirred for 30 min, and then was poured onto crushed ice. The mixture was diluted with EtOAc (15 mL) and the layers were separated. The aqueous layer was extracted with EtOAc (5 mL × 3), and the combined organic layers were washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 20:1) to give 28 (235.9 mg, 67.7%) as a colourless oil. 1H-NMR (400 MHz, CDCl3) δ 7.83 (s, 2H), 7.73 (s, 1H), 7.08 (dd, J = 8.4, 2.2 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 6.74 (d, J = 2.3 Hz, 1H), 4.11 (s, 2H), 3.70 (s, 3H), 3.50 (q, J = 15.0 Hz, 2H), 2.90–2.78 (m, 2H), 2.74–2.48 (m, 4H), 2.44–2.31 (m, 1H), 2.09–2.00 (m, 1H), 1.99–1.83 (m, 2H), 1.52 (d, J = 12.0 Hz, 1H), 1.45–1.34 (m, 12H), 1.34–1.25 (m, 2H), 1.22 (dd, J = 6.9, 1.1 Hz, 6H), 0.96 (s, 6H). HPLC: tR = 7.023 min, 96.6%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)piperidin-4-amine (30): Intermediate 28 (0.2 g, 0.3 mmol) was dissolved in a trifluoroacetic acid–dichloromethane (1:1) solution (20 mL) and stirred at room temperature overnight. After concentration, the residue was dissolved in EtOAc (10 mL), washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 2:1) to give 30 (172.1 mg, 96.1%) as a colourless oil. 1H-NMR (400 MHz, DMSO-d6) δ 8.81 (s, 1H), 8.10–7.88 (m, 3H), 7.11 (dd, J = 8.4, 2.1 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 2.0 Hz, 1H), 3.66 (s, 3H), 3.57 (d, J = 2.4 Hz, 2H), 3.29 (d, J = 11.8 Hz, 2H), 2.89–2.68 (m, 6H), 2.26 (d, J = 17.6 Hz, 1H), 2.01 (s, 1H), 1.87 (d, J = 7.9 Hz, 2H), 1.67–1.48 (m, 4H), 1.35 (t, J = 6.2 Hz, 2H), 1.17 (d, J = 6.9 Hz, 6H), 0.92 (d, J = 10.5 Hz, 6H). HPLC: tR = 13.003 min, 95.9%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)piperidin-4-amine-1-ethyl alcohol (32): Intermediate 30 (0.7 g, 1.2 mmol) was dissolved in acetonitrile (10 mL) and methyl 2-bromoacetate (0.4 g, 2.4 mmol) and triethylamine (0.6 mL, 4.7 mmol) were added. The reaction mixture was stirred at room temperature for 16 h and then poured into H2O (10 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was dissolved in THF (10 mL), and sodium borohydride (0.1 g, 2.6 mmol) and 2 drops of MeOH were added. The mixture was stirred at 70 °C for 2 h and then cooled to room temperature. After concentration, the residue was dissolved in EtOAc (10 mL), washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 1:1) to give 32 (0.4 g, 52.0%) as a colourless oil. 1H-NMR (400 MHz, DMSO-d6) δ 8.01 (s, 2H), 7.93 (s, 1H), 7.08 (dd, J = 8.4, 2.0 Hz, 1H), 6.88 (d, J = 8.5 Hz, 1H), 6.74 (d, J = 2.0 Hz, 1H), 4.39 (s, 1H), 3.64 (s, 3H), 3.55 (d, J = 18.4 Hz, 2H), 3.44 (d, J = 5.9 Hz, 2H), 2.84 (d, J = 13.3 Hz, 2H), 2.78 (dd, J = 13.7, 6.8 Hz, 1H), 2.69 (s, 2H), 2.43–2.37 (m, 3H), 2.26 (d, J = 17.8 Hz, 1H), 1.96–1.77 (m, 4H), 1.44–1.42 (m, 7H), 1.16 (d, J = 6.9 Hz, 6H), 0.91 (d, J = 7.1 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 154.49, 144.61, 140.59, 132.83, 131.67, 131.25(×2), 129.57, 128.19, 128.00, 125.01, 124.38, 122.57, 120.40, 110.56(×2), 59.29, 57.73, 55.43, 55.13, 53.47, 53.39, 52.55, 52.25, 40.99, 35.60, 33.21(×2), 29.30, 28.93, 28.28, 27.72. 27.25, 24.23, 24.12. HRMS calcd for C35H47F6N2O2, [M + H]+, 641.3463; found 641.3525. HPLC: tR = 16.772 min, 97.9%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)piperidin-4-amine-1-acetic acid (33): Intermediate 30 (0.5 g, 0.9 mmol) was dissolved in acetonitrile (10 mL), and methyl 2-bromoacetate (0.3 g, 1.8 mmol) and triethylamine (0.4 mL, 3.4 mmol) were added. The reaction mixture was stirred at room temperature for 16 h and then poured into H2O (10 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was dissolved in MeOH (5 mL) and 1 mol/L NaOH (5 mL) was added and the mixture was stirred at room temperature for 2 h. After concentration, the residue was dissolved in H2O (10 mL). Then, 1 mol/L HCl (5 mL) was added to the mixture, the mixture was extracted with EtOAc (10 mL × 3), and the combined organic layers were washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (DCM:MeOH = 5:1) to give 33 (0.4 g, 67.8%) as a colourless oil. 1H-NMR (400 MHz, DMSO-d6) δ 8.02 (s, 2H), 7.95 (s, 1H), 7.09 (dd, J = 8.5, 2.1 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.77 (d, J = 2.1 Hz, 1H), 3.65 (s, 3H), 3.63–3.51 (m, 2H), 3.20 (s, 2H), 3.14 (d, J = 10.4 Hz, 2H), 2.79 (dt, J = 13.7, 6.9 Hz, 1H), 2.70 (s, 2H), 2.62–2.51 (m, 3H), 2.26 (d, J = 17.7 Hz, 1H), 1.96 (s, 1H), 1.91–1.75 (m, 2H), 1.70–1.51 (m, 2H), 1.40 (s, 2H), 1.35 (t, J = 6.3 Hz, 2H), 1.16 (d, J = 6.8 Hz, 6H), 0.91 (d, J = 8.7 Hz, 6H). 13C-NMR (150 MHz, DMSO-d6) δ: 168.60, 154.62(×2), 145.76, 140.43, 132.53, 131.21, 130.22(×2), 129.42, 128.93, 127.74, 125.54, 124.78, 122.97, 111.48(×2), 59.03, 55.81, 54.09, 52.70, 52.43, 52.37, 51.58, 41.02, 35.50, 32.94, 29.40, 29.00, 28.53, 27.95, 24.55(×2), 24.44(×2). HRMS calcd for C35H45F6N2O3, [M + H]+, 655.3256; found 655.3345. HPLC: tR = 19.973 min, 98.1%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)piperidin-4-amine-1- methanoic acid tetrahydro-2H-pyran-4-ol ester (34): Intermediate 30 (0.2 g, 0.3 mmol) was dissolved in CH2Cl2 (5 mL), and tetrahydro-2H-pyran-4-yl carbonochloridate (0.1 g, 0.5 mmol) and triethylamine (0.2 mL, 1.4 mmol) were added. The reaction mixture was stirred at room temperature for 30 min and then poured into H2O (10 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 4:1) to give 34 (0.1 g, 45.9%) as a colourless oil. 1H-NMR (600 MHz, CDCl3) δ 7.82 (s, 2H), 7.73 (s, 1H), 7.08 (dd, J = 8.3, 2.0 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 6.73 (d, J = 2.0 Hz, 1H), 4.86–4.80 (m, 1H), 4.13 (d, J = 7.1 Hz, 2H), 3.90–3.85 (m, 2H), 3.71 (s, 3H), 3.56–3.53 (m, 3H), 3.49–3.41 (m, 1H), 2.84–2.81 (m, 2H), 2.75–2.53 (m, 4H), 2.37 (d, J = 17.7 Hz, 1H), 2.02 (s, 1H), 1.93–1.90 (m, 3H), 1.87 (s, 1H), 1.65–1.63 (m, 3H), 1.55 (d, J = 12.5 Hz, 1H), 1.46–1.37 (m, 3H), 1.35–1.29 (m, 1H), 1.22 (dd, J = 6.9, 2.4 Hz, 6H), 0.96 (s, 6H). 13C-NMR (150 MHz, CDCl3) δ: 154.47(×2), 144.35, 140.64, 131.57, 131.32, 131.10, 129.35, 128.18(×2), 125.09(×2), 124.35, 122.54, 120.51, 110.61(×2), 69.63, 65.30(×2), 55.44(×2), 55.35, 52.46, 52.23, 43.82, 43.66, 41.07, 35.57, 33.19, 32.08(×2), 29.24, 28.94, 28.33, 27.64, 24.24, 24.11(×2). HRMS calcd for C39H51F6N2O4, [M + H]+, 725.3675; found 725.3751. HPLC: tR = 15.553 min, 96.0%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((5′-isopropyl-2′-methoxy-4,4-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)azetidin-3-amine-1-methanoic acid ethyl ester (35): Intermediate 31 (0.2 g, 0.3 mmol) was dissolved in CH2Cl2 (5 mL), and ethyl chloroformate (0.1 g, 0.7 mmol) and triethylamine (0.2 mL, 1.4 mmol) were added. The reaction mixture was stirred at room temperature for 30 min and then poured into H2O (10 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were washed with H2O (10 mL × 3) and brine (10 mL × 3), dried over Na2SO4, and concentrated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether:EtOAc = 4:1) to give 35 (0.2 g, 89.2%) as a colourless oil. 1H-NMR (400 MHz, CDCl3) δ 7.78 (s, 2H), 7.76 (s, 1H), 7.09 (dd, J = 8.4, 2.1 Hz, 1H), 6.80 (d, J = 8.4 Hz, 1H), 6.74 (d, J = 2.1 Hz, 1H), 4.07 (q, J = 7.1 Hz, 2H), 3.88–3.81 (m, 1H), 3.80–3.72 (m, 3H), 3.71 (s, 3H), 3.68–3.56 (m, 2H), 3.55–3.47 (m, 1H), 2.90–2.77 (m, 2H), 2.77–2.65 (m, 1H), 2.43–2.29 (m, 1H), 2.10–2.04 (m, 1H), 2.01–1.82 (m, 2H), 1.45–1.35 (m, 2H), 1.24–1.19 (m, 9H), 0.96 (s, 6H). 13C-NMR (150 MHz, CDCl3) δ: 156.60(×2), 154.30(×2), 140.75, 131.29(×2), 131.14, 128.37, 127.94(×2), 125.39, 124.21, 122.41, 120.87, 110.55(×2), 60.92, 55.32, 54.44, 53.43, 41.59, 35.49, 33.14(×2), 29.38, 28.97, 28.22, 27.80, 24.18, 24.12(×2), 14.60(×2). HRMS calcd for C34H43F6N2O3, [M + H]+, 641.3100; found 641.3180. HPLC: tR = 11.763 min, 96.7%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((5′-isopropyl-2′-methoxy-4,4-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)azetidin-3-amine-1-methanoic acid isopropyl ester (36): Colourless oil; yield 69.3%; 1H-NMR (400 MHz, CDCl3) δ 7.77 (s, 2H), 7.76 (s, 1H), 7.09 (dd, J = 8.4, 2.2 Hz, 1H), 6.80 (d, J = 8.4 Hz, 1H), 6.74 (d, J = 2.2 Hz, 1H), 4.84 (dt, J = 12.5, 6.2 Hz, 1H), 3.83 (t, J = 8.4 Hz, 1H), 3.77 (d, J = 7.9 Hz, 1H), 3.73 (d, J = 5.4 Hz, 2H), 3.70 (s, 3H), 3.67–3.56 (m, 2H), 3.55–3.47 (m, 1H), 2.89–2.77 (m, 2H), 2.76–2.64 (m, 1H), 2.40–2.28 (m, 1H), 2.09–2.04 (m, 1H), 2.00–1.83 (m, 2H), 1.44–1.35 (m, 2H), 1.22 (d, J = 6.9 Hz, 6H), 1.19 (d, J = 6.2 Hz, 6H), 0.96 (d, J = 2.8 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 156.38(×2), 154.32(×2), 140.72, 131.48, 131.26, 131.19, 128.34, 127.93(×2), 125.35, 124.23, 122.43, 120.83, 110.53(×2), 68.17, 55.31(×2), 54.44, 53.43, 51.10, 41.56, 35.50, 33.14, 29.38, 28.96(×2), 28.23, 27.79, 24.19, 24.13, 22.11(×2). HRMS calcd for C35H45F6N2O3, [M + H]+, 655.3256; found 655.3344. HPLC: tR = 10.757 min, 96.8%.
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((5′-isopropyl-2′-methoxy-4,4-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)azetidin-3-amine-1-methanoic acid tetrahydro-2H-pyran-4-ol ester (37): Colourless oil; yield 73.6%; 1H NMR (600 MHz, CDCl3) δ 7.78 (s, 2H), 7.76 (s, 1H), 7.09 (dd, J = 8.4, 2.1 Hz, 1H), 6.80 (d, J = 8.4 Hz, 1H), 6.75 (d, J = 2.2 Hz, 1H), 4.79 (tt, J = 8.4, 4.0 Hz, 1H), 3.89–3.83 (m, 3H), 3.82–3.78 (m, 1H), 3.75 (s, 2H), 3.71 (s, 3H), 3.62 (s, 2H), 3.55–3.49 (m, 3H), 2.90–2.77 (m, 2H), 2.73 (s, 1H), 2.39–2.30 (m, 1H), 2.09–2.02 (m, 1H), 2.00–1.94 (m, 1H), 1.92–1.85 (m, 3H), 1.64–1.59 (m, 2H), 1.45–1.34 (m, 2H), 1.22 (d, J = 6.9 Hz, 6H), 0.97 (d, J = 4.2 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 155.77(×2), 154.30, 140.76, 131.53, 131.31, 131.14, 128.36, 127.90(×3), 125.40, 124.20, 122.40, 120.92, 110.57(×2), 69.44, 65.26(×2), 55.33(×2), 54.49, 53.46, 41.59, 35.48, 33.14(×2), 32.07(×2), 29.40, 28.98(×2), 28.23, 27.82, 24.20, 24.13. HRMS calcd for C37H47F6N2O4, [M + H]+, 697.3362; found 697.3459. HPLC: tR = 16.003 min, 95.8%.
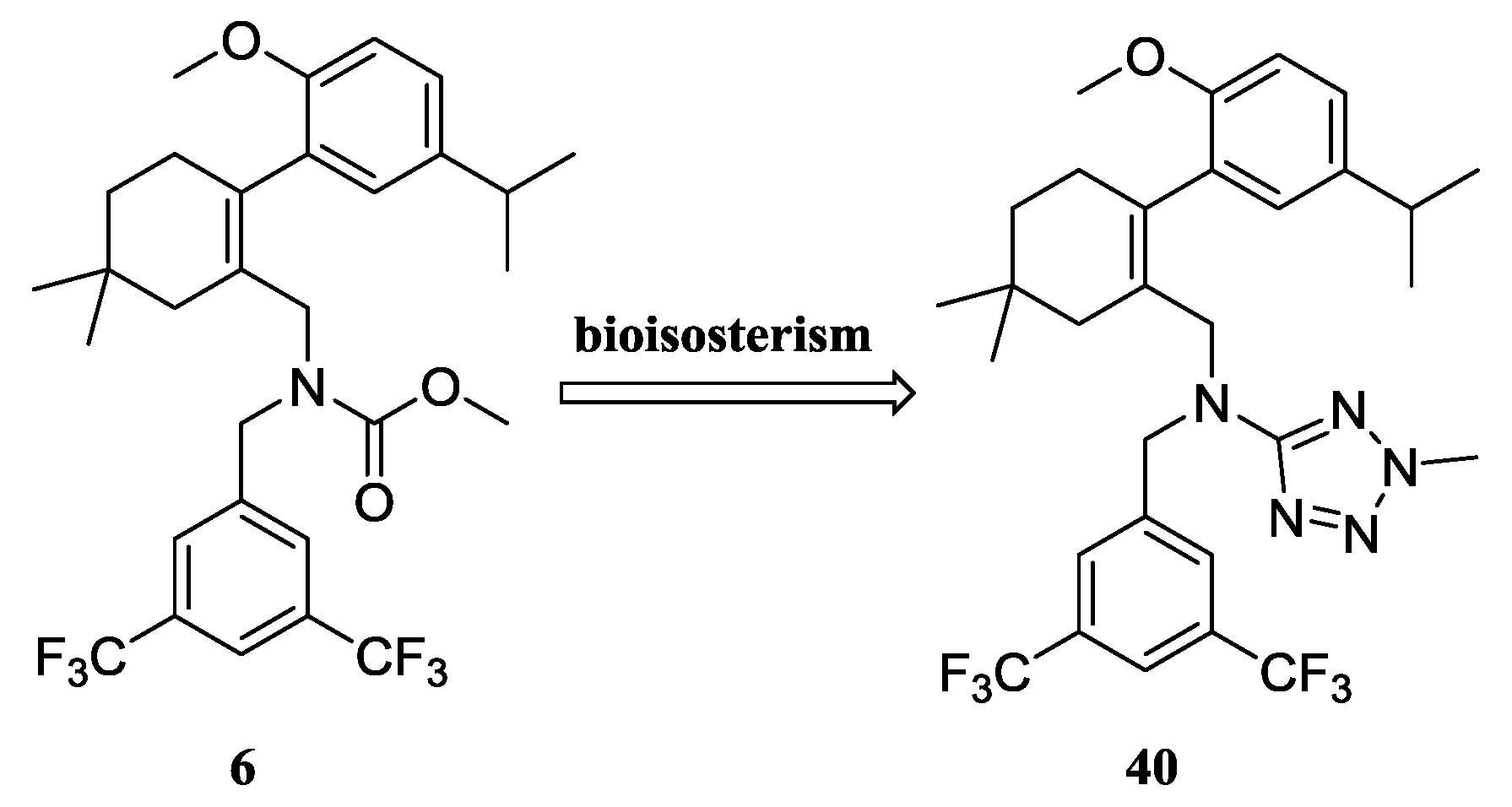
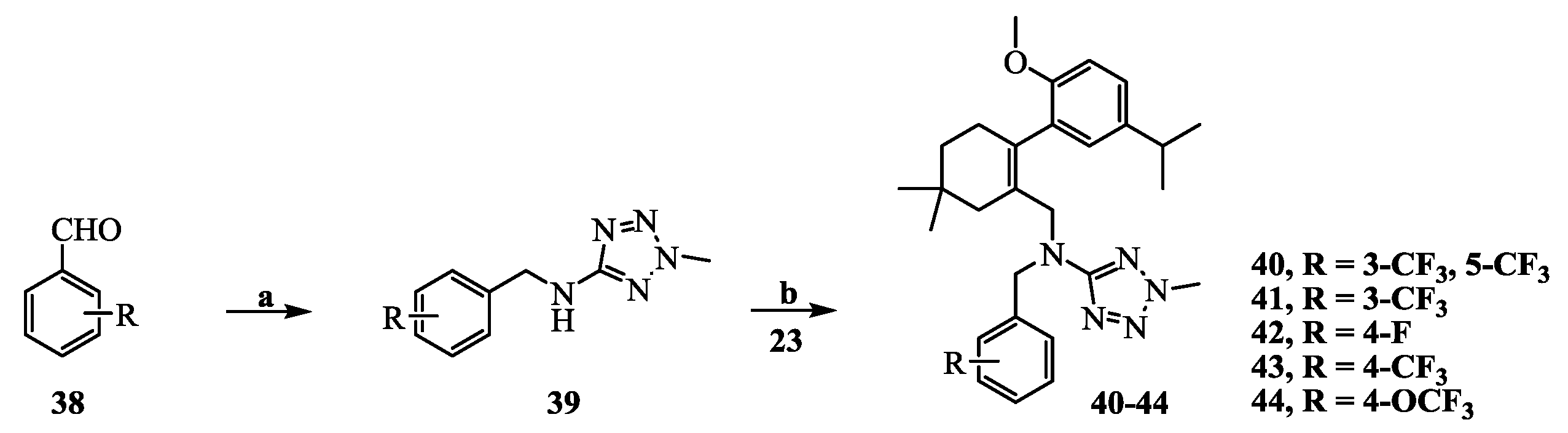
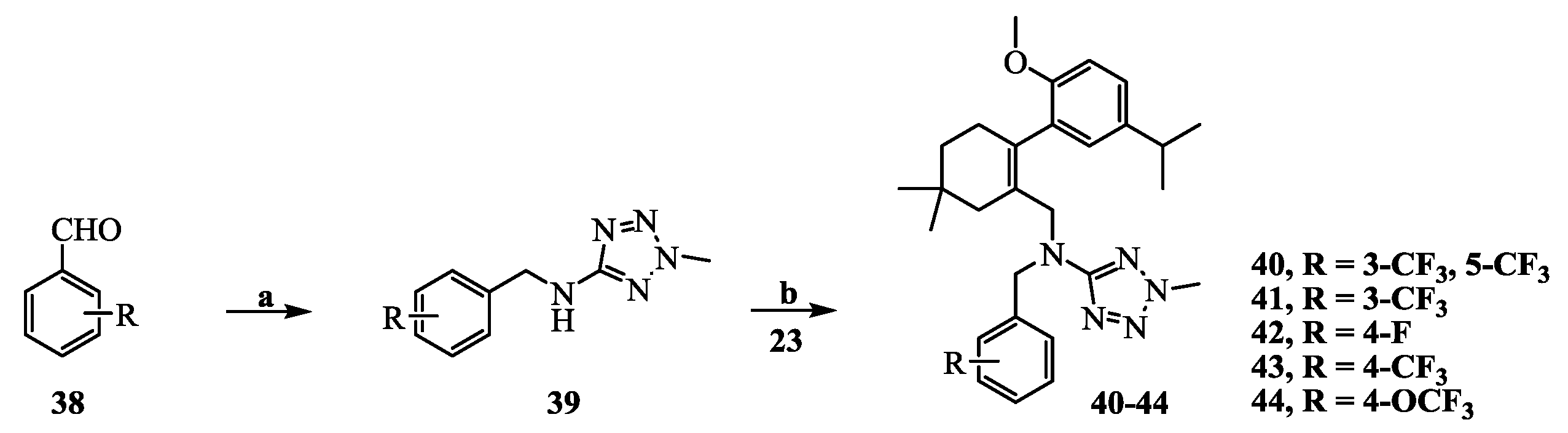
N-(3,5-Bis(trifluoromethyl)benzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)-methyl)-2-methyl-2H-tetrazol-5-amine (40): The title compound was obtained in a manner similar to that described for the preparation of intermediate 28. Colourless oil; yield 70.5%; 1H-NMR (600 MHz, DMSO-d6) δ 7.92 (s, 1H), 7.64 (s, 2H), 6.98 (dd, J = 8.4, 2.2 Hz, 1H), 6.77–6.73 (m, 2H), 4.50 (s, 2H), 4.10 (s, 3H), 3.99–3.93 (m, 2H), 3.60 (s, 3H), 2.65 (dt, J = 13.7, 6.9 Hz, 1H), 2.32 (d, J = 18.0 Hz, 1H), 2.01 (d, J = 18.0 Hz, 1H), 1.79–1.72 (m, 2H), 1.35 (t, J = 6.4 Hz, 2H), 1.02 (d, J = 6.9 Hz, 6H), 0.89 (d, J = 13.2 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 169.46, 154.12, 141.34, 140.83, 135.26, 131.26(×2), 130.54, 128.05, 127.70, 127.65, 125.61, 124.16, 122.35, 120.60, 110.59(×2), 55.21, 51.59, 49.22, 40.56, 39.20, 35.44, 33.02, 29.09, 29.02, 28.08, 27.99, 23.98(×2). HRMS calcd for C30H36F6N5O, [M + H]+, 596.2746; found 596.2847. HPLC: tR = 8.776 min, 96.1%.
N-((2-(5-Isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)methyl)-2-methyl-N-(3-(trifluoromethyl)-benzyl)-2H-tetrazol-5-amine (41): Colourless oil; yield 78.4%; 1H-NMR (600 MHz, DMSO-d6) δ 7.52 (d, J = 7.7 Hz, 1H), 7.41 (t, J = 7.7 Hz, 1H), 7.34 (s, 1H), 7.22 (d, J = 7.8 Hz, 1H), 7.02 (dd, J = 8.5, 2.3 Hz, 1H), 6.80 (d, J = 8.5 Hz, 1H), 6.77 (d, J = 2.3 Hz, 1H), 4.41 (s, 2H), 4.10 (s, 3H), 3.92 (q, J = 14.7 Hz, 2H), 3.63 (s, 3H), 2.68 (dt, J = 13.8, 6.9 Hz, 1H), 2.32 (d, J = 18.0 Hz, 1H), 2.04 (d, J = 18.1 Hz, 1H), 1.76 (s, 2H), 1.38 (t, J = 6.5 Hz, 2H), 1.05 (dd, J = 6.9, 0.8 Hz, 6H), 0.91 (d, J = 5.1 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 169.66, 154.26, 140.64, 139.40, 134.35, 130.80, 130.73, 128.32, 128.03, 125.36, 124.31, 124.29, 123.43, 123.40, 110.45(×2), 55.17, 51.16, 49.31, 40.42, 39.15, 35.54, 33.05, 29.19, 29.03, 28.20, 27.98, 24.09, 24.01. HRMS calcd for C29H37F3N5O, [M + H]+, 528.2872; found 528.2960. HPLC: tR = 8.335 min, 96.2%.
N-(4-Fluorobenzyl)-N-((2-(5-isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)methyl)-2-methyl-2H-tetrazol-5-amine (42): Colourless oil; yield 80.1%; 1H-NMR (600 MHz, DMSO-d6) δ 7.07–7.05 (m, 1H), 6.97–6.93 (m, 4H), 6.86–6.85 (m, 1H), 6.80 (d, J = 2.3 Hz, 1H), 4.30 (q, J = 15.8 Hz, 2H), 4.10 (s, 3H), 3.85 (dd, J = 44.8, 14.8 Hz, 2H), 3.66 (s, 3H), 2.72 (dt, J = 13.8, 6.9 Hz, 1H), 2.32 (d, J = 18.0 Hz, 1H), 2.08 (d, J = 18.0 Hz, 1H), 1.76 (s, 2H), 1.40 (t, J = 6.5 Hz, 2H), 1.09 (dd, J = 6.9, 0.6 Hz, 6H), 0.93 (s, 6H). 13C- NMR (150 MHz, CDCl3) δ: 169.67, 154.42, 140.66, 133.58, 130.93, 129.47, 129.41, 128.30, 128.14, 127.90, 125.25, 114.72, 114.58, 110.44(×2), 55.23, 50.53, 48.79, 40.35, 39.12, 35.64, 33.12, 29.28, 29.04, 28.25, 28.01, 24.20, 24.06. HRMS calcd for C28H37FN5O, [M + H]+, 478.2904; found 478.2999. HPLC: tR = 7.750 min, 96.7%.
N-((2-(5-Isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)methyl)-2-methyl-N-(4-(trifluoromethyl)-benzyl)-2H-tetrazol-5-amine (43): Colourless oil; yield 67.5%; 1H-NMR (600 MHz, CDCl3) δ 7.37 (d, J = 8.1 Hz, 2H), 7.10 (d, J = 8.0 Hz, 2H), 7.02 (dd, J = 8.4, 2.3 Hz, 1H), 6.76 (d, J = 2.3 Hz, 1H), 6.70 (d, J = 8.4 Hz, 1H), 4.45 (s, 2H), 4.11 (s, 3H), 4.02 (s, 2H), 3.65 (s, 3H), 2.73 (dt, J = 13.8, 6.9 Hz, 1H), 2.44 (d, J = 18.2 Hz, 1H), 2.12 (d, J = 18.1 Hz, 1H), 1.84 (d, J = 1.6 Hz, 2H), 1.48–1.42 (m, 2H), 1.14 (dd, J = 6.9, 1.8 Hz, 6H), 0.96 (s, 6H). 13C-NMR (150 MHz, CDCl3) δ: 169.62, 154.32, 142.35, 140.67, 134.24, 130.74, 128.03, 127.98, 127.70(×3), 125.30, 124.87, 124.85, 110.43(×2), 55.20, 50.82, 49.03, 40.41, 39.16, 35.60, 33.06, 29.22, 29.05, 28.30, 27.93, 24.15, 23.98. HRMS calcd for C29H37F3N5O, [M + H]+, 528.2872; found 528.2953. HPLC: tR = 8.985 min, 96.7%.
N-((2-(5-Isopropyl-2-methoxyphenyl)-5,5-dimethylcyclohex-1-enyl)methyl)-2-methyl-N-(4-(trifluoro-methoxy)benzyl)-2H-tetrazol-5-amine (44): Colourless oil; yield 66.3%; 1H-NMR (600 MHz, DMSO-d6) δ 7.14 (d, J = 8.1 Hz, 2H), 7.06–7.01 (m, 3H), 6.82 (d, J = 8.5 Hz, 1H), 6.78 (d, J = 2.2 Hz, 1H), 4.35 (s, 2H), 4.11 (s, 3H), 3.88 (m, 2H), 3.66 (s, 3H), 2.70 (dt, J = 13.8, 6.9 Hz, 1H), 2.33 (d, J = 18.4 Hz, 1H), 2.06 (d, J = 18.0 Hz, 1H), 1.76 (s, 2H), 1.39 (t, J = 6.4 Hz, 2H), 1.06 (dd, J = 6.9, 1.0 Hz, 6H), 0.92 (d, J = 1.6 Hz, 6H). 13C-NMR (150 MHz, CDCl3) δ: 169.66, 154.37, 140.66, 136.94, 133.96, 130.81, 129.00(×3), 128.15, 128.11, 125.29, 120.46(×2), 110.43(×2), 55.19, 50.66, 48.76, 40.39, 39.13, 35.61, 33.10, 29.21, 29.03, 28.25, 27.97, 24.14, 24.01. HRMS calcd for C29H37F3N5O2, [M + H]+, 544.2821; found 544.2898. HPLC: tR = 8.302 min, 97.3%.

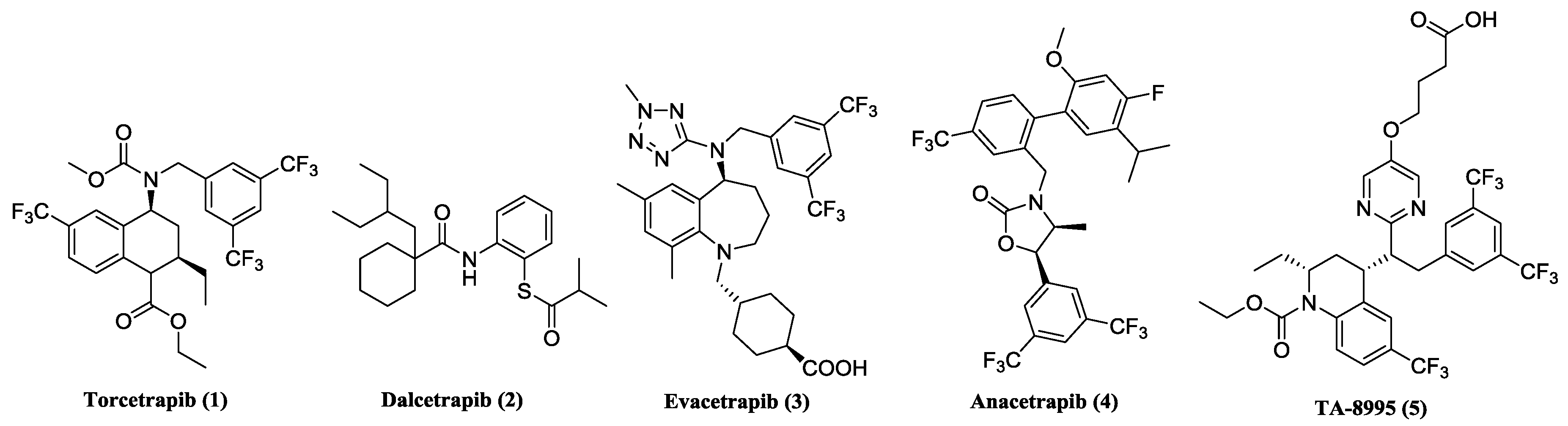
 and
and 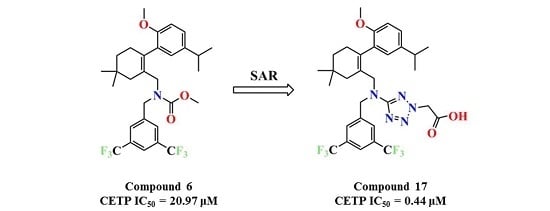

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



























