Investigation of the N-Terminus Amino Function of Arg10-Teixobactin

 ,
,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
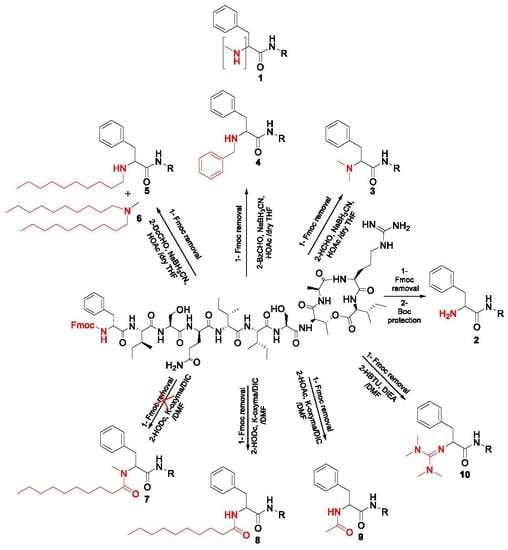
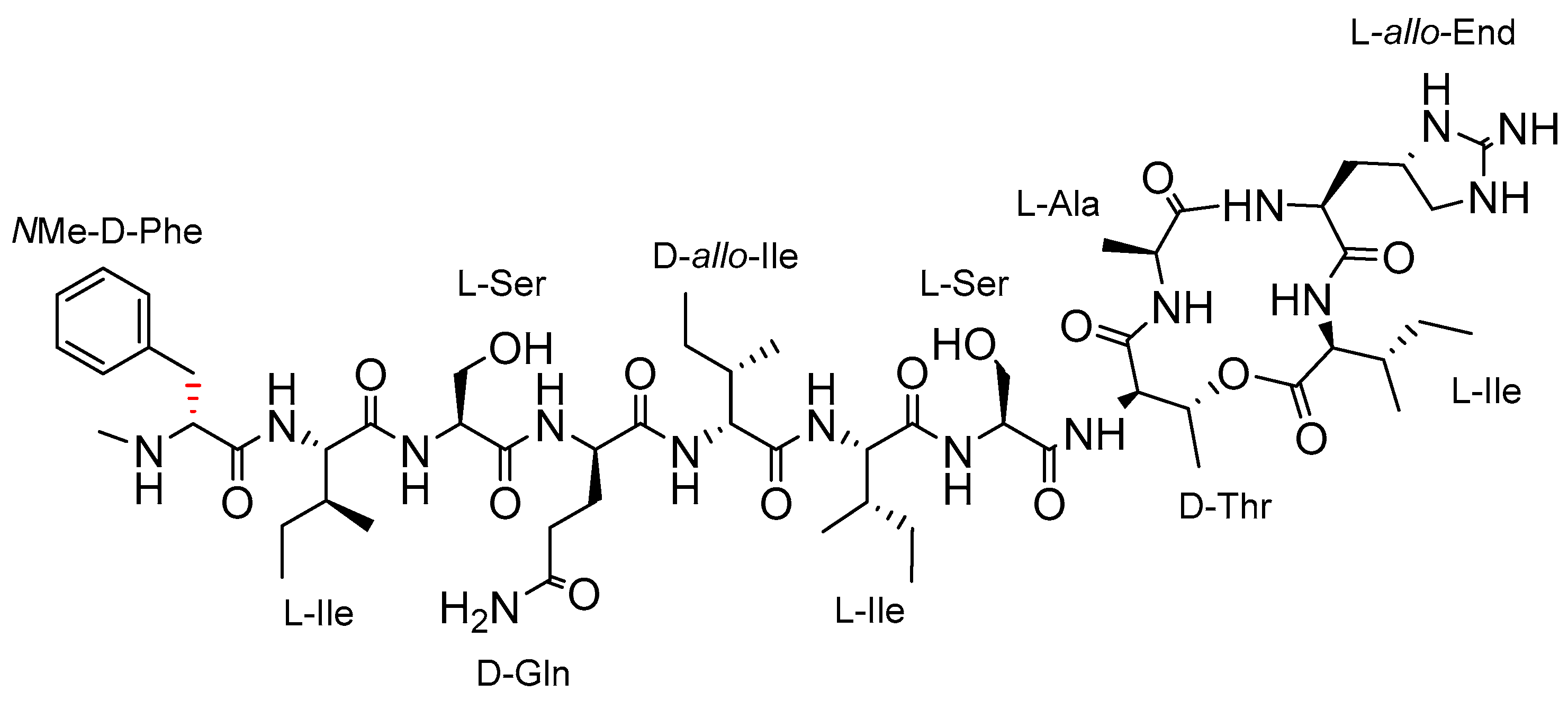
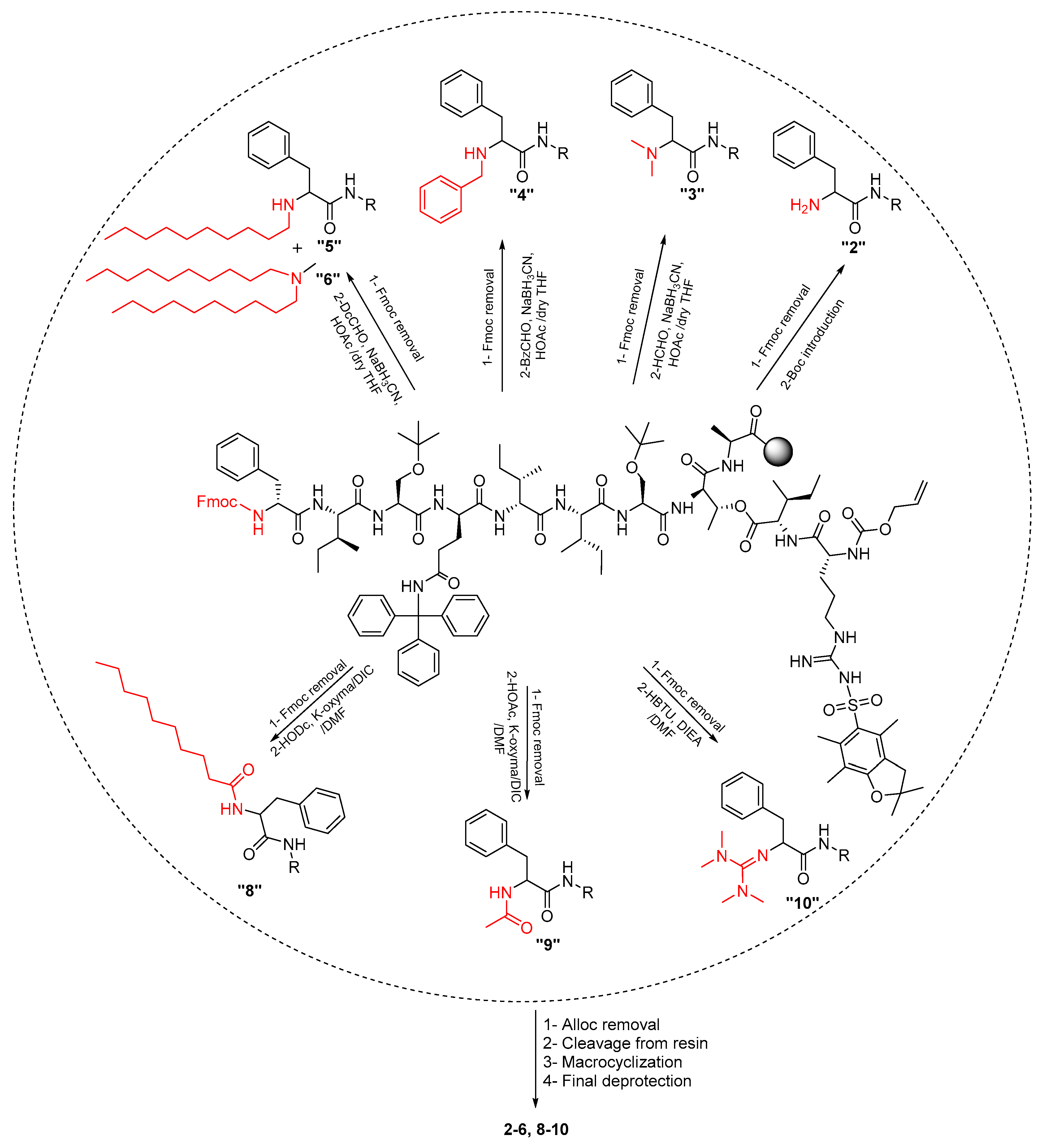
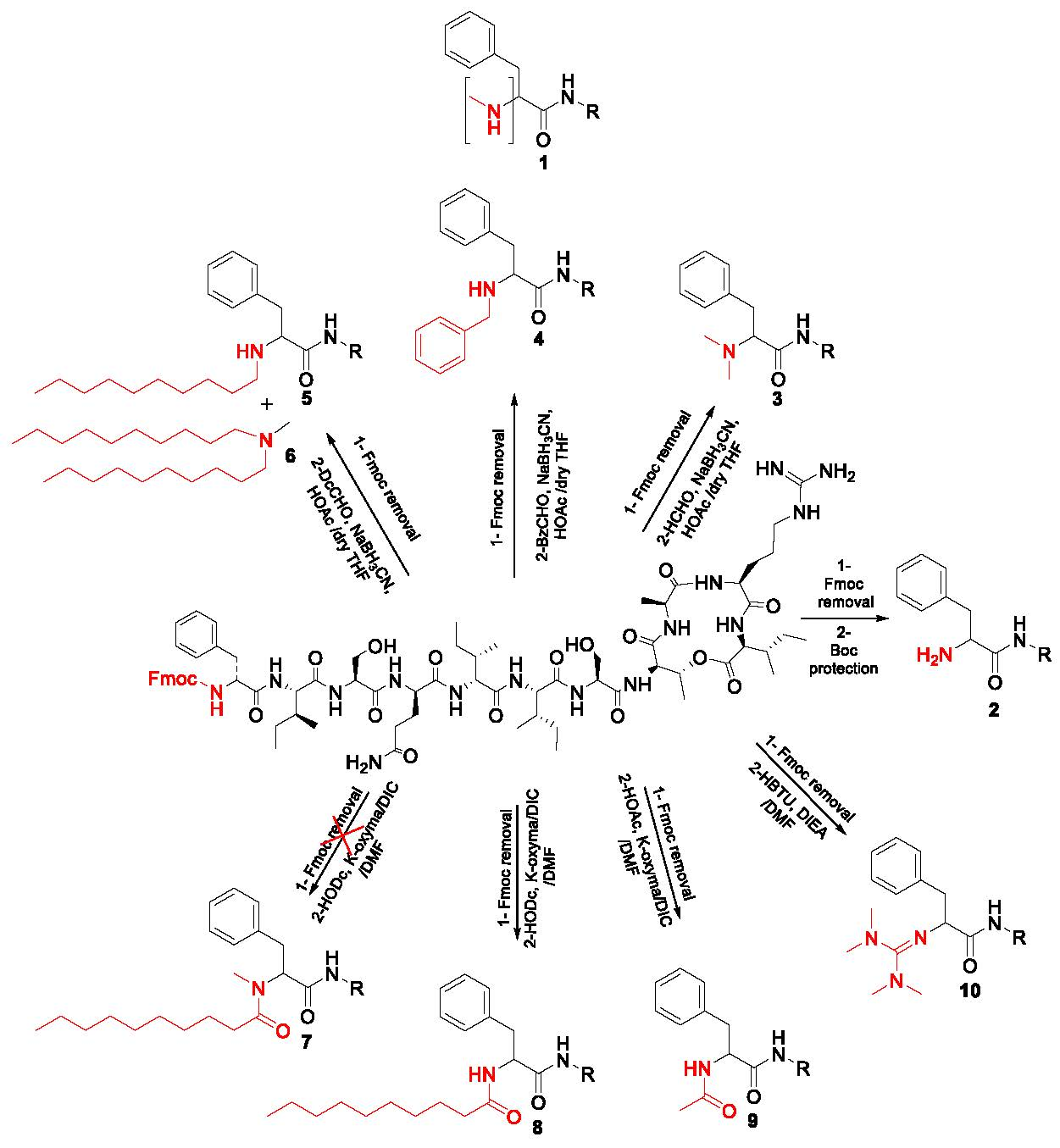
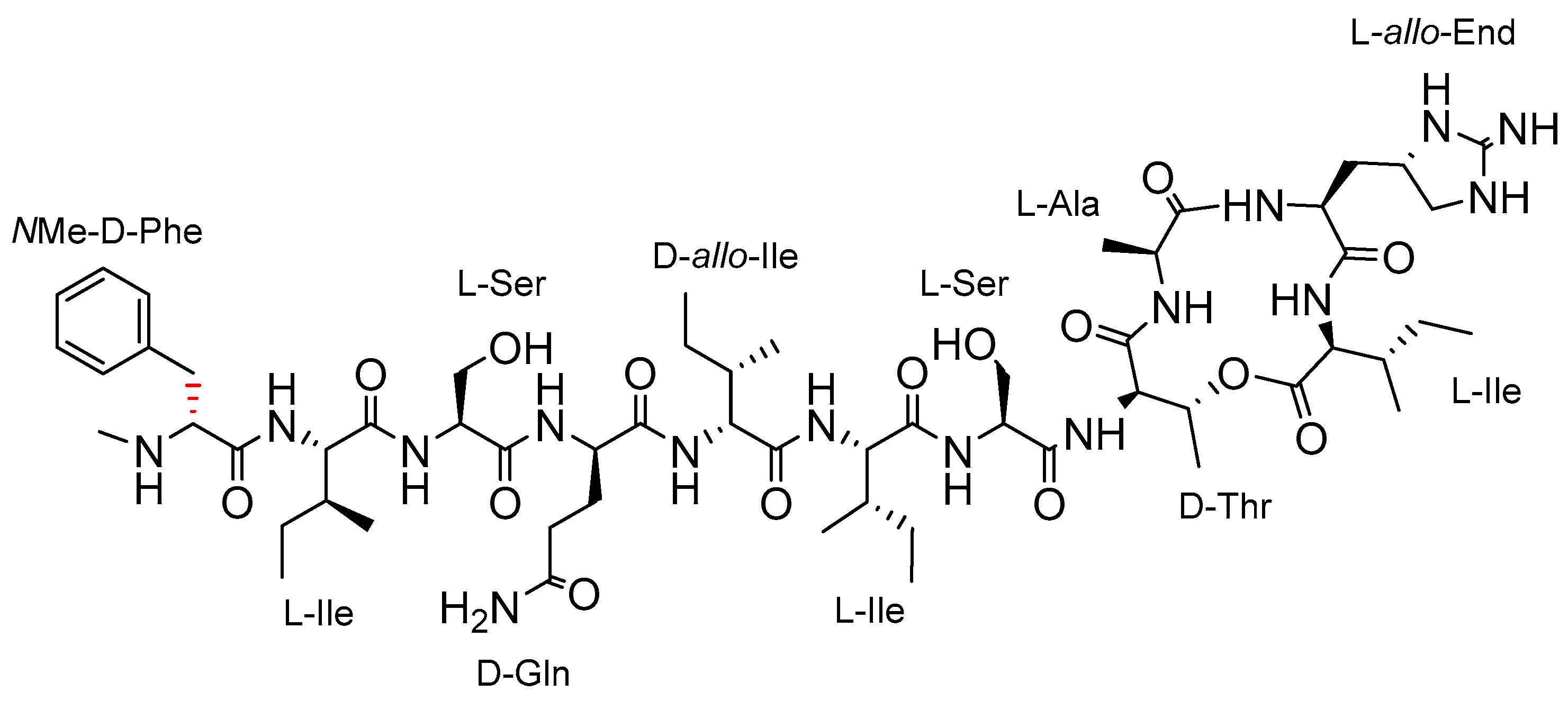
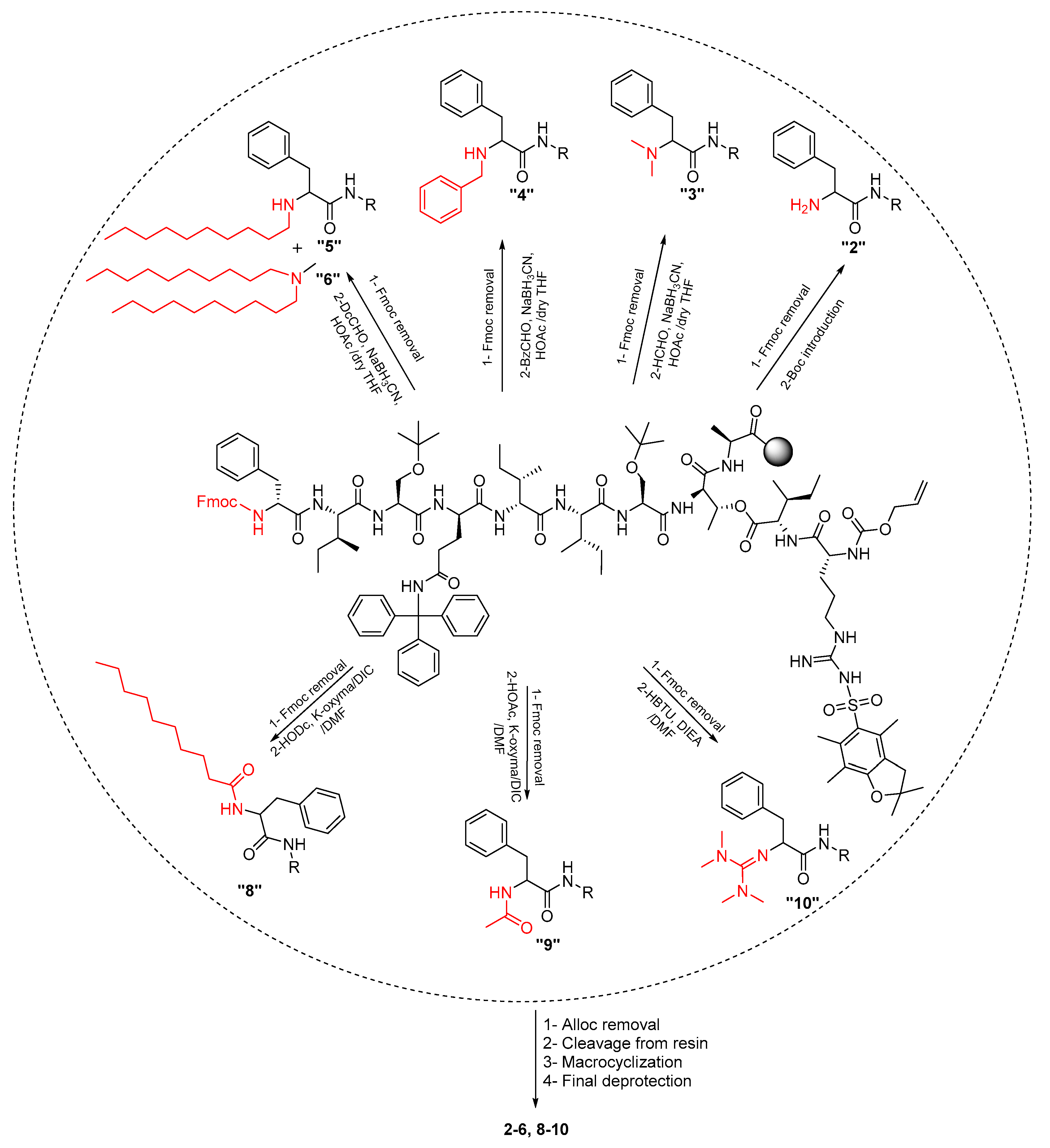
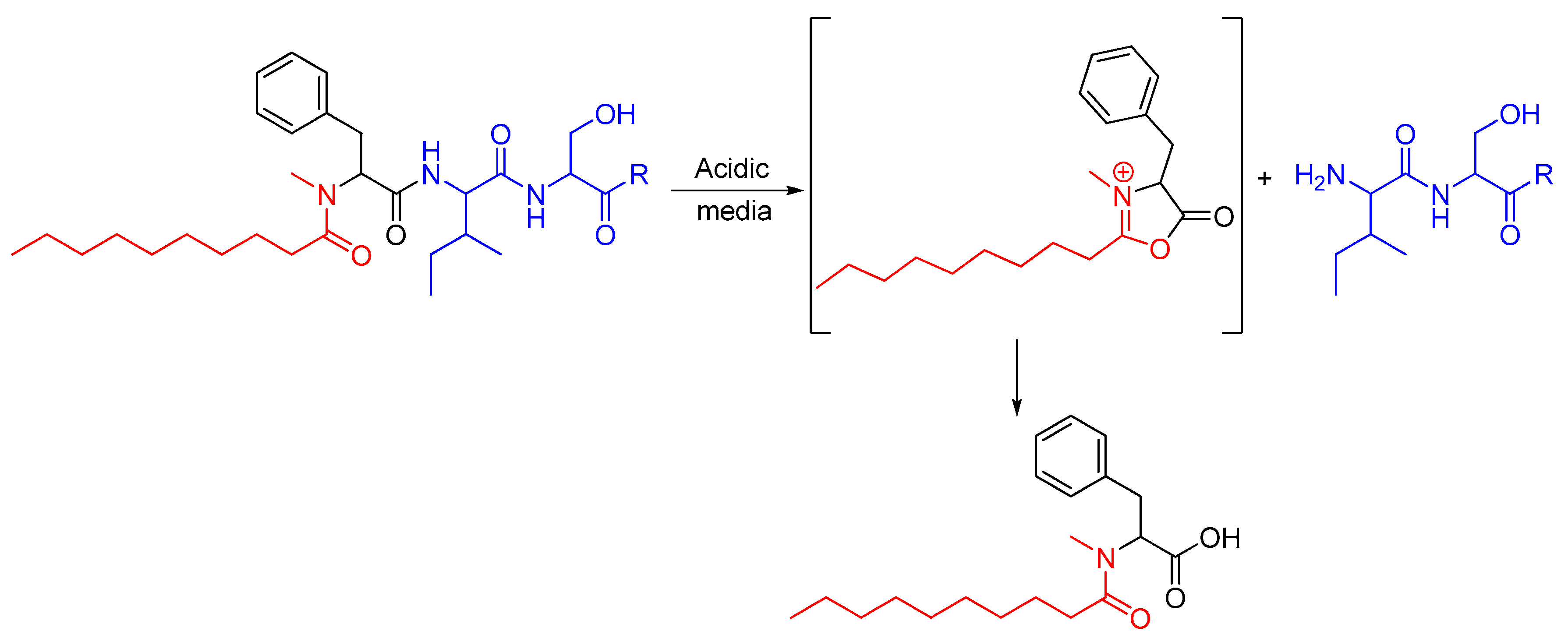
2.1. Synthetic Strategy
2.2. Microbiological Evaluation
3. Materials and Methods
3.1. Materials
3.2. Instruments
3.3. Methods
3.3.1. Chemistry
3.3.2. Determination of Minimum Inhibitory Concentration (MIC)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Jad, Y.E.; Acosta, G.A.; Naicker, T.; Ramtahal, M.; El-Faham, A.; Govender, T.; Kruger, H.G.; Torre, B.G.D.L.; Albericio, F. Synthesis and biological evaluation of a teixobactin analogue. Org. Lett. 2015, 17, 6182–6185. [Google Scholar] [CrossRef] [PubMed]
- Abdel Monaim, S.A.; Jad, Y.E.; Ramchuran, E.J.; El-Faham, A.; Govender, T.; Kruger, H.G.; de la Torre, B.G.; Albericio, F. Lysine scanning of Arg10-teixobactin: Deciphering the role of hydrophobic and hydrophilic residues. ACS Omega 2016, 1, 1262–1265. [Google Scholar] [CrossRef]
- Yang, H.; Du Bois, D.; Ziller, J.; Nowick, J. X-ray crystallographic structure of a teixobactin analogue reveals key interactions of the teixobactin pharmacophore. Chem. Commun. 2017, 53, 2772–2775. [Google Scholar] [CrossRef] [PubMed]
- Parmar, A.; Iyer, A.; Vincent, C.S.; Van Lysebetten, D.; Prior, S.H.; Madder, A.; Taylor, E.J.; Singh, I. Efficient total syntheses and biological activities of two teixobactin analogues. Chem. Commun. 2016, 52, 6060–6063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel Monaim, S.A.; Ramchuran, E.J.; El-Faham, A.; Albericio, F.; de la Torre, B.G. Converting teixobactin into a cationic antimicrobial peptide (AMP). J. Med. Chem. 2017, 60, 7476–7482. [Google Scholar] [CrossRef] [PubMed]
- Abdel Monaim, S.A.; Jad, Y.E.; Ramchuran, E.J.; El-Faham, A.; Acosta, G.A.; Naicker, T.; Govender, T.; Kruger, H.G.; de la Torre, B.G.; Albericio, F. Re-evaluation of the N-terminal substitution and the d-residues of teixobactin. RSC Adv. 2016, 6, 73827–73829. [Google Scholar] [CrossRef]
- Yang, H.; Chen, K.H.; Nowick, J.S. Elucidation of the teixobactin pharmacophore. ACS Chem. Biol. 2016, 11, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Setoh, K.; Murakami, M.; Araki, N.; Fujita, T.; Yamamoto, A.; Muranishi, S. Improvement of transdermal delivery of tetragastrin by lipophilic modification with fatty acids. J. Pharm. Pharmacol. 1995, 47, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Gulaboski, R.; Scholz, F. Lipophilicity of peptide anions: An experimental data set for lipophilicity calculations. J. Phys. Chem. B 2003, 107, 5650–5657. [Google Scholar] [CrossRef]
- Fernández-Llamazares, A.I.; Adan, J.; Mitjans, F.; Spengler, J.; Albericio, F. Tackling lipophilicity of peptide drugs: Replacement of the backbone N-methyl group of Cilengitide by N-oligoethylene glycol (N-OEG) chains. Bioconj. Chem. 2013, 25, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Orädd, G.; Schmidtchen, A.; Malmsten, M. Effects of peptide hydrophobicity on its incorporation in phospholipid membranes—An NMR and ellipsometry study. BBA Biomembr. 2011, 1808, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Govender, T.; Kruger, H.G.; Albericio, F.; Beatriz, G. An improved and efficient strategy for the total synthesis of a colistin-like peptide. Tetrahedron Lett. 2016, 57, 1885–1888. [Google Scholar] [CrossRef]
- Okimura, K.; Ohki, K.; Sato, Y.; Ohnishi, K.; Sakura, N. Semi-synthesis of polymyxin B (2–10) and colistin (2–10) analogs employing the trichloroethoxycarbonyl (Troc) group for side chain protection of α,γ-diaminobutyric acid residues. Chem. Pharm. Bull. 2007, 55, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M.; Fox, J.; Loidl, G.; Siikanen, O.; Apajalahti, J.; Hansen, F.; Frimodt-Møller, N.; Nagai, J.; Takano, M.; Vaara, T. Novel polymyxin derivatives carrying only three positive charges are effective antibacterial agents. Antimicrob. Agents Chemother. 2008, 52, 3229–3236. [Google Scholar] [CrossRef] [PubMed]
- Tsubery, H.; Ofek, I.; Cohen, S.; Fridkin, M. N-terminal modifications of polymyxin B nonapeptide and their effect on antibacterial activity. Peptides 2001, 22, 1675–1681. [Google Scholar] [CrossRef]
- Mingeot-Leclercq, M.-P.; Tulkens, P.M.; Denamur, S.; Vaara, T.; Vaara, M. Novel polymyxin derivatives are less cytotoxic than polymyxin B to renal proximal tubular cells. Peptides 2012, 35, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Teixidó, M.; Albericio, F.; Giralt, E. Solid-phase synthesis and characterization of N-methyl-rich peptides. J. Pept. Res. 2005, 65, 153–166. [Google Scholar] [CrossRef] [PubMed]
- del Fresno, M.; El-Faham, A.; Carpino, L.A.; Royo, M.; Albericio, F. Substituted guanidines: Introducing diversity in combinatorial chemistry. Org. Lett. 2000, 2, 3539–3542. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Pan, Z.; Yao, G.; Wang, W.; Fang, L.; Su, W. Synthesis and structure-activity relationship studies of teixobactin analogues. RSC Adv. 2017, 7, 1923–1926. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gram+ | Gram− | ||||
|---|---|---|---|---|---|
| S. aureus ATCC 29213 | B. subtilis ATCC 6051 | E. coli ATCC 25922 | P. aerug ATCC 27853 | ||
| Teixobactin 1 | 0.25 | 0.06 | 25 | >32 | |
| 1 | N-Me-d-Phe-Arg10-teixobactin | 2 | 0.5 | 64 | NI 1 |
| 2 | d-Phe-Arg10-teixobactin | 2 | 1 | 32 | NI |
| 3 | N-Me2-d-Phe-Arg10-teixobactin | 16 | 2 | 128 | NI |
| 4 | N-Bz-d-Phe-Arg10-teixobactin | 8 | 2 | NI | NI |
| 5 | N-De-d-Phe-Arg10-teixobactin 1 | NI | 128 | NI | NI |
| 6 | N-De2-d-Phe-Arg10-teixobactin | NI | 128 | NI | NI |
| 8 | N-Dec-d-Phe-Arg10-teixobactin 1 | NI | NI | NI | NI |
| 9 | N-Ac-d-Phe-Arg10-teixobactin 2 | NI | NI | NI | NI |
| 10 | N-Tmg-d-Phe-Arg10-teixobactin 1 | 256 | 64 | NI | NI |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monaim, S.A.H.A.; Noki, S.; Ramchuran, E.J.; El-Faham, A.; Albericio, F.; Torre, B.G.d.l. Investigation of the N-Terminus Amino Function of Arg10-Teixobactin. Molecules 2017, 22, 1632. https://doi.org/10.3390/molecules22101632
Monaim SAHA, Noki S, Ramchuran EJ, El-Faham A, Albericio F, Torre BGdl. Investigation of the N-Terminus Amino Function of Arg10-Teixobactin. Molecules. 2017; 22(10):1632. https://doi.org/10.3390/molecules22101632
Chicago/Turabian StyleMonaim, Shimaa A. H. Abdel, Sikabwe Noki, Estelle J. Ramchuran, Ayman El-Faham, Fernando Albericio, and Beatriz G. de la Torre. 2017. "Investigation of the N-Terminus Amino Function of Arg10-Teixobactin" Molecules 22, no. 10: 1632. https://doi.org/10.3390/molecules22101632