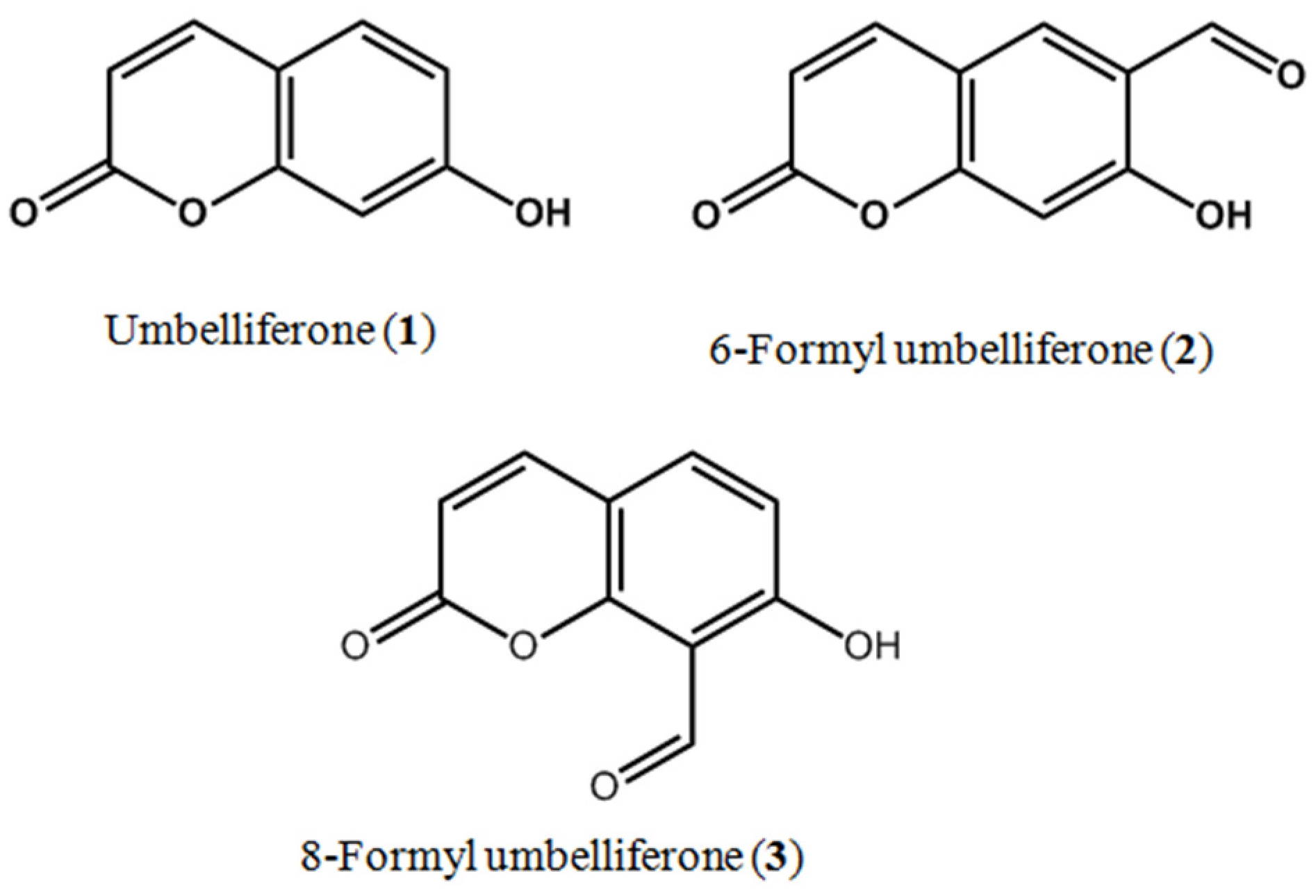
Kinetics and Molecular Docking Studies of 6-Formyl Umbelliferone Isolated from Angelica decursiva as an Inhibitor of Cholinesterase and BACE1

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Inhibitory Activity of Coumarins against AChE, BChE, and BACE1
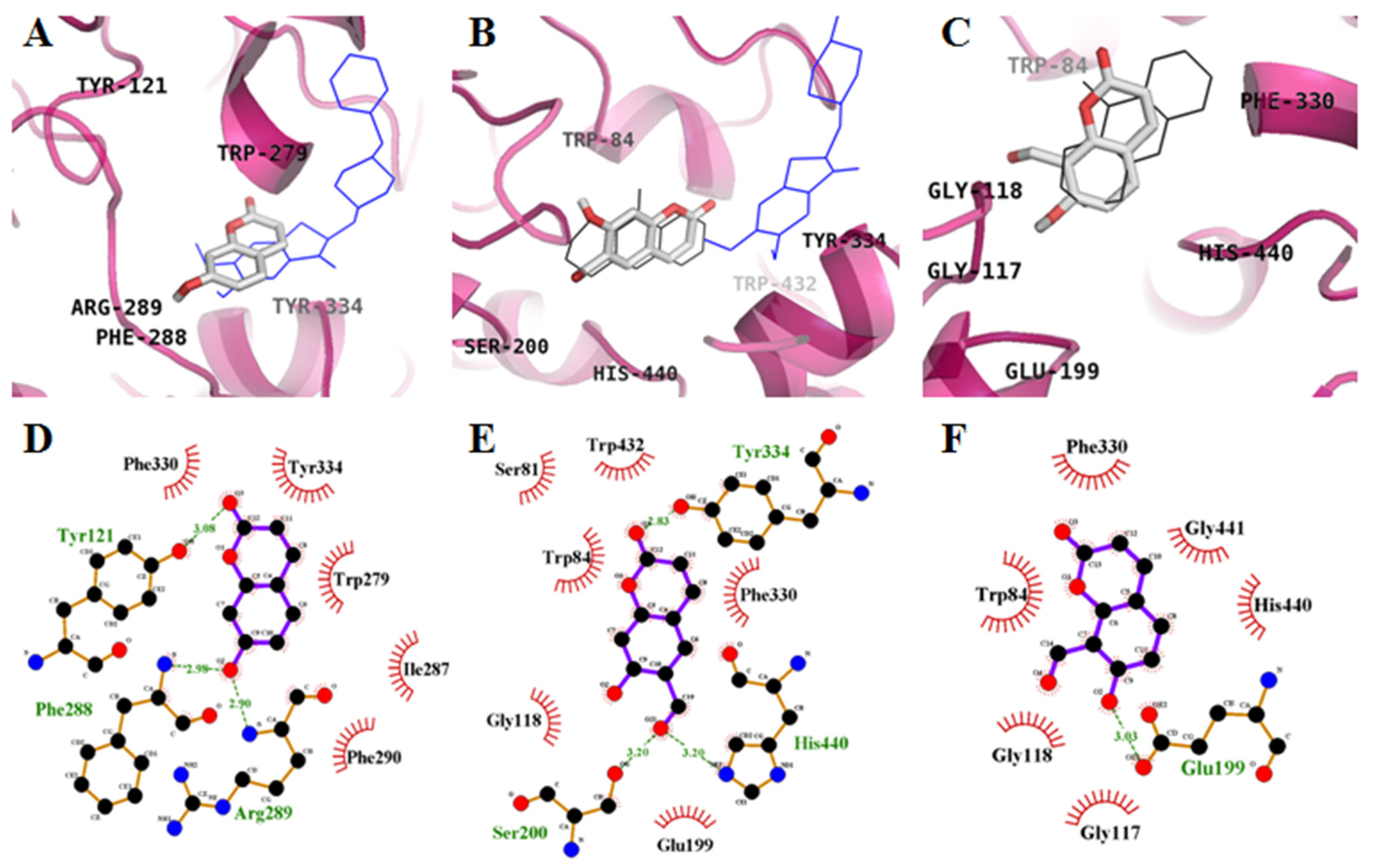
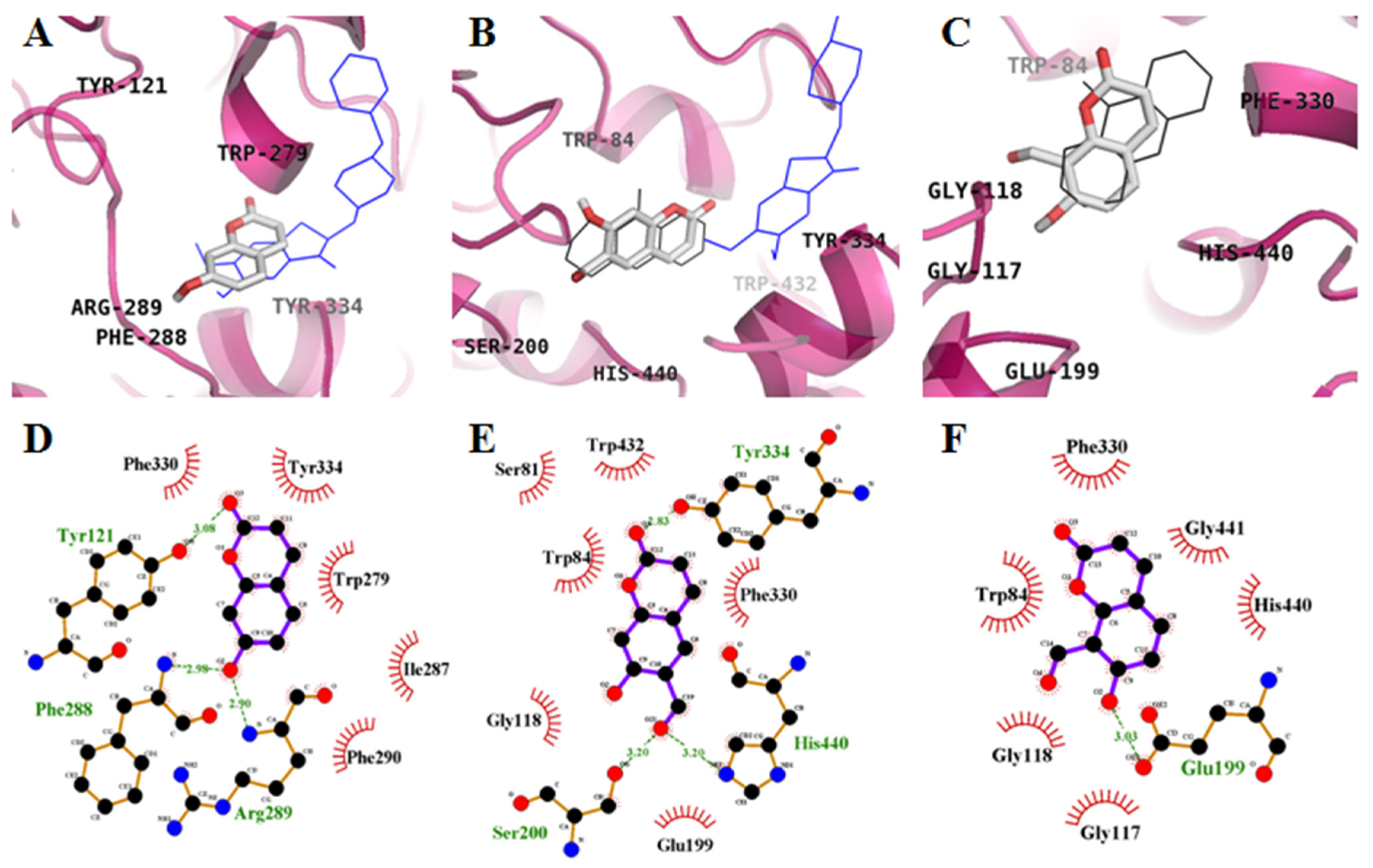
2.2. Molecular-Docking Study of the Inhibition of AChE by Coumarins
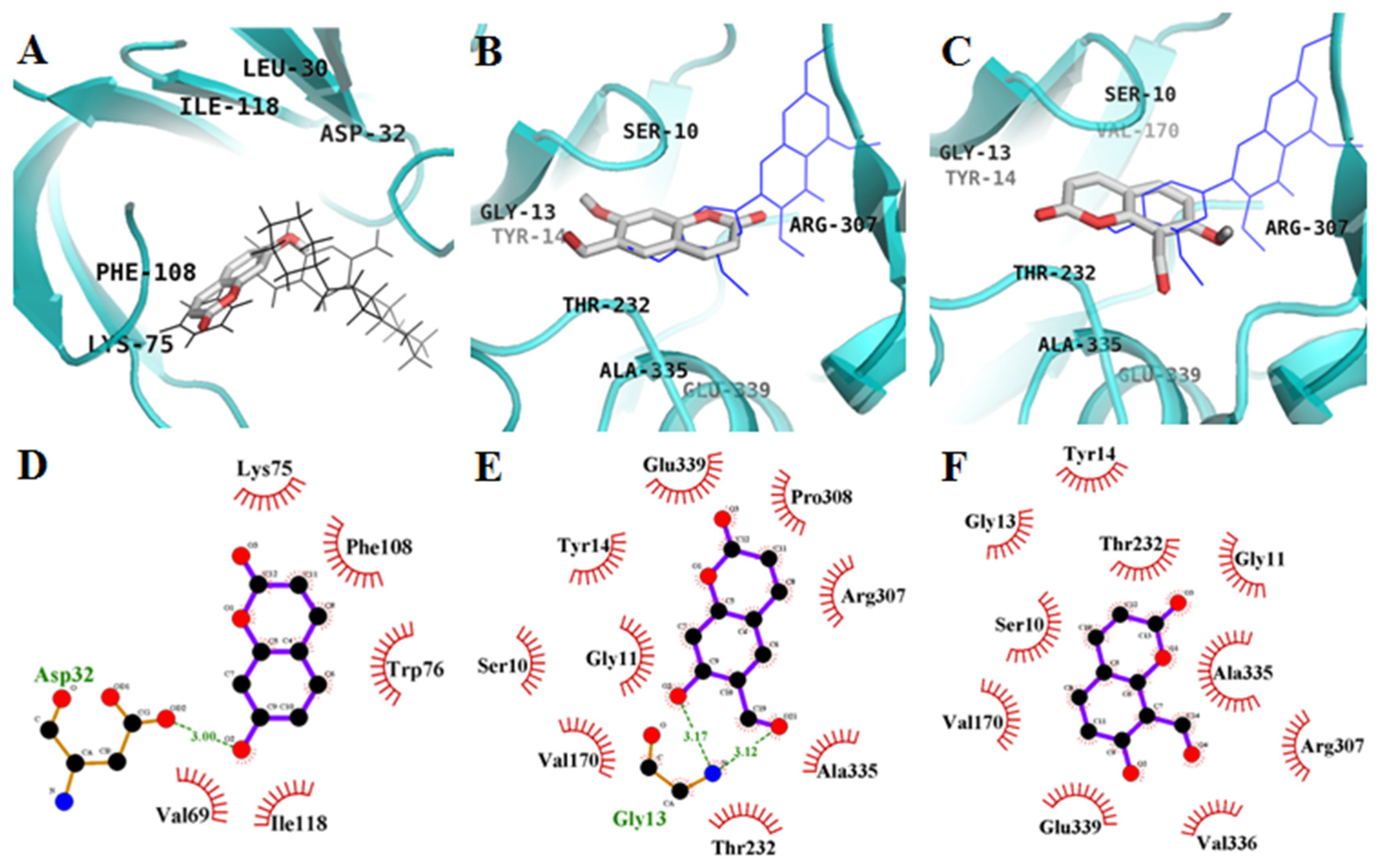
2.3. Molecular-Docking Study of the Inhibition of BACE1 by Coumarins
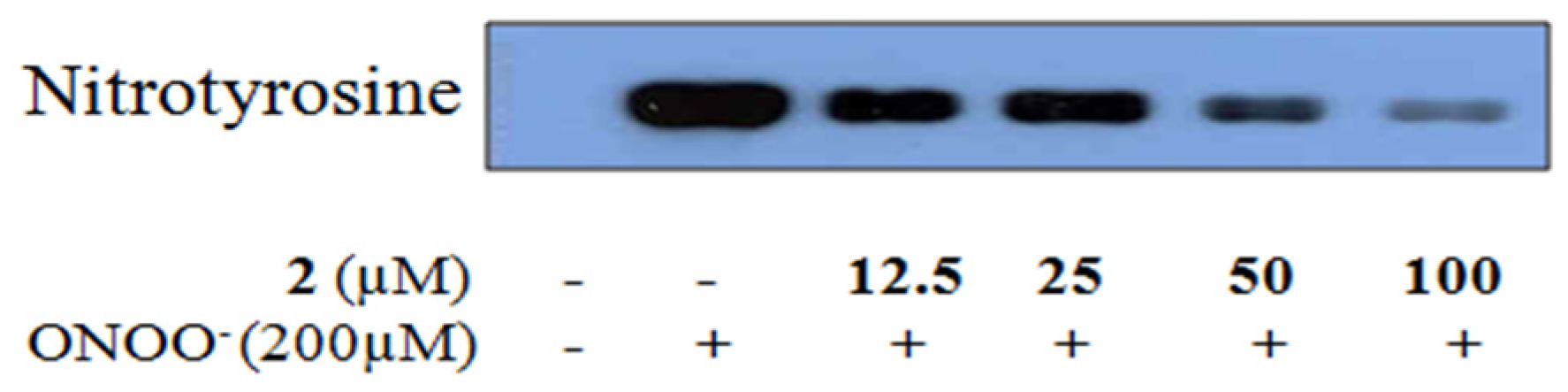
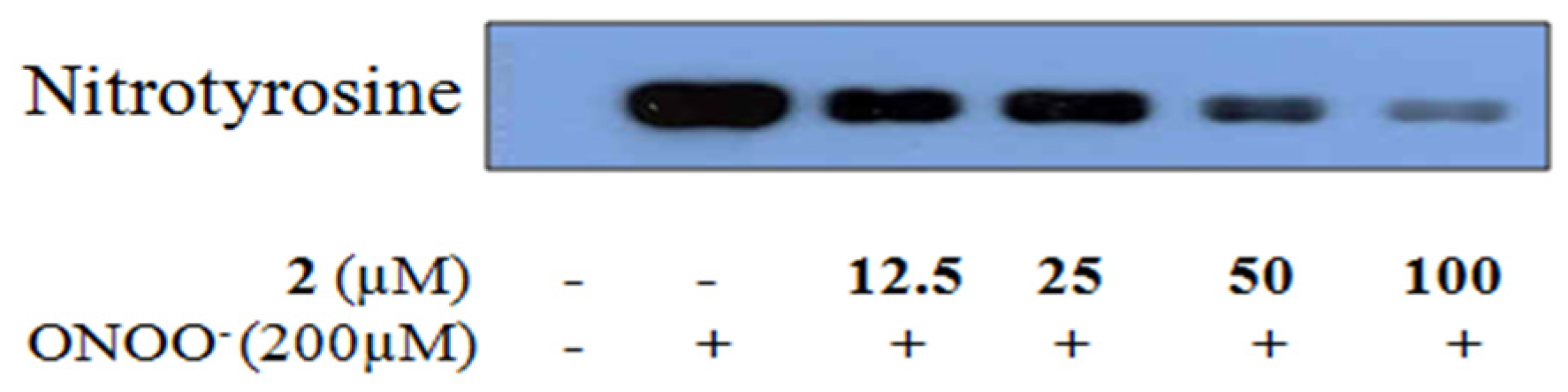
2.4. Inhibitory Activity of 6-Formyl Umbelliferone (2) against ONOO−-Mediated Tyrosine Nitration
3. Discussion
4. Material and Methods
4.1. General Experimental Procedures
4.2. Chemicals and Reagents
4.3. Isolation of Coumarins from A. Decursiva
4.4. Synthesis of 8-Formyl Umbelliferone
4.5. In Vitro BACE1 Enzyme Assay
4.6. In Vitro ChE Enzyme Assay
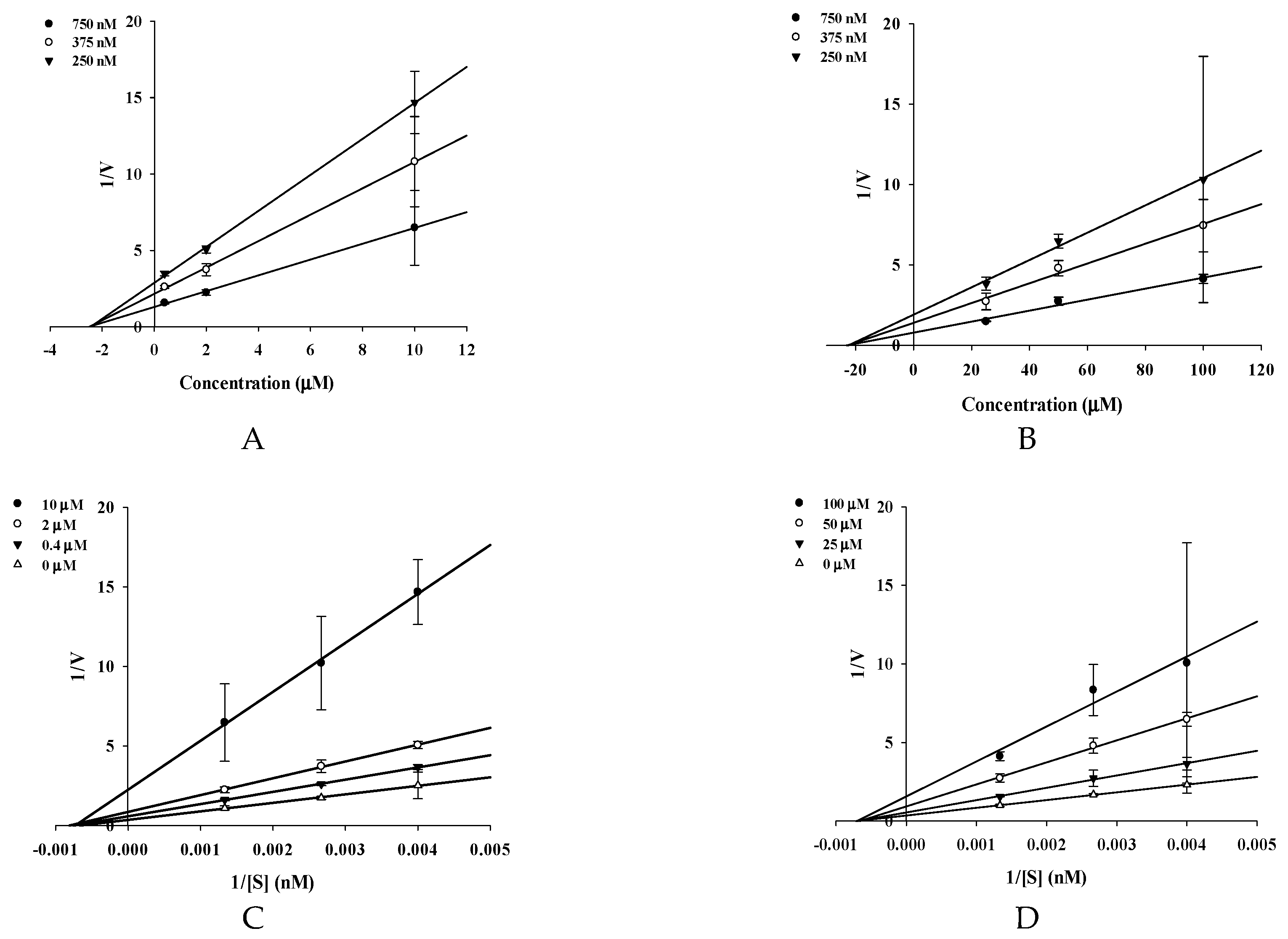
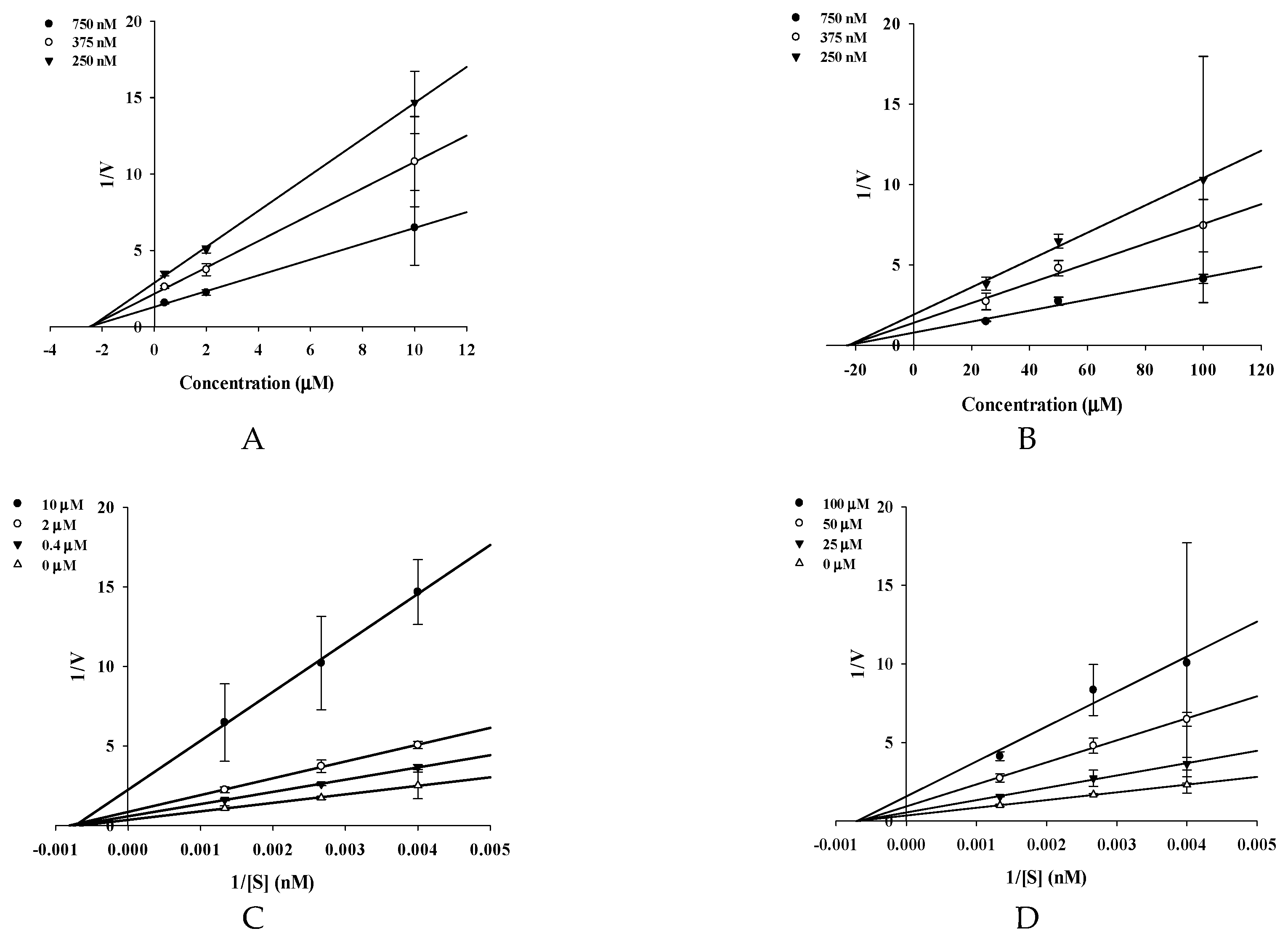
4.7. Kinetics of BACE1 Inhibition
4.8. Molecular Docking Simulation to Investigate AChE and BACE1 Inhibition Using AutoDock 4.2
4.9. Inhibition of ONOO−-Mediated Protein Tyrosine Nitration
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aisen, P.S.; Davis, K.L. The search for disease modifying treatment for Alzheimer disease. Neurology 1997, 48, 35–41. [Google Scholar] [CrossRef]
- Roberts, J.S.; Tersegno, S.M. Estimating and disclosing the risk of developing Alzheimer’s disease: Challenges, controversies and future directions. Future Neurol. 2010, 5, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Mikulca, J.A.; Nguyen, V.; Gajdosik, D.A.; Teklu, S.G.; Giunta, E.A.; Lessa, E.A.; Tran, C.H.; Terak, E.C.; Raffa, R.B. Potential novel targets for Alzheimer pharmacotherapy: II. Update on secretase inhibitors and related approaches. J. Clin. Pharm. Ther. 2014, 39, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Hemnani, T. Alzheimer’s disease pathogenesis and therapeutic interventions. J. Clin. Neurosci. 2004, 11, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Torreilles, F.; Salman-Tabcheh, S.; Guerin, M.; Torreilles, J. Neurodegenerative disorders: The role of peroxynitrite. Brain Res. Rev. 1999, 30, 153–163. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid β-peptide associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid β-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Tran, M.H.; Yamada, K.; Nakajima, A.; Mizuno, M.; He, J.; Kamei, H.; Nabeshima, T. Tyrosine nitration of a synaptic protein synaptophys in contributes to amyloid β-peptide-induced cholinergic dysfunction. Mol. Psychiatry 2003, 8, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Getchell, M.L.; Shah, D.S.; Buch, S.K.; Davis, D.G.; Getchell, T.V. 3-Nitrotyrosine immunoreactivity in olfactory receptor neurons of patients with Alzheimer’s disease: Implications for impaired odor sensitivity. Neurobiol. Aging 2003, 4, 663–673. [Google Scholar] [CrossRef]
- Vassar, R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Evin, G.; Lessene, G.; Wilkins, S. BACE inhibitors as potential drugs for the treatment of Alzheimer’s disease: Focus on bioactivity. Recent Pat. CNS Drug Discov. 2011, 6, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Schulz, V. Ginkgo extract or cholinesterase inhibitors in patients with dementia: What clinical trial and guidelines fail to consider. Phytomedicine 2003, 10, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Sarkhail, P. Traditional uses phytochemistry and pharmacological properties of the genus Peucedanum: A review. J. Ethnopharmacol. 2014, 156, 235–270. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Islam, M.N.; Ahn, B.R.; Jung, H.A.; Kim, B.W.; Choi, J.S. In vitro antioxidant and anti-inflammatory activities of Angelica decursiva. Arch. Pharm. Res. 2012, 35, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Ishita, I.J.; Islam, M.N.; Kim, Y.S.; Choi, R.J.; Sohn, H.S.; Jung, H.A.; Choi, J.S. Coumarins from Angelica decursiva inhibit lipopolysacharide-induced nitric oxide production in RAW 264.7 cells. Arch. Pharm. Res. 2016, 39, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, C.S.; Cho, S.H.; Chun, H.S.; Kim, J.K.; Kim, D.K. The effects of Angelica decursiva extract in the inhibition of cell proliferation and in the induction of apoptosis in osteogenic sarcoma cells. J. Med. Plants Res. 2009, 3, 241–245. [Google Scholar]
- Ali, M.Y.; Jung, H.A.; Choi, J.S. Anti-diabetic and anti-Alzheimer’s disease activities of Angelica decursiva. Arch. Pharm. Res. 2015, 38, 2216–2227. [Google Scholar]
- Ali, M.Y.; Jannat, S.; Jung, H.A.; Choi, R.J.; Roy, A.; Choi, J.S. Anti-Alzheimer potential of coumarins from Angelica decursiva and Artemisia capillaris and structure-activity analysis. Asian Pac. J. Trop. Med. 2016, 9, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Jannat, S.; Jung, H.A.; Jeong, H.O.; Chung, H.Y.; Choi, J.S. Coumarins from Angelica decursiva inhibit α-glucosidase activity and protein tyrosine phosphatase 1B. Chem. Biol. Interact. 2016, 252, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Choi, R.J.; Jin, S.E.; Kim, Y.S.; Ahn, B.R.; Zhao, D.; Jung, H.A.; Choi, J.S. Mechanism of anti-inflammatory activity of umbelliferone 6-carboxylic acid isolated from Angelica decursiva. J. Ethnopharmacol. 2012, 144, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Kostova, I.; Monolov, I.; Karaivonova, M. Synthesis, physicochemical characterization, and cytotoxic screening of new zirconium complexes with coumarin derivatives. Arch. Pharm. Pharm. Med. Chem. 2001, 334, 157–162. [Google Scholar] [CrossRef]
- Caffieri, S.; Lisa, F.D.; Bolesani, F.; Facco, M.; Semenzato, G.; Dall’Acqua, F.; Canton, M. The mitochondrial effects of novel apoptogenic molecules generated by psoralen photolysis as a crucial mechanism in PUVA therapy. Neoplasia 2007, 109, 4988–4994. [Google Scholar] [CrossRef] [PubMed]
- Youn, K.; Lee, J.; Ho, C.T.; Jun, M. Discovery of polymethoxyflavones from black ginger (Kaempferia parviflora) as potential β-secretase (BACE1) inhibitors. J. Funct. Food 2016, 20, 567–574. [Google Scholar] [CrossRef]
- Abascal, K.; Yarnell, E. Alzheimer’s disease: Part 2A botanical treatment plan. Altern. Complement. Ther. 2004, 10, 67–72. [Google Scholar] [CrossRef]
- Bredesen, D.E. Neurodegeneration in Alzheimer’s disease: Caspases and synaptic element interdependence. Mol. Neurodegener. 2009, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Ann. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Schliebs, R.; Arendt, T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 2011, 221, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Geula, C.; Darvesh, S. Butyrylcholinesterase, cholinergic neurotransmission and the pathology of Alzheimer’s disease. Drugs Today 2004, 40, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.; Laferla, F. Mechanisms of disease: Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D. Preventing Alzheimer’s disease. Science 2012, 337, 1488–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandy, S. Perspective: Prevention is better than cure. Nature 2011, 475, S15. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jin, L.; Peixue, L. Review of drugs for Alzheimer’s disease. Drug Discov. Ther. 2012, 6, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Kandalepas, P. The β-secretase enzyme BACE1 as a therapeutic target for Alzheimer’s disease. Alzheimers Res. Ther. 2011, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R. The role of oxidative stress in Alzheimer disease. Arch. Neurol. 1999, 56, 1449–1452. [Google Scholar] [CrossRef] [PubMed]
- Christen, Y. Oxidative stress and Alzheimer disease. Am. J. Clin. Nutr. 2000, 71, 621–629. [Google Scholar]
- Behl, C.; Moosmann, B. Antioxidant neuroprotection in Alzheimer’s disease as preventive and therapeutic approach. Free Radic. Biol. Med. 2002, 33, 182–191. [Google Scholar] [CrossRef]
- Marumoto, S.; Miyazawa, M. Structure-activity relationships for naturally occurring coumarins as β-secretase inhibitor. Bioorg. Med. Chem. 2012, 20, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; To, D.C.; Tran, M.H.; Oh, S.H.; Kim, J.A.; Ali, M.Y.; Woo, M.H.; Choi, J.S.; Min, B.S. Isolation of cholinesterase and β-secretase 1 inhibiting compounds from Lycopodiella cernua. Bioorg. Med. Chem. 2015, 23, 3126–3134. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Ali, M.Y.; Jung, H.J.; Jeong, H.O.; Chung, H.Y.; Choi, J.S. Inhibitory activities of major anthraquinones and other constituents from Cassia obtusifolia against β-secretase and cholinesterases. J. Ethnopharmacol. 2016, 191, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.M.; Lee, J.S.; Chuong, N.N.; Kim, J.A.; Oh, S.H.; Woo, M.H.; Choi, J.S.; Min, B.S. Kinetics and molecular docking studies of cholinesterase inhibitors derived from water layer of Lycopodiella cernua (L.) Pic. Serm. (II). Chem. Biol. Interact. 2016, 5, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Katsori, A.M.; Hadjipavlou-Litina, D. Coumarin derivatives: An updated patent review. Expert Opin. Ther. Pat. 2014, 24, 1323–1347. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Park, J.J.; Islam, M.N.; Jin, S.E.; Min, B.S.; Lee, J.H.; Sohn, H.S.; Choi, J.S. Inhibitory activity of coumarins from Artemisia capillaris against advanced glycation endproduct formation. Arch. Pharm. Res. 2012, 35, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Karimi, G.; Iranshahi, M.; Hosseinalizadeh, F.; Riahi, B.; Sahebkar, A. Screening of acetylcholinesterase inhibitory activity of terpenoid and coumarin derivatives from the genus Ferula. Pharmacologyonline 2010, 1, 566–574. [Google Scholar]
- Ukhov, S.V.; Kon’shin, M.E.; Odegova, T.F. Synthesis and antimicrobial activity of 2 imino coumarin-3-carboxylic acid amides. Pharm. Chem. J. 2001, 35, 364–365. [Google Scholar] [CrossRef]
- Vukovic, N.; Sukdolak, S.; Solujic, S.; Niciforovic, N. An efficient synthesis and antioxidant properties of novel imino and amino derivatives of 4-hydroxy coumarins. Arch. Pharm. Res. 2010, 33, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, N.; Dixneuf, P.H. Synthesis of triazole and coumarin compounds and their physiological activity. Top. Heterocycl. Chem. 2007, 10, 123–153. [Google Scholar]
- Qiao, Y.; Chen, B.; Yang, Y.; Wang, X.; Xu, Y.; Li, H. Rational design of a highly selective fluorescent sensor for L-histidine detection in aqueous solution. Dalton Trans. 2016, 45, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.J.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Dixon, M. The determination of enzyme inhibitor constant. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Seong, S.H.; Ali, M.Y.; Kim, H.R.; Jung, H.A.; Choi, J.S. BACE1 inhibitory activity and molecular docking analysis of meroterpenoids from Sargassum serratifolium. Bioorg. Med. Chem. 2017, 25, 3964–3970. [Google Scholar] [CrossRef] [PubMed]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds (umbelliferone, 6-formyl umbelliferone and 8-formyl umbelliferone) are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (µM) a | |||||
|---|---|---|---|---|---|---|
| AChE | BChE | SI b | BACE1 | Ki Value c | Inhibition Type d | |
| 1 | 105.48 ± 0.57 | 90.14 ± 0.02 | 0.85 | 168.54 ± 2.17 | - | - |
| 2 | 16.70 ± 1.62 | 27.90 ± 3.43 | 1.67 | 1.31 ± 0.01 | 2.27 | Noncompetitive |
| 3 | 19.13 ± 0.57 | 87.67 ± 0.48 | 4.58 | 39.82 ± 0.31 | 22.2 | Noncompetitive |
| Berberine e | 0.14 ± 0.08 | 9.81 ± 0.35 | 70.04 | - | - | - |
| Quercetin e | - | - | - | 22.08 ± 0.57 | - | - |
| Compounds | Binding Energy (kcal/mol) a | No. of H-Bond b | H-Bonds Interacting Residues c | van der Waals Interacting Residues d |
|---|---|---|---|---|
| 1 | −6.3 | 3 | Tyr121, Phe288, Arg289 | Trp279, Ile287, Phe290, Phe330, Tyr334 |
| 2 | −8.3 | 3 | Ser200, Tyr334, His440 | Ser81, Trp84, Gly118, Glu199, Phe330, Trp432 |
| 3 | −8.0 | 1 | Glu199 | Trp84, Gly117, Gly118, Phe330, His440, Gly441 |
| THA e | −9.8 | 1 | His440 | Tyr442, Phe330, Trp84, Gly118, Trp432, Gly441, Tyr334, Glu199 |
| Donepezil f | −10.6 | - | - | Tyr70, Ile275, Asp276, Trp279, Ile287, Phe288, Arg289, Tyr334, Tyr121, Ser286, Phe290, Phe330, Phe331 |
| Compounds | Binding Energy (kcal/mol) a | No. of H-Bond b | H-bonds Interacting Residues c | van der Waals Interacting Residues d |
|---|---|---|---|---|
| 1 | −5.4 | 1 | Asp32 | Lys75, Trp76, Val69, Phe108, Ile118 |
| 2 | −7.2 | 2 | Gly13 | Ser10, Gly11, Tyr14, Val170, Thr232, Arg307, Pro308, Ala335, Glu339 |
| 3 | −7.0 | - | - | Ser10, Gly11, Gly13, Tyr14, Val170, Thr232, Arg307, Ala335, Val336, Glu339 |
| QUD e | −9.3 | 4 | Asp228, Asp32, Gly230 | Lys107, Lys75, Gly74, Leu30, Thr231, Val69, Tyr198, Ile226, Thr329, Gly34, Arg235, Ser35, Tyr71, Ile118 |
| PMF f | −7.8 | 1 | Gly11 | Ser10, Tyr14, Thr232, Trp277, Glu303, Gln304, Leu306, Arg307, Pro308, Tyr320, Ala335, Val336, Glu339 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.Y.; Seong, S.H.; Reddy, M.R.; Seo, S.Y.; Choi, J.S.; Jung, H.A. Kinetics and Molecular Docking Studies of 6-Formyl Umbelliferone Isolated from Angelica decursiva as an Inhibitor of Cholinesterase and BACE1. Molecules 2017, 22, 1604. https://doi.org/10.3390/molecules22101604
Ali MY, Seong SH, Reddy MR, Seo SY, Choi JS, Jung HA. Kinetics and Molecular Docking Studies of 6-Formyl Umbelliferone Isolated from Angelica decursiva as an Inhibitor of Cholinesterase and BACE1. Molecules. 2017; 22(10):1604. https://doi.org/10.3390/molecules22101604
Chicago/Turabian StyleAli, Md Yousof, Su Hui Seong, Machireddy Rajeshkumar Reddy, Sung Yong Seo, Jae Sue Choi, and Hyun Ah Jung. 2017. "Kinetics and Molecular Docking Studies of 6-Formyl Umbelliferone Isolated from Angelica decursiva as an Inhibitor of Cholinesterase and BACE1" Molecules 22, no. 10: 1604. https://doi.org/10.3390/molecules22101604