Hydrogen-Bonding Interactions in Luminescent Quinoline-Triazoles with Dominant 1D Crystals


Abstract
:
1. Introduction
2. Results and Discussion
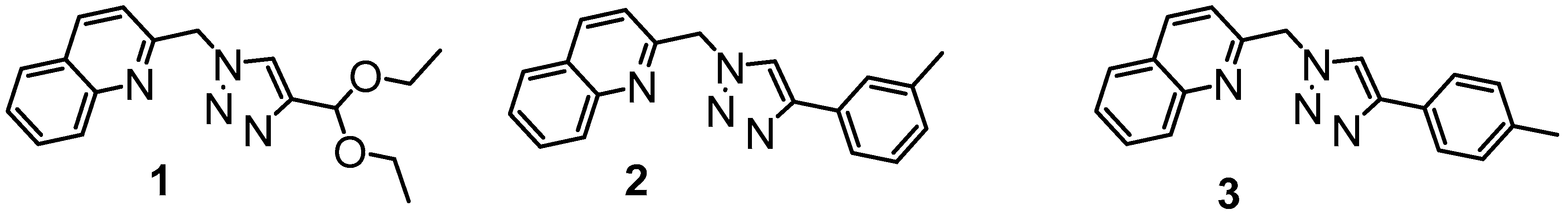
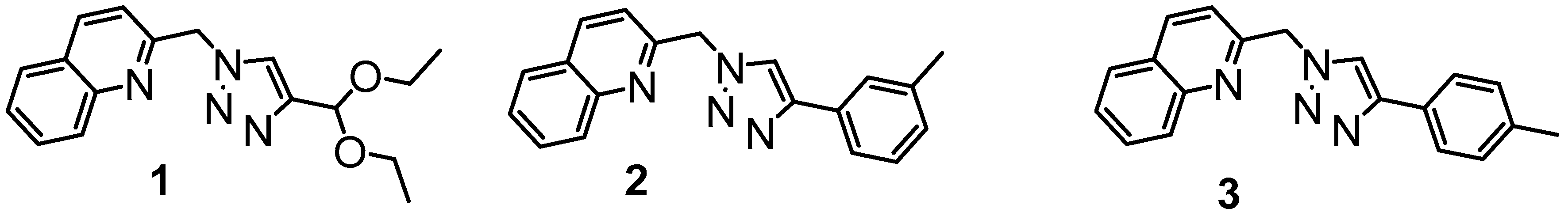
2.1. Synthesis
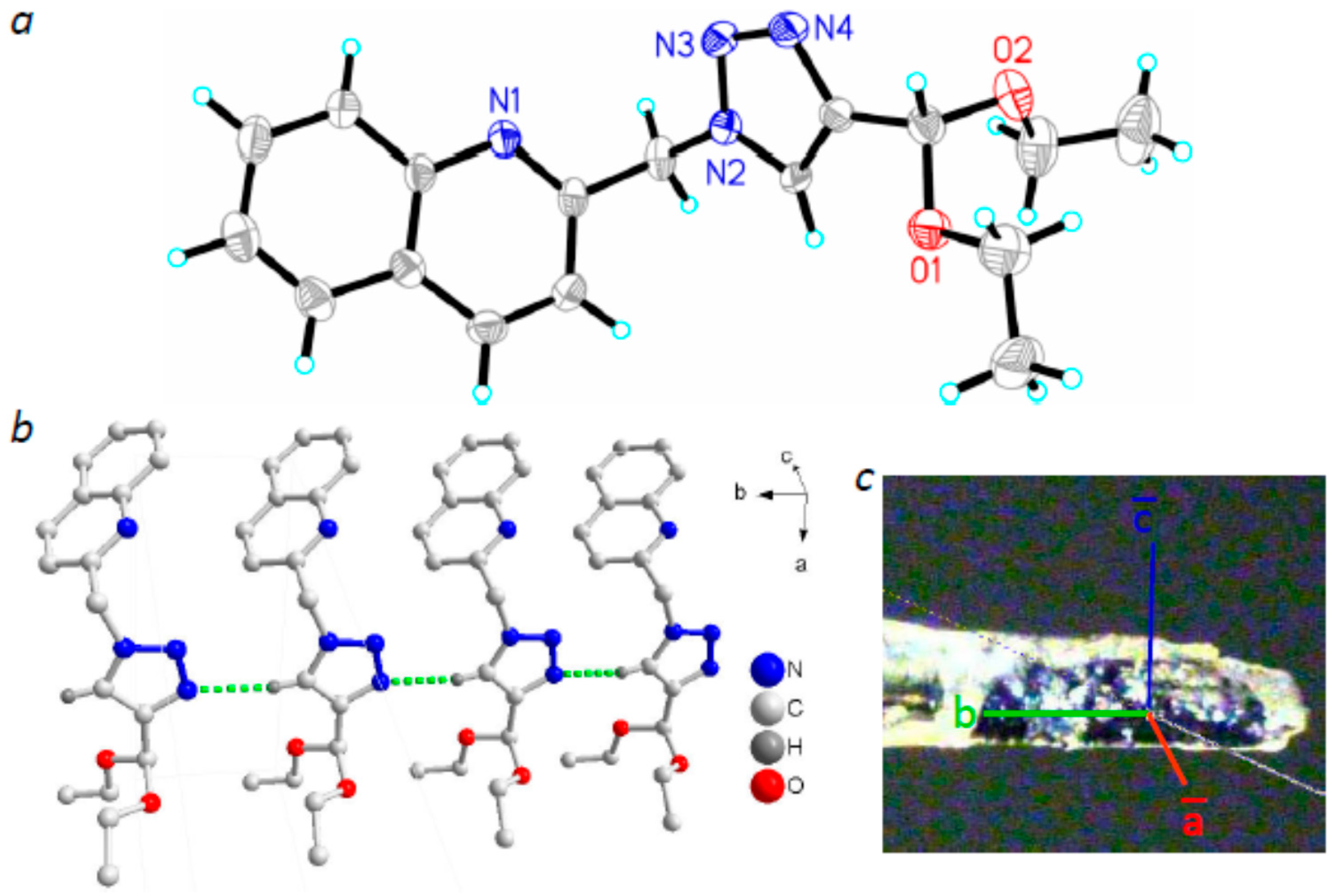
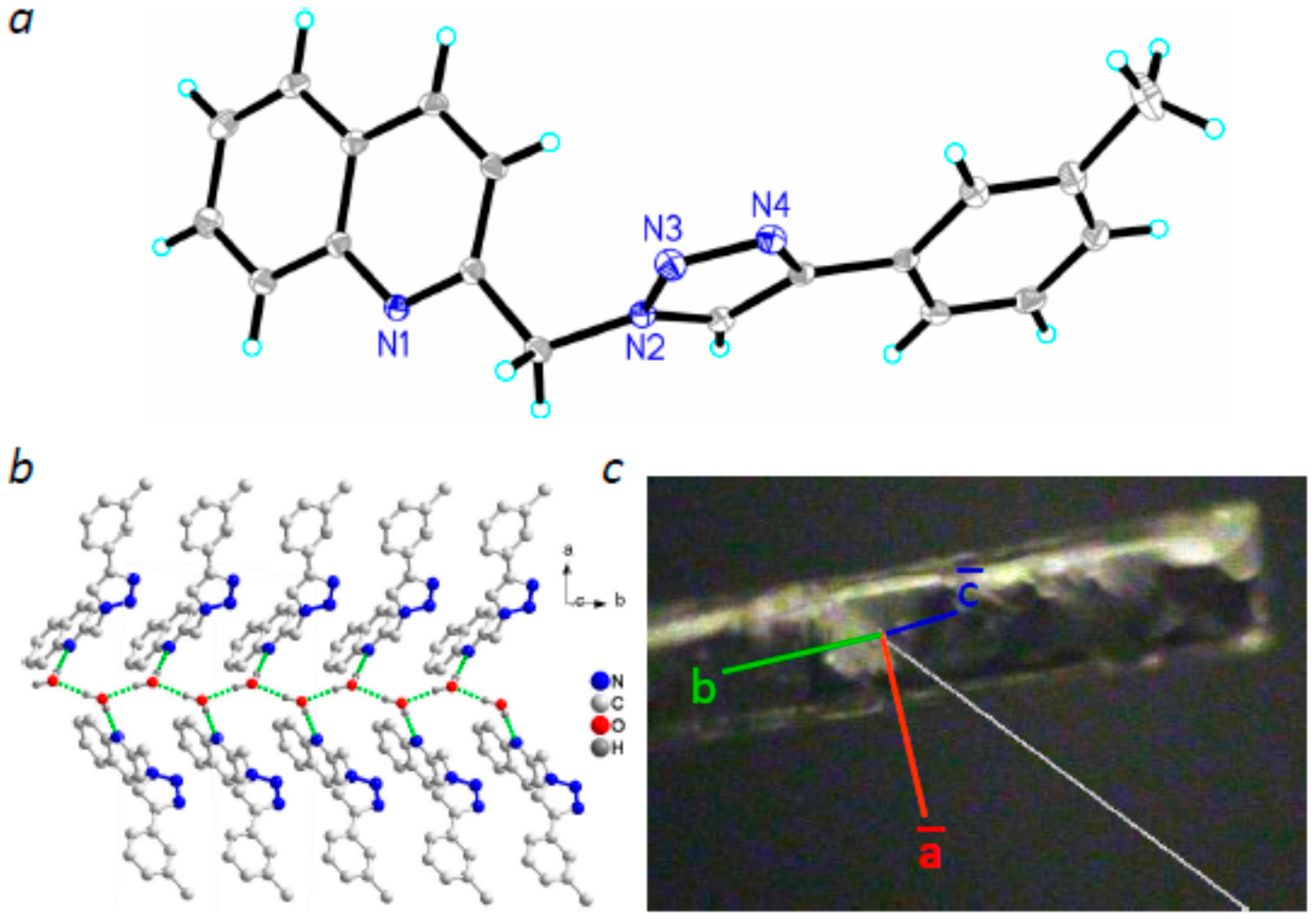
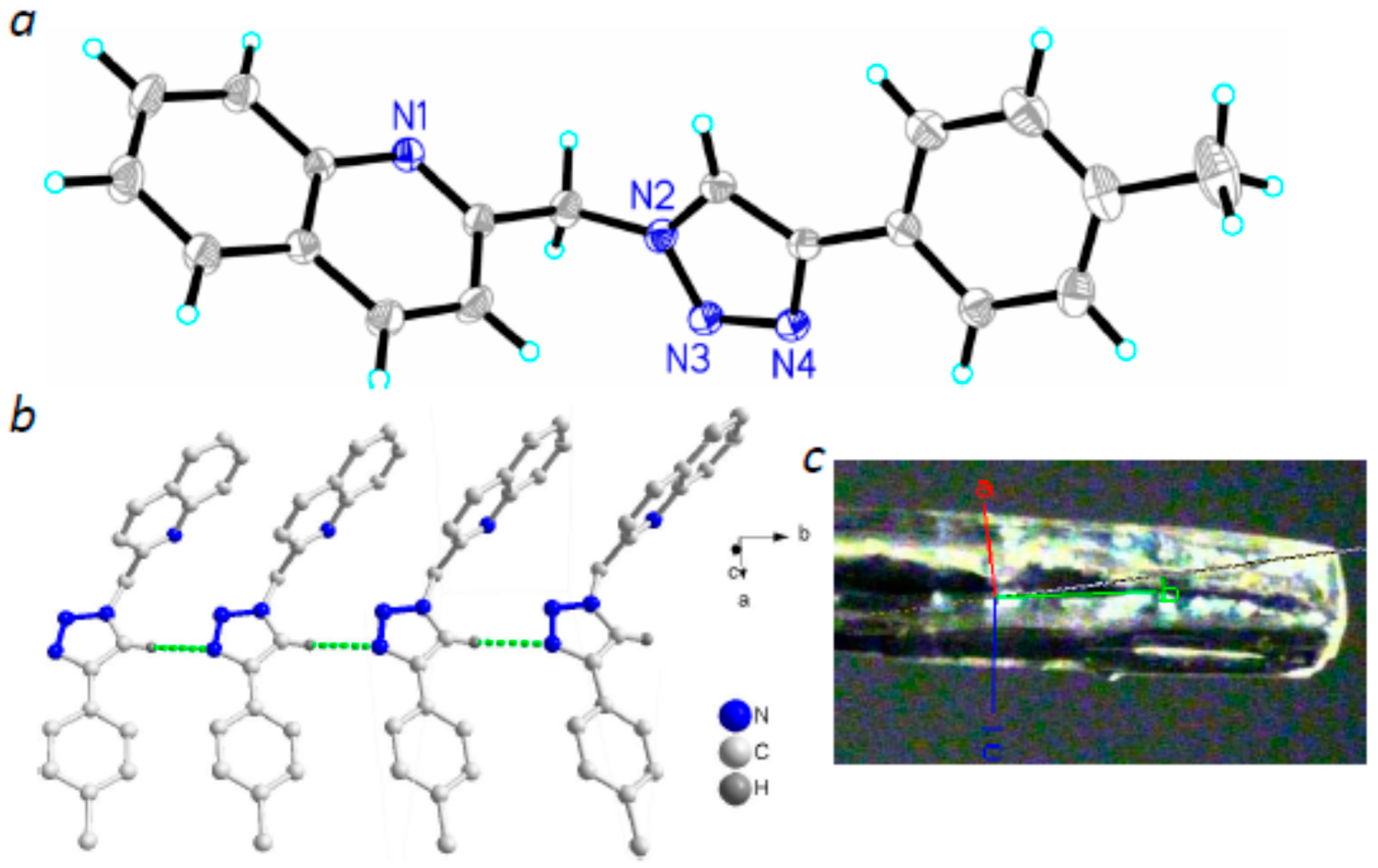
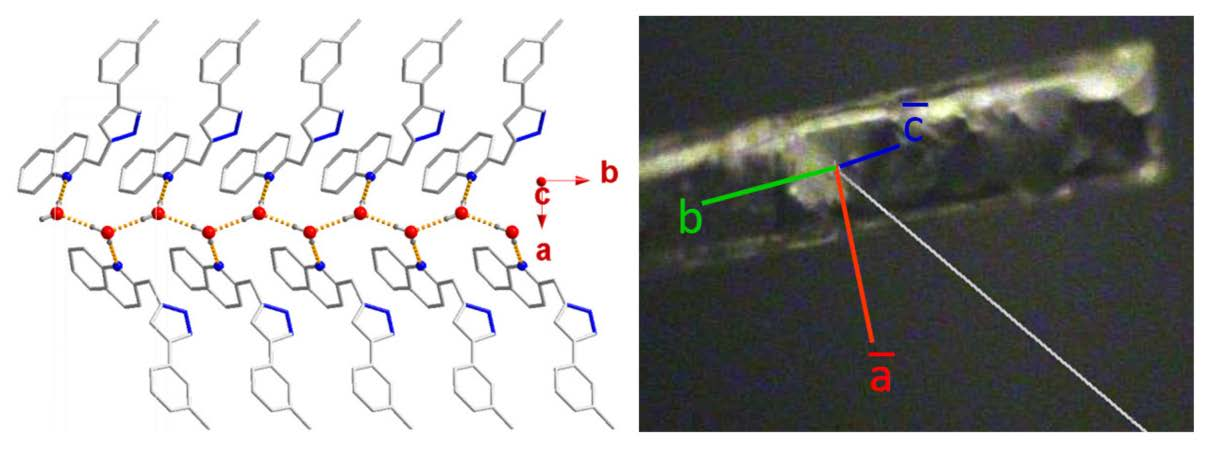
2.2. Molecular Structures
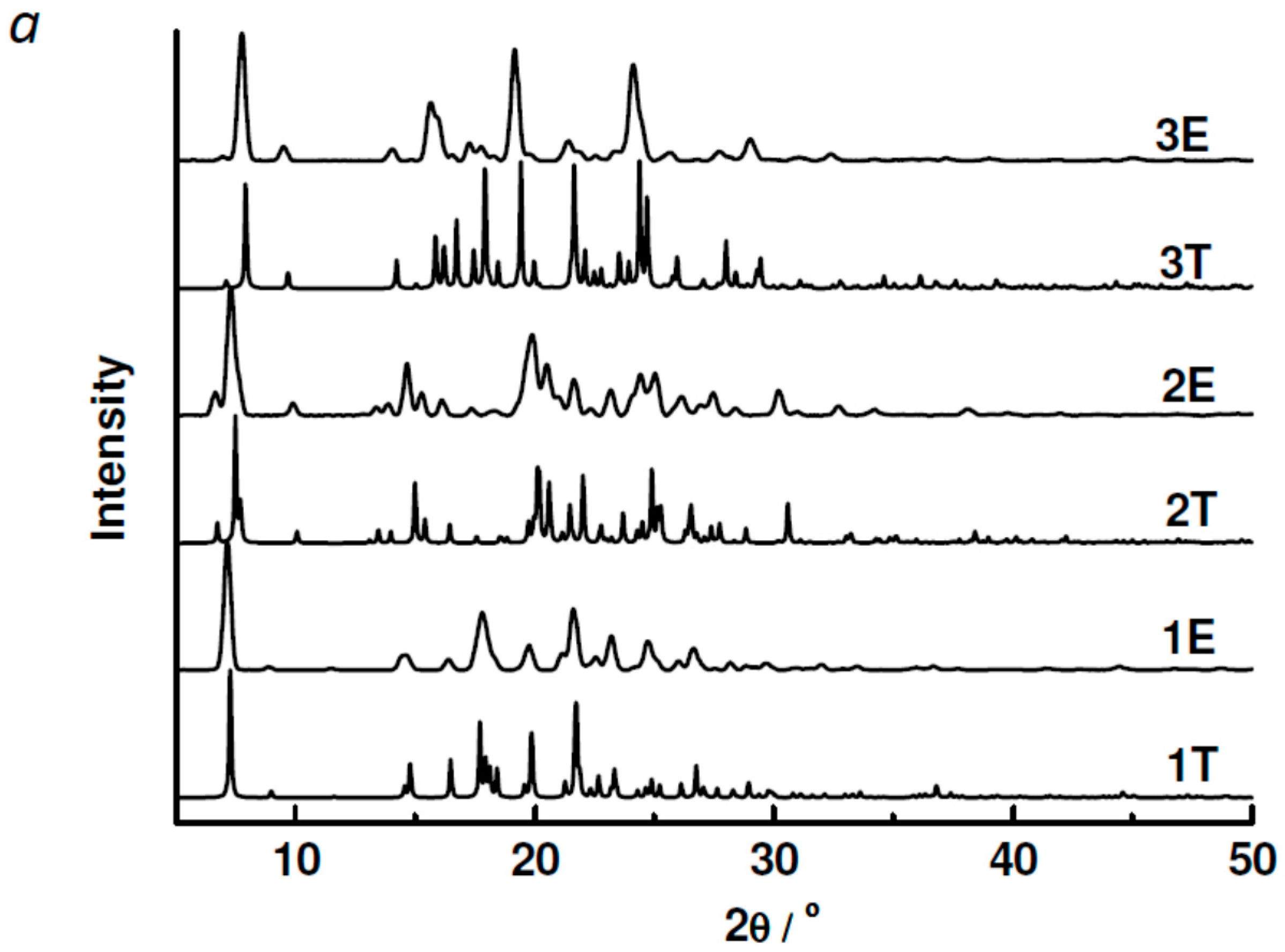
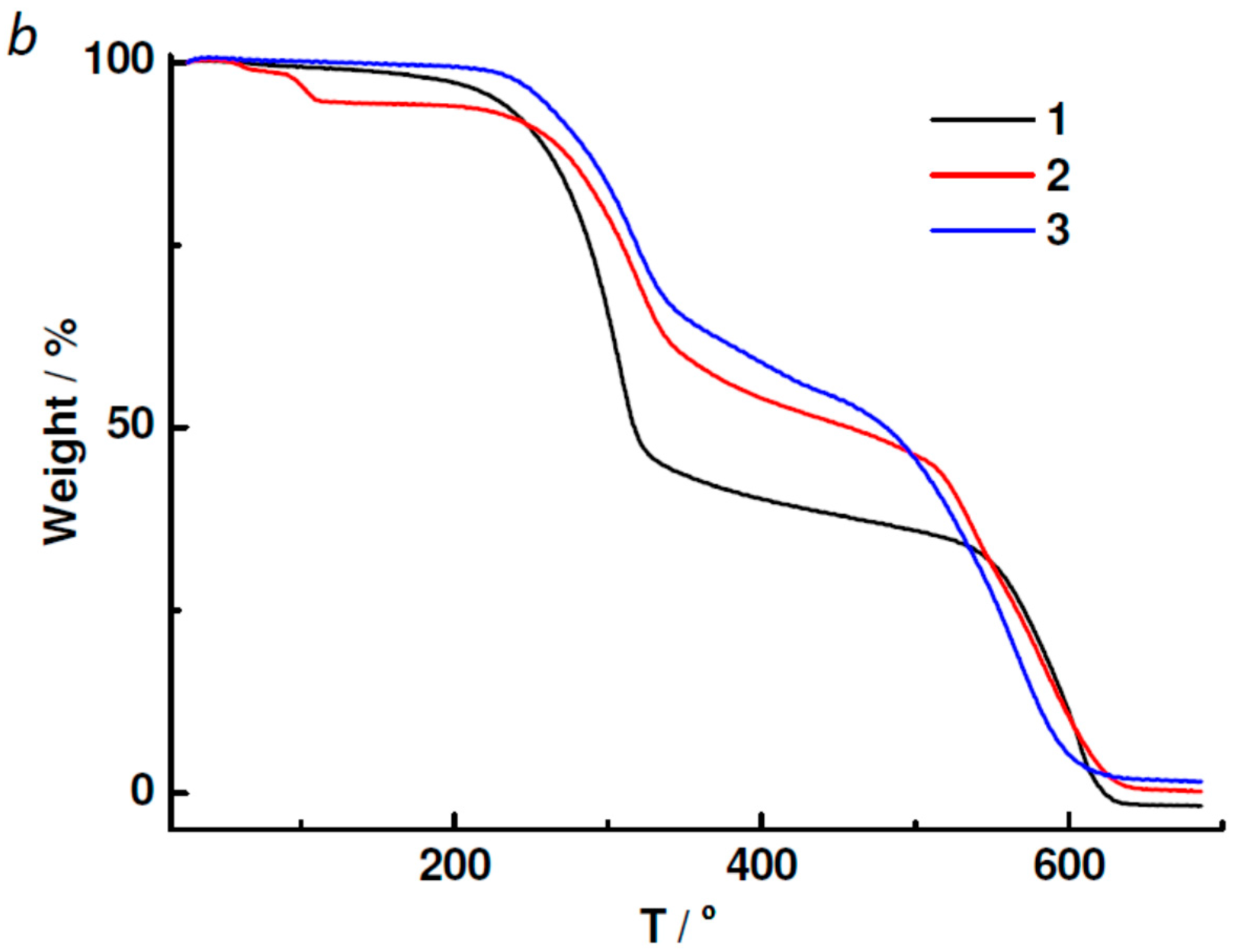
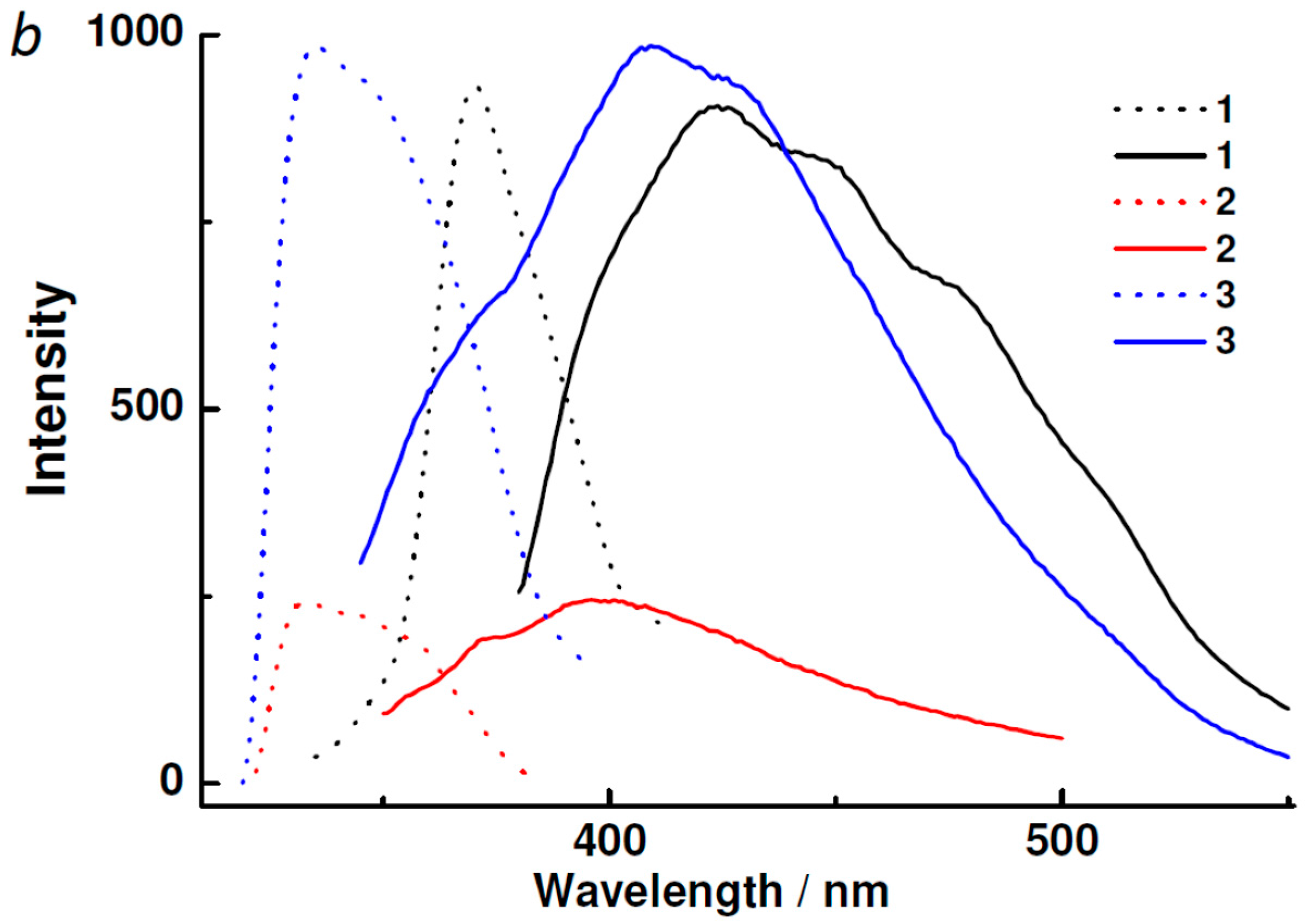
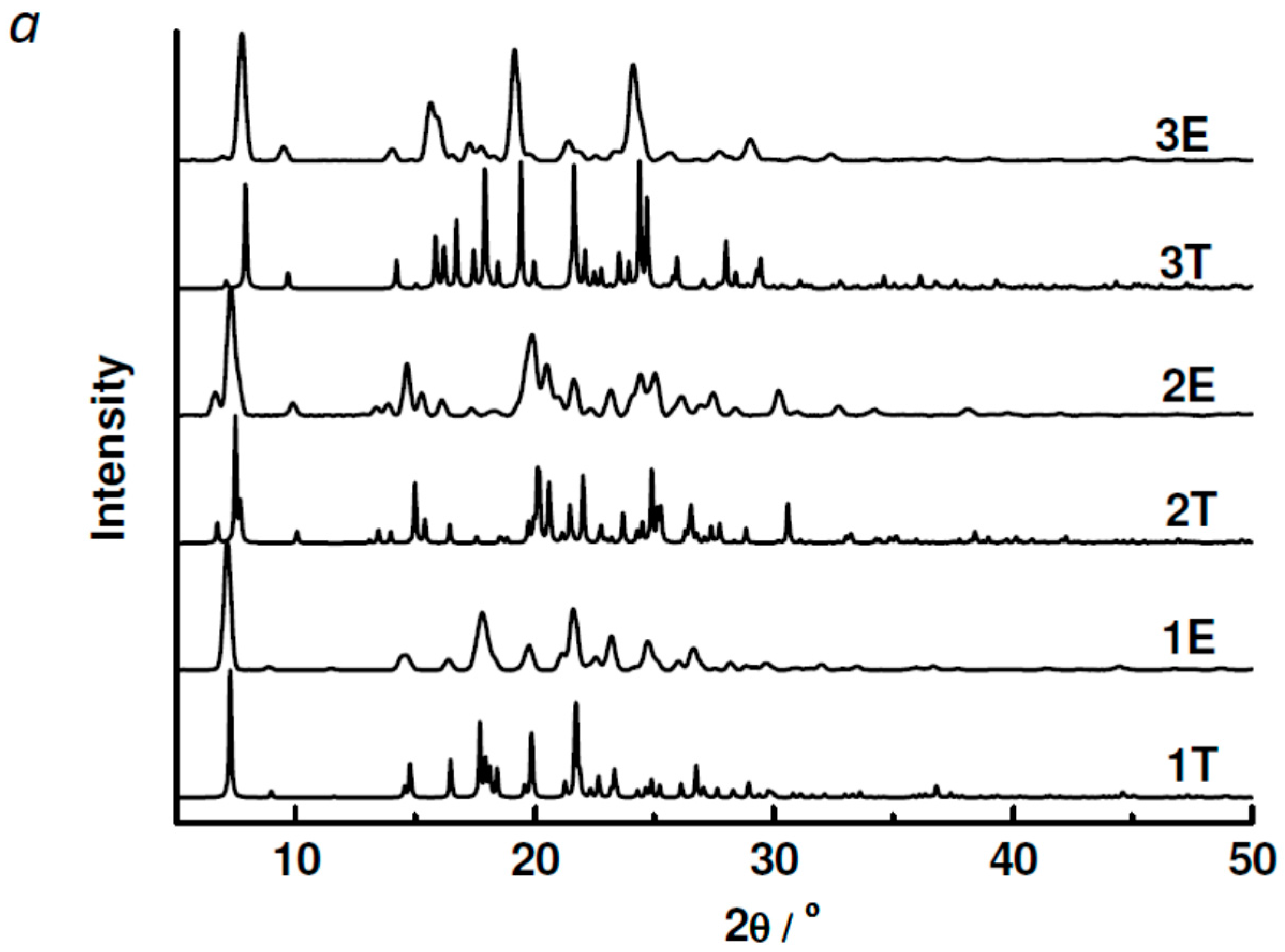
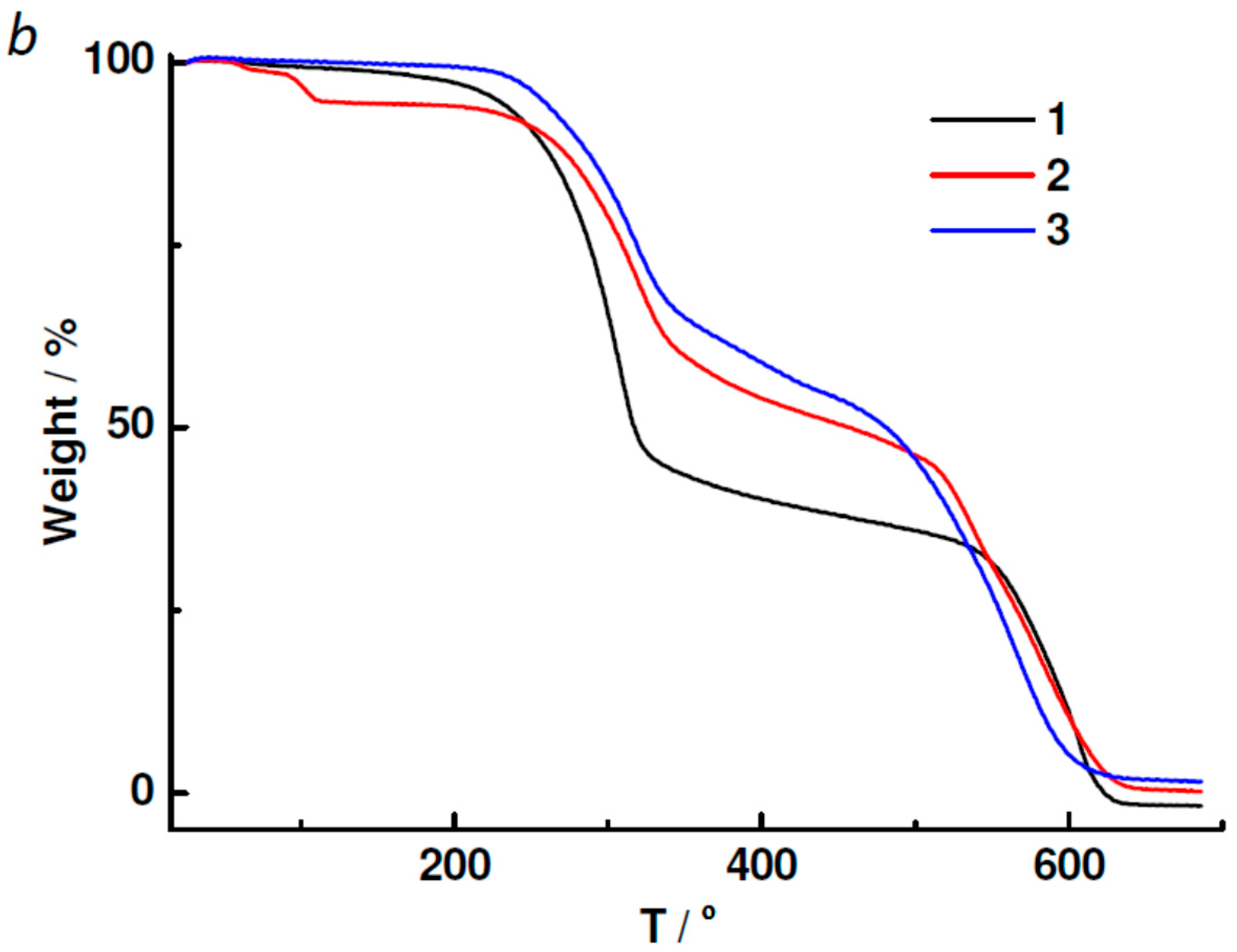
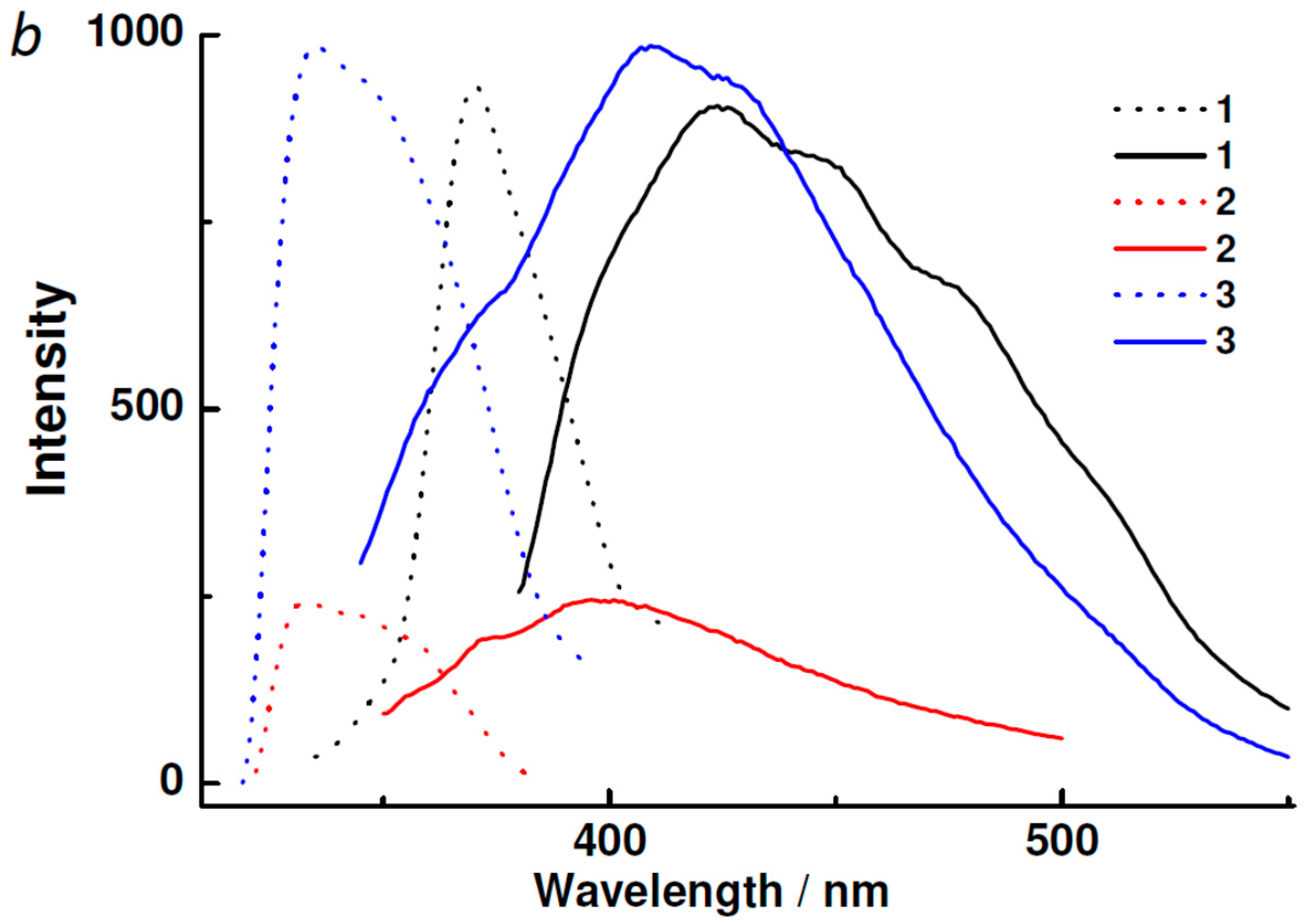
2.3. Powder XRD, TGA, UV-vis and Photoluminescent Spectroscopy
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Karmakar, A.; Illathvalappil, R.; Anothumakkool, B.; Sen, A.; Samanta, P.; Desai, A.V.; Kurungot, S.; Ghosh, S.K. Hydrogen-Bonded Organic Frameworks (HOFs): A New Class of Porous Crystalline Proton-Conducting Materials. Angew. Chem. Int. Ed. 2016, 55, 10667–10671. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Lee, J.; Ryu, J.-S. Design, synthesis, and evaluation of hinge-binder tethered 1,2,3-triazolylsalicylamide derivatives as Aurora kinase inhibitors. Bioorg. Med. Chem. 2016, 24, 2114–2124. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.-K.; Su, H.-F.; Xu, J.-H.; Wang, W.-G.; Kurmoo, M.; Lin, S.-C.; Tan, Y.-Z.; Jia, J.; Sun, D.; Zheng, L.-S. Hierarchical Assembly of a {MnII15MnIII4} Brucite Disc: Step-by-Step Formation and Ferrimagnetism. J. Am. Chem. Soc. 2016, 138, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; Fu, D.-J.; Yue, X.-X.; Liu, Y.-C.; Song, J.; Sun, H.-H.; Liu, H.-M.; Zhang, Y.-B. Design, Synthesis and Structure-Activity Relationships of Novel Chalcone-1,2,3-triazole-azole Derivates as Antiproliferative Agents. Molecules 2016, 21, 653. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, H.-Y.; Ren, Z.-G.; Lang, J.-P. Unique Deca- and Tetranuclear Halocuprate(I) Clusters of a Clamplike Ligand: Isolation, Structure, and Luminescence Properties. Eur. J. Inorg. Chem. 2014, 2014, 824–830. [Google Scholar] [CrossRef]
- Han, L.-L.; Li, Z.-H.; Chen, J.-S.; Wang, X.-P.; Sun, D. Solution and Mechanochemical Syntheses of Two Novel Cocrystals: Ligand Length Modulated Interpenetration of Hydrogen-Bonded 2D 63-hcb Networks Based on a Robust Trimeric Heterosynthon. Cryst. Growth Des. 2014, 14, 1221–1226. [Google Scholar] [CrossRef]
- Gadre, S.R.; Yeole, S.D.; Sahu, N. Quantum Chemical Investigations on Molecular Clusters. Chem. Rev. 2014, 114, 12132–12173. [Google Scholar] [CrossRef] [PubMed]
- Shishkin, O.V.; Zubatyuk, R.I.; Shishkina, S.V.; Dyakonenko, V.V.; Medviediev, V.V. Role of supramolecular synthons in the formation of the supramolecular architecture of molecular crystals revisited from an energetic viewpoint. Phys. Chem. Chem. Phys. 2014, 16, 6773–6786. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, Y.-H.; Hao, H.-J.; Liu, F.-J.; Wen, Y.-M.; Huang, R.-B.; Zheng, L.-S. Solvent-Controlled Rare Case of a Triple Helical Molecular Braid Assembled from Proton-Transferred Sebacic Acid. Cryst. Growth Des. 2011, 11, 3323–3327. [Google Scholar] [CrossRef]
- Liu, H.; Xu, J.; Li, Y.; Li, Y. Aggregate Nanostructures of Organic Molecular Materials. Acc. Chem. Res. 2010, 43, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Tiana, D.; Hendon, C.H.; Walsh, A.; Vaid, T.P. Computational screening of structural and compositional factors for electrically conductive coordination polymers. Phys. Chem. Chem. Phys. 2014, 16, 14463–14472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, J.; Reddy, P.; Dunietz, B.D.; Gavini, V. End-Group Influence on Frontier Molecular Orbital Reorganization and Thermoelectric Properties of Molecular Junctions. J. Phys. Chem. Lett. 2013, 4, 3825–3833. [Google Scholar] [CrossRef]
- Bai, S.-Q.; Young, D.J.; Hor, T.S.A. Nitrogen-rich azoles as ligand spacers in coordination polymers. Chem. Asian J. 2011, 6, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy frameworks: insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.R.; Peruffo, M.; Unwin, P.R. Quantitative Plane-Resolved Crystal Growth and Dissolution Kinetics by Coupling In Situ Optical Microscopy and Diffusion Models: The Case of Salicylic Acid in Aqueous Solution. Cryst. Growth Des. 2013, 13, 614–622. [Google Scholar] [CrossRef]
- Moh, P.Y.; Cubillas, P.; Anderson, M.W.; Attfield, M.P. Revelation of the Molecular Assembly of the Nanoporous Metal Organic Framework ZIF-8. J. Am. Chem. Soc. 2011, 133, 13304–13307. [Google Scholar] [CrossRef] [PubMed]
- Adolf, C.R.R.; Ferlay, S.; Kyritsakas, N.; Hosseini, M.W. Welding Molecular Crystals. J. Am. Chem. Soc. 2015, 137, 15390–15393. [Google Scholar] [CrossRef] [PubMed]
- Jassal, A.K.; Sharma, S.; Hundal, G.; Hundal, M.S. Structural Diversity, Thermal Studies, and Luminescent Properties of Metal Complexes of Dinitrobenzoates: A Single Crystal to Single Crystal Transformation from Dimeric to Polymeric Complex of Copper(II). Cryst. Growth Des. 2015, 15, 79–93. [Google Scholar] [CrossRef]
- Padgett, C.W.; Arman, H.D.; Pennington, W.T. Crystal Structures Elucidated from X-ray Powder Diffraction Data without Prior Indexing. Cryst. Growth Des. 2007, 7, 367–372. [Google Scholar] [CrossRef]
- Chen, T.; Sun, Z.; Song, C.; Ge, Y.; Luo, J.; Lin, W.; Hong, M. Bulk Crystal Growth and Optical and Thermal Properties of the Nonlinear Optical Crystal l-Histidinium-4-nitrophenolate 4-Nitrophenol (LHPP). Cryst. Growth Des. 2012, 12, 2673–2678. [Google Scholar] [CrossRef]
- Taylor, R. It Isn’t, It Is: The C–H···X (X = O, N, F, Cl) Interaction Really Is Significant in Crystal Packing. Cryst. Growth Des. 2016, 16, 4165–4168. [Google Scholar] [CrossRef]
- Vologzhanina, A.V.; Sokolov, A.V.; Purygin, P.P.; Zolotarev, P.N.; Blatov, V.A. Knowledge-Based Approaches to H-Bonding Patterns in Heterocycle-1-Carbohydrazoneamides. Cryst. Growth Des. 2016, 16, 6354–6362. [Google Scholar] [CrossRef]
- Paz, F.A.A.; Klinowski, J.; Vilela, S.M.F.; Tomé, J.P.C.; Cavaleiro, J.A.S.; Rocha, J. Ligand design for functional metal–organic frameworks. Chem. Soc. Rev. 2012, 41, 1088–1110. [Google Scholar] [PubMed]
- Zhao, X.-L.; Sun, W.-Y. The organic ligands with mixed N-/O-donors used in construction of functional metal—Organic frameworks. CrystEngComm 2014, 16, 3247–3258. [Google Scholar] [CrossRef]
- Liu, H.-K.; Sadler, P.J. Metal Complexes as DNA Intercalators. Acc. Chem. Res. 2011, 44, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, Z.; Bai, S.-Q.; Hor, T.S.A. “Click-and-click”—hybridised 1,2,3-triazoles supported Cu(I) coordination polymers for azide–alkyne cycloaddition. Dalton Trans. 2013, 42, 9437–9443. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, W.S.; Michaels, H.A.; Simmons, J.T.; Clark, R.J.; Dalal, N.S.; Zhu, L. Apparent Copper(II)-Accelerated Azide−Alkyne Cycloaddition. Org. Lett. 2009, 11, 4954–4957. [Google Scholar] [CrossRef] [PubMed]
- Crowley, J.D.; McMorran, D.A. “Click-Triazole” Coordination Chemistry: Exploiting 1,4-Disubstituted-1,2,3-Triazoles as Ligands. Top. Heterocycl. Chem. 2012, 28, 31–84. [Google Scholar]
- Sun, D.; Yuan, S.; Wang, H.; Lu, H.-F.; Feng, S.-Y.; Sun, D.-F. Luminescence thermochromism of two entangled copper-iodide networks with a large temperature-dependent emission shift. Chem. Commun. 2013, 49, 6152–6154. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.-Q.; Jiang, L.; Young, D.J.; Hor, T.S.A. Luminescent [Cu4I4] aggregates and [Cu3I3]-cyclic coordination polymers supported by quinolyl-triazoles. Dalton Trans. 2015, 44, 6075–6081. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.-Q.; Fang, C.-J.; He, Z.; Gao, E.-Q.; Yan, C.-H.; Hor, T.S.A. Chelating Schiff base assisted azide-bridged Mn(II), Ni(II) and Cu(II) magnetic coordination polymers. Dalton Trans. 2012, 41, 13379–13387. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.-Q.; Gao, E.-Q.; He, Z.; Fang, C.-J.; Yue, Y.-F.; Yan, C.-H. Manganese Azides Based on Co-Ligands with a Flexible Tail: Diverse Structural and Magnetic Properties. Eur. J. Inorg. Chem. 2006, 2006, 407–415. [Google Scholar] [CrossRef]
- Bai, S.-Q.; Jiang, L.; Sun, B.; Young, D.J.; Hor, T.S.A. Five Cu(I) and Zn(II) clusters and coordination polymers of 2-pyridyl-1,2,3-triazoles: Synthesis, structures and luminescence properties. CrystEngComm 2015, 17, 3305–3311. [Google Scholar] [CrossRef]
- Bai, S.-Q.; Kai, D.; Ke, K.L.; Lin, M.; Jiang, L.; Jiang, Y.; Young, D.J.; Loh, X.J.; Li, X.; Hor, T.S.A. A Triazolyl-Pyridine-Supported CuI Dimer: Tunable Luminescence and Fabrication of Composite Fibers. ChemPlusChem, 2015, 80, 1235–1240. [Google Scholar] [CrossRef]
- Bai, S.-Q.; Jiang, L.; Young, D.J.; Hor, T.S.A. Hybrid 1,2,3-Triazole Supported CuII Complexes: Tuning Assembly and Weak Interaction-Driven Crystal Growth. Aust. J. Chem. 2016, 69, 372–378. [Google Scholar] [CrossRef]
- SAINT Software Reference Manual; Version 6.0; Bruker AXS Inc.: Madison, WI, USA, 2003.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |
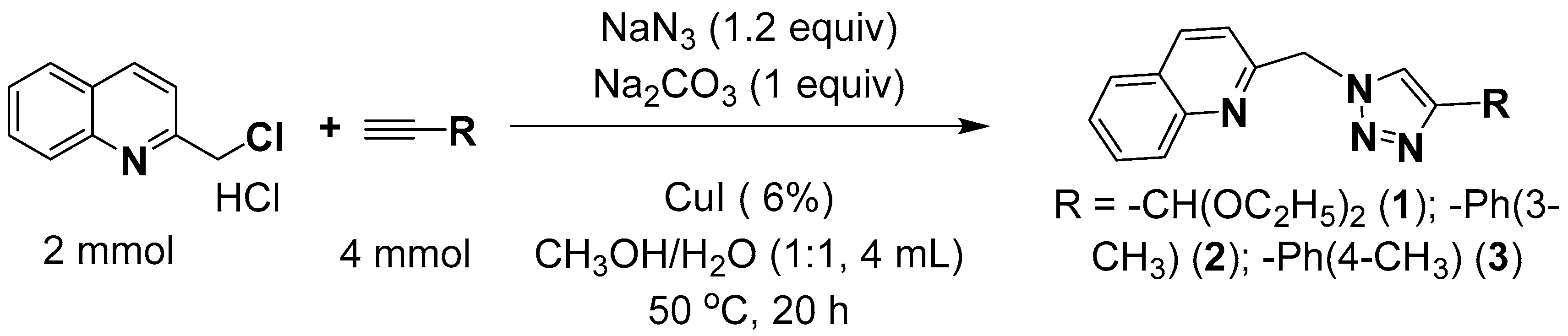


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
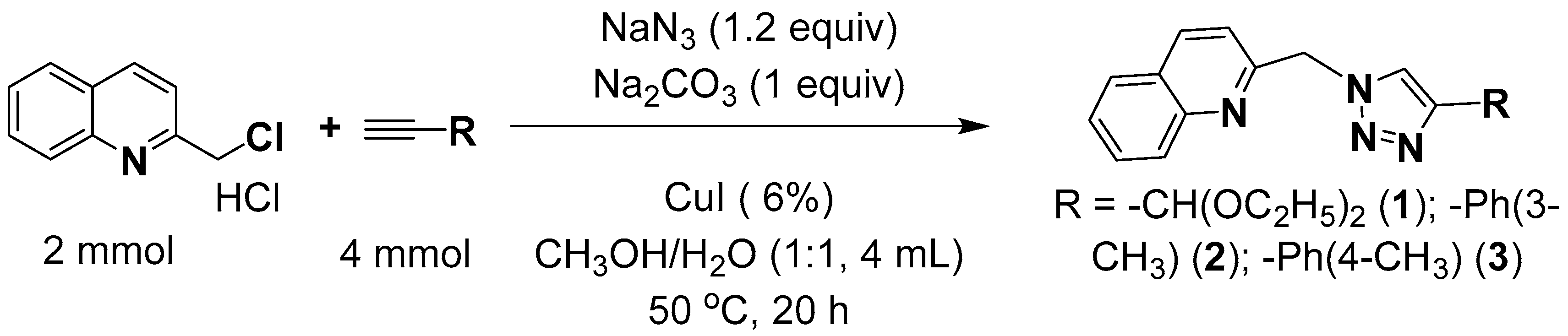{kind=link}
| D−H···A | D−H (Å) | D···A (Å) | H···A (Å) | ∠D−H···A (°) |
|---|---|---|---|---|
| 1 | ||||
| C11−H···N4A | 0.93 | 3.333(4) | 2.44 | 162 |
| Symmetry code A: x, y−1, z. | ||||
| 2 | ||||
| O1−H···N1 | 0.89 | 2.850(2) | 1.96 | 172 |
| O1−H···O1A | 0.84 | 2.793(2) | 1.96 | 177 |
| Symmetry code A: 0.5−x, 0.5+y, z. | ||||
| 3 | ||||
| C11−H···N4A | 0.95 | 3.455(2) | 2.61 | 149 |
| Symmetry code A: x, 1+y, z. | ||||
| Compound | 1 | 2·H2O | 3 |
|---|---|---|---|
| Formula | C17H20N4O2 | C19H18N4O | C19H16N4 |
| MW | 312.37 | 318.37 | 300.36 |
| T/K | 298(2) | 110(2) | 150(2) |
| Crystal size/mm3 | 0.65 × 0.16 × 0.04 | 1.10 × 0.30 × 0.05 | 1.40 × 0.20 × 0.10 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21/c | Pbca | P21/c |
| a/Å | 12.574(1) | 23.617(1) | 12.638(1) |
| b/Å | 5.5153(4) | 5.2305(2) | 5.4474(4) |
| c/Å | 24.654(2) | 26.299(1) | 22.694(2) |
| α/° | 90 | 90 | 90 |
| β/° | 104.563(2) | 90 | 99.956(2) |
| γ/° | 90 | 90 | 90 |
| V/Å3 | 1654.8(2) | 3248.6(2) | 1538.8(2) |
| Z | 4 | 8 | 4 |
| Dcalc/g cm−3 | 1.254 | 1.302 | 1.297 |
| μ/mm−1 | 0.085 | 0.084 | 0.080 |
| θ range/° | 1.67–25.88 | 1.55–26.42 | 1.64–26.38 |
| Reflections collected | 44361 | 29756 | 24940 |
| Independent reflections [Rint] | 3182 [0.0522] | 3349 [0.0392] | 3148 [0.0313] |
| Parameters | 208 | 225 | 208 |
| GOF | 1.059 | 1.025 | 1.038 |
| R1 (I > 2σ(I)) | 0.0920 | 0.0386 | 0.0429 |
| wR2 (all data) | 0.2857 | 0.1108 | 0.1217 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, S.-Q.; Young, D.J.; Hor, T.S.A. Hydrogen-Bonding Interactions in Luminescent Quinoline-Triazoles with Dominant 1D Crystals. Molecules 2017, 22, 1600. https://doi.org/10.3390/molecules22101600
Bai S-Q, Young DJ, Hor TSA. Hydrogen-Bonding Interactions in Luminescent Quinoline-Triazoles with Dominant 1D Crystals. Molecules. 2017; 22(10):1600. https://doi.org/10.3390/molecules22101600
Chicago/Turabian StyleBai, Shi-Qiang, David James Young, and T. S. Andy Hor. 2017. "Hydrogen-Bonding Interactions in Luminescent Quinoline-Triazoles with Dominant 1D Crystals" Molecules 22, no. 10: 1600. https://doi.org/10.3390/molecules22101600