Molecular Dynamic Simulation of Space and Earth-Grown Crystal Structures of Thermostable T1 Lipase Geobacillus zalihae Revealed a Better Structure

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussions
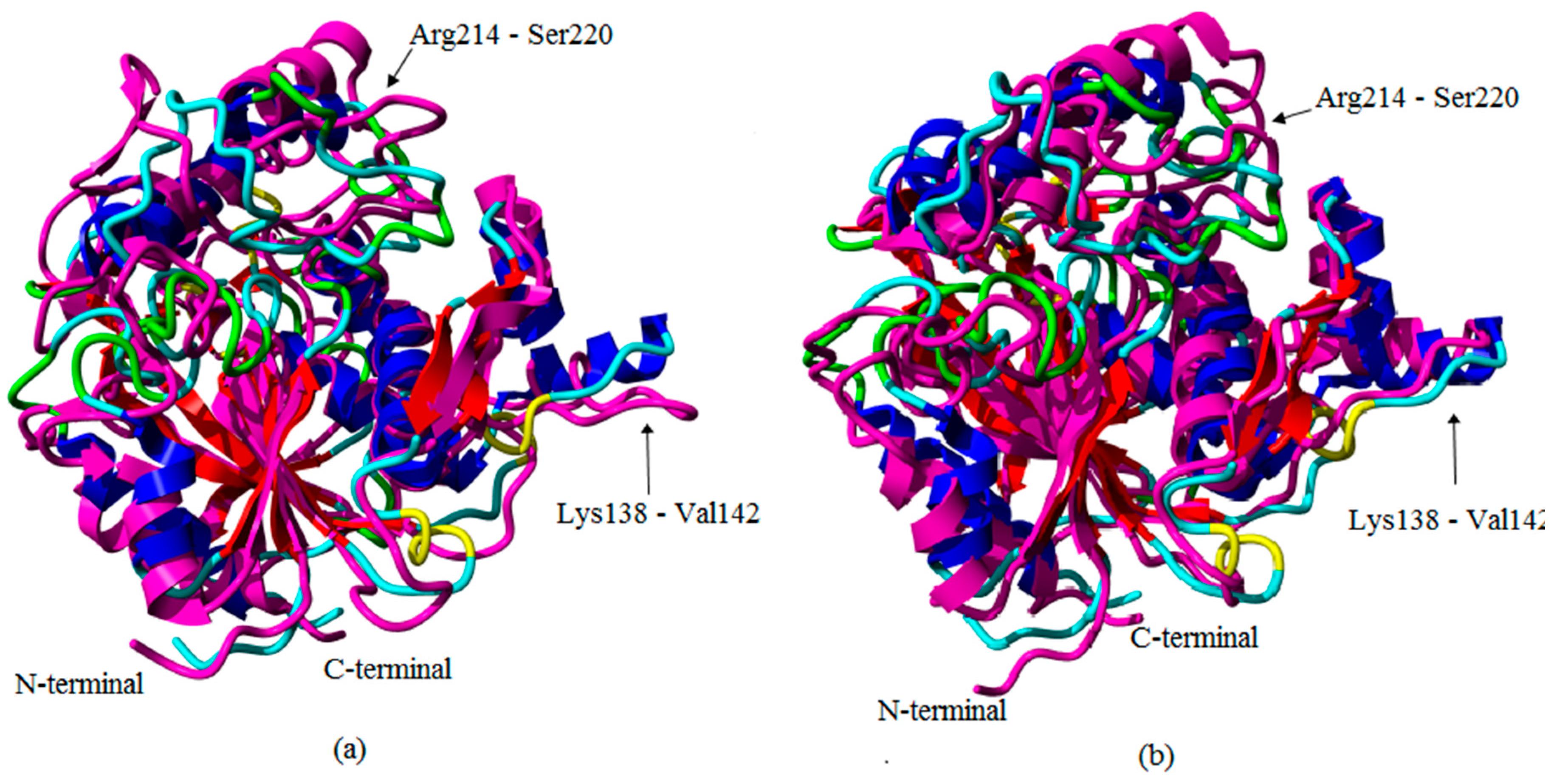
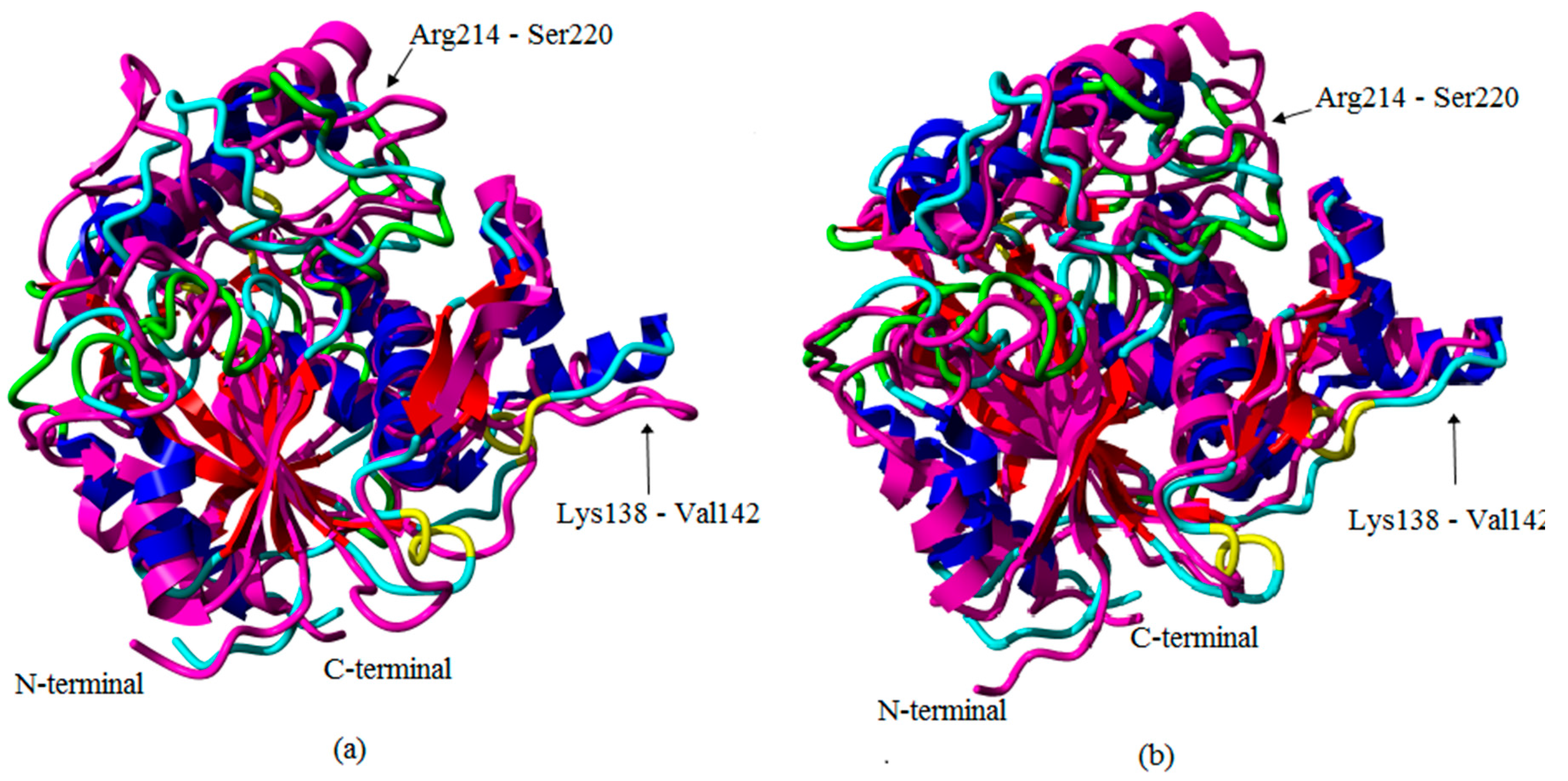
2.1. Preliminary in Silico Analyses of T1 Lipase Crystal Structures of Space- and Earth-Grown
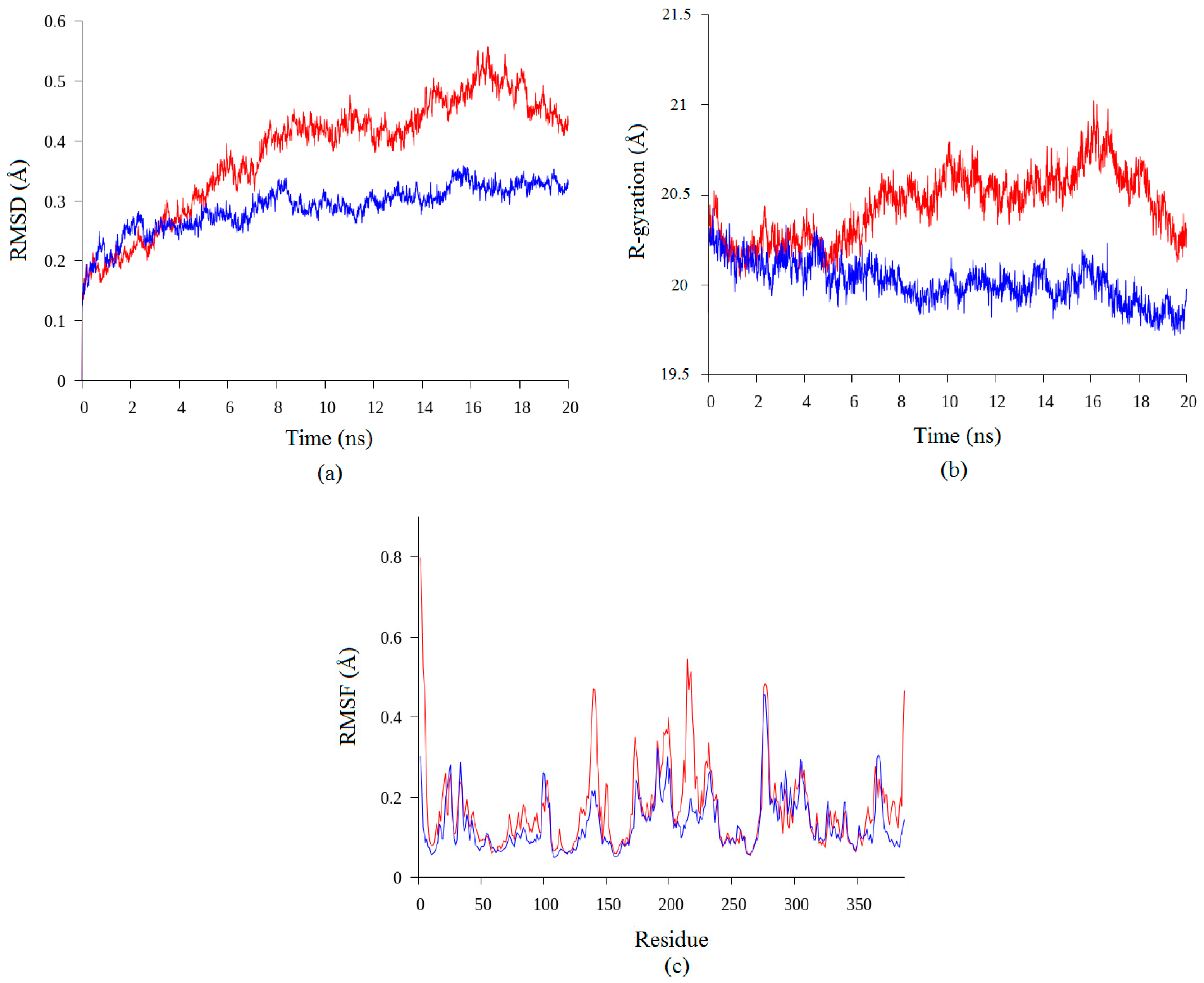
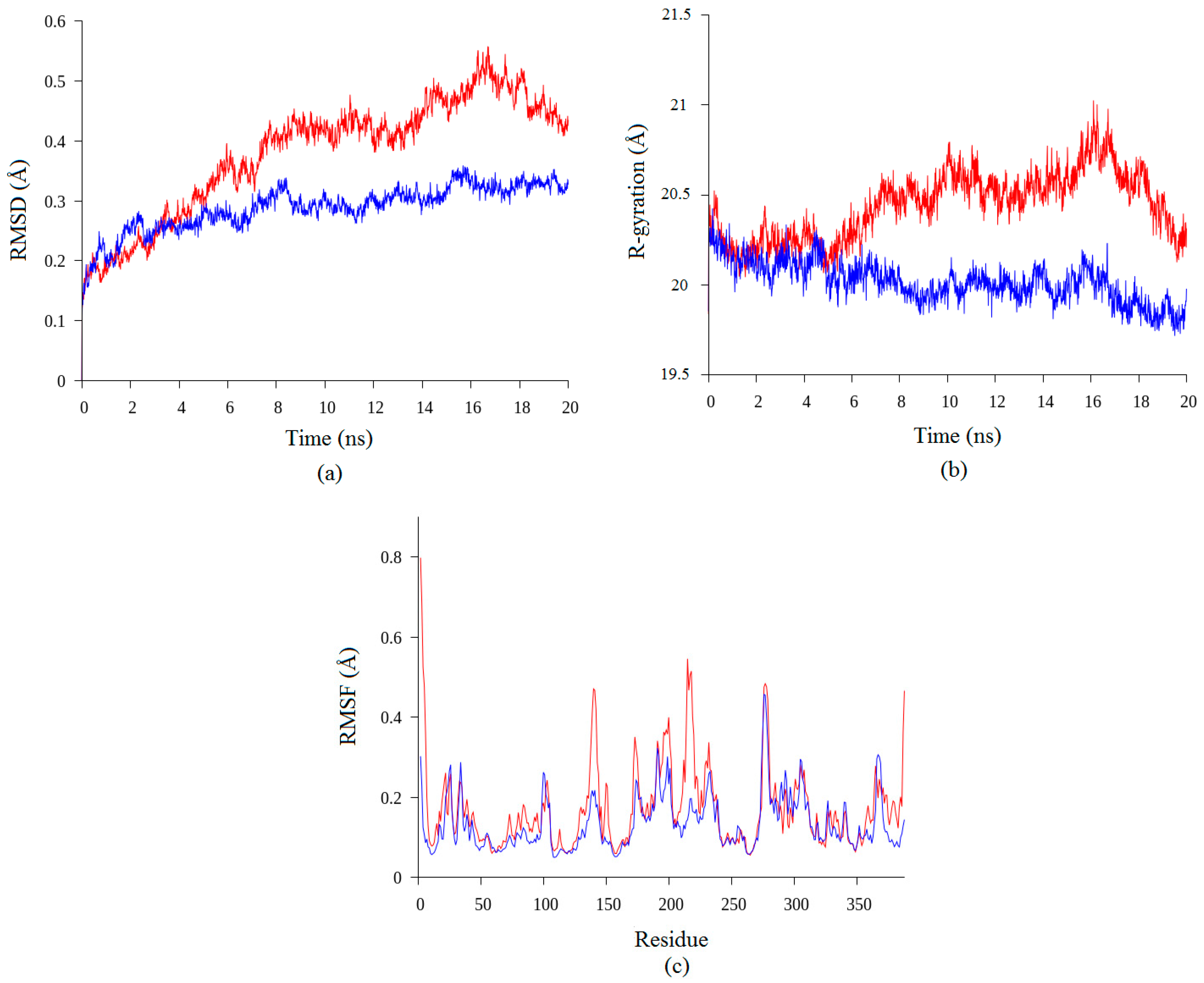
2.2. Molecular Dynamic Study of Space- and Earth-Grown Structures
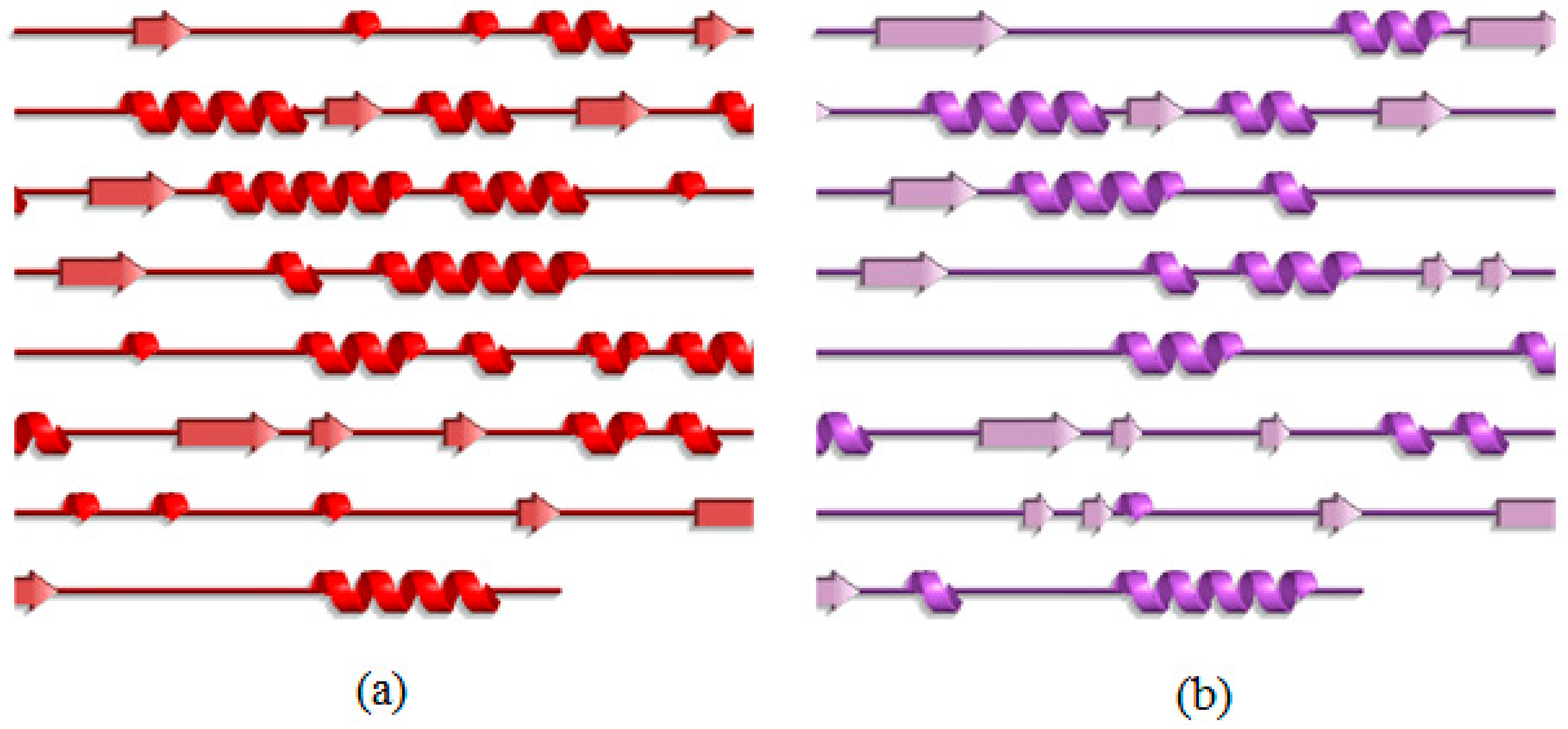
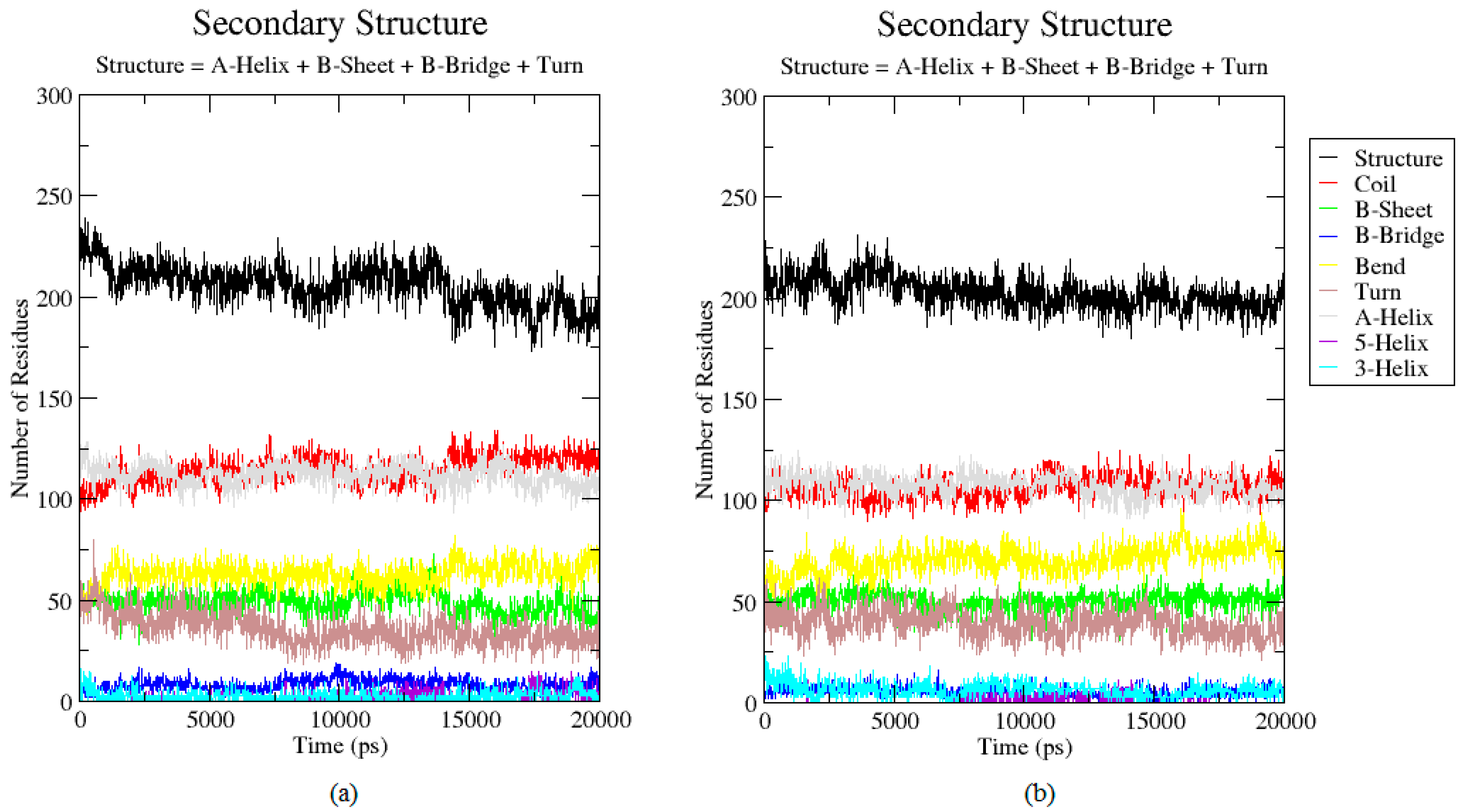
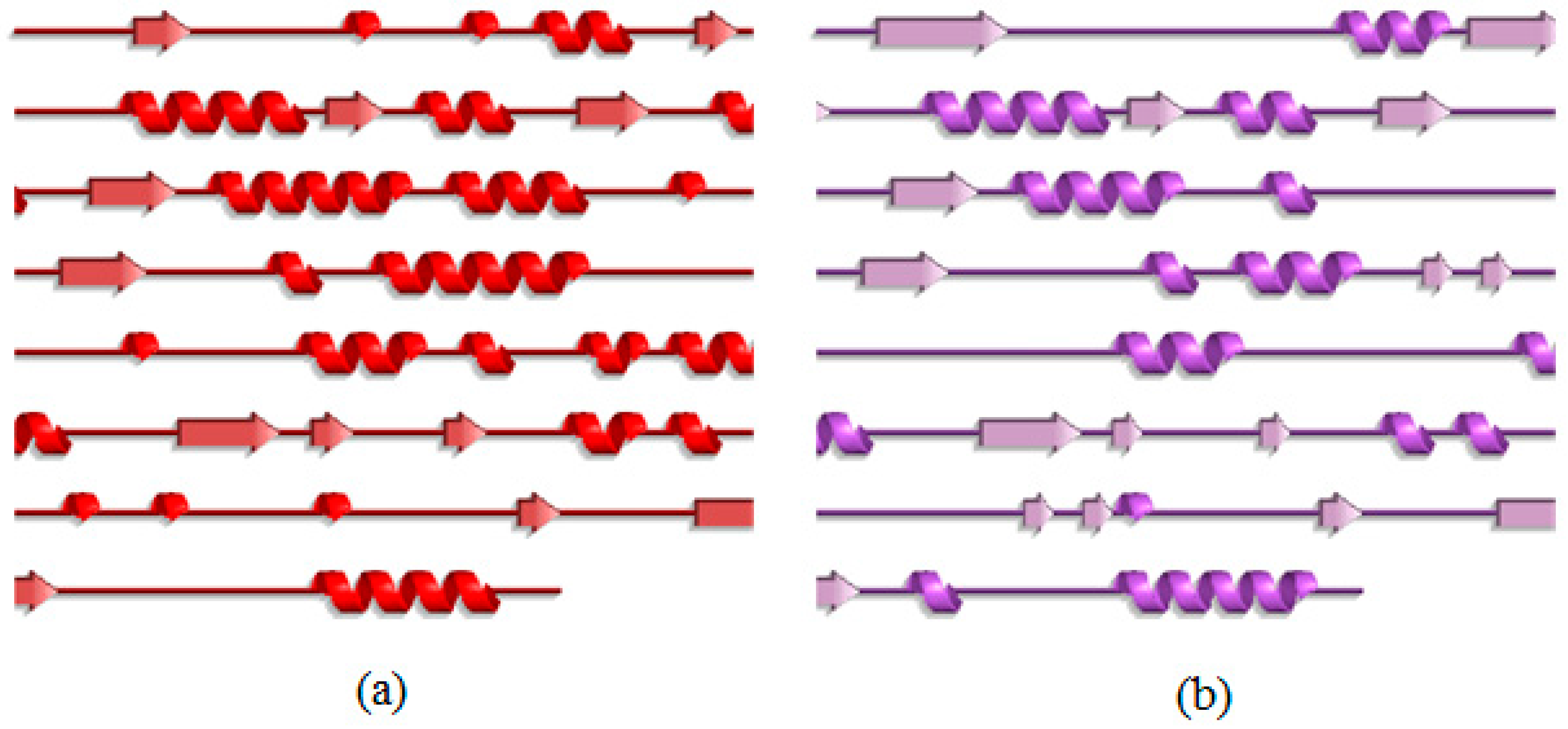
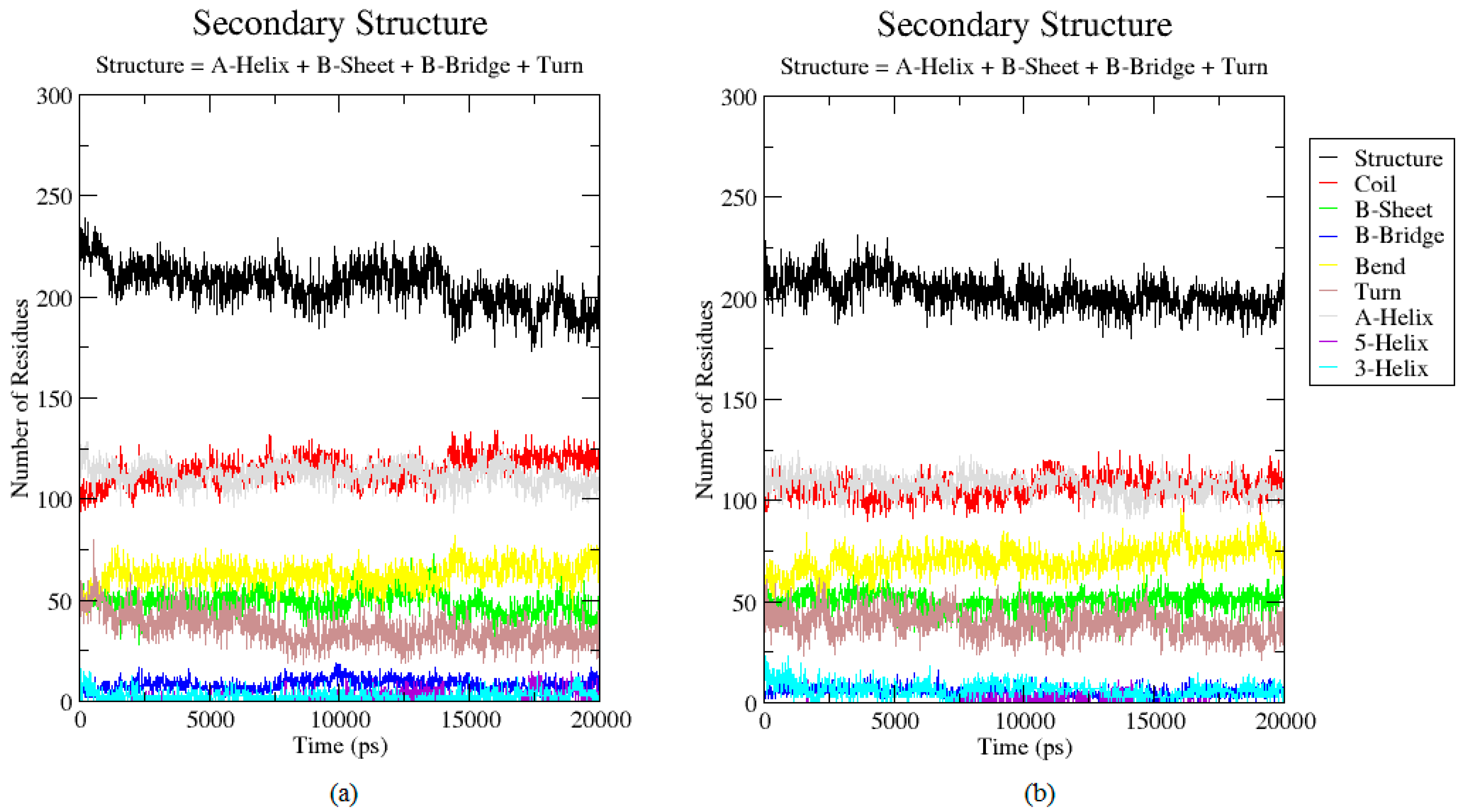
2.3. Analysis of the Secondary Structure
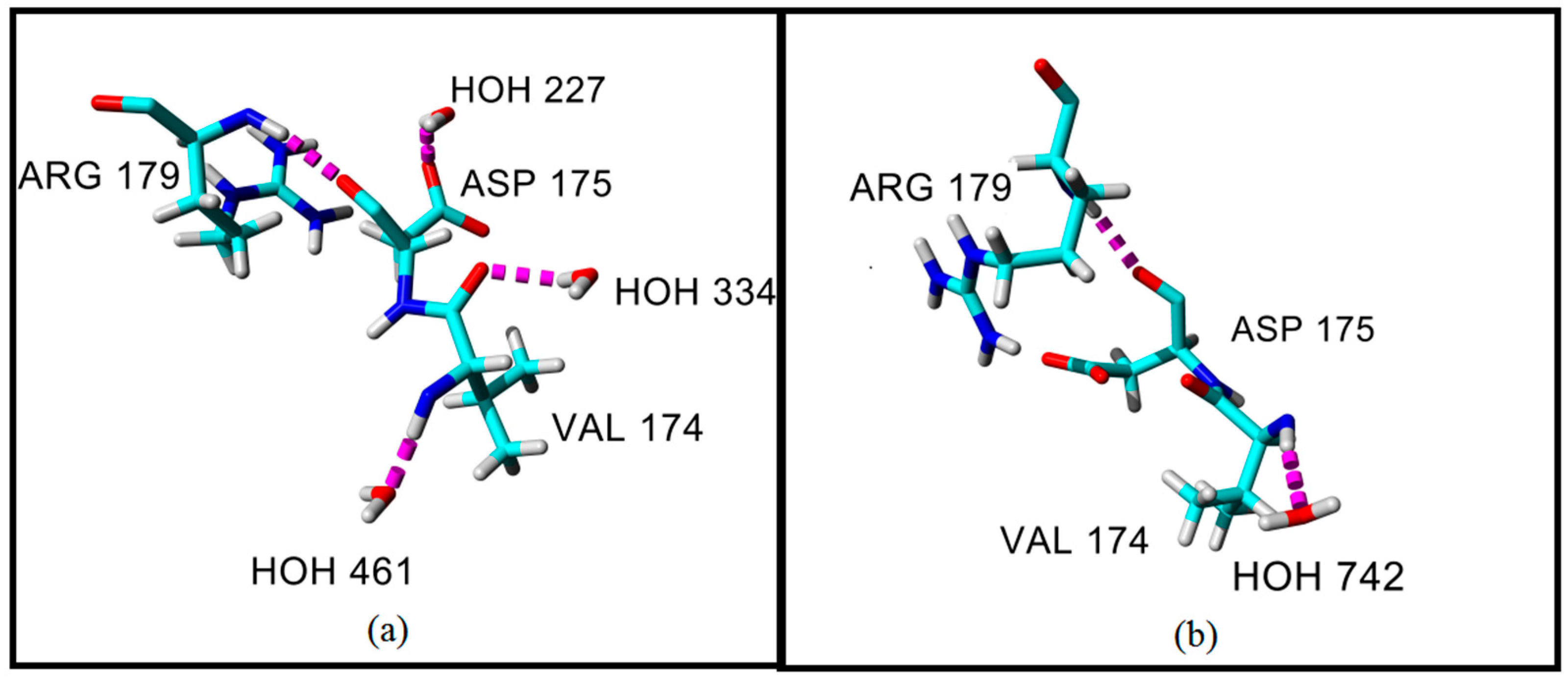
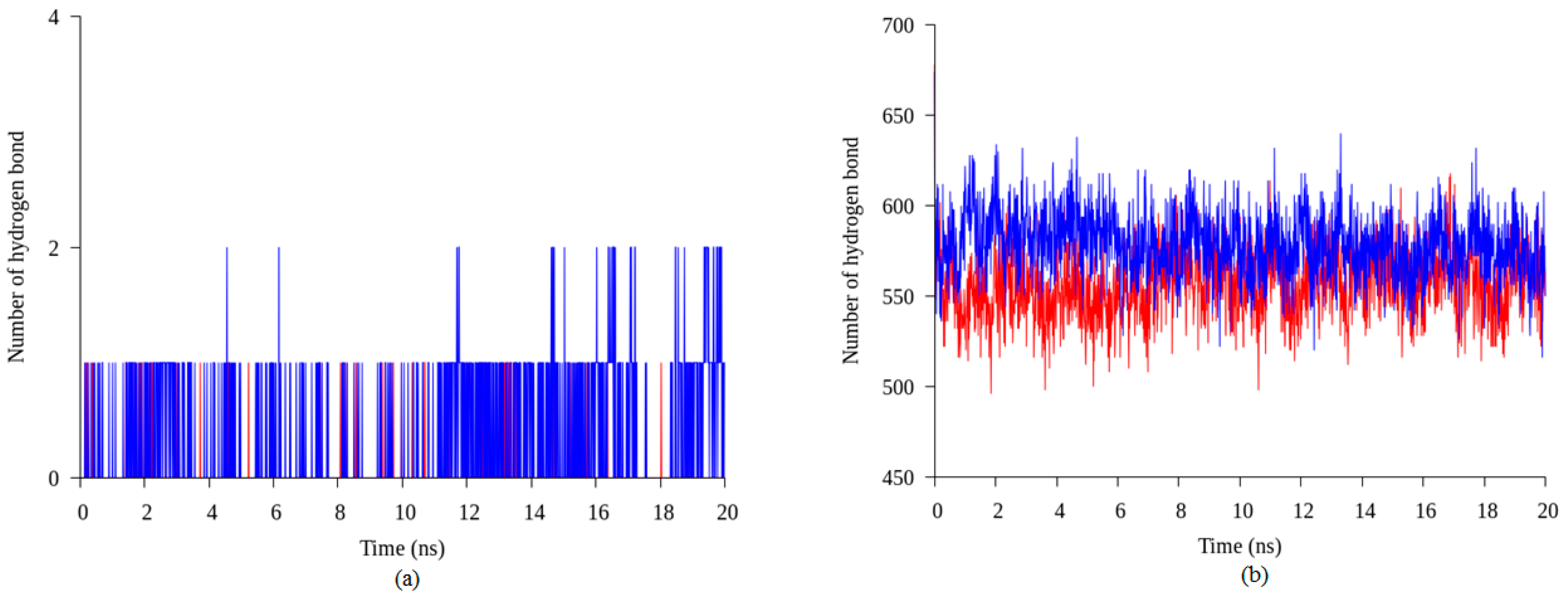
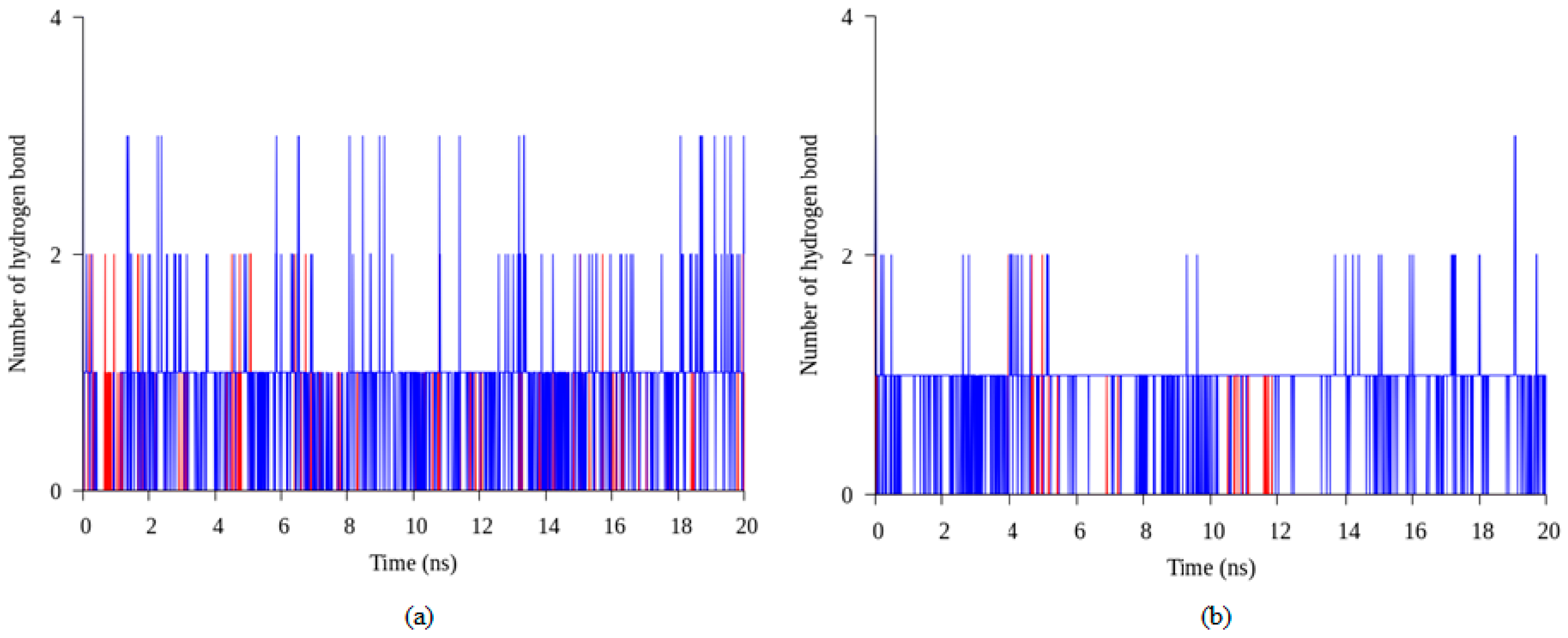
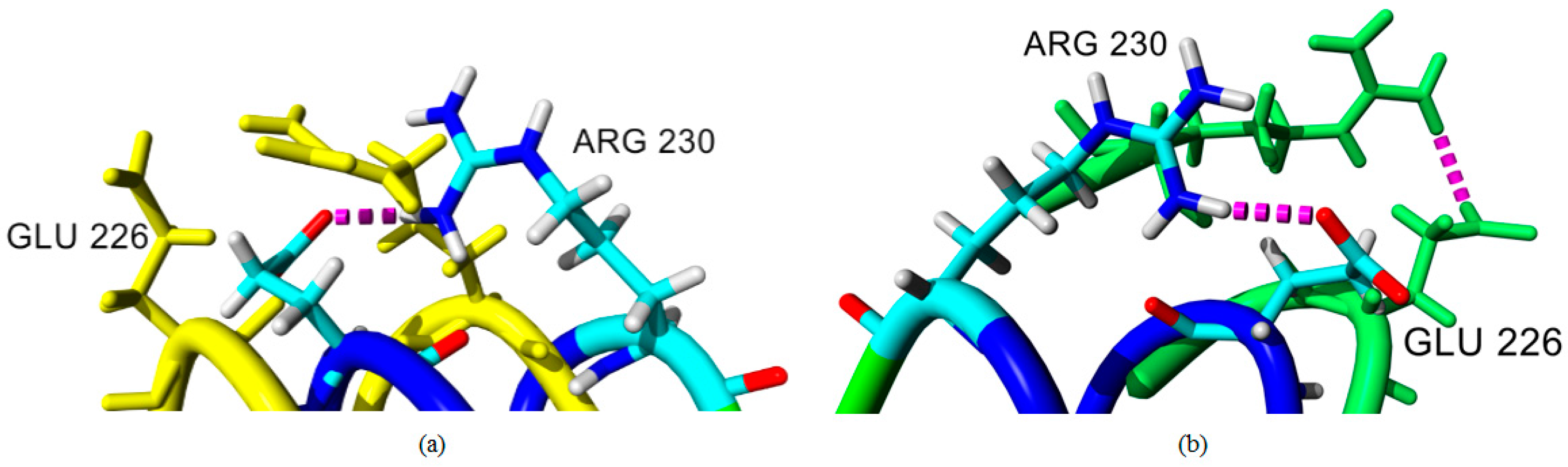
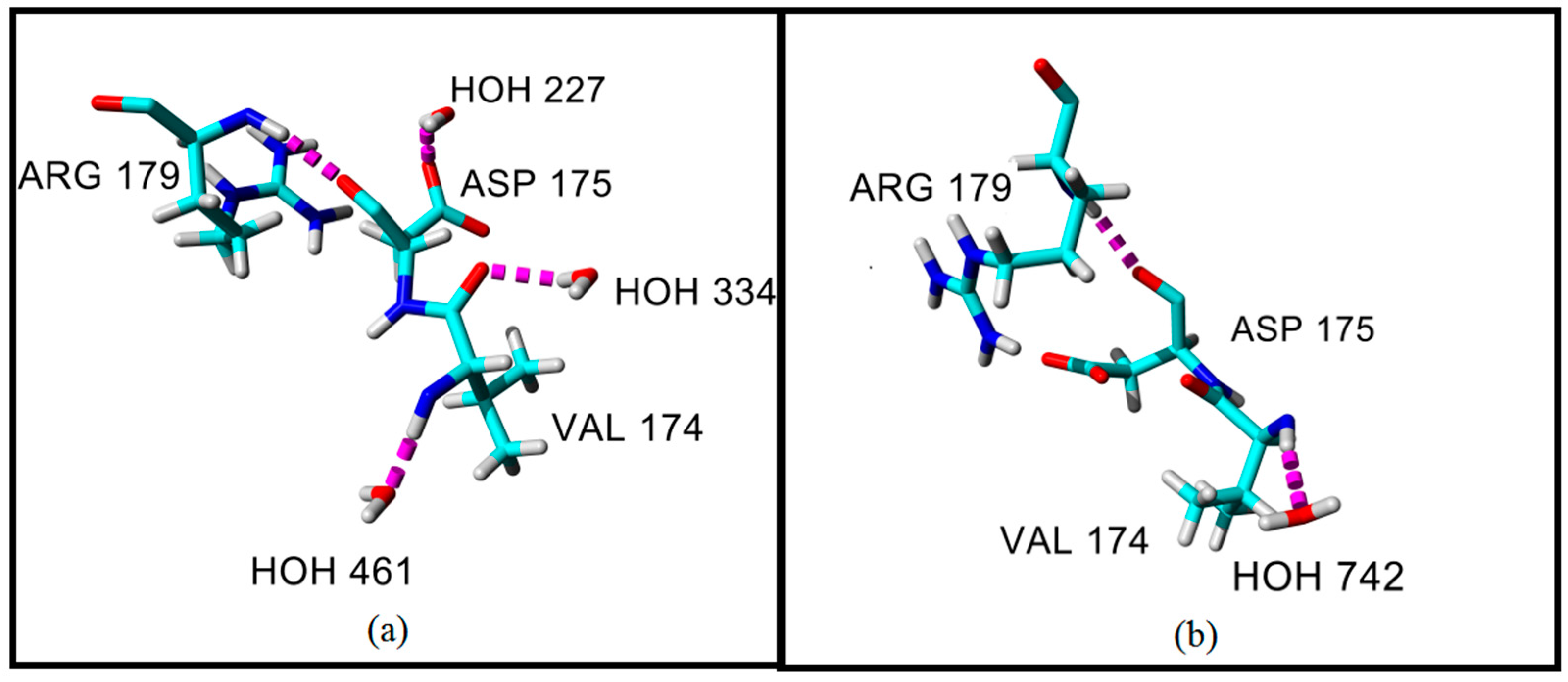
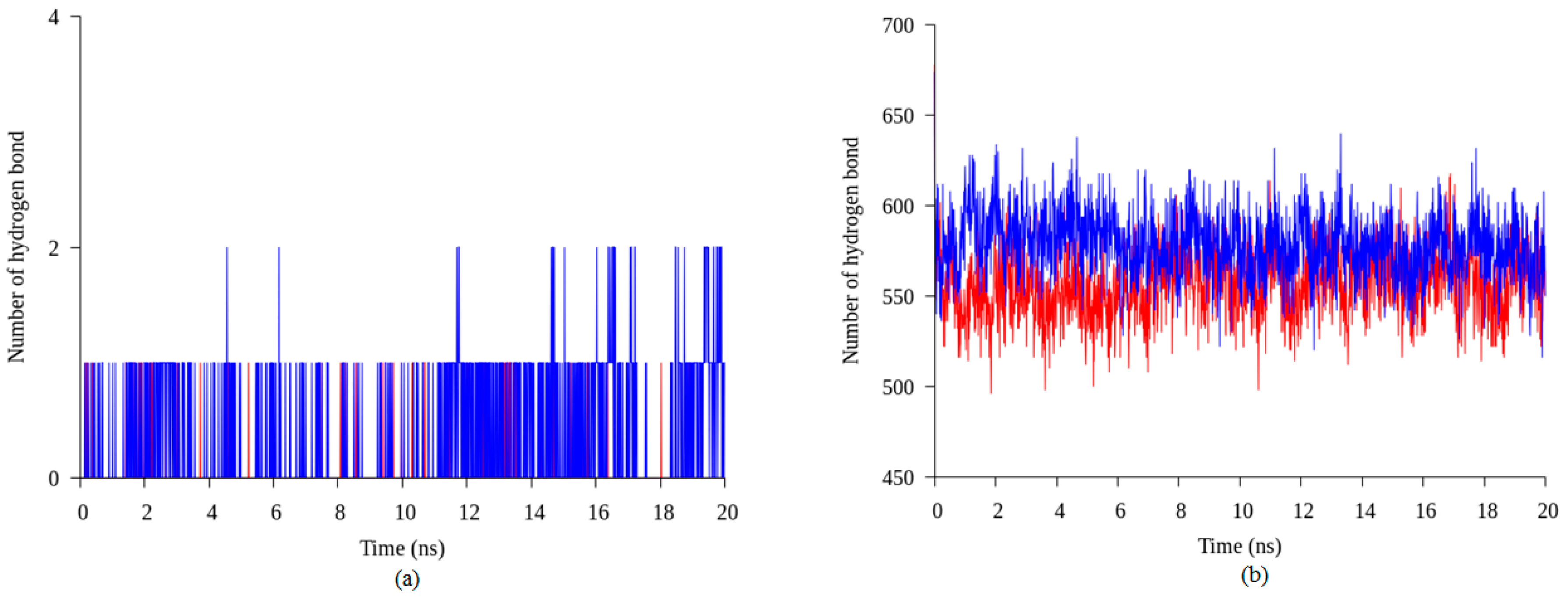
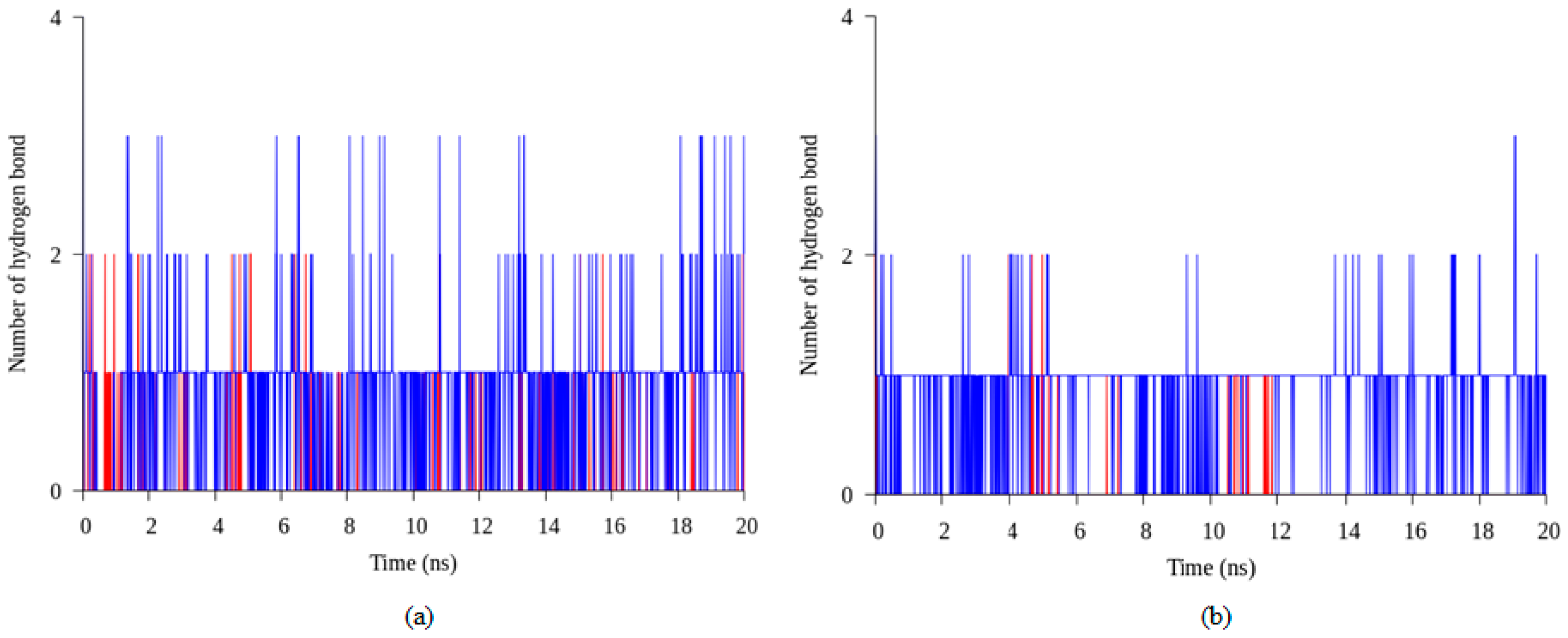
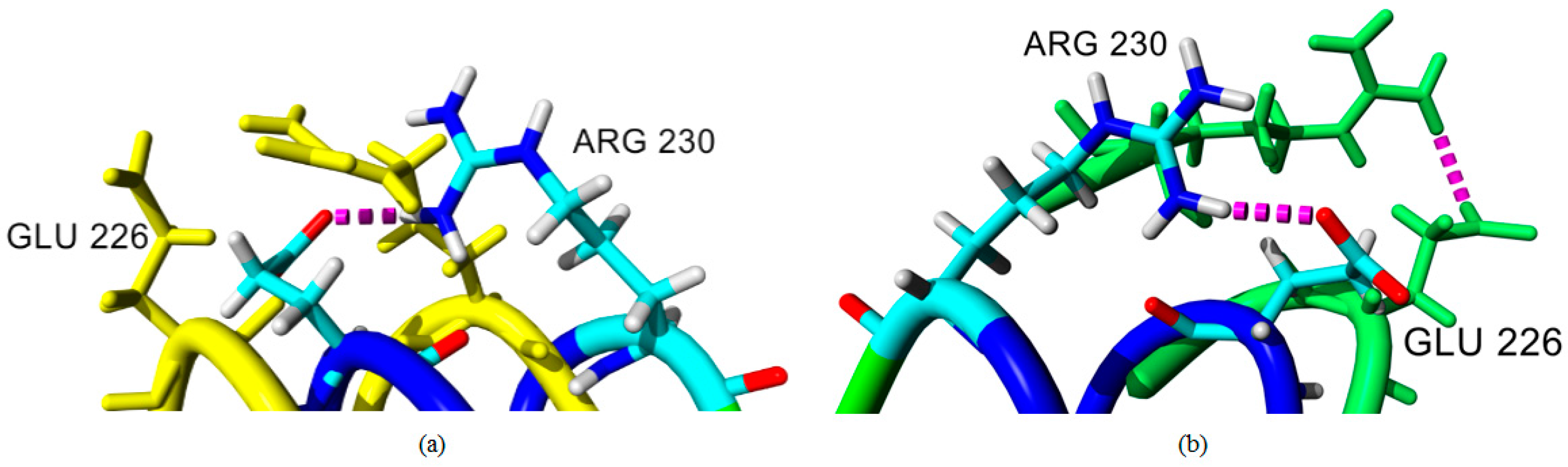
2.4. Hydrogen Bond and Ionic Interaction Formation and Deformation
3. Materials and Methods
3.1. Preliminary in Silico Study of Space- and Earth-Grownspace- and Earth-Grown Crystal Structures of T1 Lipase
3.2. Molecular Dynamic Simulation
3.3. Analysis of MD Simulation Trajectories
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ng, J.D.; Gavira, J.A.; Garcia-Ruiz, J.M. Protein crystallization by capillary counter diffusion for applied crystallographic structure determination. J. Struct. Biol. 2003, 142, 218–231. [Google Scholar] [CrossRef]
- Snell, E.H.; Helliwell, J.R. Macromolecular crystallization in microgravity. Rep. Prog. Phys. 2005, 68, 799–853. [Google Scholar] [CrossRef]
- Law, R.J.; Capener, C.; Baaden, M.; Bond, P.J.; Campbell, J.; Patargias, G.; Arinaminpathy, Y.; Sansom, M.S.P. Membrane protein structure quality in molecular dynamics simulation. J. Mol. Graph. Model. 2005, 24, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Mark, A.E. Refinement of homology-based protein structures by molecular dynamics simulation techniques. Protein Sci. 2003, 13, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Leow, T.C.; Rahman, R.N.Z.A.; Basri, M.; Salleh, A.B. A thermoalkaliphilic lipase of Geobacillus sp. T1. Extremophiles 2007, 11, 527–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, H.; Yamamoto, T.; Leow, T.C.; Mori, T.; Salleh, A.B.; Basri, M.; Inoeu, T.; Kai, Y.; Rahman, R.N.Z.R.A. Novel cation-π interaction revealed by crystal structure of thermoalkalophilic lipase. Proteins 2008, 70, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Aris, S.N.A.M.; Leow, T.C.; Ali, M.S.M.; Basri, M.; Salleh, A.B.; Rahman, R.N.Z.A. Crystallographic analysis of ground and space thermostable T1 lipase crystal obtained via counter diffusion method approach. BioMed Res. Int. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Miele, A.E.; Federici, L.; Sciara, G.; Draghi, F.; Brunori, M.; Vallone, B. Analysis of the effect of microgravity on protein crystal quality: The case of a myoglobin triple mutant. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 982. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.D.; Sauter, C.; Lorber, B.; Kirkland, N.; Arnez, J.; Giege, R. Comparative analysis of space-grown and earth- grown crystals of an aminoacyl-tRNA synthetase: Space-grown crystals are more useful for structural determination. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Pan, J.; Niu, X.; Zhou, Y.; Bi, R. Influence of microgravity on protein crystal structures. Chin. Sci. Bull. 2000, 45, 1002–1006. [Google Scholar] [CrossRef]
- Groot, B.L.; Hayward, S.; van Aalten, D.M.F.; Amadei, A.; Berendsen, H.J.C. Domain motions in bacteriophage T4 lysozyme: A comparison between molecular dynamics and crystallographic data. Proteins 1998, 31, 116–127. [Google Scholar] [CrossRef]
- McCarthy, A.N.; Grigera, J.R. Effect of pressure on the conformation of proteins. A molecular dynamics simulation of lysozyme. J. Mol. Graph. Model. 2006, 24, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, K.E.; Alemasov, N.A.; Vorobjev, Y.N.; Boldyreva, E.V.; Kolchanov, N.A.; Afonnikov, D.A. Molecular dynamics simulations of the Nip7 proteins from the marine deep- and shallow-water Pyrococcus species. BMC Struct. Biol. 2014, 14, 23. [Google Scholar]
- Pikkemaat, M.G.; Linssen, A.B.; Berendsen, H.J.; Janssen, D.B. Molecular dynamics simulations as a tool for improving protein stability. Protein Eng. 2002, 15, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [PubMed]
- Galzitskaya, O.V.; Garbuzynskiy, S.O. Entropy capacity determines protein folding. Proteins 2006, 63, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Baweja, M.; Singh, P.K.; Sadaf, A.; Tiwari, R.; Nain, L.; Khare, S.K.; Shukla, P. Cost effective characterization process and molecular dynamic simulation of detergent compatible alkaline protease from Bacillus pumilus strain MP27. Process Biochem. 2017, 58, 199–203. [Google Scholar] [CrossRef]
- Vogt, G.; Woell, S.; Argos, P. Protein thermal Stability, hydrogen bonds, and ion pairs. J. Mol. Biol. 1997, 269, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.K.; Pace, C.N. Hydrogen Bonding Stabilizes Globular Proteins. Biophys. J. 1996, 71, 2033–2039. [Google Scholar] [CrossRef]
- Efimov, A.V.; Brazhnikov, E.V. Relationship between intramolecular hydrogen bonding and solvent accessibility of side-chain donors and acceptors in proteins. FEBS Lett. 2003, 554, 389–393. [Google Scholar] [CrossRef]
- Szilagyi, A.; Zavodsky, P. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: Results of a comprehensive survey. Structure 2000, 8, 493–504. [Google Scholar] [CrossRef]
- Kar, T.; Scheiner, S. Comparison of cooperativity in CH-O and OH-O hydrogen bonds. J. Phys. Chem. A 2006, 108, 9161–9168. [Google Scholar] [CrossRef]
- Ragone, R. Hydrogen-bonding classes in proteins and their contribution to the unfolding reaction. Protein Sci. 2001, 10, 2075–2082. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nussinov, R. Salt Bridge Stability in Monomeric Proteins. J. Mol. Biol. 1999, 293, 1241–1255. [Google Scholar] [CrossRef] [PubMed]
- Makhatadze, G.I.; Loladze, V.V.; Ermolenko, D.N.; Chen, X.F.; Thomas, S.T. Contribution of Surface salt bridges to protein stability: Guidelines for protein engineering. J. Mol. Biol. 2003, 327, 1135–1148. [Google Scholar] [CrossRef]
- Strop, P.; Mayo, S.L. Contribution of surface salt bridges to protein stability. Biochemistry 2000, 39, 1251–1255. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.N.Z.R.A.; Fujiwara, S.; Nakamura, H.; Takagi, M.; Imanaka, T. Ion pairs involved in maintaining a thermostable structure of glutamate dehydrogenase from a hyperthermophilic archaeon. Biochem. Biophys. Res. Commun. 1998, 248, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nussinov, R. Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys. J. 2002, 83, 1595–1612. [Google Scholar] [CrossRef]
- Karshikoff, A.; Ladenstein, R. Ion pairs and the thermotolerance of proteins from hyperthermophiles: A ‘traffic rule’ for hot roads. Trends Biochem. Sci. 2001, 26, 550–556. [Google Scholar] [CrossRef]
- Pack, S.P.; Yoo, Y.J. Protein thermostability: Structure-based difference of amino acid between thermophilic and mesophilic proteins. J. Biotechnol. 2004, 111, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.P.; Li, M.; Zhou, Y.; Yang, L.R.; Xu, G. Introducing a salt bridge into the lipase of Stenotrophomonas maltophilia results in a very large increase in thermal stability. Biotechnol. Lett. 2015, 37, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Habash, J.; Boggon, T.J.; Raftery, J.; Chayen, N.E.; Zagalskyc, P.F.; Helliwell, J.R. Apocrustacyanin C1 crystals grown in space and on earth using vapour-diffusion geometry: Protein structure refinements and electron-density map comparisons. Acta Cryst. 2003, 59, 1117–1123. [Google Scholar]
- Giege, R.; Drenth, J.; Ducruix, A.; McPherson, A.; Saenger, W. Crystallogenesis of biological macromolecules. Biological, microgravity and other physico-chemical aspects. Progress Cryst. Growth Characteriz. Mater. 1995, 30, 237–281. [Google Scholar] [CrossRef]
- Bogon, T.J.; Chayen, N.E.; Snell, E.H.; Dong, J.; Lautenschlager, P.; Potthast, L.; Siddons, D.P.; Stojanoff, V.; Gordon, E.; Thompson, A.W.; et al. Protein crystal movements and fluid flows during microgravity growth. Philos. Trans. Mathem. Phys. Eng. Sci. 1998, 356, 1045–1061. [Google Scholar] [CrossRef]
- Lorber, B.; Giege, R. Nucleation and growth of thaumatin crystals within a gel under microgravity on STS-95 mission vs. under Earth’s gravity. J. Cryst. Growth 2001, 231, 252–261. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F.A. Biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishak, S.N.H.; Aris, S.N.A.M.; Halim, K.B.A.; Ali, M.S.M.; Leow, T.C.; Kamarudin, N.H.A.; Masomian, M.; Rahman, R.N.Z.R.A. Molecular Dynamic Simulation of Space and Earth-Grown Crystal Structures of Thermostable T1 Lipase Geobacillus zalihae Revealed a Better Structure. Molecules 2017, 22, 1574. https://doi.org/10.3390/molecules22101574
Ishak SNH, Aris SNAM, Halim KBA, Ali MSM, Leow TC, Kamarudin NHA, Masomian M, Rahman RNZRA. Molecular Dynamic Simulation of Space and Earth-Grown Crystal Structures of Thermostable T1 Lipase Geobacillus zalihae Revealed a Better Structure. Molecules. 2017; 22(10):1574. https://doi.org/10.3390/molecules22101574
Chicago/Turabian StyleIshak, Siti Nor Hasmah, Sayangku Nor Ariati Mohamad Aris, Khairul Bariyyah Abd Halim, Mohd Shukuri Mohamad Ali, Thean Chor Leow, Nor Hafizah Ahmad Kamarudin, Malihe Masomian, and Raja Noor Zaliha Raja Abd Rahman. 2017. "Molecular Dynamic Simulation of Space and Earth-Grown Crystal Structures of Thermostable T1 Lipase Geobacillus zalihae Revealed a Better Structure" Molecules 22, no. 10: 1574. https://doi.org/10.3390/molecules22101574