
Lead Discovery of Type II BRAF V600E Inhibitors Targeting the Structurally Validated DFG-Out Conformation Based upon Selected Fragments


Abstract
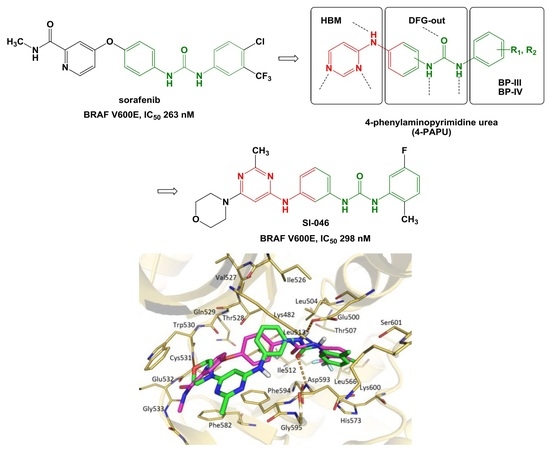
:
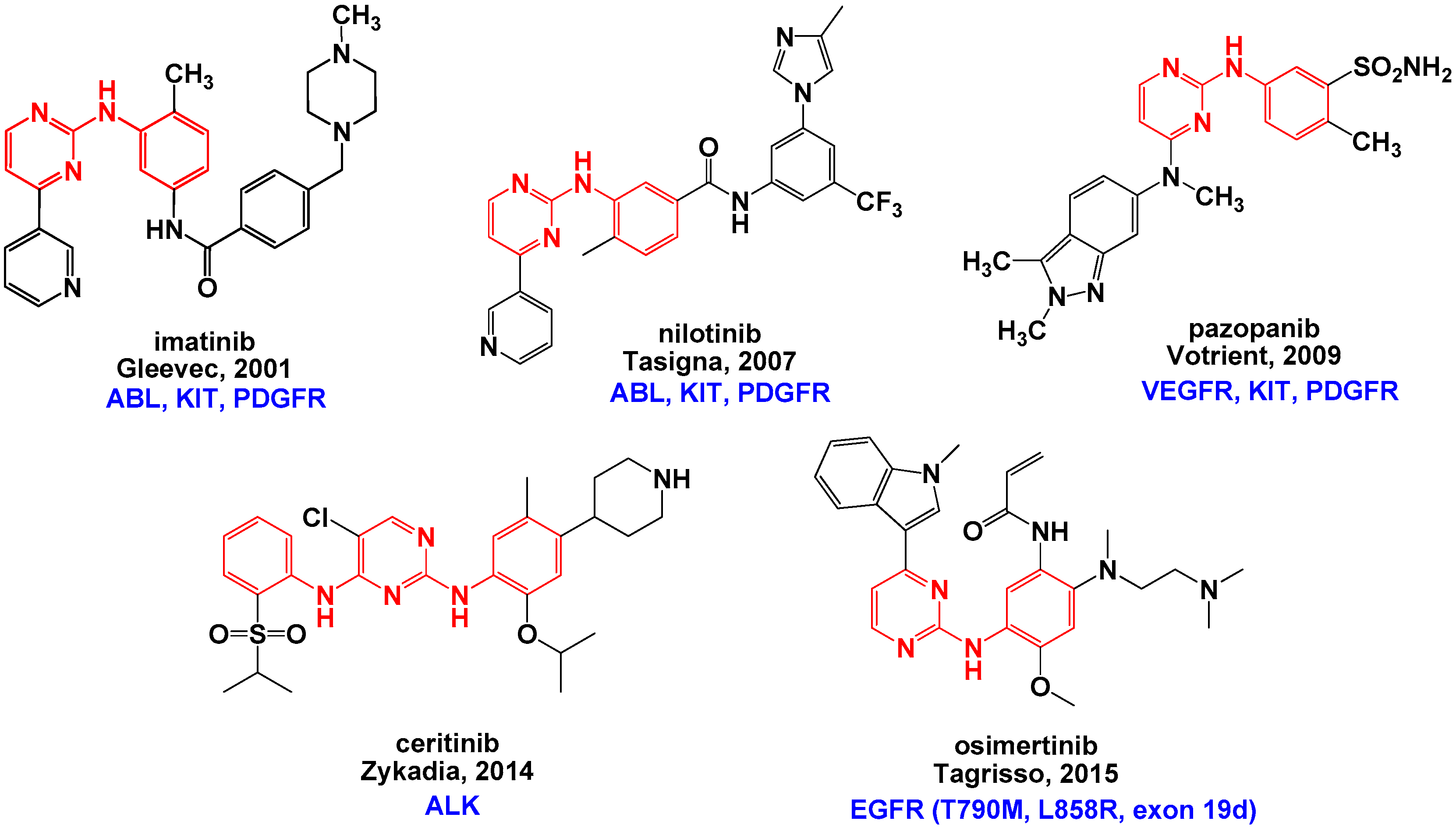
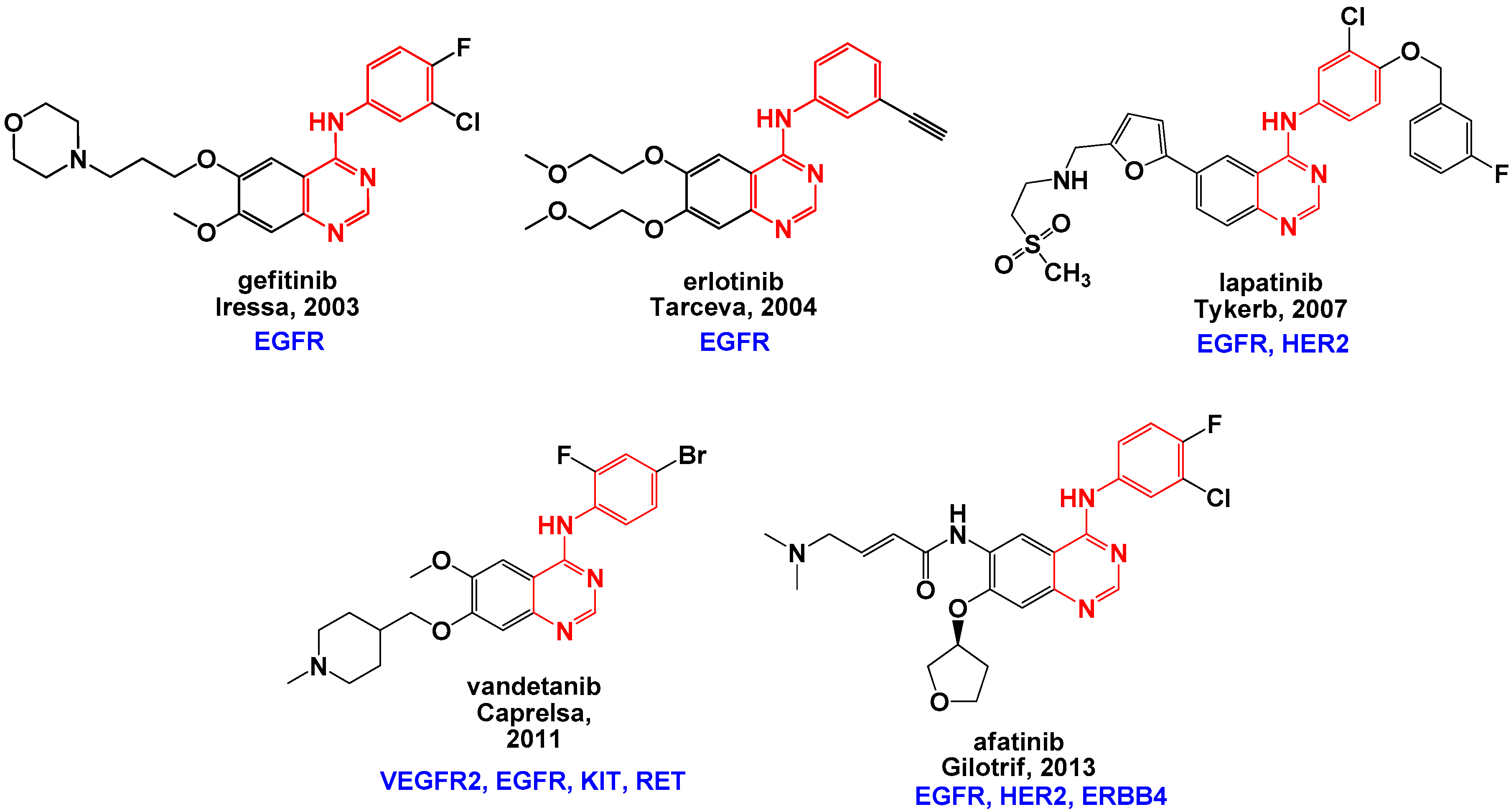
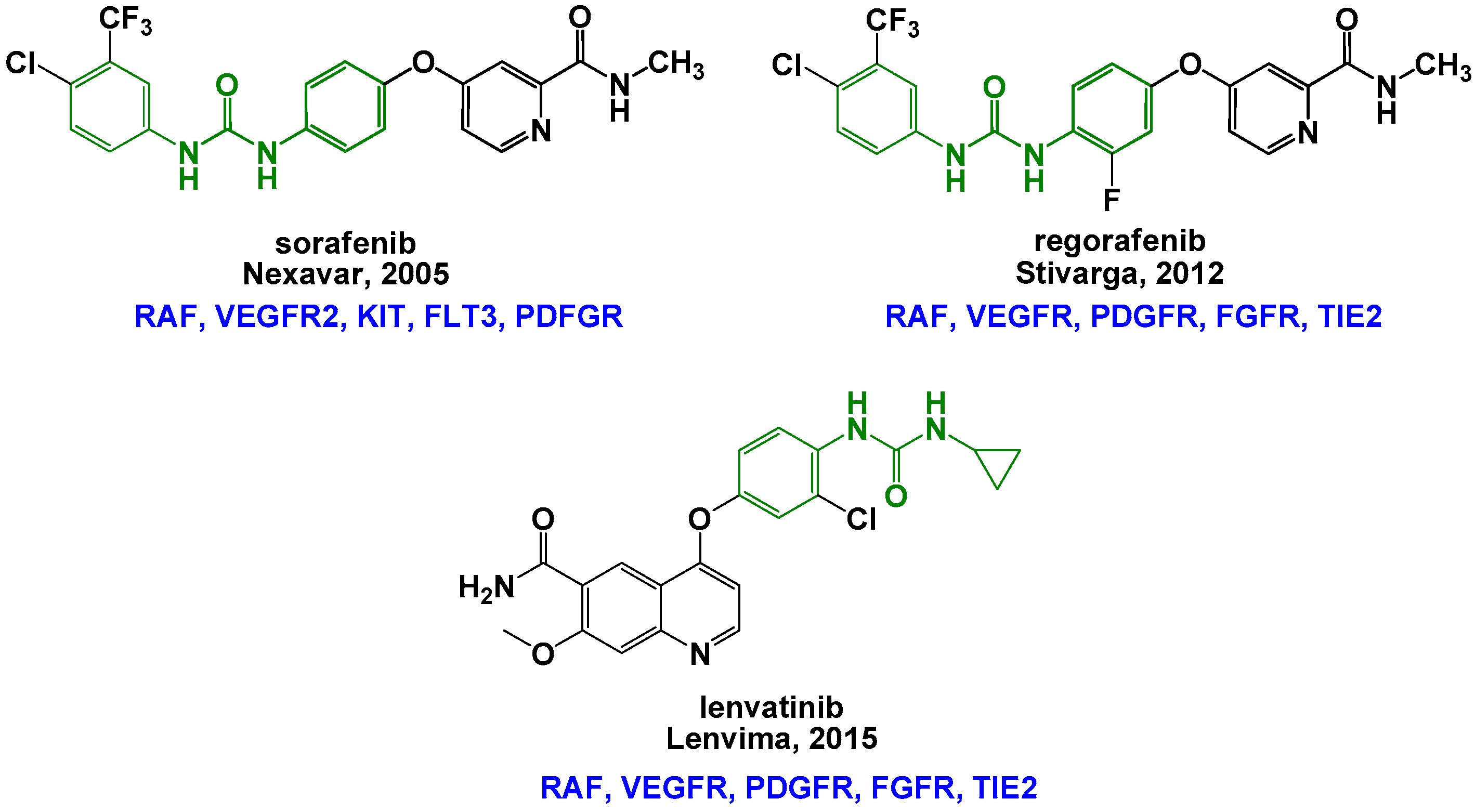
1. Introduction
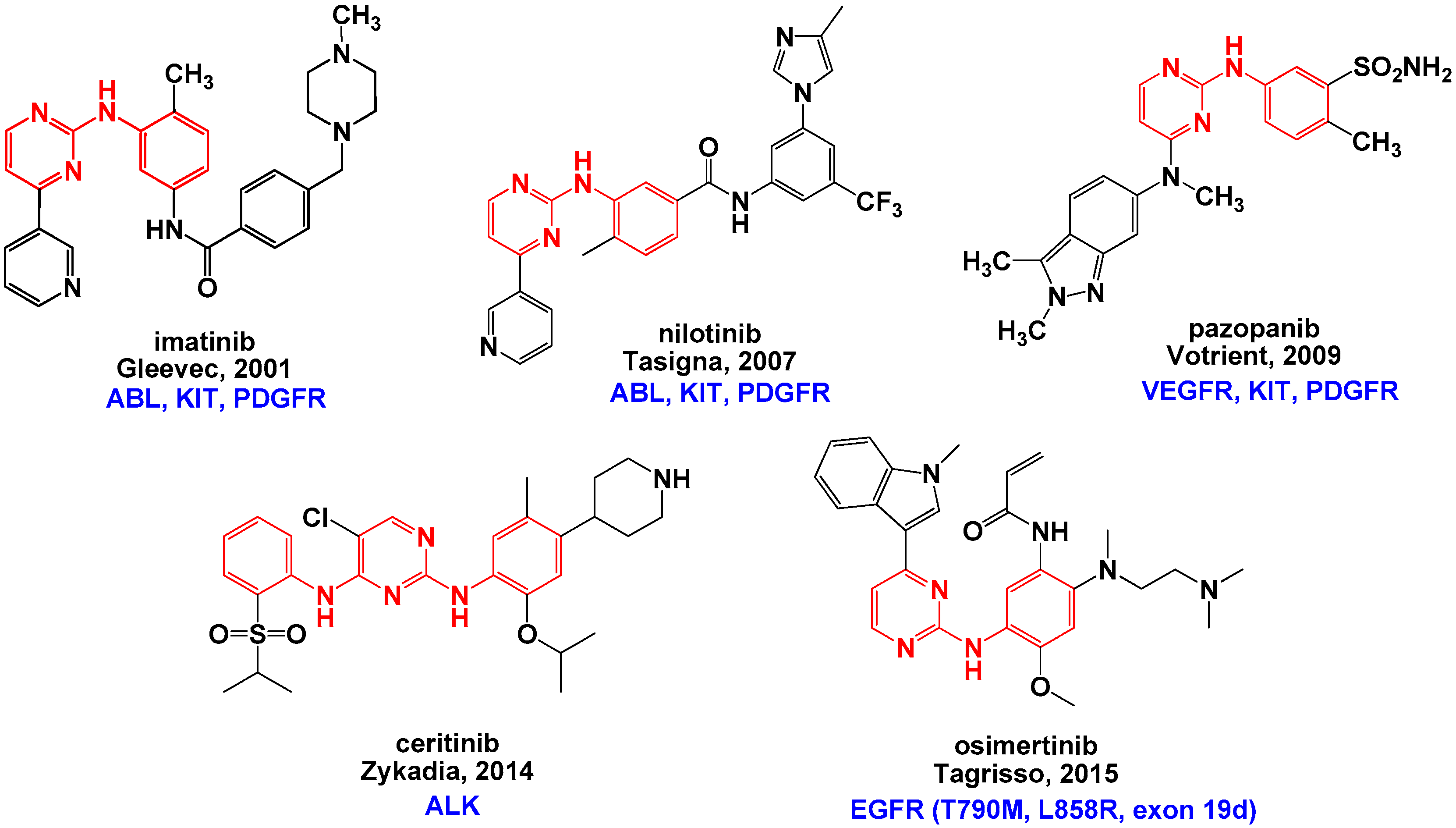
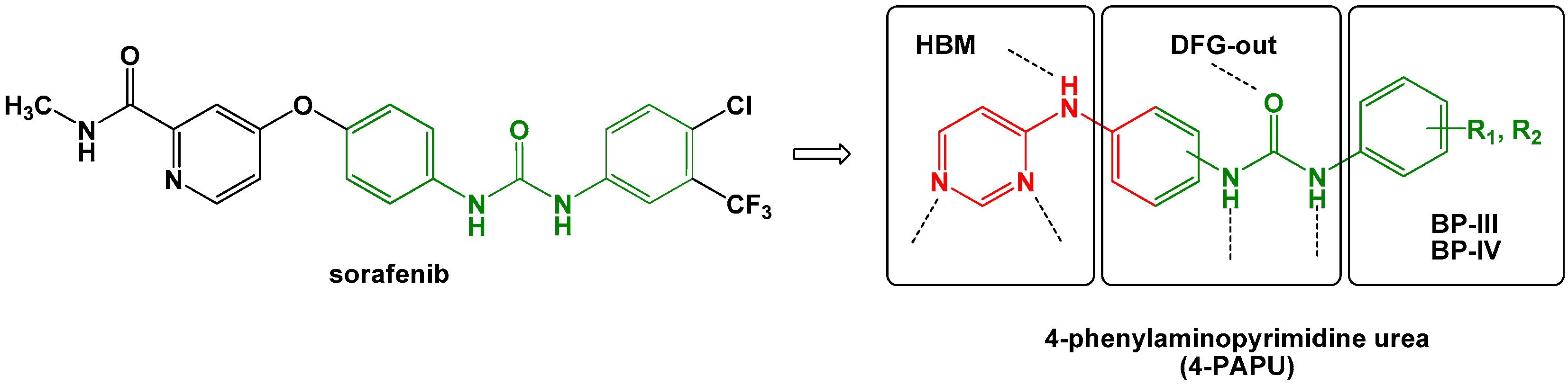
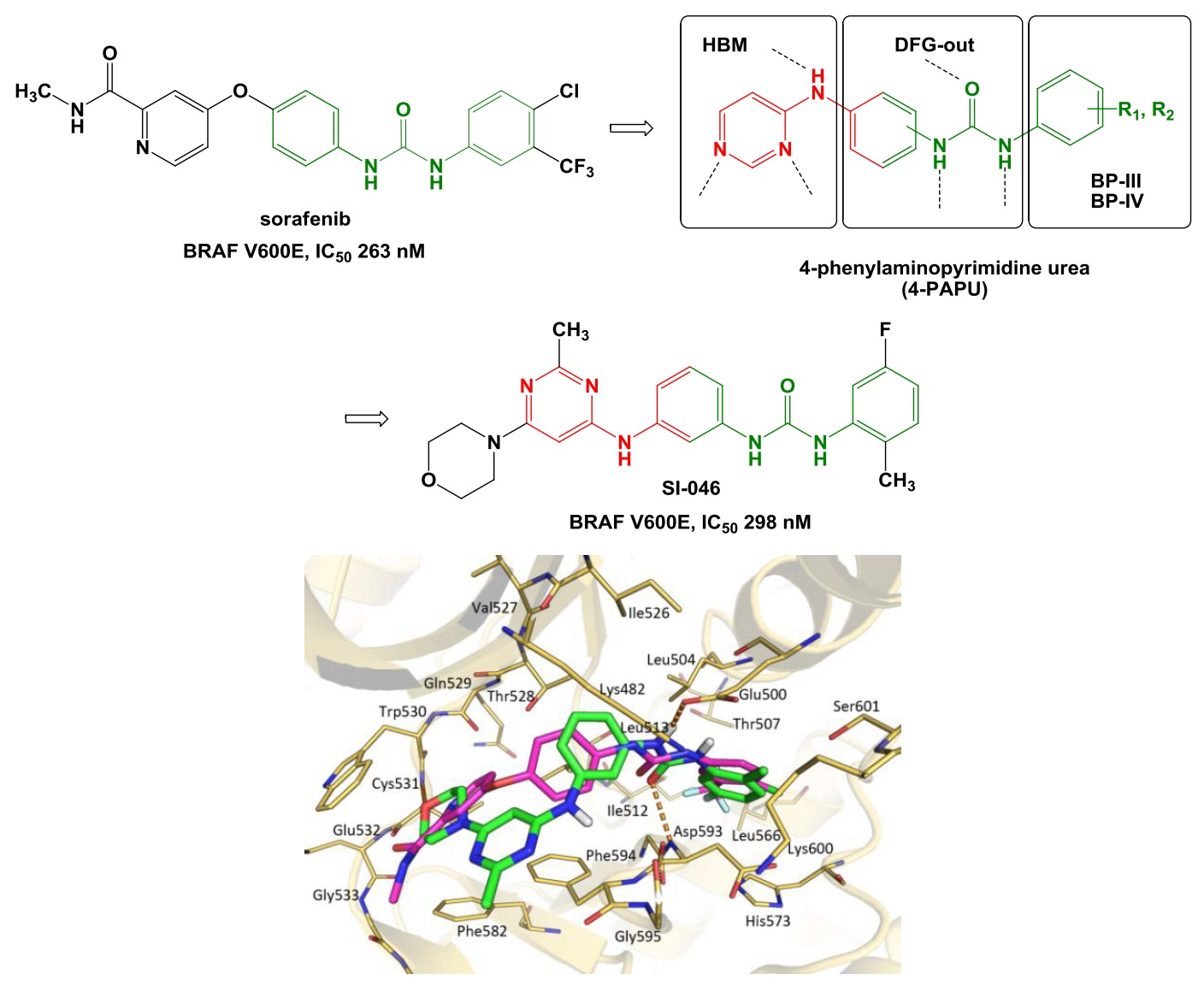
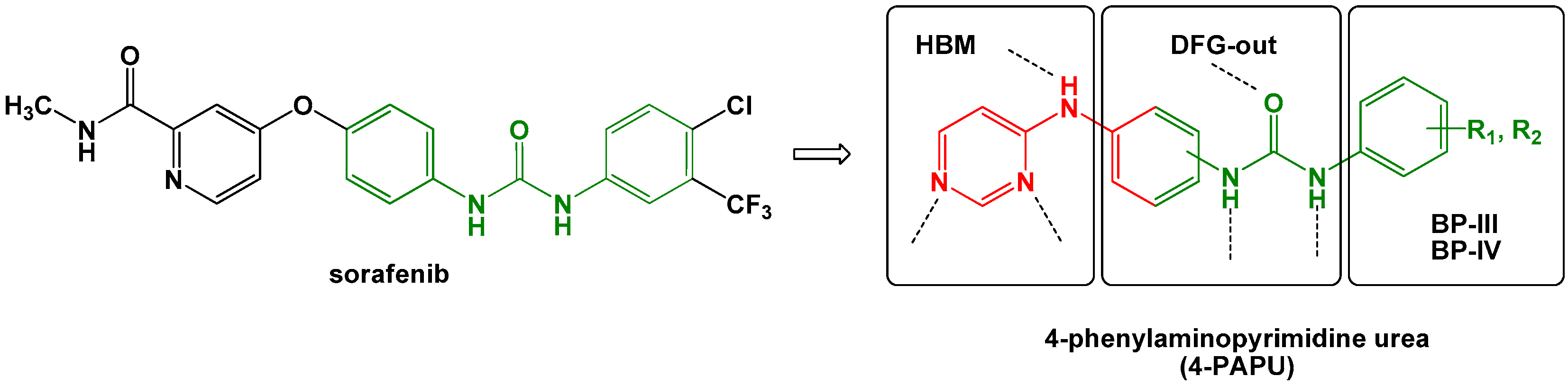
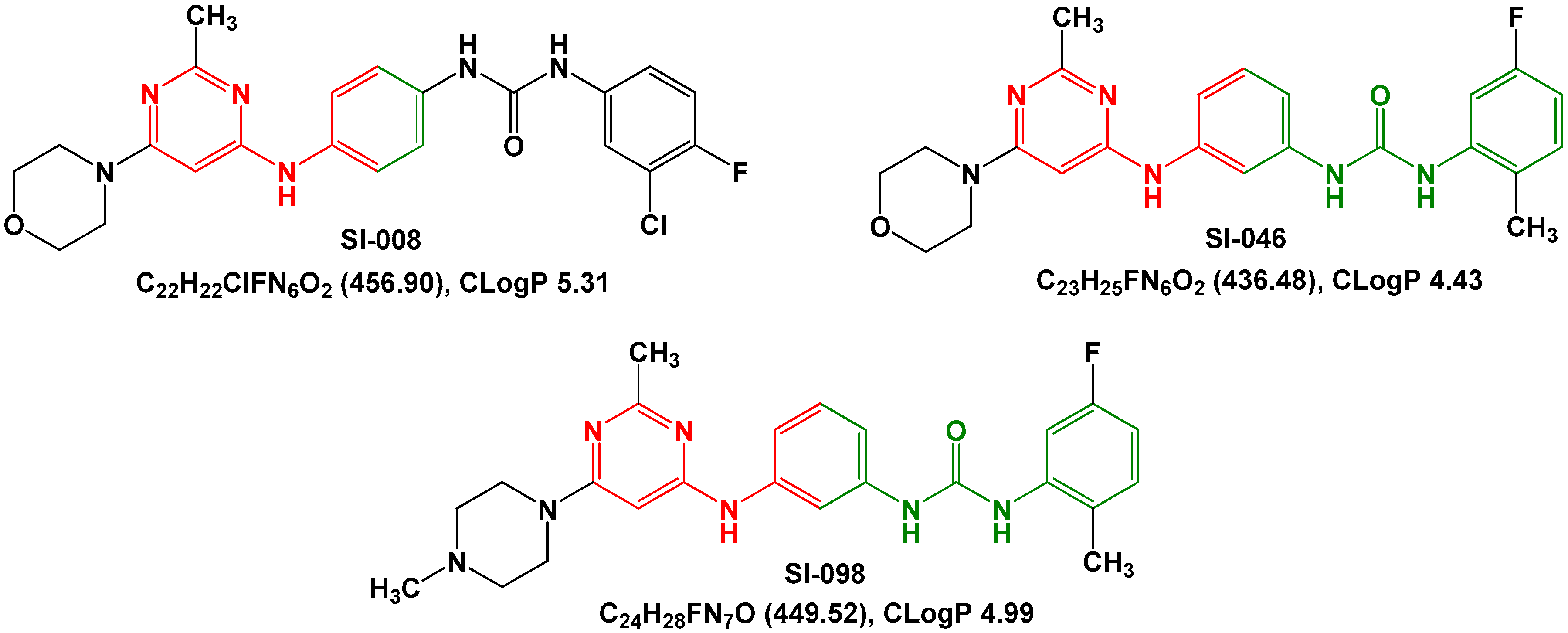
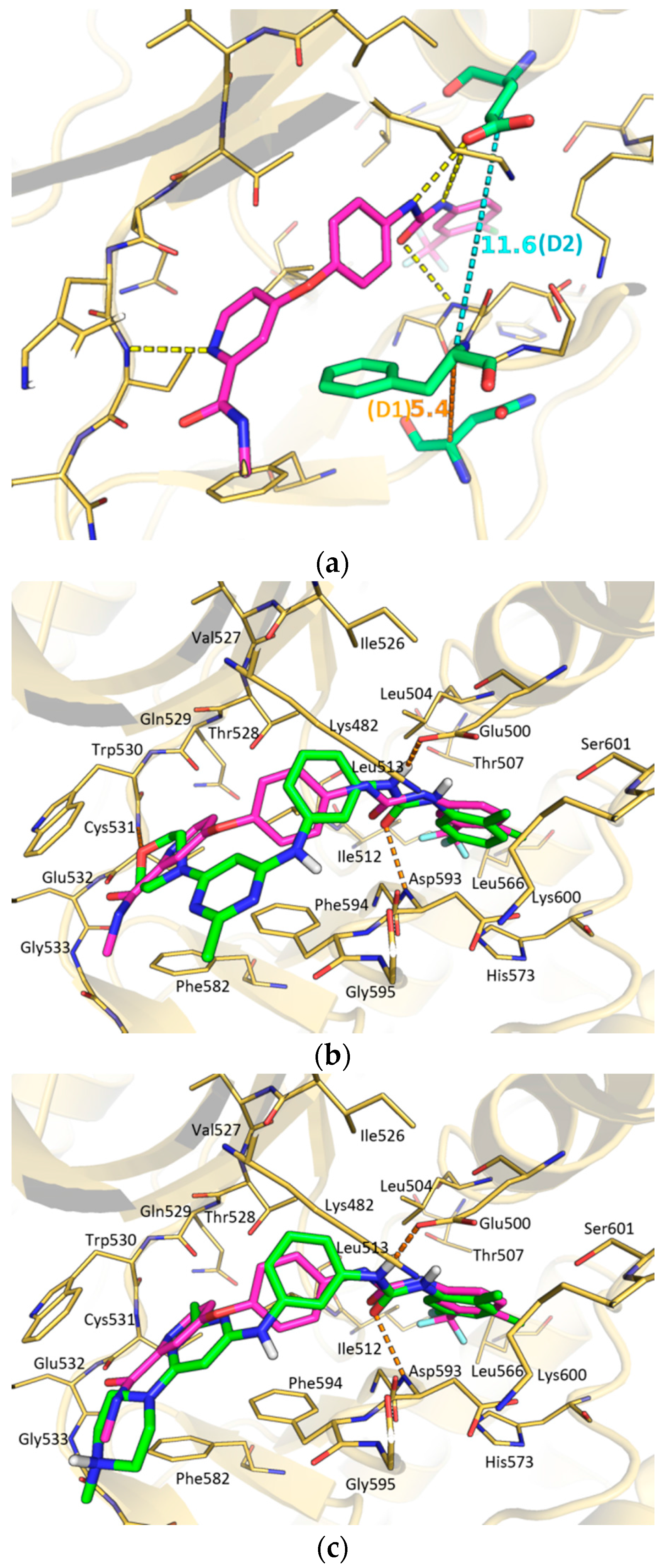
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.2. Biochemical Assays
3.3. Docking
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| DFG | Asp-Phe-Gly |
| FDA | Food and Drug Administration |
| HRD | His-Arg-Asp |
| INN | International Nonproprietary Names |
| PDB | Protein Data Bank |
| SAR | structure-activity relationship |
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Bridges, A.J. Chemical inhibitors of protein kinases. Chem. Rev. 2001, 101, 2541–2571. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Alessi, D.R. Kinase drug discovery—What’s next in the field? ACS Chem. Biol. 2012, 8, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Grütter, C.; Rauh, D. Strategies for the selective regulation of kinases with allosteric modulators: Exploiting exclusive structural features. ACS Chem. Biol. 2012, 8, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, R.S.K.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.; Peterson, J.R.; Dunbrack, R.L.; Levy, R.M. Conformational Analysis of the DFG-out kinase motif and biochemical profiling of structurally validated type II inhibitors. J. Med. Chem. 2014, 58, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, H.; Wang, L.; Liu, Y.; Knapp, S.; Liu, Q.; Gray, N.S. Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? ACS Chem. Biol. 2014, 9, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Clausen, M.H.; Nielsen, T.E. Allosteric small-molecule kinase inhibitors. Pharmacol. Ther. 2015, 156, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Schindler, T.; Bornmann, W.; Pellicena, P.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 2000, 289, 1938–1942. [Google Scholar] [CrossRef] [PubMed]
- Nagar, B.; Bornmann, W.G.; Pellicena, P.; Schindler, T.; Veach, D.R.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002, 62, 4236–4243. [Google Scholar] [PubMed]
- Manley, P.W.; Cowan-Jacob, S.W.; Buchdunger, E.; Fabbro, D.; Fendrich, G.; Furet, P.; Meyer, T.; Zimmermann, J. Imatinib: A selective tyrosine kinase inhibitor. Eur. J. Cancer 2002, 38, S19–S27. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Uehling, D.E.; Harris, P.A. Recent progress on MAP kinase pathway inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 4047–4056. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.H.; Brage, S.E. BRAF inhibitors in cancer therapy. Pharmacol. Ther. 2014, 142, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Rheault, T.R.; Stellwagen, J.C.; Adjabeng, G.M.; Hornberger, K.R.; Petrov, K.G.; Waterson, A.G.; Dickerson, S.H.; Mook, R.A.; Laquerre, S.G.; King, A.J.; et al. Discovery of dabrafenib: A selective inhibitor of Raf kinases with antitumor activity against B-Raf-driven tumors. ACS Med. Chem. Lett. 2013, 4, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.Y.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.M.; Vincent, P.; McHugh, M.; et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.-H.; Zopf, D. Regorafenib (BAY 73–4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.N.; Adelinet, C.; Berdini, V.; Beke, L.; Bonnet, P.; Brehmer, D.; Calo, F.; Coyle, J.E.; Day, P.J.; Frederickson, M.; et al. Structure-based design of type II inhibitors applied to Maternal Embryonic Leucine Zipper Kinase. ACS Med. Chem. Lett. 2015, 6, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Bosc, N.; Wroblowski, B.; Aci-Sèche, S.; Meyer, C.; Bonnet, P. A proteometric analysis of human kinome: Insight into discriminant conformation-dependent residues. ACS Chem. Biol. 2015, 10, 2827–2840. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Liao, J.J.L. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 2007, 50, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Niculescu-Duvaz, D.; Gaulon, C.; Dijkstra, H.P.; Niculescu-Duvaz, I.; Zambon, A.; Menard, D.; Suijkerbuijk, B.; Nourry, A.; Davies, L.; Manne, H.; et al. Pyridoimidazolones as novel potent inhibitors of V-RAF murine sarcoma viral oncogene homologue B1 (BRAF). J. Med. Chem. 2009, 52, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, J.; Wang, F.; Chen, X.; He, Y.; You, Q.; Zhou, H. Identification of type II inhibitors targeting BRAF using privileged pharmacophores. Chem. Biol. Drug Des. 2014, 83, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Manley, P.W.; Breitenstein, W.; Bruggen, J.; Cowan-Jacob, S.W.; Furet, P.; Mestan, J.; Meyer, T. Urea derivatives of STI571 as inhibitors of Bcr-Abl and PDCFR kinases. Bioorg. Med. Chem. Lett. 2004, 14, 5793–5797. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhou, H. Urea Derivatives and Preparation, Intermediates and Uses Thereof. WO 2012/139499 A1. Available online: http://worldwide.espacenet.com/publicationDetails/originalDocument?CC=WO&NR=2012139499A1&KC=A1&FT=D&ND=3&date=20121018&DB=EPODOC&locale=en_EP (accessed on 28 March 2015).
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Harder, E; Damm, W.; Friesner, R.A.; Sherman, W. Improving the prediction of absolute solvation free energies using the next generation OPLS force field. J. Chem. Theory Comput. 2012, 8, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
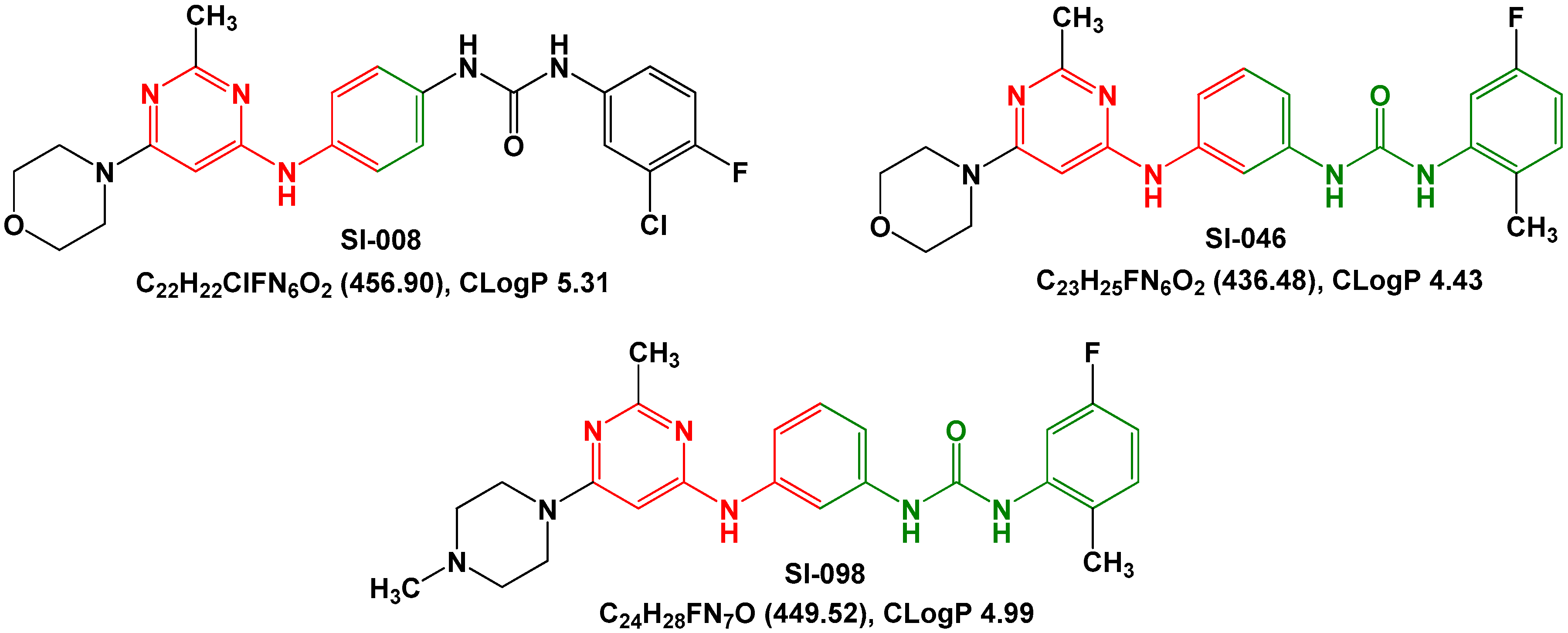
| Compound | IC50 (nM) | GlideScore (kJ/mol) |
|---|---|---|
| SI-046 | 298 | 11.3 |
| SI-008 | 685 | 10.3 |
| SI-098 | >10,000 | 10.2 |
| sorafenib | 263 | 12.1 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Zhang, X.; You, Q. Lead Discovery of Type II BRAF V600E Inhibitors Targeting the Structurally Validated DFG-Out Conformation Based upon Selected Fragments. Molecules 2016, 21, 879. https://doi.org/10.3390/molecules21070879
Zhang Q, Zhang X, You Q. Lead Discovery of Type II BRAF V600E Inhibitors Targeting the Structurally Validated DFG-Out Conformation Based upon Selected Fragments. Molecules. 2016; 21(7):879. https://doi.org/10.3390/molecules21070879
Chicago/Turabian StyleZhang, Qingwen, Xuejin Zhang, and Qidong You. 2016. "Lead Discovery of Type II BRAF V600E Inhibitors Targeting the Structurally Validated DFG-Out Conformation Based upon Selected Fragments" Molecules 21, no. 7: 879. https://doi.org/10.3390/molecules21070879
APA StyleZhang, Q., Zhang, X., & You, Q. (2016). Lead Discovery of Type II BRAF V600E Inhibitors Targeting the Structurally Validated DFG-Out Conformation Based upon Selected Fragments. Molecules, 21(7), 879. https://doi.org/10.3390/molecules21070879