
Design and Stereochemical Research (DFT, ECD and Crystal Structure) of Novel Bedaquiline Analogs as Potent Antituberculosis Agents

Abstract
:
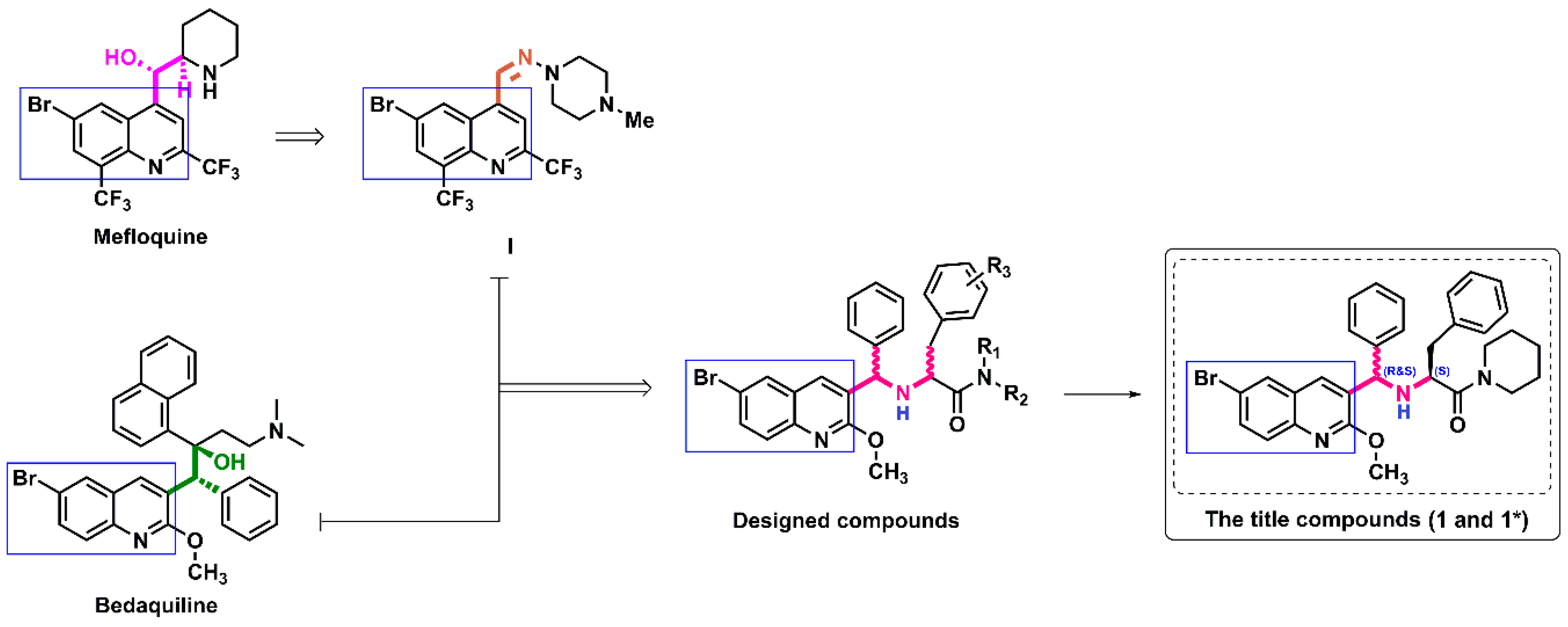
1. Introduction
2. Results and Discussion
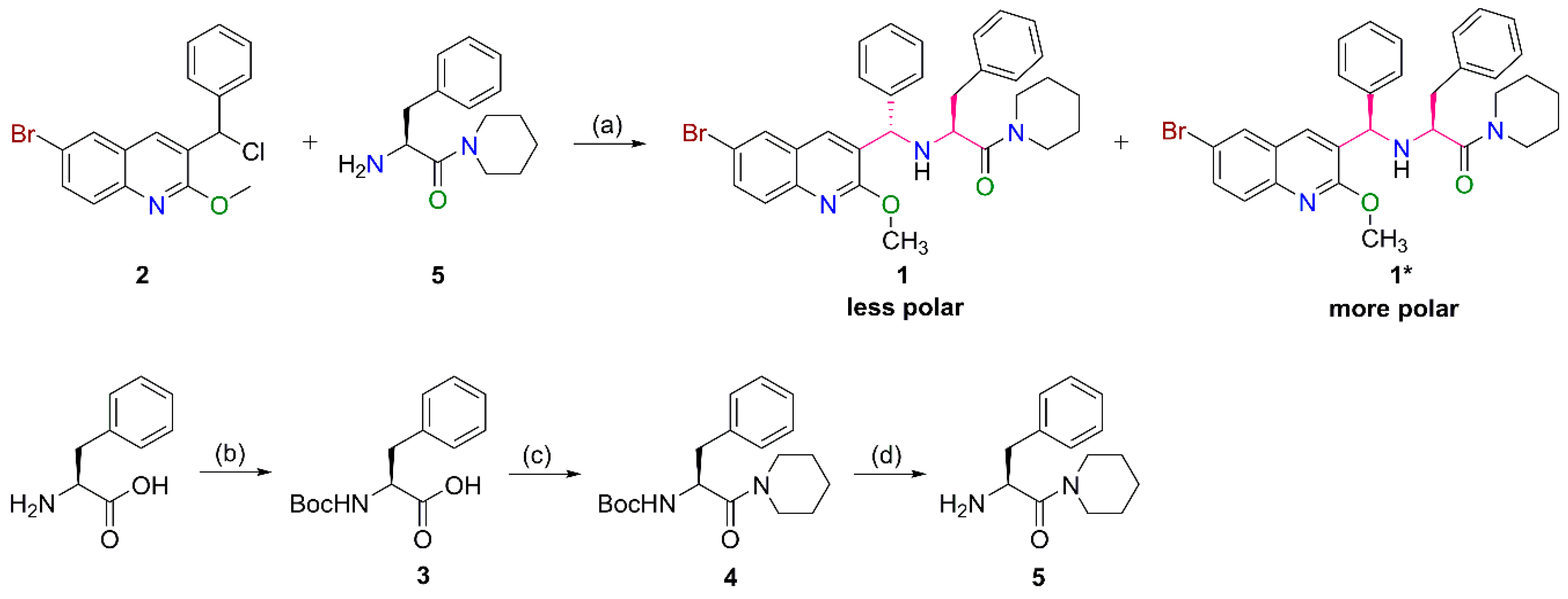
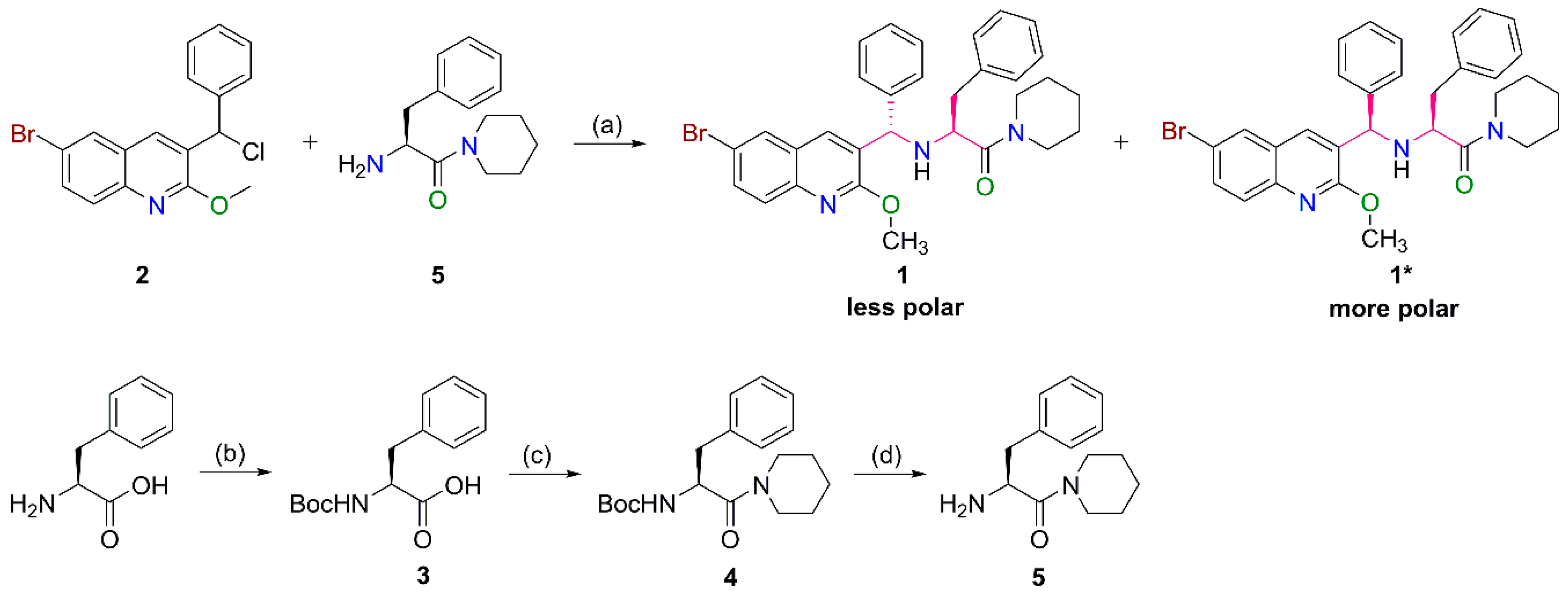
2.1. Preparation of the Target Compounds
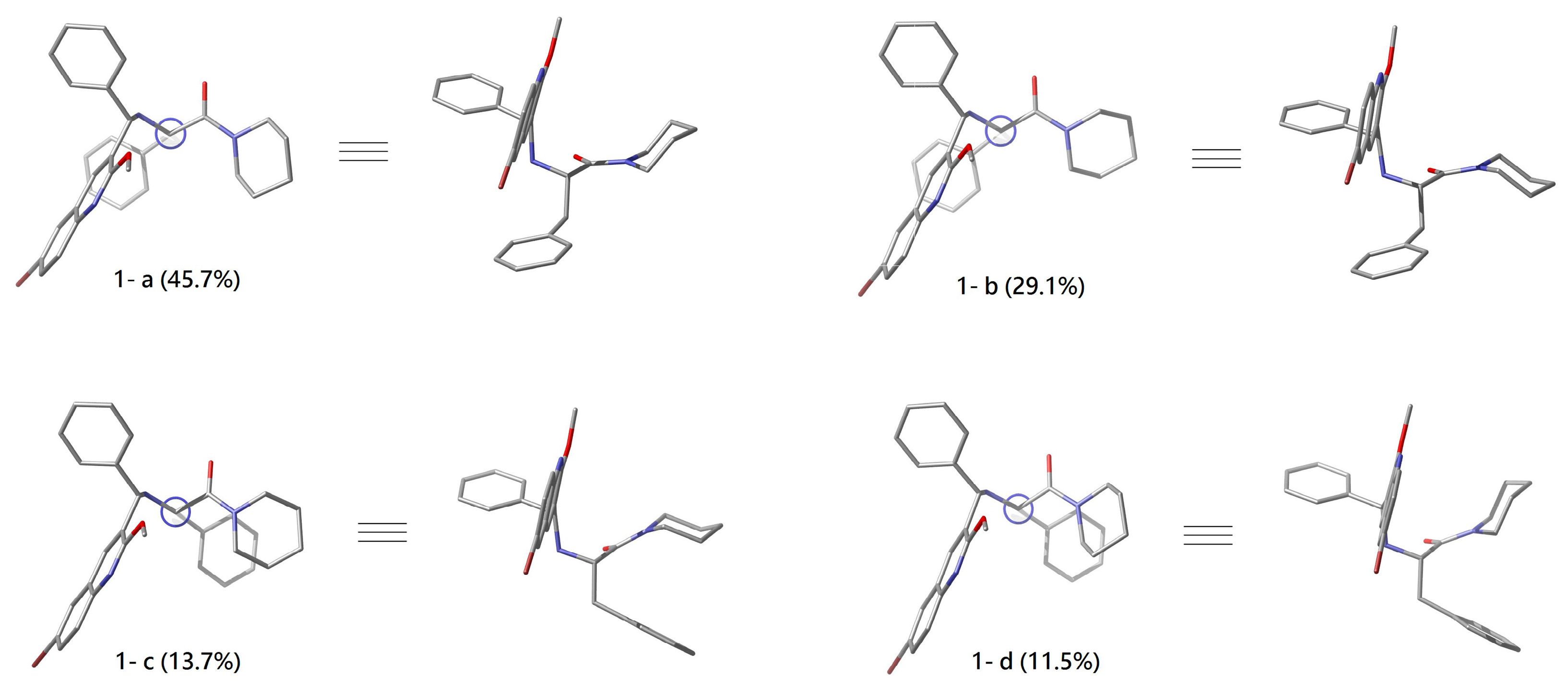
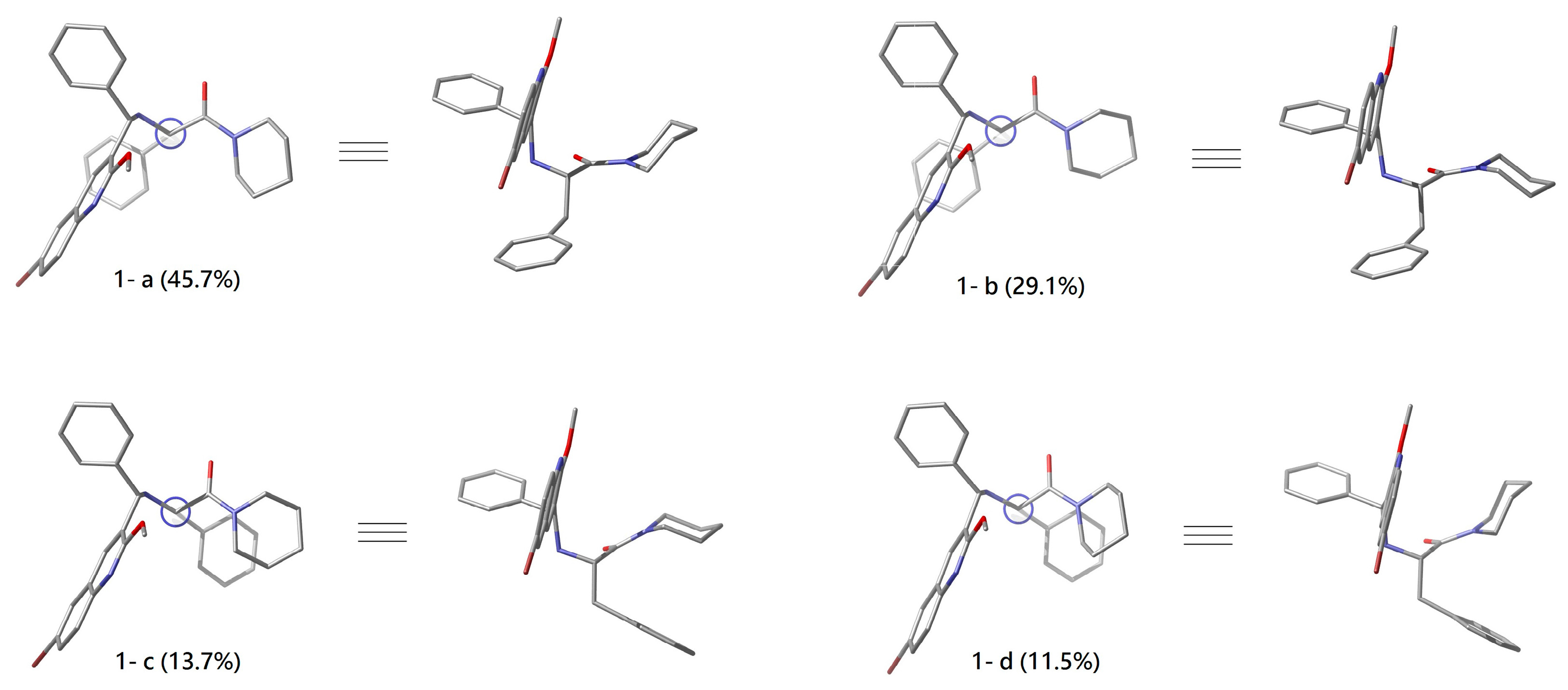
2.2. Conformational Analyses of 1 and 1*
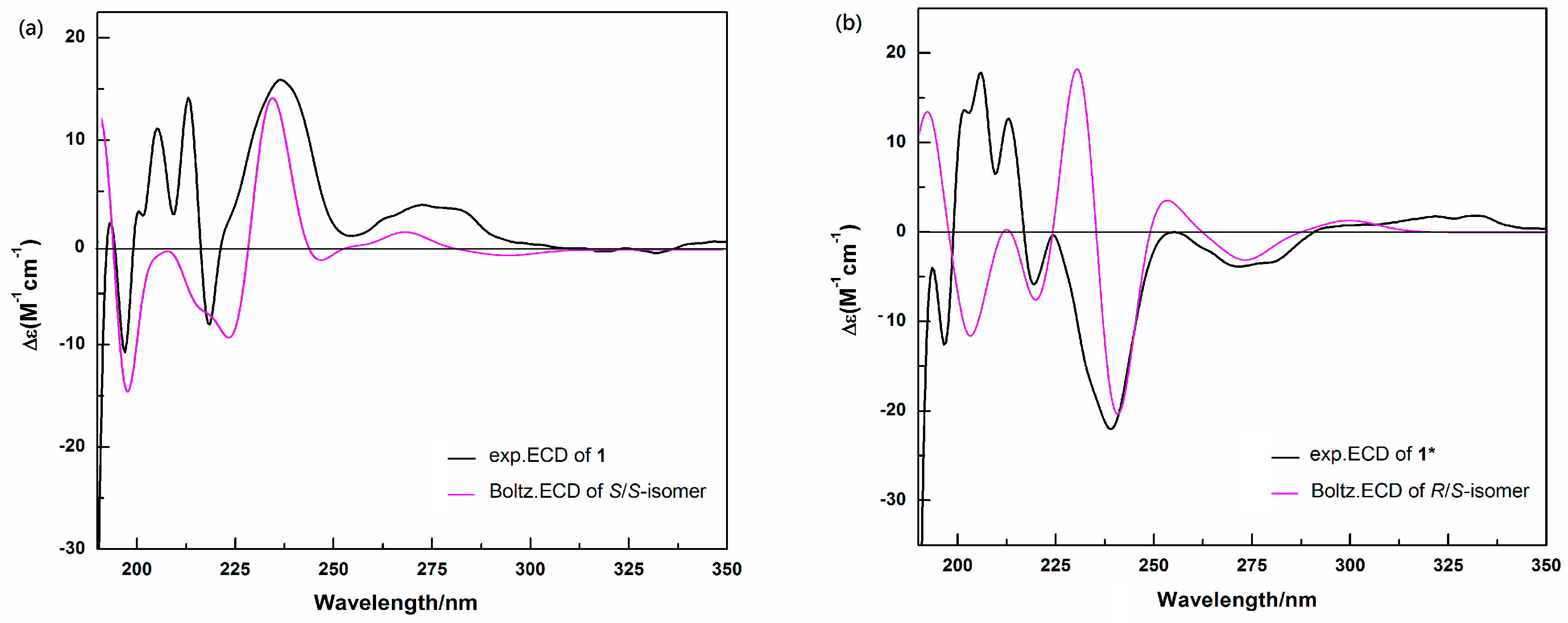
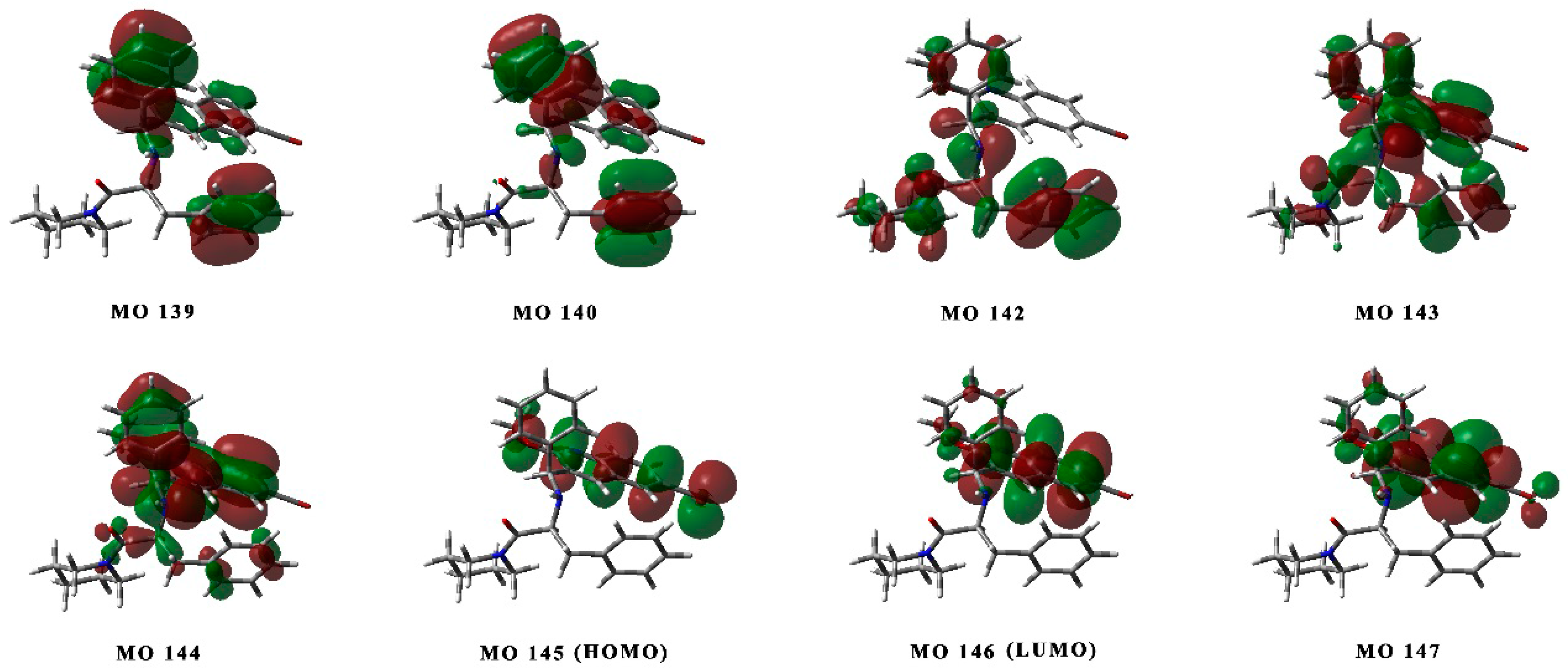
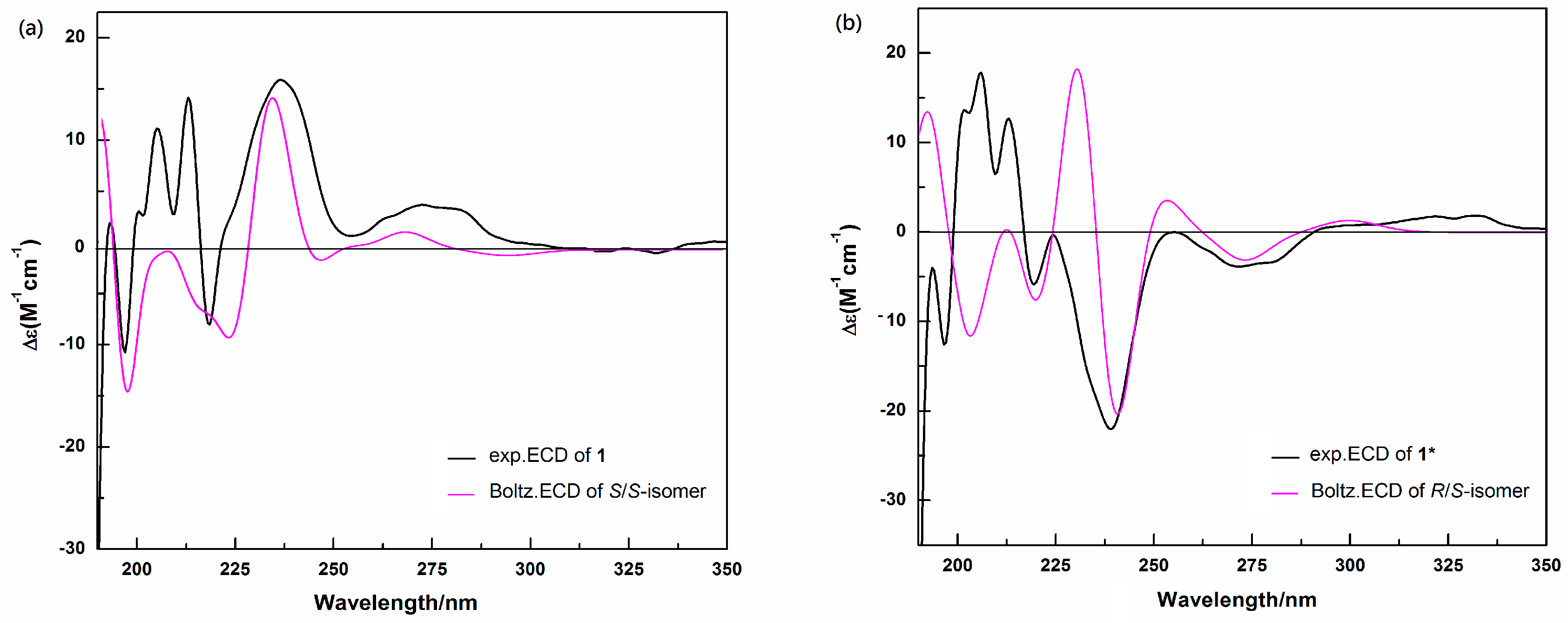
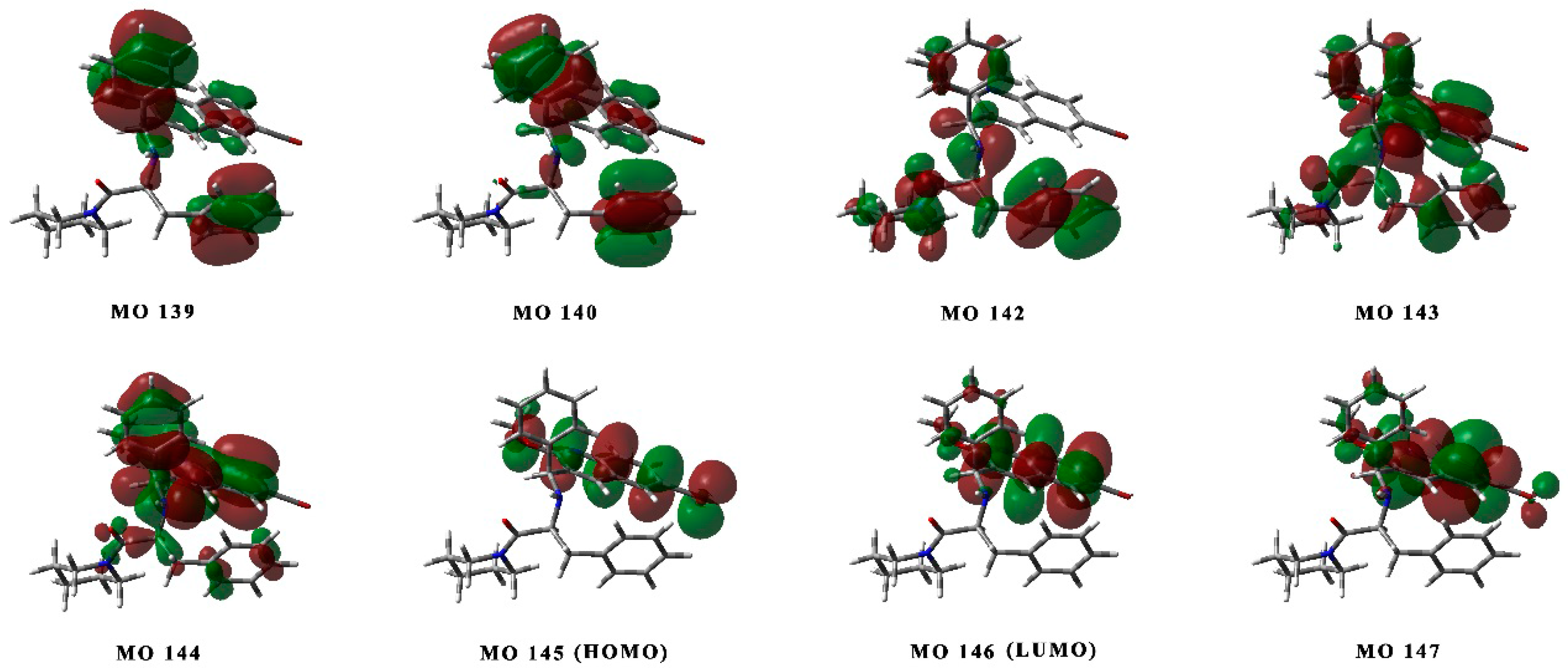
2.3. Determination of the Absolute Configurations of 1 and 1* by ECD
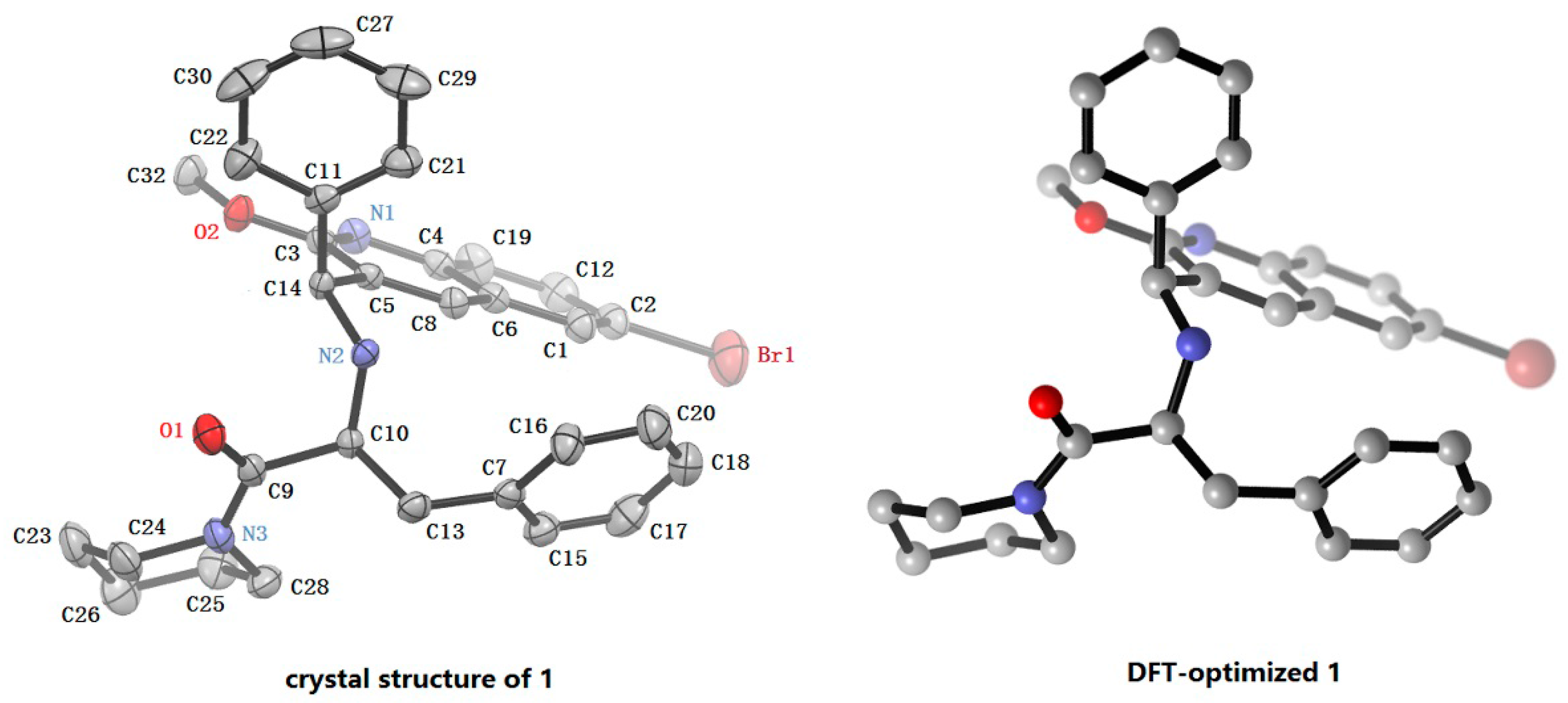
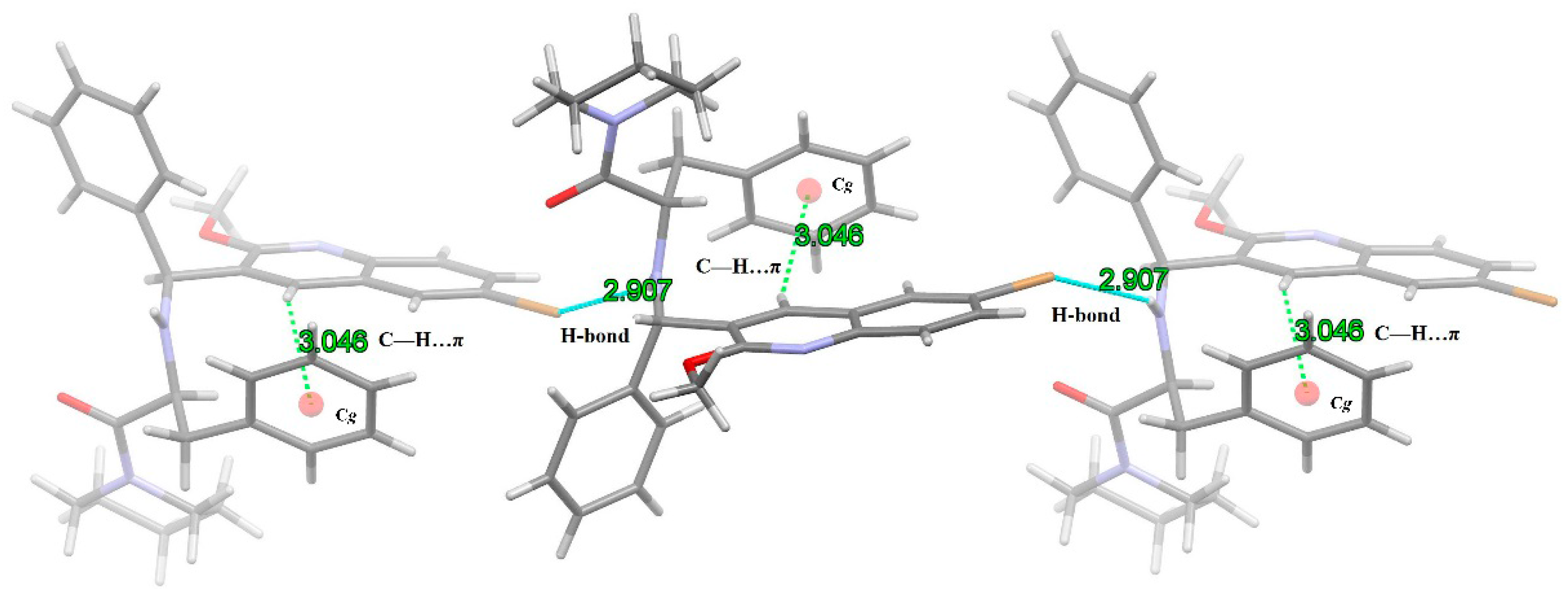
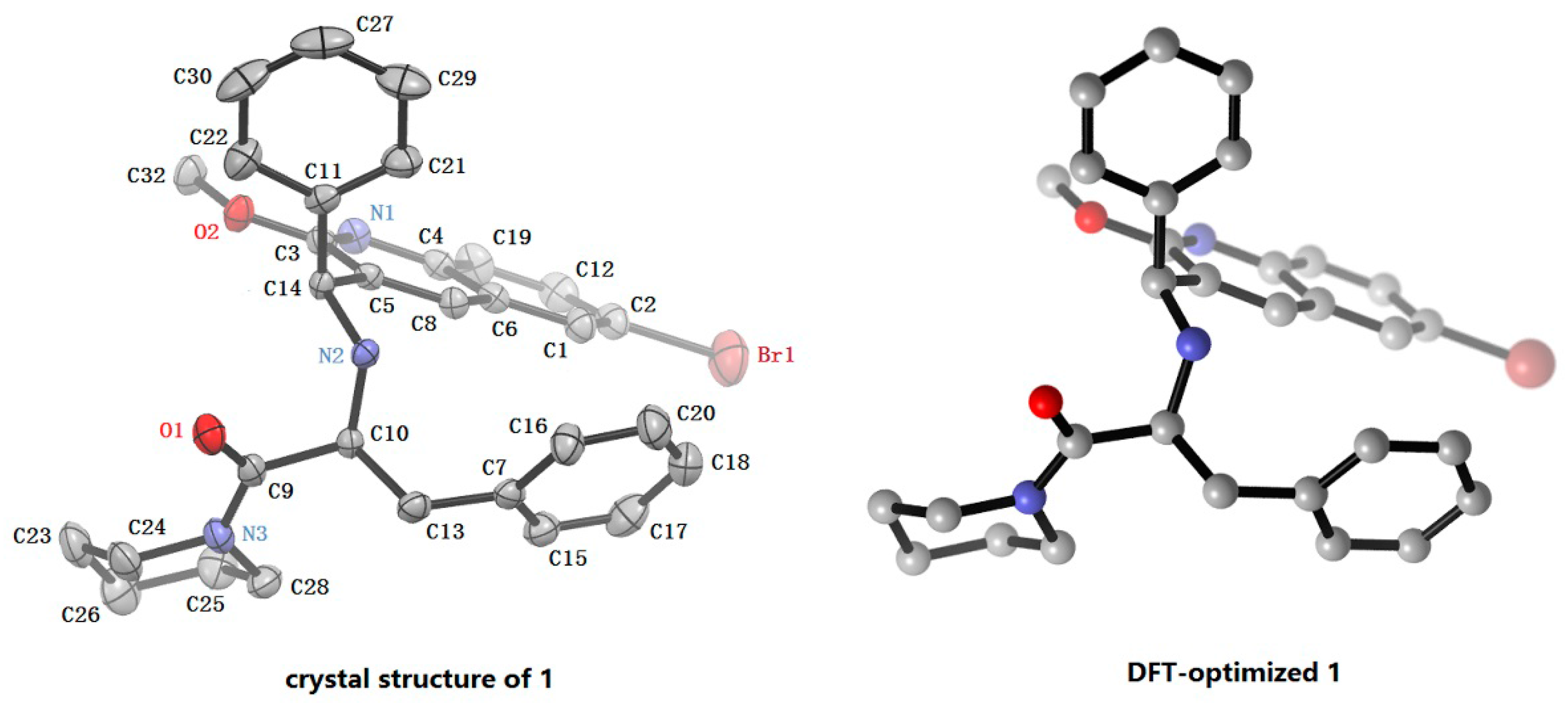
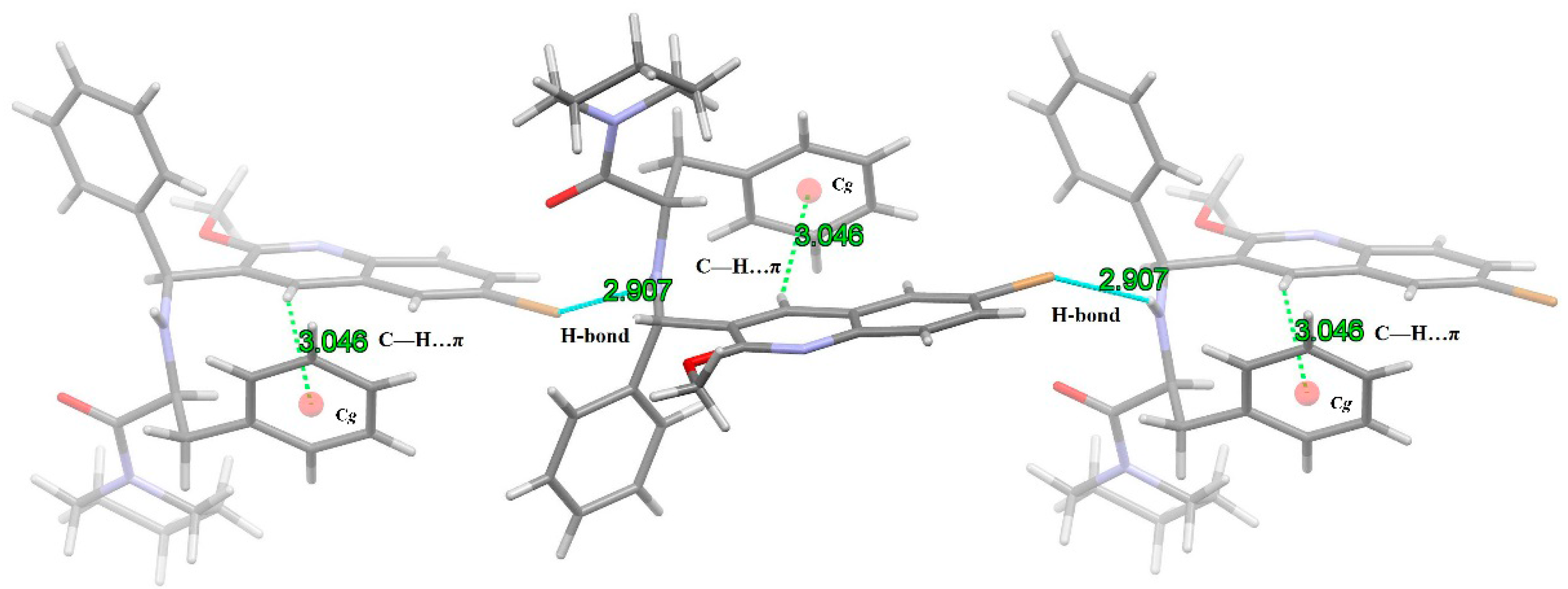
2.4. X-ray Crystal Structure Analysis of 1
2.5 Anti-Tuberculosis Activity
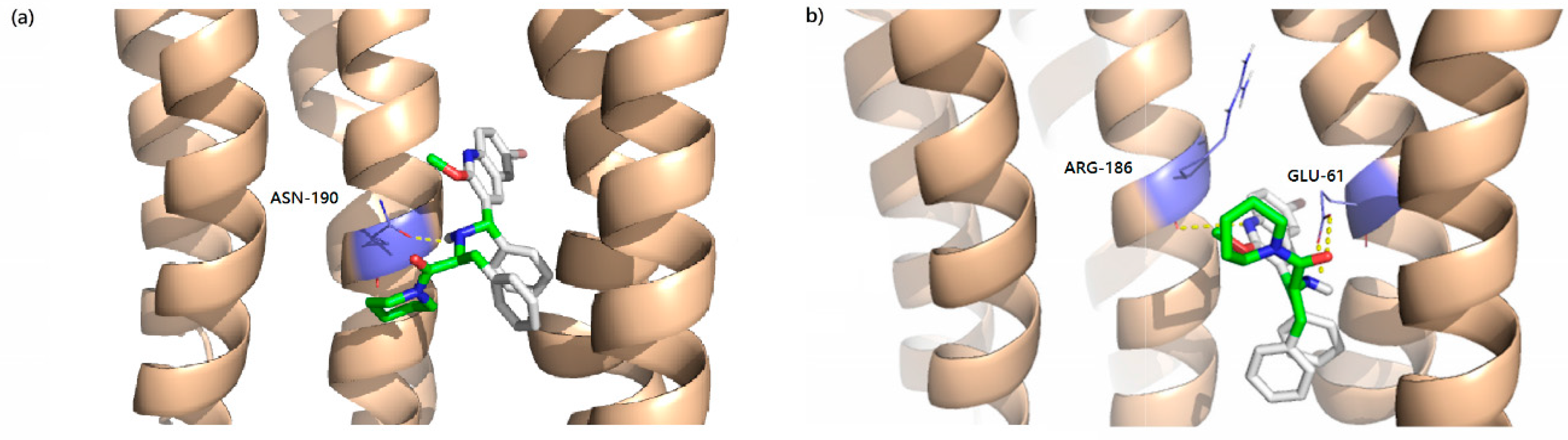
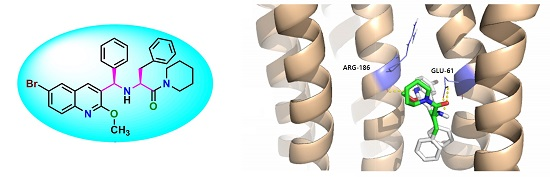
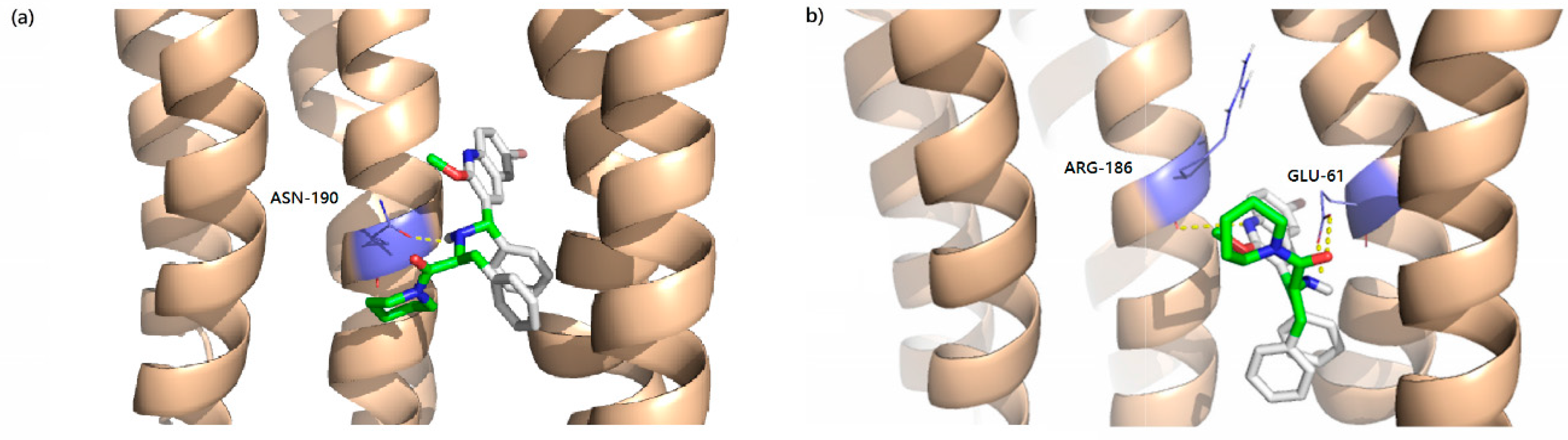
2.6 Docking Analysis
3. Materials and Methods
3.1. General Information
3.2. Materials
3.2.1. General Procedure for the Synthesis of 5
3.2.2. General Procedure for the Synthesis of 1 and 1*
3.3. Computational Details
3.3.1. Quantum Chemical Calculations
3.3.2. Mycobacterium tuberculosis ATPase Structural Prediction
3.3.3. Molecular Docking
3.4. Antimycobacterial Test for 1 and 1*
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Organization, W.H. Global Tuberculosis Report; WHO Report; WHO Press: Geneva, Switzerland, 2015. [Google Scholar]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.; Neefs, J.M.; Winkler, H.; van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Tucker, M.E. FDA Approves Bedaquiline for Resistant TB Treatment. Available online: http://www.medscape.com/viewarticle/776901 (accessed on 1 July 2016).
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL. Nucleic Acids Res. 1997, 25, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Boeckmann, B.; Ferro, S.; Gasteiger, E. Swiss-Prot.: Juggling between evolution and stability. Brief. Bioinform. 2004, 5, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Cross, R.L. Molecular Motors: Turning the ATP motor. Nature 2004, 427, 407–408. [Google Scholar] [CrossRef] [PubMed]
- Kluge, C.; Dimroth, P. Kinetics of inactivation of the F1F0 ATPase of Propionigenium modestum by dicyclohexylcarbodiimide in relationship to hydrogen ion and sodium concentration: Probing the binding site for the coupling ions. Biochemistry 1993, 32, 10378–10386. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, M.R.; Koymans, L.H.; Guillemont, J.E.; Koul, A.; Andries, K. A computational model of the inhibition of Mycobacterium tuberculosis ATPase by a new drug candidate R207910. Proteins Struct. Funct. Bioinform. 2007, 67, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Diacon, A.H.; Pym, A.; Grobusch, M.; Patientia, R.; Rustomjee, R.; Page-Shipp, L.; Pistorius, C.; Krause, R.; Bogoshi, M.; Churchyard, G. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 2009, 360, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Guillemont, J.; Van Gestel, J.; Venet, M.; Poignet, H.; Decrane, L.; Vernier, D. Quinoline Derivatives and Their Use as Mycobacterial Inhibitors. WO Patent 2004/011436, 5 February 2004. [Google Scholar]
- Upadhayaya, R.S.; Kulkarni, G.M.; Vasireddy, N.R.; Vandavasi, J.K.; Dixit, S.S.; Sharma, V.; Chattopadhyaya, J. Design, synthesis and biological evaluation of novel triazole, urea and thiourea derivatives of quinoline againstMycobacterium tuberculosis. Bioorg. Med. Chem. 2009, 17, 4681–4692. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Vandavasi, J.K.; Vasireddy, N.R.; Sharma, V.; Dixit, S.S.; Chattopadhyaya, J. Design, synthesis, biological evaluation and molecular modelling studies of novel quinoline derivatives against Mycobacterium tuberculosis. Bioorg. Med. Chem. 2009, 17, 2830–2841. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Lahore, S.V.; Sayyed, A.Y.; Dixit, S.S.; Shinde, P.D.; Chattopadhyaya, J. Conformationally-constrained indeno[2,1-c]quinolines—A new class of anti-mycobacterial agents. Org. Biomol. Chem. 2010, 8, 2180–2197. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Shinde, P.D.; Sayyed, A.Y.; Kadam, S.A.; Bawane, A.N.; Poddar, A.; Plashkevych, O.; Foldesi, A.; Chattopadhyaya, J. Synthesis and structure of azole-fused indeno[2,1-c]quinolines and their anti-mycobacterial properties. Org. Biomol. Chem. 2010, 8, 5661–5673. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Vandavasi, J.K.; Kardile, R.A.; Lahore, S.V.; Dixit, S.S.; Deokar, H.S.; Shinde, P.D.; Sarmah, M.P.; Chattopadhyaya, J. Novel quinoline and naphthalene derivatives as potent antimycobacterial agents. Eur. J. Med. Chem. 2010, 45, 1854–1867. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhong, W.; Liu, P.; Xiao, J.; Zheng, Z.; Xie, Y.; Zhao, G.; Wang, X.; Wang, L.; Li, X. Aromatic Butan-2-ol Compounds, Preparation Methods and Uses Thereof. U.S. Patent 8,674,136, 18 March 2014. [Google Scholar]
- Tantry, S.J.; Shinde, V.; Balakrishnan, G.; Markad, S.D.; Gupta, A.K.; Bhat, J.; Narayan, A.; Raichurkar, A.; Jena, L.K.; Sharma, S.; et al. Scaffold morphing leading to evolution of 2,4-diaminoquinolines and aminopyrazolopyrimidines as inhibitors of the ATP synthesis pathway. Med. Chem. Comm. 2016, 7, 1022–1032. [Google Scholar] [CrossRef]
- Qiao, C.; Wang, X.; Xie, F.; Zhong, W.; Li, S. Asymmetric Synthesis and Absolute Configuration Assignment of a New Type of Bedaquiline Analogue. Molecules 2015, 20, 22272–22285. [Google Scholar] [CrossRef] [PubMed]
- Guillemont, J.E.G.; Lancois, D.F.A.; Pasquier, E.T.J.; Andries, K.J.L.M.; Koul, A. Antibicaterial Quinoline Dervatives, WO. Patent 2007/014885/A1, 26 July 2006. [Google Scholar]
- Mao, J.; Wang, Y.; Wan, B.; Kozikowski, A.P.; Franzblau, S.G. Design, Synthesis, and Pharmacological Evaluation of Mefloquine-Based Ligands as Novel Antituberculosis Agents. Chem. Med. Chem. 2007, 2, 1624–1630. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Wang, L.; Chen, Y.; Yuan, L.; Xu, W.; Sun, T. Synthesis, crystal, computational study and in vitro anti-tuberculosis activity of N-(furan-2-yl-methyl)-N-(phenyl (quinolin-3-yl) methyl) acetamide derivatives. J. Mol. Struct. 2011, 1005, 113–120. [Google Scholar] [CrossRef]
- Bai, Y.-F.; Yuan, L.; Chen, Y.; Wang, L.-J.; Wang, C.; Sun, T.-M. Synthesis, Crystal and Calculated Structure, and Biological Activity of 2-((6-Bromo-2-methoxyquinolin-3-yl)(phenyl) methyl)-2,3,7,7a-tetrahydro-3a,6-epoxyisoindol-1(6H)-one. J. Chem. Crystallogr. 2012, 42, 318–322. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, L.; Chen, Y.; Xu, W.; Sun, T. Determination of Absolute Configurations of Bedaquiline Analogs by Quantum Chemical Electronic Circular Dichroism Calculations and an X-ray Diffraction Study. Eur. J. Org. Chem. 2014, 18, 3814–3821. [Google Scholar] [CrossRef]
- Wang, Z.; Li, L.; Zhou, Z.; Geng, Y.; Chen, Y.; Sun, T. Design, Synthesis, Configuration Research, and in vitro Antituberculosis Activities of two Chiral Naphthylamine Substituted Analogs of Bedaquiline. J. Heterocycl. Chem. 2016, in press. [Google Scholar] [CrossRef]
- Polavarapu, P.L.; Donahue, E.A.; Shanmugam, G.; Scalmani, G.; Hawkins, E.K.; Rizzo, C.; Ibnusaud, I.; Thomas, G.; Habel, D.; Sebastian, D. A Single Chiroptical Spectroscopic Method May Not Be Able To Establish the Absolute Configurations of Diastereomers: Dimethylesters of Hibiscus and Garcinia Acids. J. Phys. Chem. A 2011, 115, 5665–5673. [Google Scholar] [CrossRef] [PubMed]
- Krykunov, M.; Kundrat, M.D.; Autschbach, J. Calculation of circular dichroism spectra from optical rotatory dispersion, and vice versa, as complementary tools for theoretical studies of optical activity using time-dependent density functional theory. J. Chem. Phys. 2006, 125. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Inoue, Y.; Grimme, S. Time Dependent Density Functional Theory Calculations for Electronic Circular Dichroism Spectra and Optical Rotations of Conformationally Flexible Chiral Donor—Acceptor Dyad. J. Org. Chem. 2006, 71, 9797–9806. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.-M.; Yan, L.-K.; Wang, J.-P.; Su, Z.-M. TDDFT Studies on the Electronic Structures and Chiroptical Properties of Mono-Tin-Substituted Wells-Dawson Polyoxotungstates. J. Phys. Chem. A 2012, 116, 4152–4158. [Google Scholar] [CrossRef] [PubMed]
- Bosnich, B. Molecular mechanics force fields for cyclopentadienyl complexes. Chem. Soc. Rev. 1994, 23, 387–395. [Google Scholar] [CrossRef]
- Huang, N.; Kalyanaraman, C.; Bernacki, K.; Jacobson, M.P. Molecular mechanics methods for predicting protein-ligand binding. Phys. Chem. Chem. Phys. 2006, 8, 5166–5177. [Google Scholar] [CrossRef] [PubMed]
- Tanak, H. Crystal Structure, Spectroscopy, and Quantum Chemical Studies of (E)-2-[(2-Chlorophenyl) iminomethyl]-4-trifluoromethoxyphenol. J. Phys. Chem. A 2011, 115, 13865–13876. [Google Scholar] [CrossRef] [PubMed]
- Tanak, H. Density functional computational studies on 2-[(2,4-Dimethylphenyl)iminomethyl]-3,5-dimethoxyphenol. Int. J. Quantum Chem. 2012, 112, 2392–2402. [Google Scholar] [CrossRef]
- Fitzgerald, G.; Andzelm, J. Chemical applications of density functional theory: Comparison to experiment, Hartree-Fock, and perturbation theory. J. Phys. Chem. 1991, 95, 10531–10534. [Google Scholar] [CrossRef]
- Andzelm, J.; Wimmer, E. Density functional Gaussian-type-orbital approach to molecular geometries, vibrations, and reaction energies. J. Chem. Phys. 1992, 96, 1280–1303. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO-MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Cole, S.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.; Eiglmeier, K.; Gas, S.; Barry, C.R. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Šali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Ponder, J.W.; Richards, F.M. An efficient newton-like method for molecular mechanics energy minimization of large molecules. J. Comput. Chem. 1987, 8, 1016–1024. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Bruker, A. Saint and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2005. [Google Scholar]
- Sheldrick, G. SHELXT 97. In Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G. SHELXTL Version 5.1; Bruker Analytical X-ray Instruments Inc.: Madison, WI, USA, 1998. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Streek, J.V.; Wood, P.A. Mercury CSD 2.0–new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Gaussian 09, Revision C. 01; Gaussian: Wallingford, CT, USA, 2010.
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Rastogi, V.K.; Girvin, M.E. Structural changes linked to proton translocation by subunit c of the ATP synthase. Nature 1999, 402, 263–268. [Google Scholar] [PubMed]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. Arch. Biochem. Biophys. 1978, 185, 584–591. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Bowie, J.U.; Luthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Liithy, R.; Bowie, J.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Huitric, E.; Verhasselt, P.; Andries, K.; Hoffner, S.E. In vitro Antimycobacterial Spectrum of a Diarylquinoline ATP Synthase Inhibitor. Antimicrob. Agents Chemother. 2007, 51, 4202–4204. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds reported herein are available from the authors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conf. | G (kcal/mol) | △G (kcal/mol) | Pi% |
|---|---|---|---|
| 1-a | −2566110.908 | 0 | 45.7 |
| 1-b | −2566110.645 | 0.263 | 29.1 |
| 1-c | −2566110.203 | 0.705 | 13.7 |
| 1-d | −2566110.099 | 0.809 | 11.5 |
| Empirical Formula | C31H32BrN3O2 |
|---|---|
| Formula weight | 558.5 |
| CCDC number | 1419767 |
| Temperature (K) | 296 |
| Crystal size | 0.32 × 0.22 × 0.15 |
| Crystal color | colourless |
| Crystal system | monoclinic |
| Space group | P 21 21 21 |
| a (Å) | 9.2390(3) |
| b (Å) | 15.4237(5) |
| c (Å) | 19.6921(6) |
| α (°) | 90.00 |
| β (°) | 90.00 |
| γ (°) | 90.00 |
| V (Å3) | 2806.12(15) |
| Z | 4.00 |
| Theta (max) | 25.65 |
| R1, wR2 | 0.0600, 0.1987 |
| Data completeness | 1.75/0.99 |
| Max. and min. transmission | 0.749, 0.741 |
| Wavelength | 0.71073 |
| D-H···A | d(D-H) | d(H…A) | d(D…A) | < (DHA) |
|---|---|---|---|---|
| N(2)-H(2)···Br(1)i | 0.86 | 2.907 | 3.871 | 149.03 |
| Br(1)···H(2)-N(2)ii | 0.86 | 2.907 | 3.871 | 149.03 |
| Bond Distances (Å) | Exp.(1) | Cal.(1-a) | Diff. | Bond Angle (°) | Exp.(1) | Cal.(1-a) | Diff. |
|---|---|---|---|---|---|---|---|
| Br(1)–C(2) | 1.861(4) | 1.921 | 0.060 | Br(1)–C(2)–C(12) | 119.6(3) | 118.6 | −0.1 |
| C(3)–O(2) | 1.332(5) | 1.351 | 0.019 | N(2)–C(10)–C(13) | 109.8(3) | 110.3 | 0.5 |
| C(5)–C(14) | 1.518(5) | 1.521 | 0.003 | N(3)–(9)–C(10) | 117.7(4) | 119.7 | 2.0 |
| C(9)–O(1) | 1.204(5) | 1.226 | 0.022 | C(3)–O(2)–C(32) | 117.1(4) | 117.3 | 0.2 |
| C(9)–N(3) | 1.347(6) | 1.365 | 0.018 | C(5)–C(14)–C(11) | 112.2(3) | 112.2 | 0.0 |
| C(9)–C(10) | 1.546(6) | 1.551 | 0.005 | C(10)–N(2)–C(14) | 114.1(3) | 114.4 | 0.3 |
| C(10)–N(2) | 1.449(5) | 1.457 | 0.008 | ||||
| C(10)–C(13) | 1.543(6) | 1.551 | 0.008 | ||||
| C(11)–C(14) | 1.498(6) | 1.526 | 0.028 | ||||
| C(14)–N(2) | 1.472(5) | 1.476 | 0.004 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geng, Y.; Li, L.; Wu, C.; Chi, Y.; Li, Z.; Xu, W.; Sun, T. Design and Stereochemical Research (DFT, ECD and Crystal Structure) of Novel Bedaquiline Analogs as Potent Antituberculosis Agents. Molecules 2016, 21, 875. https://doi.org/10.3390/molecules21070875
Geng Y, Li L, Wu C, Chi Y, Li Z, Xu W, Sun T. Design and Stereochemical Research (DFT, ECD and Crystal Structure) of Novel Bedaquiline Analogs as Potent Antituberculosis Agents. Molecules. 2016; 21(7):875. https://doi.org/10.3390/molecules21070875
Chicago/Turabian StyleGeng, Yiding, Linwei Li, Chengjun Wu, Yumeng Chi, Zhen Li, Wei Xu, and Tiemin Sun. 2016. "Design and Stereochemical Research (DFT, ECD and Crystal Structure) of Novel Bedaquiline Analogs as Potent Antituberculosis Agents" Molecules 21, no. 7: 875. https://doi.org/10.3390/molecules21070875
APA StyleGeng, Y., Li, L., Wu, C., Chi, Y., Li, Z., Xu, W., & Sun, T. (2016). Design and Stereochemical Research (DFT, ECD and Crystal Structure) of Novel Bedaquiline Analogs as Potent Antituberculosis Agents. Molecules, 21(7), 875. https://doi.org/10.3390/molecules21070875