
Targeting Bacterial Dsb Proteins for the Development of Anti-Virulence Agents


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
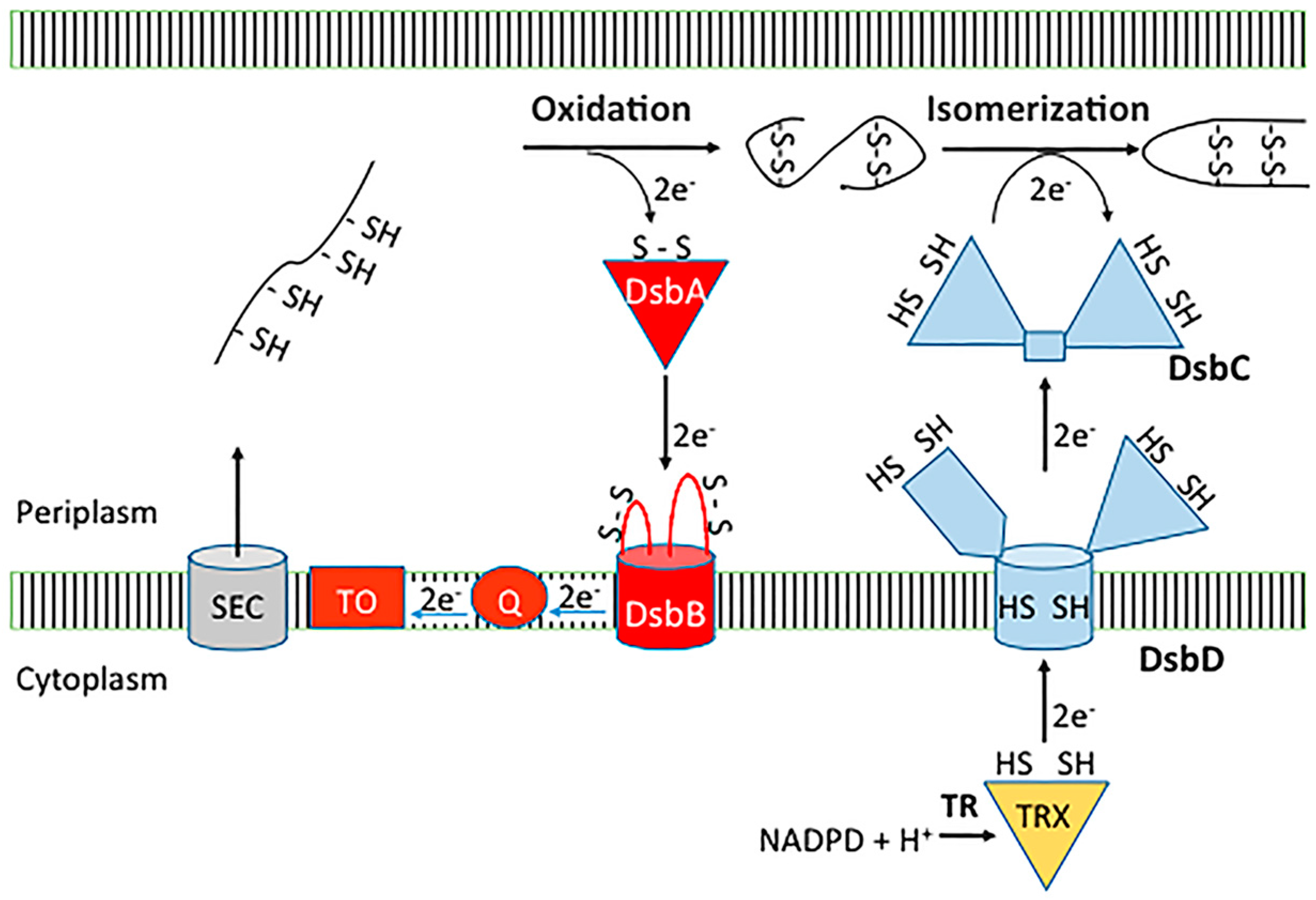
2. Dsb Disulfide Forming Pathways in the Model Organism E. coli K-12
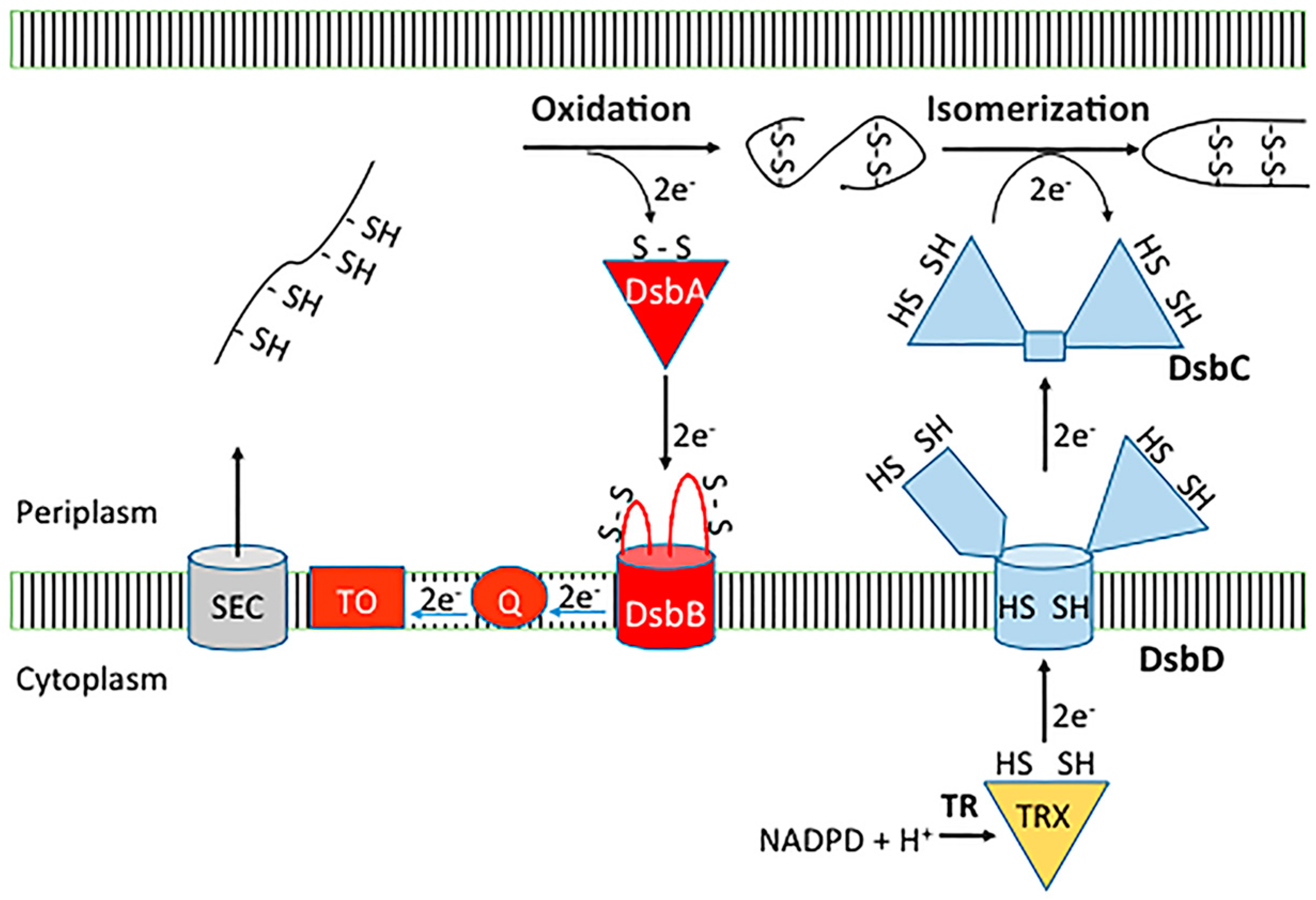
2.1. Dsb Oxidative Pathway
2.2. Dsb Isomerase Pathway
3. Distribution of Dsb Systems across Bacteria
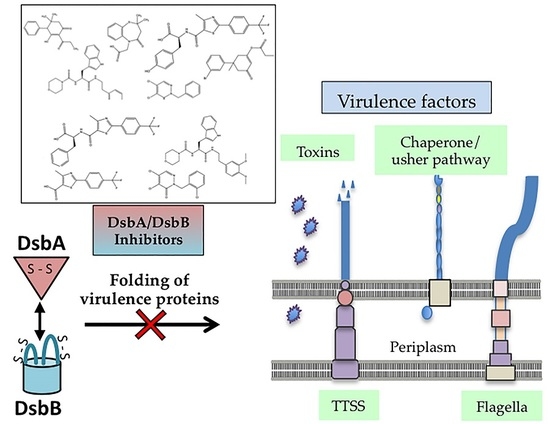
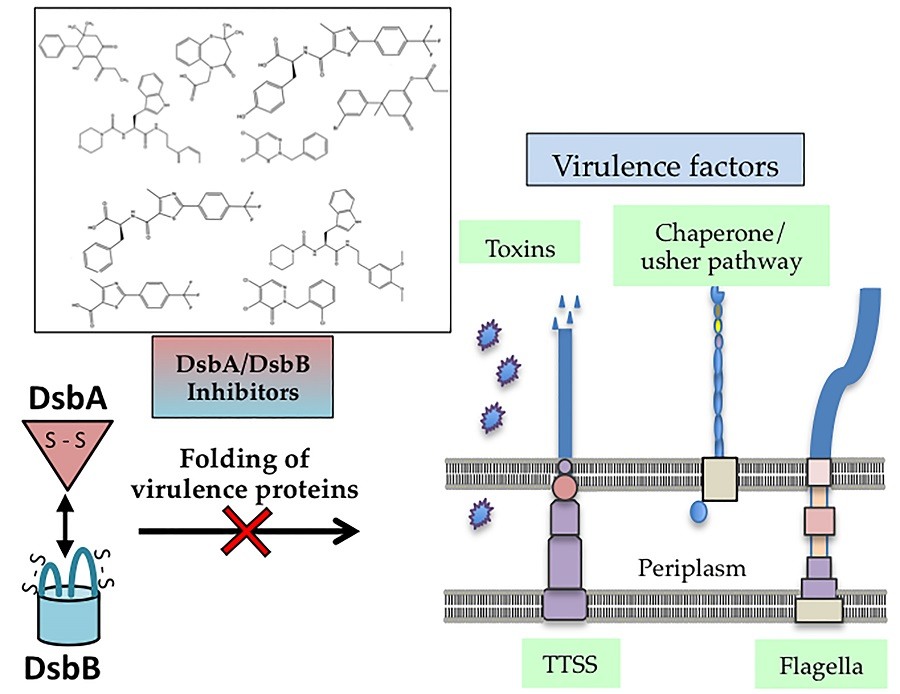
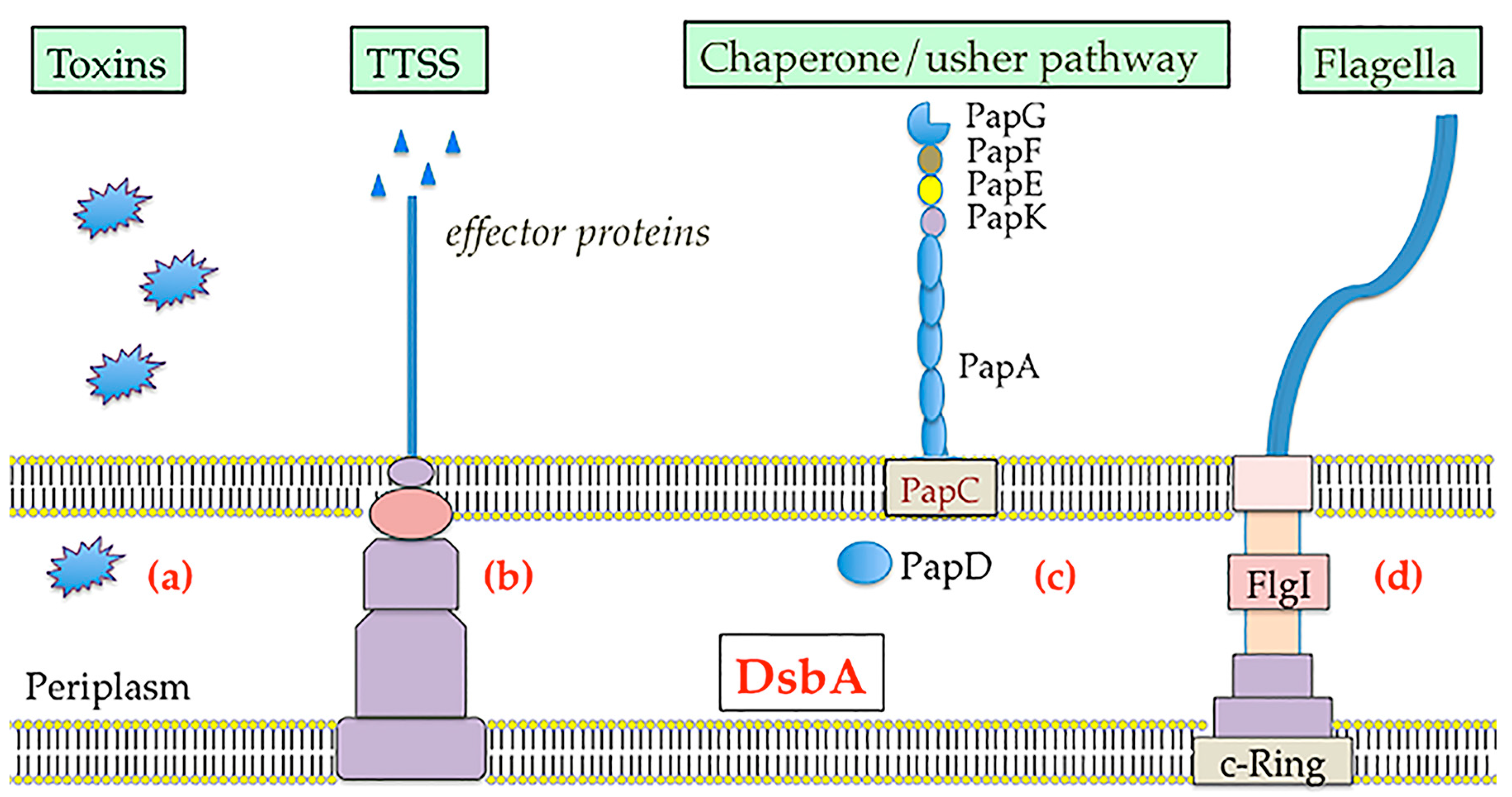
4. Dsb Systems and Bacterial Virulence
5. Targeting Dsb Proteins for the Development of Anti-virulence Agents
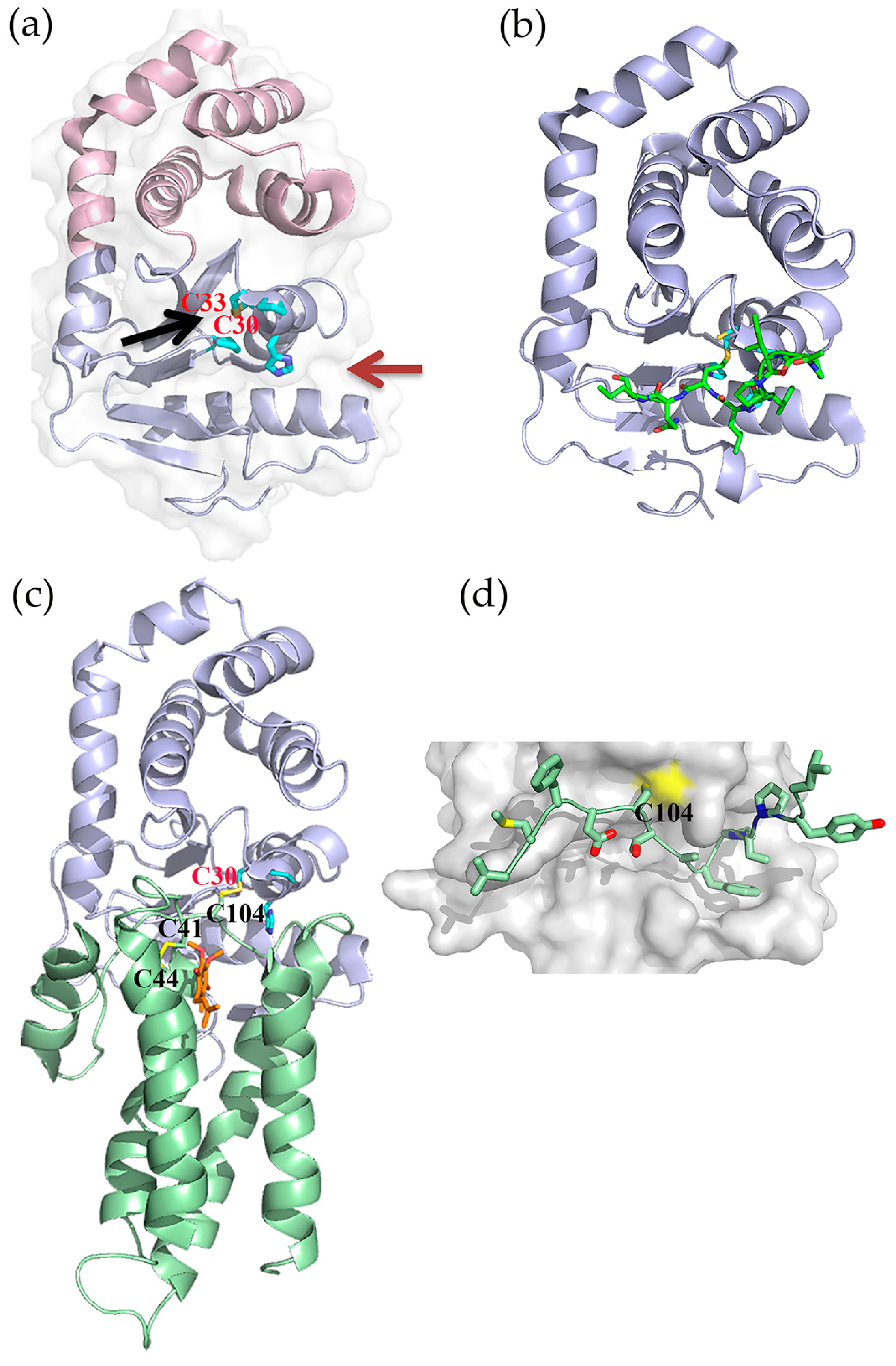
5.1. Fragment-Based Drug Discovery Targeting E. coli DsbB
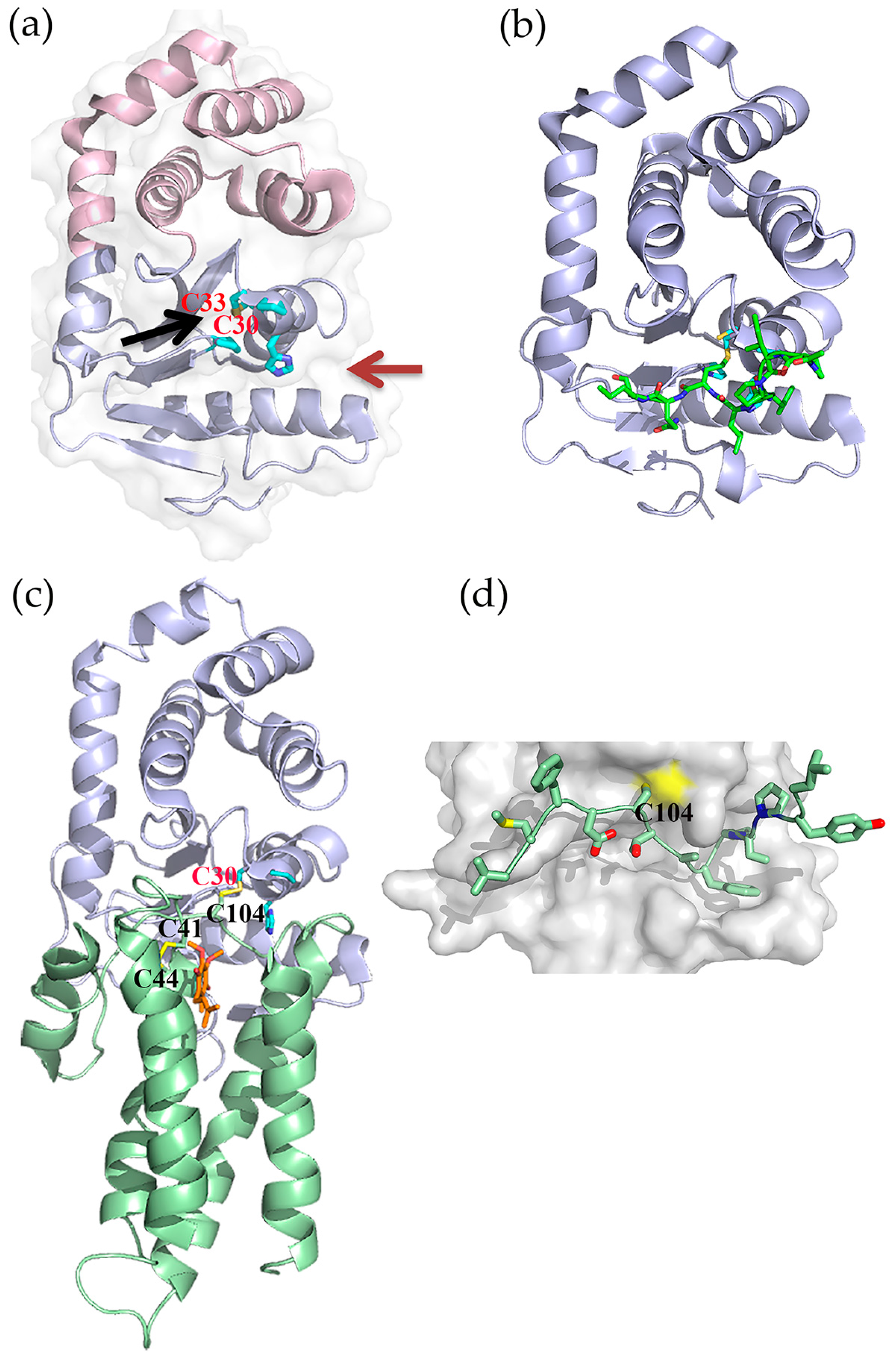
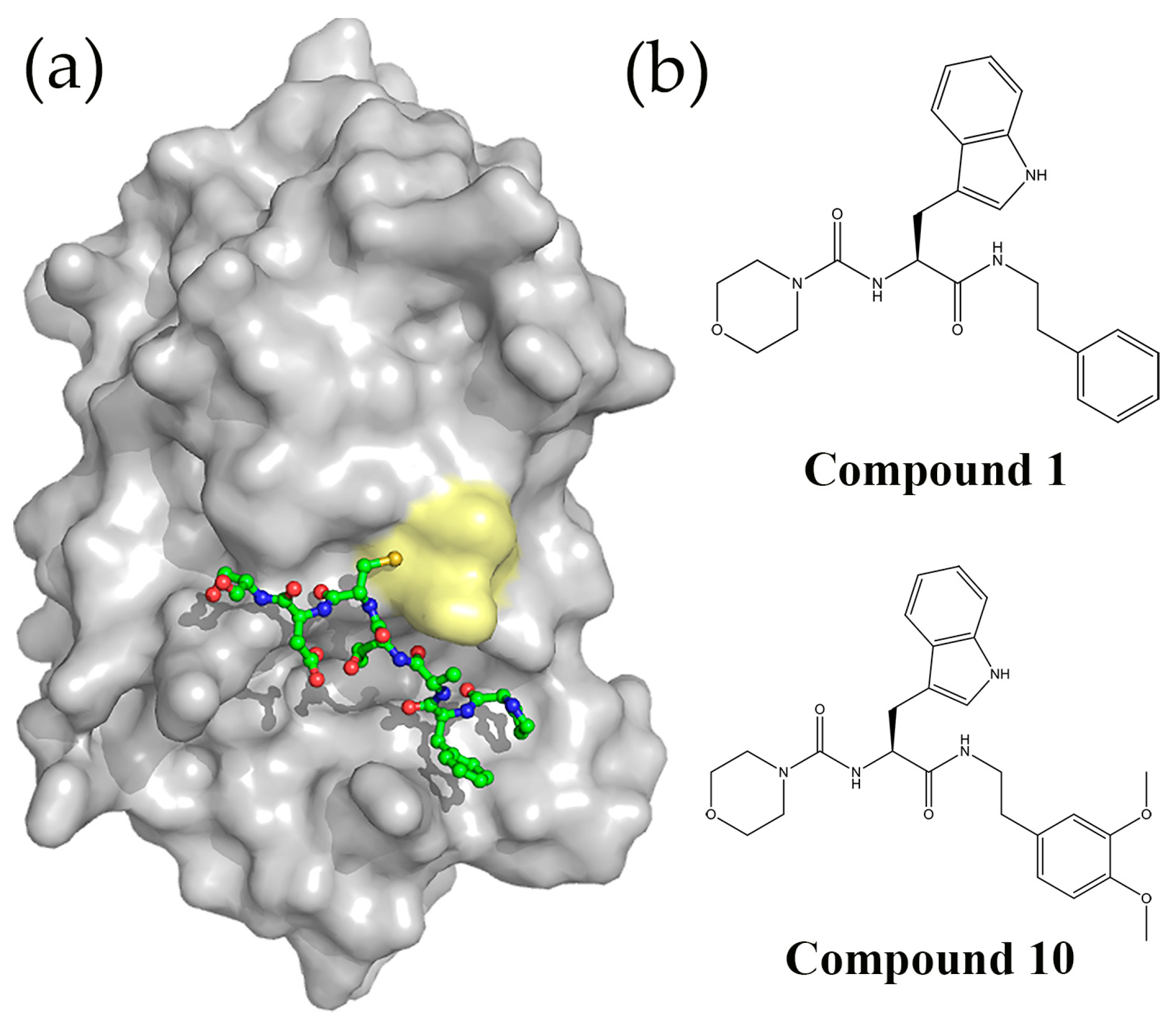
5.2. Fragment-Based Drug Discovery Targeting E. coli DsbA
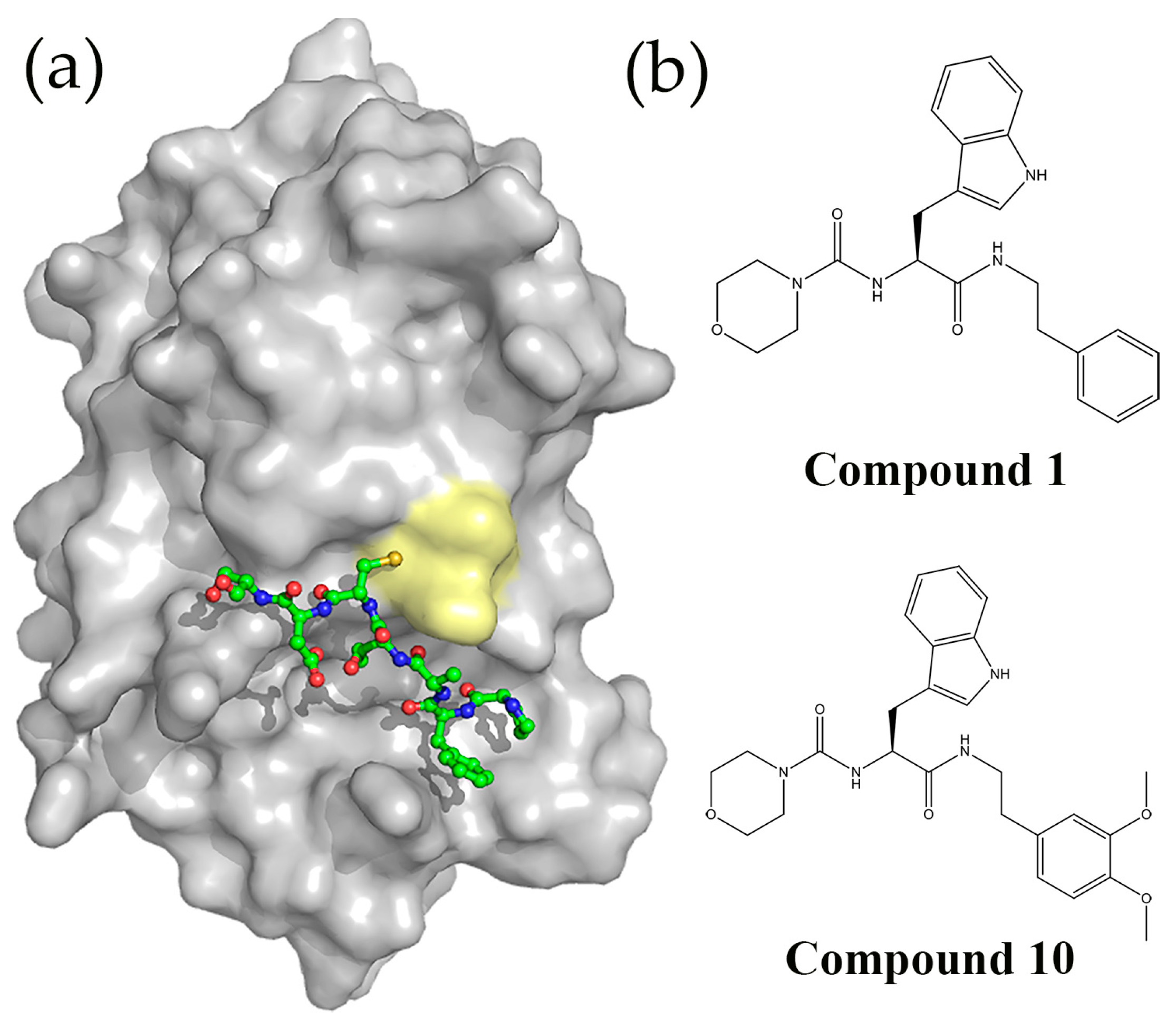
5.3. Peptides and Peptidomimetics Targeting DsbA
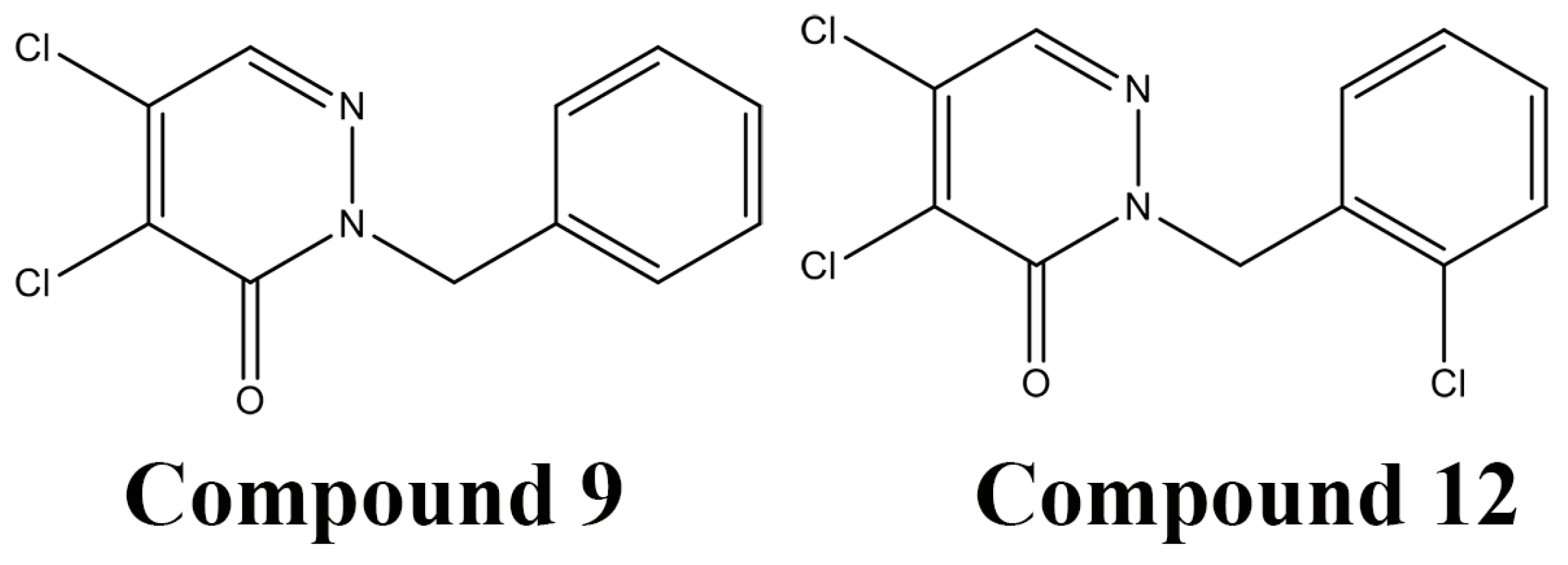
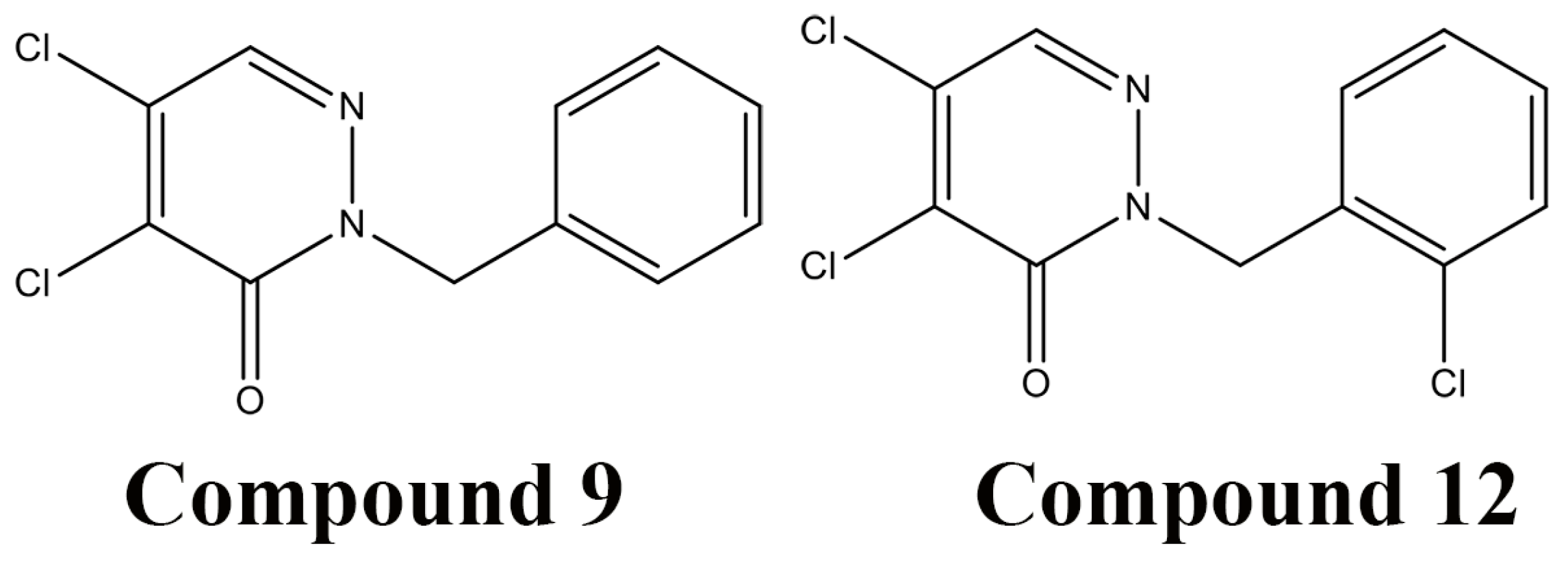
5.4. High-throughout Screening for DsbB Inhibitors
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CSP | Chemical Shift Perturbations |
| Dsb | Disulfide Bond |
| ESI-MS | Electrospray Ionization Mass Spectrometry |
| EPEC | Enteropathogenic E. coli |
| FBDD | Fragment-Based Drug Discovery |
| ITC | Isothermal Titration Calorimetry |
| LC-MS/MS | Liquid Chromatography-Mass Spectrometry |
| HSQC | Heteronuclear Single Quantum Coherence |
| MDR | Multi Drug Resistant |
| NMR | Nuclear Magnetic Resonance |
| Q | Quinones |
| SAR | Structure-Activity Relationship |
| TINS | Target Immobilized NMR Screening |
| TO | Terminal Oxidases |
| TR | Thioredoxin Reductase |
| UPEC | Uropathogenic E. coli |
References
- Spellberg, B.; Guidos, R.; Gilbert, D.; Bradley, J.; Boucher, H.W.; Scheld, W.M.; Bartlett, J.G.; Edwards, J., Jr. Infectious Diseases Society of America. The epidemic of antibiotic-resistant infections: A call to action for the medical community from the infectious diseases society of america. Clin. Infect. Dis. 2008, 46, 155–164. [Google Scholar] [CrossRef] [PubMed]
- WHO. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Conly, J.; Johnston, B. Where are all the new antibiotics? The new antibiotic paradox. Can. J. Infect. Dis. Med. Microbiol. 2005, 16, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Powers, J.H.; Brass, E.P.; Miller, L.G.; Edwards, J.E., Jr. Trends in antimicrobial drug development: Implications for the future. Clin. Infect. Dis. 2004, 38, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- The Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations; HM Goverment and Wellcome Trust: London, UK, 2014.
- Roca, I.; Akova, M.; Baquero, F.; Carlet, J.; Cavaleri, M.; Coenen, S.; Cohen, J.; Findlay, D.; Gyssens, I.; Heure, O.E.; et al. The global threat of antimicrobial resistance: Science for intervention. New Microbes New Infect. 2015, 6, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Shlaes, D. Fix the antibiotics pipeline. Nature 2011, 472. [Google Scholar] [CrossRef] [PubMed]
- Zucca, M.; Scutera, S.; Savoia, D. New antimicrobial frontiers. Mini Rev. Med. Chem. 2011, 11, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Escaich, S. Novel agents to inhibit microbial virulence and pathogenicity. Exp. Opin. Ther. Pat. 2010, 20, 1401–1418. [Google Scholar] [CrossRef] [PubMed]
- Cegelski, L.; Marshall, G.R.; Eldridge, G.R.; Hultgren, S.J. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 2008, 6, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Heras, B.; Scanlon, M.J.; Martin, J.L. Targeting virulence not viability in the search for future antibacterials. Br. J. Clin. Pharmacol. 2015, 79, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.C.; Popat, R.; Diggle, S.P.; Brown, S.P. Targeting virulence: Can we make evolution-proof drugs? Nat. Rev. Microbiol. 2014, 12, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Heras, B.; Shouldice, S.R.; Totsika, M.; Scanlon, M.J.; Schembri, M.A.; Martin, J.L. DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 2009, 7, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, S.; Akiyama, Y.; Ito, K. Identification and characterization of an Escherichia coli gene required for the formation of correctly folded alkaline phosphatase, a periplasmic enzyme. EMBO J. 1992, 11, 57–62. [Google Scholar] [PubMed]
- Depuydt, M.; Messens, J.; Collet, J.F. How proteins form disulfide bonds. Antioxid. Redox Signal. 2011, 15, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Shouldice, S.R.; Heras, B.; Walden, P.M.; Totsika, M.; Schembri, M.A.; Martin, J.L. Structure and function of DsbA, a key bacterial oxidative folding catalyst. Antioxid. Redox Signal. 2011, 14, 1729–1760. [Google Scholar] [CrossRef] [PubMed]
- Hiniker, A.; Bardwell, J.C. In vivo substrate specificity of periplasmic disulfide oxidoreductases. J. Biol. Chem. 2004, 279, 12967–12973. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, Y.; Kamitani, S.; Kusukawa, N.; Ito, K. In vitro catalysis of oxidative folding of disulfide-bonded proteins by the Escherichia coli dsbA (ppfA) gene product. J. Biol. Chem. 1992, 267, 22440–22445. [Google Scholar] [PubMed]
- Bardwell, J.C.; Lee, J.O.; Jander, G.; Martin, N.; Belin, D.; Beckwith, J. A pathway for disulfide bond formation in vivo. Proc. Natl. Acad. Sci. USA 1993, 90, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L.; Bardwell, J.C.; Kuriyan, J. Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature 1993, 365, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Guddat, L.W.; Bardwell, J.C.; Zander, T.; Martin, J.L. The uncharged surface features surrounding the active site of Escherichia coli DsbA are conserved and are implicated in peptide binding. Protein Sci. 1997, 6, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Paxman, J.J.; Borg, N.A.; Horne, J.; Thompson, P.E.; Chin, Y.; Sharma, P.; Simpson, J.S.; Wielens, J.; Piek, S.; Kahler, C.M.; et al. The structure of the bacterial oxidoreductase enzyme DsbA in complex with a peptide reveals a basis for substrate specificity in the catalytic cycle of DsbA enzymes. J. Biol. Chem. 2009, 284, 17835–17845. [Google Scholar] [CrossRef] [PubMed]
- Missiakas, D.; Georgopoulos, C.; Raina, S. Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc. Natl. Acad. Sci. USA 1993, 90, 7084–7088. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K.; Murakami, S.; Suzuki, M.; Nakagawa, A.; Yamashita, E.; Okada, K.; Ito, K. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell 2006, 127, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Ito, K. Respiratory chain strongly oxidizes the CXXC motif of DsbB in the Escherichia coli disulfide bond formation pathway. EMBO J. 1999, 18, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K.; Takahashi, Y.H.; Ito, K. Reactivities of quinone-free DsbB from Escherichia coli. J. Biol. Chem. 2005, 280, 33035–33044. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K.; Ito, K. Structure and mechanisms of the DsbB-DsbA disulfide bond generation machine. Biochim. Biophys. Acta 2008, 1783, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Regeimbal, J.; Bardwell, J.C. DsbB catalyzes disulfide bond formation de novo. J. Biol. Chem. 2002, 277, 32706–32713. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.H.; Inaba, K.; Ito, K. Characterization of the menaquinone-dependent disulfide bond formation pathway of Escherichia coli. J. Biol. Chem. 2004, 279, 47057–47065. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, M.; Otto, A.; Seckler, R.; Glockshuber, R. Bacterial protein disulfide isomerase: Efficient catalysis of oxidative protein folding at acidic pH. Biochemistry 1993, 32, 12251–12256. [Google Scholar] [CrossRef] [PubMed]
- Zapun, A.; Creighton, T.E. Effects of DsbA on the disulfide folding of bovine pancreatic trypsin inhibitor and alpha-lactalbumin. Biochemistry 1994, 33, 5202–5211. [Google Scholar] [CrossRef] [PubMed]
- Zapun, A.; Missiakas, D.; Raina, S.; Creighton, T.E. Structural and functional characterization of DsbC, a protein involved in disulfide bond formation in Escherichia coli. Biochemistry 1995, 34, 5075–5089. [Google Scholar] [CrossRef] [PubMed]
- Bessette, P.H.; Cotto, J.J.; Gilbert, H.F.; Georgiou, G. In vivo and in vitro function of the Escherichia coli periplasmic cysteine oxidoreductase DsbG. J. Biol. Chem. 1999, 274, 7784–7792. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.A.; Haebel, P.W.; Torronen, A.; Rybin, V.; Baker, E.N.; Metcalf, P. Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli. Nat. Struct. Biol. 2000, 7, 196–199. [Google Scholar] [PubMed]
- Heras, B.; Edeling, M.A.; Schirra, H.J.; Raina, S.; Martin, J.L. Crystal structures of the DsbG disulfide isomerase reveal an unstable disulfide. Proc. Natl. Acad. Sci. USA 2004, 101, 8876–8881. [Google Scholar] [CrossRef] [PubMed]
- Rietsch, A.; Bessette, P.; Georgiou, G.; Beckwith, J. Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J. Bacteriol. 1997, 179, 6602–6608. [Google Scholar] [PubMed]
- Stewart, E.J.; Katzen, F.; Beckwith, J. Six conserved cysteines of the membrane protein DsbD are required for the transfer of electrons from the cytoplasm to the periplasm of Escherichia coli. EMBO J. 1999, 18, 5963–5971. [Google Scholar] [CrossRef] [PubMed]
- Haebel, P.W.; Goldstone, D.; Katzen, F.; Beckwith, J.; Metcalf, P. The disulfide bond isomerase DsbC is activated by an immunoglobulin-fold thiol oxidoreductase: Crystal structure of the DsbC-DsbDalpha complex. EMBO J. 2002, 21, 4774–4784. [Google Scholar] [CrossRef] [PubMed]
- Rozhkova, A.; Stirnimann, C.U.; Frei, P.; Grauschopf, U.; Brunisholz, R.; Grutter, M.G.; Capitani, G.; Glockshuber, R. Structural basis and kinetics of inter- and intramolecular disulfide exchange in the redox catalyst DsbD. EMBO J. 2004, 23, 1709–1719. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Porat, A.; Ye, J.; Beckwith, J. Redox-active cysteines of a membrane electron transporter DsbD show dual compartment accessibility. EMBO J. 2007, 26, 3509–3520. [Google Scholar] [CrossRef] [PubMed]
- Dutton, R.J.; Boyd, D.; Berkmen, M.; Beckwith, J. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. USA 2008, 105, 11933–11938. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, J.P.; Stirnimann, C.U.; Brozzo, M.S.; Malojcic, G.; Grutter, M.G.; Capitani, G.; Glockshuber, R. DsbL and DsbI form a specific dithiol oxidase system for periplasmic arylsulfate sulfotransferase in uropathogenic Escherichia coli. J. Mol. Biol. 2008, 380, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Heras, B.; Totsika, M.; Jarrott, R.; Shouldice, S.R.; Guncar, G.; Achard, M.E.; Wells, T.J.; Argente, M.P.; McEwan, A.G.; Schembri, M.A. Structural and functional characterization of three DsbA paralogues from Salmonella enterica serovar typhimurium. J. Biol. Chem. 2010, 285, 18423–18432. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, C.W.; Kohli, M.; Killoran, A.; Touchie, G.A.; Kadner, R.J.; Martin, N.L. Characterization of SrgA, a Salmonella enterica serovar typhim Neisseria urium virulence plasmid-encoded paralogue of the disulfide oxidoreductase DsbA, essential for biogenesis of plasmid-encoded fimbriae. J. Bacteriol. 2003, 185, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Langford, P.R.; Kroll, J.S. Functional diversity of three different DsbA proteins from Neisseria meningitidis. Microbiology 2004, 150, 2993–3000. [Google Scholar] [CrossRef] [PubMed]
- Vivian, J.P.; Scoullar, J.; Rimmer, K.; Bushell, S.R.; Beddoe, T.; Wilce, M.C.; Byres, E.; Boyle, T.P.; Doak, B.; Simpson, J.S.; et al. Structure and function of the oxidoreductase DsbA1 from Neisseria meningitidis. J. Mol. Biol. 2009, 394, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Kouwen, T.R.; van der Goot, A.; Dorenbos, R.; Winter, T.; Antelmann, H.; Plaisier, M.C.; Quax, W.J.; van Dijl, J.M.; Dubois, J.Y. Thiol-disulphide oxidoreductase modules in the low-GC Gr Bordetella am-positive bacteria. Mol. Microbiol. 2007, 64, 984–999. [Google Scholar] [CrossRef] [PubMed]
- McMahon, R.M.; Premkumar, L.; Martin, J.L. Four structural subclasses of the antivirulence drug target disulfide oxidoreductase DsbA provide a platform for design of subclass-specific inhibitors. Biochim. Biophys. Acta 2014, 1844, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Kroll, J.S. DsbA: A protein-folding catalyst contributing to bacterial virulence. Microbes Infect. 1999, 1, 1221–1228. [Google Scholar] [CrossRef]
- Lasica, A.M.; Jagusztyn-Krynicka, E.K. The role of Dsb proteins of Gram-negative bacteria in the process of pathogenesis. FEMS Microbiol. Rev. 2007, 31, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, J.C.A.; McGovern, K.; Beckwith, J. Identification of a protein required for disulfide bond formation in vivo. Cell 1991, 67, 581–589. [Google Scholar] [CrossRef]
- Yu, J.; Edwards-Jones, B.; Neyrolles, O.; Kroll, J.S. Key role for DsbA in cell-to-cell spread of Shigella flexneri, permitting secretion of Ipa proteins into interepithelial protrusions. Infect. Immun. 2000, 68, 6449–6456. [Google Scholar] [CrossRef] [PubMed]
- Yu, J. Inactivation of DsbA, but not DsbC and DsbD, affects the intracellular survival and virulence of shigella flexneri. Infect. Immun. 1998, 66, 3909–3917. [Google Scholar] [PubMed]
- Totsika, M.; Heras, B.; Wurpel, D.J.; Schembri, M.A. Characterization of two homologous disulfide bond systems involved in virulence factor biogenesis in uropathogenic Escherichia coli CFT073. J. Bacteriol. 2009, 191, 3901–3908. [Google Scholar] [CrossRef] [PubMed]
- Peek, J.A.; Taylor, R.K. Characterization of a periplasmic thiol: Disulfide interchange protein required for the functional maturation of secreted virulence factors of Vibrio cholerae. Proc. Natl. Acad. Sci. USA 1992, 89, 6210–6214. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Webb, H.; Hirst, T.R. A homologue of the Escherichia coli DsbA protein involved in disulphide bond formation is required for enterotoxin biogenesis in Vibrio cholerae. Mol. Microbiol. 1992, 6, 1949–1958. [Google Scholar] [CrossRef] [PubMed]
- Stenson, T.H.; Weiss, A.A. DsbA and DsbC are required for secretion of pertussis toxin by Bordetella pertussis. Infect. Immun. 2002, 70, 2297–2303. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, G.R. The type III secretion injectisome. Nat. Rev. Microbiol. 2006, 4, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Galan, J.E.; Wolf-Watz, H. Protein delivery into eukaryotic cells by type III secretion machines. Nature 2006, 444, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Watarai, M.; Tobe, T.; Yoshikawa, M.; Sasakawa, C. Disulfide oxidoreductase activity of shigella flexneri is required for release of Ipa proteins and invasion of epithelial cells. Proc. Natl. Acad. Sci. USA 1995, 92, 4927–4931. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.W.; Plano, G.V. DsbA is required for stable expression of outer membrane protein YscC and for efficient Yop secretion in Yersinia pestis. J. Bacteriol. 1999, 181, 5126–5130. [Google Scholar] [PubMed]
- Miki, T.; Okada, N.; Danbara, H. Two periplasmic disulfide oxidoreductases, DsbA and SrgA, target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type III secretion system. J. Biol. Chem. 2004, 279, 34631–34642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Z.; Donnenberg, M.S. DsbA is required for stability of the type IV pilin of enteropathogenic Escherichia coli. Mol. Microbiol. 1996, 21, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Jacob-Dubuisson, F.; Pinkner, J.; Xu, Z.; Striker, R.; Padmanhaban, A.; Hultgren, S.J. PapD chaperone function in pilus biogenesis depends on oxidant and chaperone-like activities of DsbA. Proc. Natl. Acad. Sci. USA 1994, 91, 11552–11556. [Google Scholar] [CrossRef] [PubMed]
- Dailey, F.E.; Berg, H.C. Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli. Proc. Natl. Acad. Sci. USA 1993, 90, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Turcot, I.; Ponnampalam, T.V.; Bouwman, C.W.; Martin, N.L. Isolation and characterization of a chromosomally encoded disulphide oxidoreductase from Salmonella enterica serovar typhimurium. Can. J. Microbiol. 2001, 47, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Burall, L.S.; Harro, J.M.; Li, X.; Lockatell, C.V.; Himpsl, S.D.; Hebel, J.R.; Johnson, D.E.; Mobley, H.L. Proteus mirabilis genes that contribute to pathogenesis of urinary tract infection: Identification of 25 signature-tagged mutants attenuated at least 100-fold. Infect. Immun. 2004, 72, 2922–2938. [Google Scholar] [CrossRef] [PubMed]
- Ireland, P.M.; McMahon, R.M.; Marshall, L.E.; Halili, M.; Furlong, E.; Tay, S.; Martin, J.L.; Sarkar-Tyson, M. Disarming burkholderia pseudomallei: Structural and functional characterization of a disulfide oxidoreductase (DsbA) required for virulence in vivo. Antioxid. Redox Signal. 2014, 20, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.W.; Creighton, T.E. Reactivity and ionization of the active site cysteine residues of DsbA, a protein required for disulfide bond formation in vivo. Biochemistry 1994, 33, 5974–5983. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Fragment-based lead discovery grows up. Nat. Rev. Drug Discov. 2013, 12, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Quintyne, K.I.; Baker, S.; Wallis, F.; Gupta, R. Good clinical and radiological response to BRAF inhibitor in patient with metastatic thin melanoma. BMJ Case Rep. 2012. [Google Scholar] [CrossRef] [PubMed]
- Fruh, V.; Zhou, Y.; Chen, D.; Loch, C.; Ab, E.; Grinkova, Y.N.; Verheij, H.; Sligar, S.G.; Bushweller, J.H.; Siegal, G. Application of fragment-based drug discovery to membrane proteins: Identification of ligands of the integral membrane enzyme DsbB. Chem. Biol. 2010, 17, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Halili, M.A.; Bachu, P.; Lindahl, F.; Bechara, C.; Mohanty, B.; Reid, R.C.; Scanlon, M.J.; Robinson, C.V.; Fairlie, D.P.; Martin, J.L. Small molecule inhibitors of disulfide bond formation by the bacterial DsbA-DsbB dual enzyme system. ACS Chem. Biol. 2015, 10, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Sharma, P.; Mohanty, B.; Ilyichova, O.V.; Mulcair, M.D.; Williams, M.L.; Gleeson, E.C.; Totsika, M.; Doak, B.C.; Caria, S.; et al. Application of fragment-based screening to the design of inhibitors of Escherichia coli DsbA. Angew. Chem. 2015, 54, 2179–2184. [Google Scholar] [CrossRef] [PubMed]
- Dailey, F.E.; Berg, H.C. Change in direction of flagellar rotation in Escherichia coli mediated by acetate kinase. J. Bacteriol. 1993, 175, 3236–3239. [Google Scholar] [PubMed]
- Duprez, W.; Premkumar, L.; Halili, M.A.; Lindahl, F.; Reid, R.C.; Fairlie, D.P.; Martin, J.L. Peptide inhibitors of the Escherichia coli DsbA oxidative machinery essential for bacterial virulence. J. Med. Chem. 2015, 58, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Duprez, W.; Bachu, P.; Stoermer, M.J.; Tay, S.; McMahon, R.M.; Fairlie, D.P.; Martin, J.L. Virtual screening of peptide and peptidomimetic fragments targeted to inhibit bacterial dithiol oxidase DsbA. PLoS ONE 2015, 10, e0133805. [Google Scholar] [CrossRef] [PubMed]
- Kurth, F.; Duprez, W.; Premkumar, L.; Schembri, M.A.; Fairlie, D.P.; Martin, J.L. Crystal structure of the dithiol oxidase DsbA enzyme from proteus mirabilis bound non-covalently to an active site peptide ligand. J. Biol. Chem. 2014, 289, 19810–19822. [Google Scholar] [CrossRef] [PubMed]
- Landeta, C.; Blazyk, J.L.; Hatahet, F.; Meehan, B.M.; Eser, M.; Myrick, A.; Bronstain, L.; Minami, S.; Arnold, H.; Ke, N.; et al. Compounds targeting disulfide bond forming enzyme DsbB of Gram-negative bacteria. Nat. Chem. Biol. 2015, 11, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.P.; Fernandes, P.A.; Ramos, M.J. Similarities and differences in the thioredoxin superfamily. Prog. Biophys. Mol. Biol. 2006, 91, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Sannigrahi, S.; Scoullar, J.; Kahler, C.M.; Tzeng, Y.L. Characterization of DsbD in Neisseria meningitidis. Mol. Microbiol. 2011, 79, 1557–1573. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, R.P.; Paxman, J.J.; Scanlon, M.J.; Heras, B. Targeting Bacterial Dsb Proteins for the Development of Anti-Virulence Agents. Molecules 2016, 21, 811. https://doi.org/10.3390/molecules21070811
Smith RP, Paxman JJ, Scanlon MJ, Heras B. Targeting Bacterial Dsb Proteins for the Development of Anti-Virulence Agents. Molecules. 2016; 21(7):811. https://doi.org/10.3390/molecules21070811
Chicago/Turabian StyleSmith, Roxanne P., Jason J. Paxman, Martin J. Scanlon, and Begoña Heras. 2016. "Targeting Bacterial Dsb Proteins for the Development of Anti-Virulence Agents" Molecules 21, no. 7: 811. https://doi.org/10.3390/molecules21070811
APA StyleSmith, R. P., Paxman, J. J., Scanlon, M. J., & Heras, B. (2016). Targeting Bacterial Dsb Proteins for the Development of Anti-Virulence Agents. Molecules, 21(7), 811. https://doi.org/10.3390/molecules21070811