
Synthesis of Chiral, Enantiopure Allylic Amines by the Julia Olefination of α-Amino Esters

 ,
,  ,
, 
Abstract
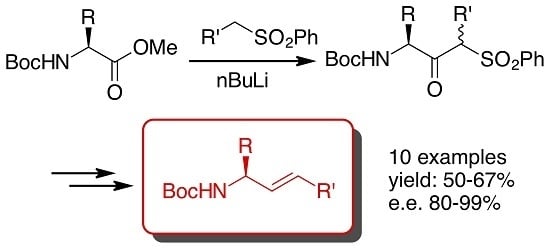
:
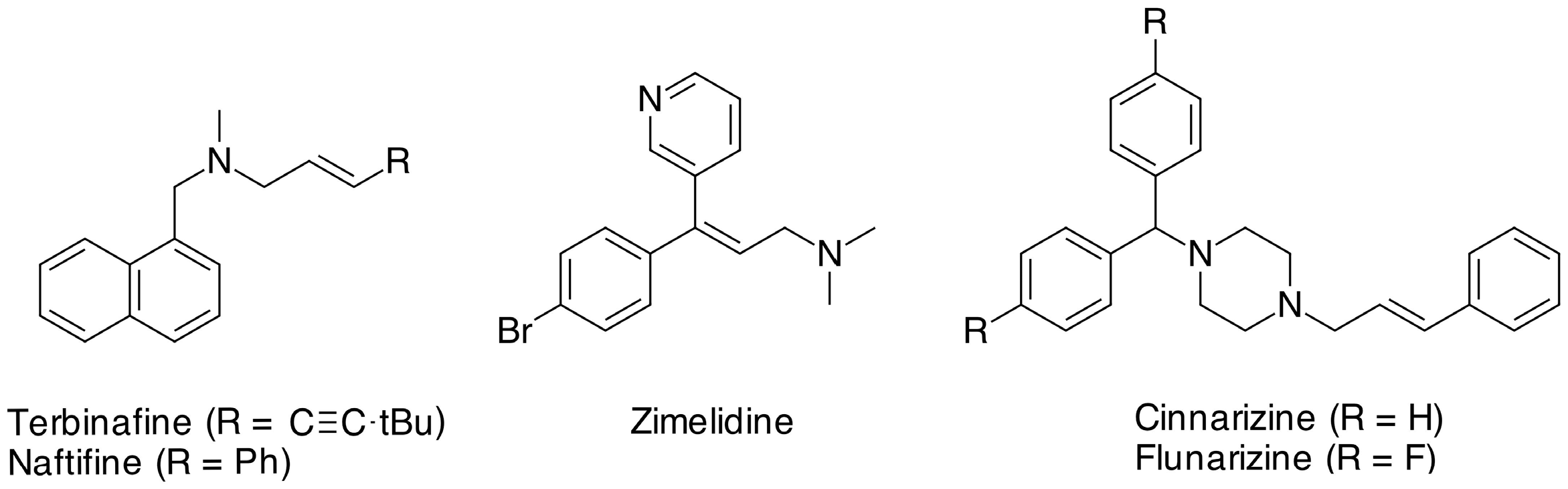
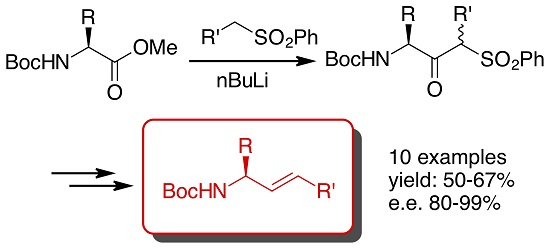
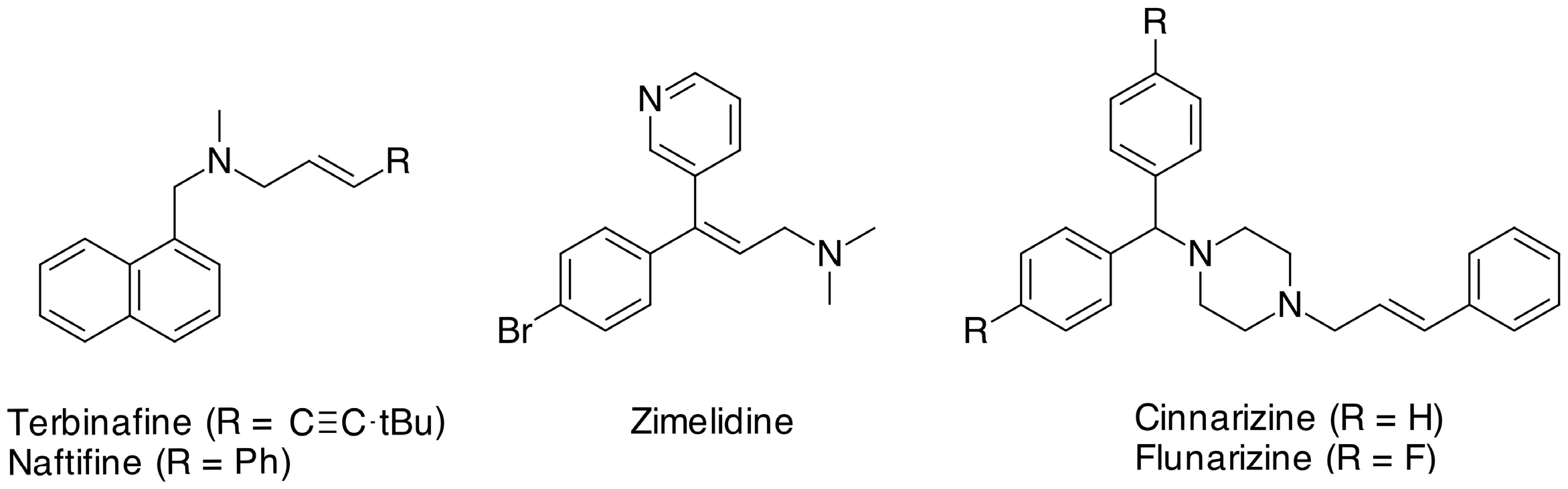
1. Introduction
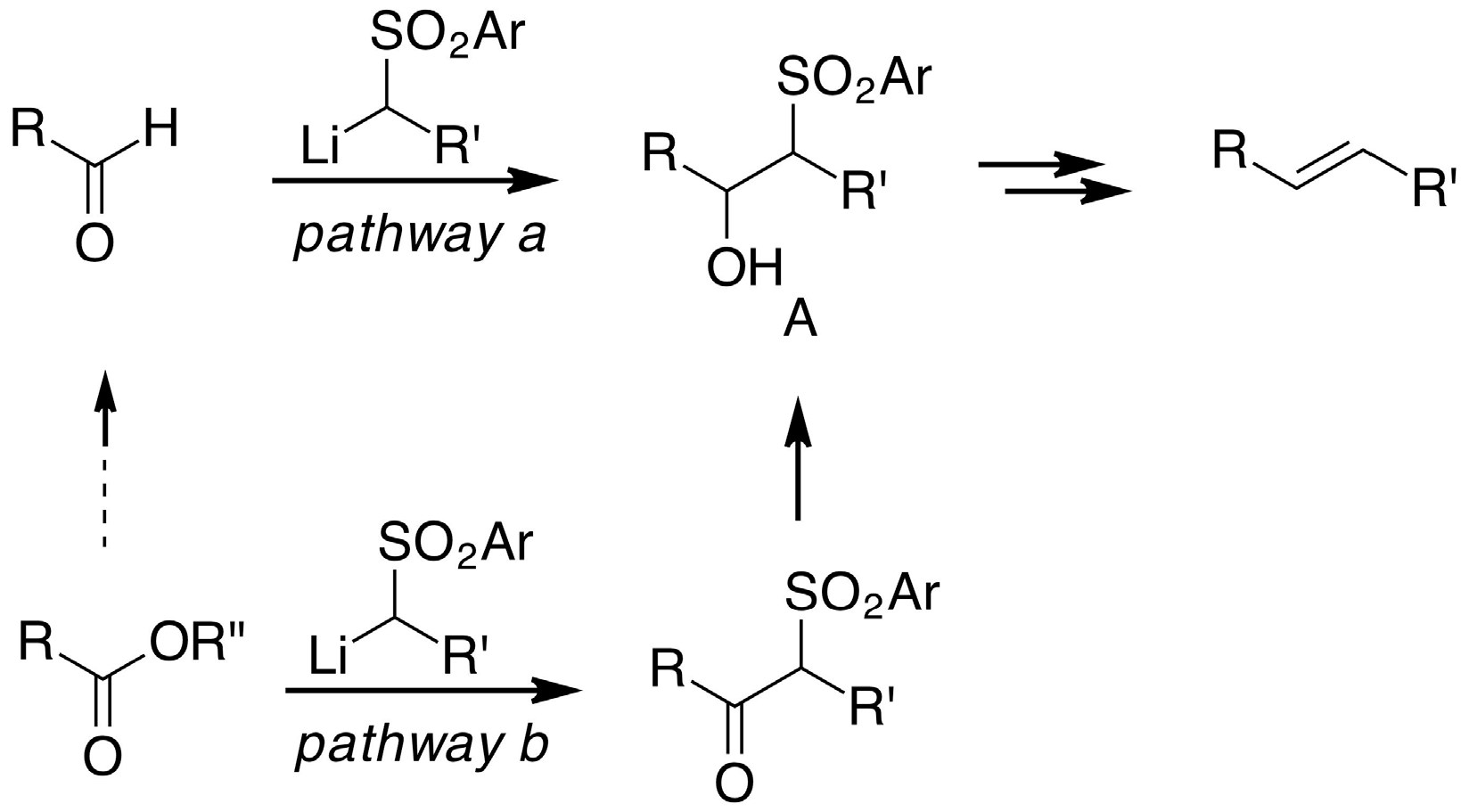
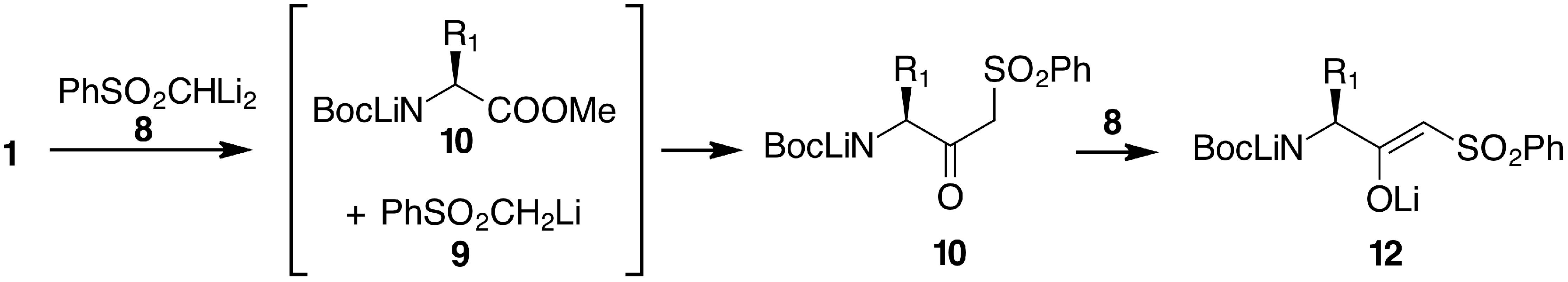
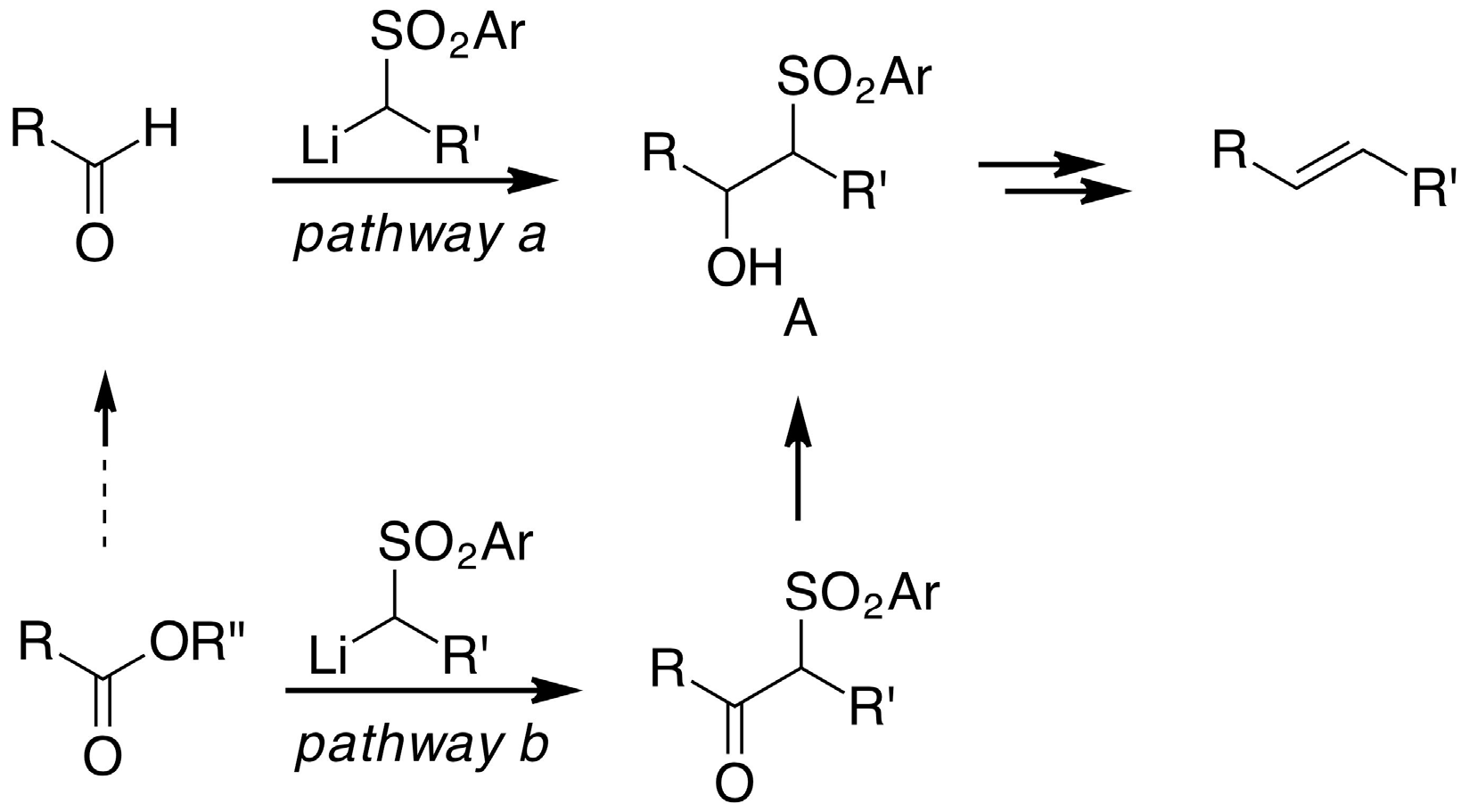
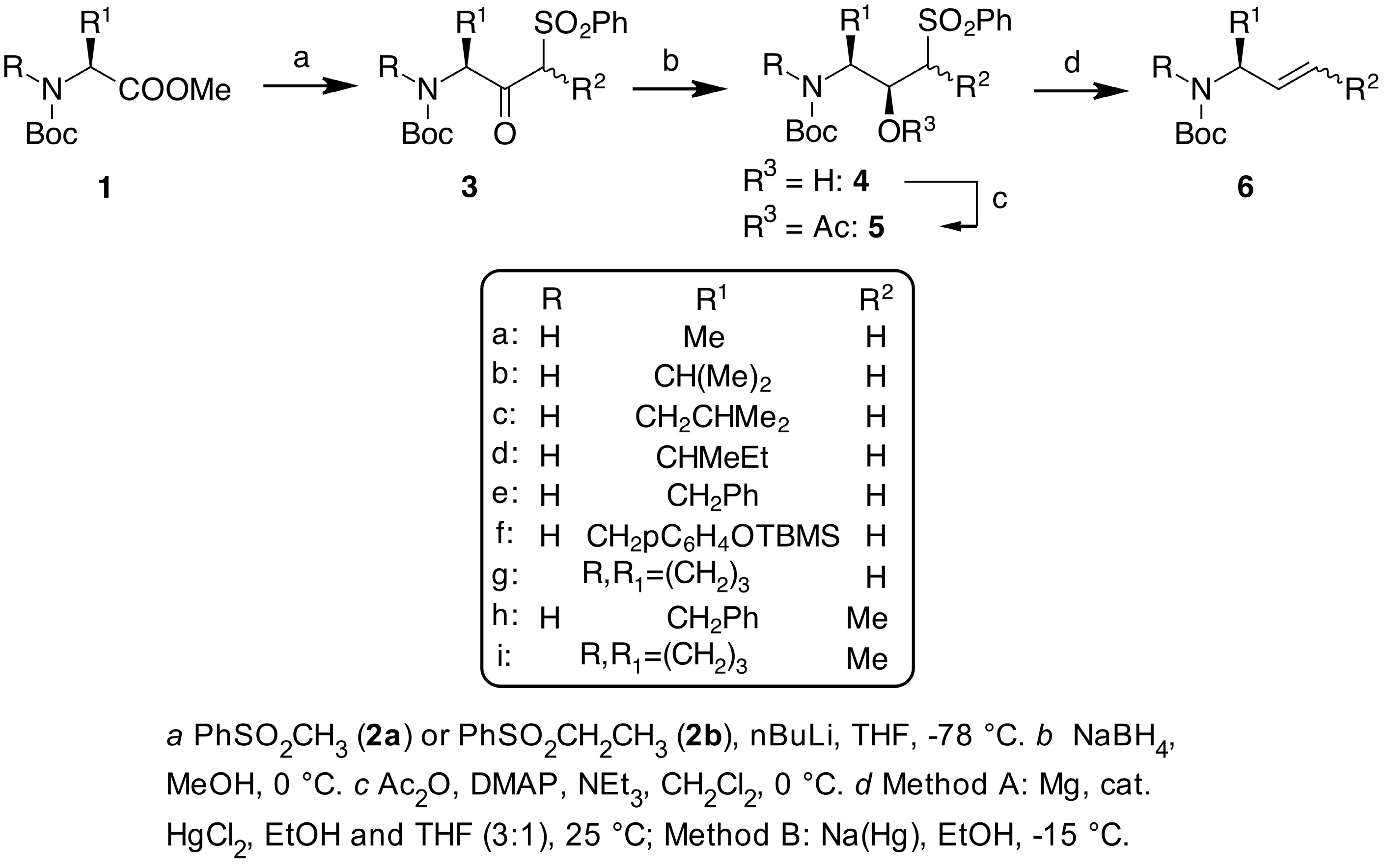
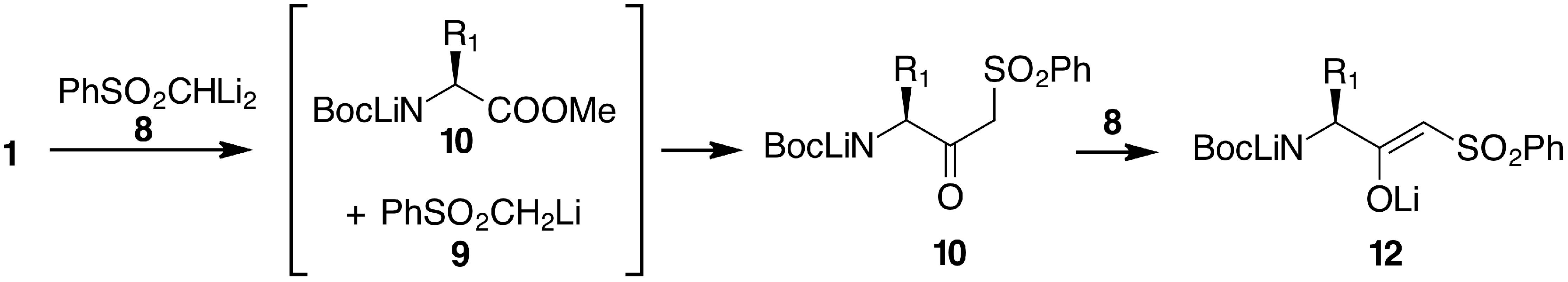
2. Results and Discussion
3. Experimental Section
3.1. General Procedure for the Synthesis of α-Ketosulfones 3
3.2. Spectroscopic and Analytical Data for α-Ketosulfones 3
3.3. General Procedure for the Synthesis of Allylamines 6 from Ketosulfones 3
3.4. Spectroscopic and Analytical Data for Alcohols 4, Acetates 5 and Allylamines 6
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Boc | t-Butoxycarbonyl |
| n-BuLi | n-Butyllithium |
| COSY | Correlation Spectroscopy |
| DEPT | Distortionless Enhancement by Polarization Transfer |
| ee | Enantiomeric Excess |
| ESI | Electrospray Ionization |
| HRGC | High Resolution Gas Chromatography |
| HPLC | High Performance Liquid Chromatography |
| HRMS | High Resolution Mass Spectrometry |
| HSQC | Heteronuclear Single Quantum Coherence Spectroscopy |
| IR | Infrared |
| KHMDS | Potassium Hexamethyldisilazanide |
| MS | Mass Spectrometry |
| NMR | Nuclear Magnetic Resonance |
| TBDMS | t-Butyldimethylsilyl |
| THF | Tetrahydrofuran |
| TLC | Thin Layer Chromatography |
| TMEDA | Teramethylethylenediamine |
References and Note
- Skoda, E.M.; Davis, G.C.; Wipf, P. Allylic Amines as Key Building Blocks in the Synthesis of (E)-Alkene Peptide Isosters. Org. Process Res. Dev. 2012, 16, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Batra, S. Applications of Allylamines for the Synthesis of Aza-Heterocycles. Tetrahedron 2011, 67, 8959–9061. [Google Scholar] [CrossRef]
- Johannsen, M.; Jørgensen, K.A. Allylic Amination. Chem. Rev. 1998, 98, 1689–1708. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Stephenson, C.R. Total Synthesis of Syringolin A. J. Org. Lett. 2010, 12, 3453–3455. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Burke, A.M.; Kotani, S.; Ziller, J.W.; Rychnovsky, S.D. Total Synthesis of (−)-Lycoperine A. Org. Lett. 2009, 12, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.; Khan, I.A. Alkaloids and Saponins from Blue Cohosh. Phytochemistry 2008, 69, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Brust, T.F.; Watts, V.J.; Dai, M. Palladium-Catalyzed Regio- and Stereoselective γ-Arylation of Tertiary Allylic Amines: Identification of Potent Adenylyl Cyclase Inhibitors. Org. Lett. 2015, 17, 892–895. [Google Scholar] [CrossRef] [PubMed]
- Foot, J.S.; Deodhar, M.; Turner, C.I.; Yin, P.; Van Dam, E.M.; Silva, D.G.; Olivieri, A.; Holt, A.; McDonald, I.A. The discovery and development of selective 3-fluoro-4-aryloxyallylamine inhibitors of the amine oxidase activity of semicarbazide-sensitive amine oxidase/vascular adhesion protein-1 (SSAO/VAP-1). Bioorga. Med. Chem. Lett. 2012, 22, 3935–3940. [Google Scholar] [CrossRef] [PubMed]
- Kitahata, N.; Han, S.-Y.; Noji, N.; Saito, T.; Kobayashi, M.; Nakano, T.; Kuchitsu, K.; Shinozaki, K.; Yoshida, S.; Matsumoto, S.; Tsujimoto, M.; Asami, T. A 9-cis-epoxycarotenoid dioxygenase inhibitor for use in the elucidation of abscisic acid action mechanisms. Bioorg. Med. Chem. 2006, 14, 5555–5561. [Google Scholar] [CrossRef] [PubMed]
- Stütz, A. Allylamine Derivatives—A New Class of Active Substances in Antifungal Chemotherapy. Angew. Chem. Int. Ed. Engl. 1987, 26, 320–328. [Google Scholar] [CrossRef]
- Carlsson, A. A paradigm shift in brain research. Science 2001, 294, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Towse, G. Cinnarizine—a labyrinthine sedative. J. Laryngol. Otol. 1980, 94, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Bebin, M.; Bleck, T.P. New Anticonvulsant Drugs. Focus on Flunarizine, Fospheniotoyn, Midazolam, Stiripentol. Drugs 1994, 48, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Szcześniak, P.; Stecko, S. An approach to asymmetric synthesis of β-aryl alanines by Pd(0)-catalyzed cross-coupling and cyanate-to-isocyanate rearrangement. RSC Adv. 2015, 5, 30882–30888. [Google Scholar] [CrossRef]
- Fustero, S.; Cuñat, A.C.; Flores, S.; Báez, C.; Oliver, J.; Cynamon, M.; Gütschow, M.; Mertens, M.D.; Delgado, O.; Tresadern, G.; et al. Design, Synthesis, and Biological Evaluation of Novel Fluorinated Ethanolamines. Chem. Eur. J. 2011, 17, 14772–14784. [Google Scholar] [CrossRef] [PubMed]
- Blacker, A.J.; Roy, M.; Hariharan, S.; Headley, C.; Upare, A.; Jagtap, A.; Wankhede, K.; Mishra, S.K.; Dube, D.; Bhise, S.; et al. Convenient Method for Synthesis of N-Protected α-Amino Epoxides: Key Intermediates for HIV Protease Inhibitors. Org. Process Res. Dev. 2011, 15, 331–338. [Google Scholar] [CrossRef]
- Martín, R.; Alcón, M.; Pericàs, M.A.; Riera, A. Ring-Closing Metathesis of Chiral Allylamines. Enantioselective Synthesis of (2S,3R,4S)-3,4-Dihydroxyprolin. J. Org. Chem. 2002, 67, 6896–6901. [Google Scholar] [CrossRef] [PubMed]
- Marvin, C.C. Synthesis of amines and ammonium salts. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Molander, G.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 6, pp. 34–99. [Google Scholar]
- Hartwig, J.F.; Stanley, L.M. Mechanistically driven development of iridium catalysts for asymmetric allylic substitution. Acc. Chem. Res. 2010, 43, 1461–1475. [Google Scholar] [CrossRef] [PubMed]
- Helmchen, G. Iridium-Catalyzed Asymmetric Allylic Substitutions. In Iridium Complexes in Organic Synthesis; Oro, L.A., Claver, C., Eds.; Wiley-VCH: Weinheim, Germany, 2009; pp. 145–172. [Google Scholar]
- Robak, M.T.; Herbage, M.A.; Ellman, J.A. Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev. 2010, 110, 3600–3740. [Google Scholar] [CrossRef] [PubMed]
- Overman, L.E.; Carpenter, N.E. The Allylic Trihaloacetimidate Rearrangment. Org. React. 2005, 66. [Google Scholar] [CrossRef]
- Liu, T.-L.; Wang, C.-J.; Zhang, X. Synthesis of Chiral Aliphatic Amines through Asymmetric Hydrogenation. Angew. Chem. Int. Ed. Engl. 2013, 52, 8416–8419. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.A.; Zhao, B.; Shi, Y. Recent advances in transition metal-catalyzed sp3 C–H amination adjacent to double bonds and carbonyl groups. Chem. Soc. Rev. 2012, 41, 931–9412. [Google Scholar] [CrossRef] [PubMed]
- Baidya, M.; Yamamoto, H. Advancements in the Nascent Nitroso-Ene Reaction. Synthesis 2013, 45, 1931–1938. [Google Scholar] [CrossRef]
- Gryko, D.; Chalko, J.; Jurczak, J. Synthesis and reactivity of N-protected-α-amino aldehydes. Chirality 2003, 15, 514–541. [Google Scholar] [CrossRef]
- Jurczak, J.; Gołebiowski, A. Optically active N-protected alpha-amino aldehydes in organic synthesis. Chem. Rev. 1989, 89, 149–164. [Google Scholar] [CrossRef]
- Reetz, M.T. Synthesis and Diastereoselective Reactions of N,N-Dibenzylamino Aldehydes and Related Compounds. Chem. Rev. 1999, 99, 1121–1162. [Google Scholar] [CrossRef] [PubMed]
- Stallforth, P.; Matthies, S.; Adibekian, A.; Gillingham, D.G.; Hilvert, D.; Seeberger, P.H. De novo chemoenzymatic synthesis of sialic acid. Chem. Commun. 2012, 48, 11987–11989. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.-Y.; Knaus, E.E. Improved Synthesis of Chiral N-Protected Allylic Amines. Synthesis 1994, 1463–1466. [Google Scholar] [CrossRef]
- Saari, W.S.; Fisher, T.E. A Convenient and Versatile Synthesis of Chiral Aliphatic and Allylic Amines. Synthesis 1990, 453–454. [Google Scholar] [CrossRef]
- Mirilashvili, S.; Chasid-Rubinstein, N.; Albeck, A. Optically Active γ-Hydroxy Sulfone Julia Reagents for the Synthesis of Peptidyl Olefin Peptidomimetics. Eur. J. Org. Chem. 2008, 2008, 3461–3464. [Google Scholar] [CrossRef]
- Shirota, O.; Nakanishi, K.; Berova, N. Phytosphingosines—A facile synthesis and spectroscopic protocol for configurational assignment. Tetrahedron 1999, 55, 13643–13658. [Google Scholar] [CrossRef]
- Gurjar, M.K.; Pal, S.; Rama Rao, A.V. Synthesis of Novel C2-Symmetric and Pseudo C2-Symmetric Based Diols, Epoxides and Dideoxy Derivatives of HIV Protease Inhibitors. Tetrahedron 1997, 53, 4769–4778. [Google Scholar] [CrossRef]
- Wei, G.; Cohen, T. A Highly Efficient Conversion of a Simple Derivative of the Amino Acid Proline into a Nearly Enantiomerically Pure N-Protected Allyl Amine: Use of Thionyl Chloride to Promote the Peterson Olefination. Synlett 2011, 2011, 2697–2700. [Google Scholar] [CrossRef]
- Julia, M.; Paris, J.-M. Syntheses a l’aide de sulfones v(+)-methode de synthese generale de doubles liaisons. Tetrahedron Lett. 1973, 14, 4833–4836. [Google Scholar] [CrossRef]
- Markó, I.E.; Pospíšil, J. Science of Synthesis; de Meijere, A., Ed.; Thieme: Stuttgart, Germany, 2009; Volume 47a, pp. 105–160. [Google Scholar]
- Lee, G.H.; Lee, H.K.; Choi, E.B.; Kim, B.T.; Pak, C.S. An efficient Julia olefination mediated by magnesium in ethanol. Tetrahedron Lett. 1995, 36, 5607–5609. [Google Scholar] [CrossRef]
- Keck, G.E.; Savin, K.A.; Weglarz, M.A. Use of Samarium Diiodide as an Alternative to Sodium/Mercury Amalgam in the Julia-Lythgoe Olefination. J. Org. Chem. 1995, 60, 3194–3204. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Cheng, D.; Li, J. Benzotriazole-Mediated Stereoselective Olefination of Carboxylic Esters: Transformation of α-Amino Acid Esters into Chiral Allylamines. J. Org. Chem. 1998, 63, 3438–3444. [Google Scholar] [CrossRef]
- Charrier, C.; Ettouati, L.; Paris, J. New application of the Julia olefination for the synthesis of Tyr-Gly E-alkene and carba isostere pseudopeptides. Tetrahedron Lett. 1999, 40, 5705–5707. [Google Scholar] [CrossRef]
- Lygo, B. Use of an Alanine Derived ß-Ketosulfone in the Synthesis of Peptide Isosteres. Synlett 1992, 793–795. [Google Scholar] [CrossRef]
- As it was not possible to separate the enantiomeric ketosulfones 3, the ee were measured after conversion of 3 into the corresponding allylamine 6 (Scheme 1), except for 3f whose ee was determined by chiral HPLC.
- Lygo, B.; Rudd, C.N. Synthesis of Xaa-Gly-Xaa′ keto-methylene tri-peptide isosteres incorporating phenylalanine, tyrosine and valine units. Tetrahedron Lett. 1995, 36, 3577–3580. [Google Scholar] [CrossRef]
- Řehová, L.; Císařová, I.; Ullrich, J. Divergent Reactivity of Alkyl Aryl Sulfones with Bases: Selective Functionalization of ortho-Aryl and α-Alkyl Units Enabled by a Unique Carbanion Transmetalation. Eur. J. Org. Chem. 2014, 1461–1476. [Google Scholar] [CrossRef]
- Rönn, R.; Sabnis, Y.A.; Gossas, T.; Akerblom, E.; Danielson, U.H.; Hallberg, A.; Johansson, A. Exploration of acyl sulfonamides as carboxylic acid replacements in protease inhibitors of the hepatitis C virus full-length NS3. Bioorg. Med. Chem. 2006, 14, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Severi, D.; Ettouati, L.; Paris, J. Synthesis of 2-methyl-5-amino-4-oxo-3-sulfonyl esters as precursors of pseudodipeptides. Lett. Pept. Sci. 2002, 9, 11–14. [Google Scholar] [CrossRef]
- Benedetti, F.; Miertus, S.; Norbedo, S.; Tossi, A.; Zlatoidzky, P. Versatile and Stereoselective Synthesis of Diamino Diol Dipeptide Isosteres, Core Units of Pseudopeptide HIV Protease Inhibitors. J. Org. Chem. 1997, 62, 9348–9353. [Google Scholar] [CrossRef]
- Hwang, S.H.; Kurth, M.J. 1,3-Dipolar Cycloaddition of Nitrile Oxides to 1-Phenylsulfonyl-1,3-butadienes: Synthesis of 3-(4,5-Dihydroisoxazol-5-yl)pyrroles. Tetrahedron Lett. 2002, 43, 53–56. [Google Scholar] [CrossRef]
- Fujisawa, T.; Odake, S.; Ogawa, Y.; Yasuda, J.; Morita, Y.; Morikawa, T. Design and synthesis of sulfur based inhibitors of matrix metalloproteinase-1. Chem. Pharm. Bull. 2002, 50, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Moriwake, T.; Hamano, S.; Saito, S.; Torii, S. A Straightforward Synthesis of Allyl Amines from α-Amino Acids without Racemization. Chem. Lett. 1987, 2085–2088. [Google Scholar] [CrossRef]
- Luly, J.R.; Dellaria, J.F.; Plattner, J.J.; Soderquist, J.; Yi, N. A synthesis of protected aminoalkyl epoxides from alpha.-amino acids. J. Org. Chem. 1987, 52, 1487–1492. [Google Scholar] [CrossRef]
- Fu, G.; Zou, X.-M.; Fu, Y.-Q.; Mou, K.; Ma, C.; Lu, Y.; Xu, P. Synthesis of protected aminoalkyl sulfinyl dilactones from α-amino acids. J. Chin. Pharm. Sci. 2007, 16, 119–124. [Google Scholar]
- Ramu, E.; Venkateswara Rao, B. A short approach to the synthesis of the ritonavir and lopinavir core and its C-3 epimer via cross metathesis. Tetrahedron: Asymmetry 2009, 20, 2201–2204. [Google Scholar] [CrossRef]
- Blakemore, P.R. The modified Julia olefination: alkene synthesis via the condensation of metallated heteroarylalkylsulfones with carbonyl compounds. J. Chem. Soc. Perkin Trans. 1 2002, 2563–2585. [Google Scholar] [CrossRef]
- Pfund, E.; Lequeux, T.; Gueyrard, D. Synthesis of Fluorinated and Trifluoromethyl-Substituted Alkenes through the Modified Julia Olefination: An Update. Synthesis 2015, 47, 1534–1546. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3a–i, 6a–f are available from the authors.


| Entry | 2a (equiv.) | Base (equiv.) | T (°C) | Yield a % | ee a,b % |
|---|---|---|---|---|---|
| 1 | 1 | n-BuLi (2) | −78 | 55 | <70 |
| 2 | 1 | n-BuLi (2) | −30 | 66 | <70 |
| 3 | 1 | n-BuLi (3) | −78 | 86 | <70 |
| 4 | 2 | n-BuLi (4) | −78 | 89 | 65 |
| 5 | 2 | n-BuLi (4) | −30 | 42 | <70 |
| 6 | 2 | n-BuLi/TMEDA (4) | −78 | - | - |
| 7 | 2 | KHMDS | −78 | - | - |
| 8 | 2 | NaH | −78 | - | - |
| 9 | 2 | n-BuLi (4) | −78 | 89 c | 95 c |

| Entry | Aminoester | Sulfone | R2 | Product | Yield% |
|---|---|---|---|---|---|
| 1 | Ala (1a) | 2a | H | 3a | 89 |
| 2 | Val (1b) | 2a | H | 3b | 84 |
| 3 | Leu (1c) | 2a | H | 3c | 89 |
| 4 | Ile (1d) | 2a | H | 3d | 86 |
| 5 | Phe (1e) | 2a | H | 3e | 94 |
| 6 | Tyr (1f) b | 2a | H | 3f | 80 |
| 7 | Pro (1g) | 2a | H | 3g | 60 c |
| 8 | Phe (1e) | 2b | Me | 3h | 77 d |
| 9 | Pro (1g) | 2b | Me | 3i | 80 c,e |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allylamine | Method a | Yield b % | ee c % | Allylamine | Method a | Yield b % | ee c % | ||
|---|---|---|---|---|---|---|---|---|---|
| 6a |  | A (1h) | 68 (60) | 95 | 6f |  | A d (2h) | 65 (52) | 80 e |
| 6b |  | A (2h) | 60 (50) | >99 | 6f′ |  | B d (4h) | 71 (57) | ND |
| 6c |  | A (2h) | 67 (62) | >99 | 6g |  | B (2h) | 84 (50) | >99 |
| 6d |  | A (3h) | 69 (59) | >99 | 6h |  | B (4h) | 71 f (55) | ND |
| 6e |  | A (2h) | 68 (64) | >99 | 6i |  | B (12h) | 65 g (52) | ND |
| B (4h) | 71 (67) | >99 | |||||||
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benedetti, F.; Berti, F.; Fanfoni, L.; Garbo, M.; Regini, G.; Felluga, F. Synthesis of Chiral, Enantiopure Allylic Amines by the Julia Olefination of α-Amino Esters. Molecules 2016, 21, 805. https://doi.org/10.3390/molecules21060805
Benedetti F, Berti F, Fanfoni L, Garbo M, Regini G, Felluga F. Synthesis of Chiral, Enantiopure Allylic Amines by the Julia Olefination of α-Amino Esters. Molecules. 2016; 21(6):805. https://doi.org/10.3390/molecules21060805
Chicago/Turabian StyleBenedetti, Fabio, Federico Berti, Lidia Fanfoni, Michele Garbo, Giorgia Regini, and Fulvia Felluga. 2016. "Synthesis of Chiral, Enantiopure Allylic Amines by the Julia Olefination of α-Amino Esters" Molecules 21, no. 6: 805. https://doi.org/10.3390/molecules21060805